

Abstract
Rationale
The recent development of cavity ring‐down laser spectroscopy (CRDS) instruments capable of measuring 17O‐excess in water has created new opportunities for studying the hydrologic cycle. Here we apply this new method to studying the triple oxygen (17O/16O, 18O/16O) and hydrogen (2H/1H) isotope ratios of gypsum hydration water (GHW), which can provide information about the conditions under which the mineral formed and subsequent post‐depositional interaction with other fluids.
Methods
We developed a semi‐automated procedure for extracting GHW by slowly heating the sample to 400°C in vacuo and cryogenically trapping the evolved water. The isotopic composition (δ17O, δ18O and δ2H values) of the GHW is subsequently measured by CRDS. The extraction apparatus allows the dehydration of five samples and one standard simultaneously, thereby increasing the long‐term precision and sample throughput compared with previous methods. The apparatus is also useful for distilling brines prior to isotopic analysis. A direct comparison is made between results of 17O‐excess in GHW obtained by CRDS and fluorination followed by isotope ratio mass spectrometry (IRMS) of O2.
Results
The long‐term analytical precision of our method of extraction and isotopic analysis of GHW by CRDS is ±0.07‰ for δ17O values, ±0.13‰ for δ18O values and ±0.49‰ for δ2H values (all ±1SD), and ±1.1‰ and ±8 per meg for the deuterium‐excess and 17O‐excess, respectively. Accurate measurement of the 17O‐excess values of GHW, of both synthetic and natural samples, requires the use of a micro‐combustion module (MCM). This accessory removes contaminants (VOCs, H2S, etc.) from the water vapour stream that interfere with the wavelengths used for spectroscopic measurement of water isotopologues. CRDS/MCM and IRMS methods yield similar isotopic results for the analysis of both synthetic and natural gypsum samples within analytical error of the two methods.
Conclusions
We demonstrate that precise and simultaneous isotopic measurements of δ17O, δ18O and δ2H values, and the derived deuterium‐excess and 17O‐excess, can be obtained from GHW and brines using a new extraction apparatus and subsequent measurement by CRDS. This method provides new opportunities for the application of water isotope tracers in hydrologic and paleoclimatologic research. © 2015 The Authors. Rapid Communications in Mass Spectrometry Published by John Wiley & Sons Ltd.
The isotopic composition of crystallization water of gypsum (CaSO4·2H2O) has been utilized as a palaeoclimatic proxy to trace geological and hydrogeological processes (see, amongst others,1, 2, 3, 4, 5, 6, 7, 8). To date, investigations have focused mainly on the analysis and interpretation of the 18O and 2H values of gypsum hydration water (GHW) and the derived deuterium excess values (d‐excess). This derived parameter is defined as:
where δ2H and δ18O denote 2H/1H and 18O/16O in water standardized with respect to V‐SMOW.9
Conversely, the relationship between the δ17O‐ δ18O pair (known as the 17O‐excess) in GHW has not been applied yet in hydrology and paleoclimatology. The excess (or depletion) of 17O in water is defined as:
where δ17O and δ18O denote 17O/16O and 18O/16O in water standardized with respect to V‐SMOW.10
The addition of the 17O‐excess to the isotopic measurements of GHW can provide additional information on the environmental conditions under which the gypsum formed, as well as its post‐depositional interactions with other fluids. For example, the 17O‐excess has been shown to be less sensitive to temperature effects than the d‐excess during evaporation.11 Thus, combining the 17O‐excess and d‐excess recorded by GHW may provide information about the relative effects of humidity and temperature change at the time of gypsum formation.
The most common method for extracting GHW involves step heating a sample in a vacuum line and cryogenically trapping the water.12, 13 Following extraction, the isotopic composition of the hydration water is determined by isotope ratio mass spectrometry (IRMS, e.g.14) or, more recently, by cavity ring‐down laser spectroscopy (CRDS).6, 15 The latter method allows the simultaneous measurement of oxygen and hydrogen isotopic ratios without the need to convert the water into another gas, thereby minimizing the opportunity for isotopic fractionation.
Here we describe a modification of the extraction procedure introduced by Hodell et al.6 and report triple oxygen (16O, 17O and 18O) and hydrogen (1H and 2H) isotope analysis using a new generation CRDS analyzer (Picarro L2140‐i).16 To validate the 17O‐excess values obtained by CRDS, a synthetic gypsum standard and a variety of natural gypsum samples were measured for their δ17O and δ18O values by fluorination followed by IRMS of O2.17
Experimental
Description of the vacuum assembly
We developed a semi‐automated procedure for extracting hydration water from gypsum by slowly heating the sample to 400°C in vacuo and cryogenically trapping the evolved water. The Water Analyzer Sample Preparation (WASP) system comprises an adapted, programmable gas chromatography (GC) oven and six individual extraction lines attached to a common backing line, coupled to a two‐stage rotary pump (Edwards® E2M2, Crawley, UK) (Fig. 1). A liquid nitrogen (LN2) trap is fitted before the vacuum pump to improve pumping efficiency and avoid backstreaming of pump oil.
Figure 1.
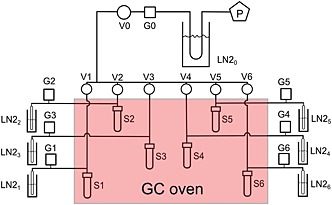
Schematic of the WASP (Water Analyzer Sample Preparation) device developed for the extraction of gypsum hydration water. P: Two‐stage rotation pump; G: Pirani gauges; V: Swagelok® values; S: 12‐mm Pyrex tubes for samples; LN2: Cryogenic traps and 6‐mm OD tubes for freezing GHW.
Each line is ~1 m in length and composed of ¼‐inch O.D. stainless steel tubing with an I.D. of ~4 mm (Fig. 1). The six lines can be isolated individually by means of Swagelok valves (Swagelok SS‐DSS4, London, UK). Samples are loaded into disposable 12‐mm Pyrex tubes located in the GC oven (ThermoFinnigan TRACE GC; Thermo Scientific, Bremen, Germany) (see Supporting Information). The programmable oven provides complete control over heating steps and rapid cooling following the analysis; this permits several consecutive runs of the apparatus per day. The hydration water is recovered outside the oven in 6‐mm break‐seal tubes by cryogenic trapping in LN2 (Fig. 1). The Pyrex and break‐seal tubes are connected to the stainless steel tubing via ¼‐inch Swagelock Ultra‐torr unions fitted with VitonTM O‐rings, which are replaced after every five or six runs of the apparatus. All the stainless steel lines outside the GC oven are wrapped in heating tape and held at 70°C to prevent condensation of water in the extraction lines.
A Pirani gauge (Edwards® APG100 Active Pirani vacuum gauge) is fitted to each of the six lines for monitoring pressure via an Edwards TIC 6 head instrument controller. The pressure in each line is continuously monitored during the extraction using a computer interface to the WASP system. The change in pressure as a function of time allows identification of any abnormalities (e.g. leaks) during the GHW extraction procedure (Fig. 2).
Figure 2.
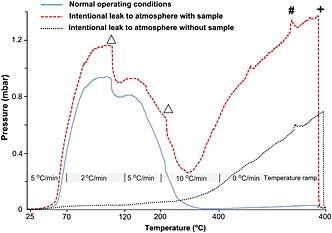
Typical pressure profile recorded during the extraction of GHW. The temperature ramp used along the dehydration process is given. The final pressure differs depending on whether the line is leaking or functioning properly. Vacuum leaks can affect the isotopic composition of the extracted GHW. Δ = raise LN2 trap; # = heat cold spots; + = pump non‐condensable gases.
Extraction of gypsum hydration water
Gypsum samples (~200 mg for CRDS analyses and ~800 mg for IRMS) were loaded into 12‐mm OD Pyrex tubes and topped with 3 cm of quartz wool to prevent loss of gypsum powder during dehydration. The experiments utilized analytical‐grade powdered gypsum (C/2360/50; Fisher Scientific, Hemel Hempstead, UK) (denoted as NEWGYP standard in this work). In addition, seven natural samples of gypsum were ground to a homogenous powder in an agate mortar and the GHW was extracted using the WASP for subsequent analysis by CRDS and IRMS.
The presence of organic matter and other contaminants in gypsum, including gases derived from microbial activity such as H2S and CH4 trapped in the mineral lattice, may produce spectral interferences in the CRDS measurements when GHW is analyzed, as has also been reported for the analysis of water extracted from the xylem of plants.18, 19 Such contamination is likely to be greater in gypsum deposited in environments associated with organic matter degradation, such as lakes and marine settings. To evaluate gypsum formed in a wide variety of depositional environments, our natural sample set included gypsum formed by evaporation in a lake (PI 6A‐13H‐2 8 cm and PI 6C‐7H‐2 26 cm6), gypsum derived from seawater evaporation in a coastal setting (Salina 18), gypsum speleothems generated by drip water evaporation in a gypsum cave (SBL‐2.3 and SBL‐820), and two samples of natural gypsum formed in hydrothermal environments (CRI‐017 and BG‐1021).
The samples were dried at 45°C for 24 h, and then placed under vacuum to a pressure of ~5 × 10−3 mbar for at least 3 h at room temperature to remove adsorbed water. This low‐vacuum pumping (10−2–10−3 mbar) is effective at removing adsorbed water with no detectable loss of hydration water.22 To improve sample throughput, some samples were pumped offline for 3 h in a desiccator, connected to a two‐stage rotary pump (Edwards) and a LN2 trap fitted in‐line between the pump and the desiccator. No difference was found in the isotopic results between the two pumping methods. This allows up to 18 gypsum samples (15 unknowns and 3 standards) to be extracted each day, thereby considerably increasing the sample throughput compared with earlier methods (e.g.6).
After the lines had been completely evacuated (pressure ~5 × 10−3 mbar), each branch was isolated from the vacuum and the glass 6‐mm break‐seal tubes at the end of each of the lines were immersed in LN2 for water trapping. GHW was released from the samples by slowly heating the sample to 400°C (Fig. 2). The temperature ramp was 5°C/min, between 25°C and 70°C, and reduced to 2°C/min between 70°C and 120°C. In this temperature range gypsum dehydration occurs under low‐to‐medium vacuum conditions.23 The slower heating during the second step is required to avoid rapid dehydration of the sample that can lead to gypsum powder blowing through the line. At 110°C, the cryogenic traps were raised (~3 cm) to increase the surface area for freezing the water. The ramp was 5°C/min between 120°C and 200°C. At 200°C, the cryogenic traps were again raised (~5 cm). After this step, the temperature ramp increased to 10°C/min between 200°C and 400°C. Once the oven reached 400°C, this temperature was held constant for 40 min to ensure complete recovery of all water in the lines by the cryogenic traps. Finally, non‐condensable gases were pumped away for 30 s before the 6‐mm break‐seal tubes were flame sealed. The GHW extraction process takes approximately 2 h per run. The pressure in the lines typically reaches 1.2 mbar at 130°C (after 30 min) and then gradually decreases to ~10−2 mbar at the end of the heating stage (Fig. 2).
The system was tested for memory effects by extracting the hydration water of isotopically depleted and enriched gypsum in two consecutive runs of the apparatus. These gypsum standards (DPL‐gyp and ENR‐gyp) were produced by hydrating anhydrous CaSO4 with isotopically depleted (−8.6‰, −16.1‰ and −131.4‰ for δ17O, δ18O and δ2H values, respectively) and enriched (11.2‰, 21.7‰ and 57.8‰ for δ17O, δ18O and δ2H, respectively) waters, following the procedure of Conley and Bundy.24 Prior to the first extraction, the lines were evacuated to a pressure of ~5 × 10−3 mbar for 4 days before the first experiments to remove potential adsorbed water remaining from previous samples. The system was evacuated for 30 min between the two consecutive runs.
During GHW extraction, the pressure changes with time can be utilized to identify anomalies caused by vacuum leaks (Fig. 2), water adsorption in the lines, or low amounts of water released by the sample. The system was tested for vacuum leaks that might occur due to deterioration of the Viton O‐rings by intentionally using compromised O‐rings in some of the lines and comparing those results with those from uncompromised O‐rings.
Distillation of brines
Routine isotopic analysis of brines can produce significant contamination problems in CRDS analyzers due to the accumulation of salts in the valves and pipework and problems with the needle used to inject water into the vaporizer. To solve this issue, we utilized the WASP for the distillation of saline waters prior to CRDS analysis.
Evaporated marine waters from two different brine pools from a natural salt factory (Cabo de Gata Salina, SE Spain, 36°45'32''N, 2°13'08''W) were utilized for the experiments. The water density measured in situ using a hydrometer, ranged from 1.13 g cm−3 (DEPO‐03) to 1.09 g cm−3 (DEPO‐06). Volume of 200 μL of water were loaded into 12‐mm OD tubes and then topped with 3 cm of quartz wool in order to prevent salts from moving through the vacuum line. The samples were loaded into the WASP and a LN2 trap was placed on each sample tube to freeze the water sample and avoid evaporation during vacuum pumping. Subsequently, the assembly was evacuated for 2 min (to a pressure of ~5 × 10−3 mbar) and the lines again isolated by closing the Swagelok valves. The LN2 traps were then removed from the sample tubes and placed on the 6‐mm break‐seal tubes. The heating stage was set to a temperature ramp of 5°C/min, with a final temperature of 150°C (maximum pressure ~8 mbar), and held isothermally for 40 min to facilitate complete sample recovery in the traps (final pressure was typically ~2 × 10−2 mbar). The traps were raised 5 cm during this final stage. Finally, the lines were evacuated for 30 s (pressure ~5 × 10−3 mbar) prior to flame sealing the 6‐mm tubes. In addition to the brines, four internal water standards (JRW, BOTTY, SPIT and ENR) were distilled using the same methodology in order to assess any isotopic fractionation during the extraction procedure. The brines were run in duplicate and analyzed by CRDS, along with the water standards.
Isotopic measurement of GHW by CRDS
Isotope analyses were conducted at the Godwin Laboratory for Palaeoclimate Research (Department of Earth Sciences) at the University of Cambridge (Cambridge, UK).
The water oxygen and hydrogen isotopes were measured simultaneously by CRDS using a L2140‐i Picarro water isotope analyzer for triple oxygen isotope analysis,16 interfaced with an A0211 high‐precision vaporizer (both from Picarro, Santa Clara, CA, USA). A Picarro micro‐combustion module (MCM®) was used in the majority of the experiments. The MCM comprises an 8‐cm‐long cylindrical cartridge filled with metallic wool coated with a proprietary catalyst, and it was placed in‐line between the vaporizer and the water isotope analyzer. The MCM has been shown to remove combustible compounds from water samples.18, 19 For experiments that utilized the MCM (denoted as 'MCM On' hereafter) the cartridge was held at a constant temperature of 200°C. For experiments that did not utilize the accessory (denoted as 'MCM Off' hereafter), the WARM mode (~80°C) configuration was used. This configuration avoids water vapour condensation in the MCM transfer line, thereby reducing the memory effect between injections; however, this MCM mode does not remove organics from the vapor stream. Both the MCM On and MCM Off experiments used dry air (containing 21% O2) as the carrier gas.
After water extraction, the 6‐mm OD tubes containing 40–60 μL of hydration water were stored until ready for analysis. Prior to analysis, water was frozen into the base of the break‐seal tube by immersion in LN2. Subsequently, the tube was scored with a diamond cutter, broken to a fixed height of 25 mm, and then quickly inserted into the 2‐mL septum‐capped vials used by an A0325 autosampler (Picarro). Each sample was injected ten times into the vaporizer, which was heated to 110°C. Memory effects from previous samples were avoided by rejecting the first three analyses. The values for the final seven injections were averaged with a typical in‐sample precision (±1SD) of ±0.02‰ for δ17O values, ±0.04‰ for δ18O values, and ±0.19‰ for δ2H values, as observed from repeated analysis of an in‐house water standard (SPIT, n = 23) over the period of study.
The results were normalized against V‐SMOW by analyzing internal standards before and after each set of 10 to 12 samples. To this end, four internal water standards (JRW, BOTTY, SPIT and ENR) were calibrated against V‐SMOW and SLAP, using δ17O values of 0.0‰ and −29.6986‰, respectively, and δ18O values of 0.0‰ and −55.5‰, respectively.25, 26 All the 17O‐excess values are given in per meg units (0.001‰).
The GISP standard water was analyzed during the calibration as an unknown, yielding an average 17O‐excess value of 25±12 per meg during the course of the study. This value is in good agreement with the results reported by Schoenemann et al.26 (22±11 per meg). No measurable differences were observed in the calibration of the liquid water standards when the MCM was utilized. The in‐run drift of the instrument was corrected (when necessary) by the analysis of an internal standard every 3 or 4 samples. The 17O‐excess and d‐excess were calculated for each injection using the corrected δ17O, δ18O and δ2H values. The 17O‐excess and d‐excess from the seven repeated analyses were then averaged. Typical in‐sample 17O‐excess and d‐excess precisions (±1SD) in water standards (SPIT, n = 23) were 13 per meg and 0.2‰, respectively. This agrees with the guaranteed precision given by the manufacturer (15 per meg for 17O‐excess). No 17O‐excess or d‐excess values were rejected.
Isotopic measurement of GHW by fluorination‐IRMS method
Fluorination‐IRMS analyses were conducted at the Institute for Geology and Mineralogy at the University of Cologne (Cologne, Germany). An autosampler injects water samples (2.8 μL) into a CoF3 reactor held at 375°C which converts H2O into O2 and HF, following the method developed by Barkan and Luz.17 Helium carries the products through a double cold trap immersed in LN2 to freeze out the HF. The O2 is then trapped using 5Ǻ molecular sieve held at LN2 temperature. Helium is pumped away after disconnecting the oxygen trap from the He stream and sample O2 is transferred to one tube in a 12‐fold sample manifold immersed in LN2. For this study a manifold typically contained four reference water extractions, leaving eight sample tubes for O2‐extractions from two or three unknown water samples. To minimize memory effects at least three sample injections were discarded when switching between waters of different isotopic values. The CoF3 line is LabView (National Instruments, Austin, TX, USA) controlled and runs fully automated overnight.
The sample O2 was then analyzed on a MAT253 isotope ratio mass spectrometer (Thermo Scientific) in dual‐inlet mode at m/z 32, 33 and 34. Each measurement comprises 66 samples to reference comparisons divided into 3 blocks of 22 cycles each. Each block begins with a peak centre and adjustment of bellow pressure, such that the ion beam intensity at m/z 32 is 8.5 V. The idle time was set to 17 s and the integration time to 12 s. The baseline was measured for 240 s prior to the first sample. The δ17Osample and δ18Osample values were normalized to V‐SMOW via our internal lab standards that were measured with the samples. As for the CRDS data, SLAP of δ17O = −29.6986‰ and δ18O = −55.5‰ was used for SMOW‐SLAP scaling.26 Analysis of SMOW (n = 15) and SLAP (n = 15) during the course of this study gave −28.699 ± 0.011‰ (1SD) and −53.622 ± 0.024‰ (1SD) for the δ17Omeasured‐SLAP and δ18Omeasured‐SLAP values, respectively. This slightly contracted scale relative to the IAEA‐scale appears to be typical for MAT253 instruments.26
Results and Discussion
GHW extraction and δ17O, δ18O and δ2H measurements
Our extraction procedure for GHW has several advantages over previous methods, including higher sample throughput and greater precision. The ability to extract five unknowns and a gypsum standard in the same run improves the long‐term reproducibility of the method. In addition, our system allows continuous monitoring of the pressure to detect anomalies in the mineral water extraction procedure.
By monitoring the pressure of the WASP during GHW extraction, vacuum leaks can be detected in individual lines (Fig. 2). When operating correctly, the pressure in the lines displayed similar profiles, first increasing up to 1 mbar at around 120°C, and then decreasing to below 5 × 10−2 mbar at the end of the extraction process. These profiles for the lines with compromised O‐rings showed slightly greater pressures (up to 1.2 mbar) at 120°C (Fig. 2). The pressure decreased by as much as 0.2 mbar when the LN2 trap was raised for a second time, but subsequently increased to around 1.4 mbar by the end of the run. In the line run as a blank with no sample, the pressure increased constantly to reach a final value of 0.7 mbar, and no drop in pressure occurred during the run (Fig. 2).
The isotopic composition of the NEWGYP standard extracted in the leaky line showed no difference within error (0.03‰ for the δ17O value, 0.03‰ for the δ18O value and −51.24‰ for the δ2H value) from that obtained from the properly functioning line (0.07‰ for the δ17O value, 0.09‰ for the δ18O value, and −51.39‰ for the δ2H value) (Table 1). However, slight differences were observed in the results of the ENR‐gyp samples. The enriched gypsum extracted in the leaking line showed slightly lower isotope values (12.78‰ for the δ17O value, 24.58‰ for the δ18O value, and 33.56‰ for the δ2H value) than those from the properly functioning line in the same run (12.96‰ for the δ17O value, 24.93‰ for the δ18O value, and 34.63‰ for the δ2H value) (Table 1). This suggests that vacuum leaks can alter the isotopic ratio values of samples extracted using the WASP, especially for hydration waters with an isotopic composition significantly different from that of the ambient water vapour in the laboratory atmosphere. Therefore, it is vital to monitor the pressure in the WASP during GHW extraction to ensure that no leaks occur during extraction.
Table 1.
Memory effect tests and experiments of vacuum leakage in the WASP lines during GHW extraction
| Sample/Line | Line status | δ17O (‰) | 1σ (‰) | δ18O (‰) | 1σ (‰) | δ2H (‰) | 1σ (‰) | 17O‐excess (per meg) | 1σ (per meg) |
|---|---|---|---|---|---|---|---|---|---|
| ENR‐gyp L5 | “Fresh” line | 12.96 | 0.04 | 24.93 | 0.05 | 34.73 | 0.19 | −126 | 9 |
| ENR‐gyp L6 | “Fresh” leaking line | 12.78 | 0.04 | 24.58 | 0.05 | 33.56 | 0.39 | −120 | 8 |
| ENR‐gyp L1 | After depleted sample | 13.12 | 0.03 | 25.24 | 0.05 | 35.10 | 0.38 | −125 | 14 |
| ENR‐gyp L2 | After depleted sample | 13.08 | 0.03 | 25.14 | 0.05 | 34.66 | 0.36 | −115 | 13 |
| NEWGYP‐ L3 | “Fresh” line | 0.07 | 0.02 | 0.09 | 0.02 | −51.39 | 0.18 | 20 | 14 |
| NEWGYP‐L4 | “Fresh” leaking line | 0.03 | 0.02 | 0.03 | 0.02 | −51.24 | 0.18 | 17 | 14 |
| DPL‐gyp L1 | “Fresh” line | −6.79 | 0.04 | −12.83 | 0.06 | −149.66 | 0.24 | 1 | 14 |
| DPL‐gyp L2 | “Fresh” line | −6.89 | 0.02 | −13.04 | 0.03 | −150.35 | 0.16 | 10 | 9 |
| DPL‐gyp L5 | After Enriched sample | −6.75 | 0.03 | −12.77 | 0.03 | −150.75 | 0.04 | 11 | 19 |
| DPL‐gyp L6 | After Enriched sample | −6.78 | 0.02 | −12.79 | 0.02 | −150.80 | 0.11 | −10 | 21 |
The term “fresh” refers to a line in which the previous sample analyzed was close in isotopic value to the next sample and memory effects should be minimized.
Monitoring the water yield from each sample is also necessary to ensure data quality. The water yield of the sample, measured as the weight loss after the extraction process, can allow incomplete gypsum dehydration and sample contamination (i.e., <100% gypsum) to be detected. The percentage of water in the gypsum samples in our experiments was on average 20.7 ± 0.4 (Table 2). This range is consistent with the stoichiometric percentage of water in gypsum (20.9), which suggests that complete gypsum dehydration took place during the extractions.
Table 2.
Isotopic analysis (δ17O, δ18O and δ2H values) of hydration water of a gypsum standard (NEWGYP) extracted using the WASP and analyzed by CRDS and IRMS
| Line/date | Method | δ17O (‰) | 1σ (±) | δ18O (‰) | 1σ (±) | δ2H (‰) | 1σ (±) | d‐excess (‰) | 1σ (±) | 17O‐excess (per meg) | 1σ (per meg) | H2O (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L3‐28/1/15 | CRDS | 0.12 | 0.05 | 0.21 | 0.07 | −51.23 | 0.14 | −52.3 | 0.4 | 21 | 15 | 21.3 |
| L4‐29/1/15 | CRDS | 0.20 | 0.04 | 0.35 | 0.04 | −50.83 | 0.12 | −52.8 | 0.3 | 27 | 15 | 20.8 |
| L5‐30/1/15 | CRDS | 0.23 | 0.04 | 0.42 | 0.10 | −50.91 | 0.23 | −53.3 | 0.6 | 19 | 11 | 20.9 |
| L4‐30/1/15 | IRMS | 0.36 | 0.05 | 0.67 | 0.09 | ‐ | ‐ | ‐ | ‐ | 7 | 3 | 20.5 |
| L5‐30/1/15 | IRMS | 0.35 | 0.01 | 0.65 | 0.02 | ‐ | ‐ | ‐ | ‐ | 10 | 8 | 20.6 |
| L2‐30/1/15 | IRMS | 0.29 | 0.03 | 0.52 | 0.05 | ‐ | ‐ | ‐ | ‐ | 11 | 3 | 20.9 |
| L1‐30/1/15 | IRMS | 0.34 | 0.09 | 0.62 | 0.17 | ‐ | ‐ | ‐ | ‐ | 10 | 4 | 20.7 |
| L2‐30/1/15 | IRMS | 0.29 | 0.10 | 0.53 | 0.19 | ‐ | ‐ | ‐ | ‐ | 9 | 7 | 20.6 |
| L3‐30/1/15 | IRMS | 0.17 | 0.11 | 0.32 | 0.20 | ‐ | ‐ | ‐ | ‐ | 0 | 8 | 20.8 |
| L4‐30/1/15 | IRMS | 0.17 | 0.03 | 0.31 | 0.06 | ‐ | ‐ | ‐ | ‐ | 10 | 4 | 20.8 |
| L5‐30/1/15 | IRMS | 0.28 | 0.06 | 0.51 | 0.10 | ‐ | ‐ | ‐ | ‐ | 9 | 4 | 20.4 |
| L6‐30/1/15 | CRDS | 0.16 | 0.04 | 0.25 | 0.07 | −50.89 | 0.52 | −54.2 | 0.5 | 18 | 7 | 20.9 |
| L1‐03/2/15 | CRDS | 0.13 | 0.05 | 0.16 | 0.09 | −50.34 | 0.32 | −51.6 | 0.6 | 33 | 8 | 20.8 |
| L2‐03/2/15 | CRDS | 0.14 | 0.02 | 0.25 | 0.03 | −51.28 | 0.17 | −53.3 | 0.2 | 8 | 14 | 21.9 |
| L4‐04/2/15 | CRDS | 0.17 | 0.02 | 0.30 | 0.05 | −51.48 | 0.29 | −53.9 | 0.4 | 20 | 16 | 20.4 |
| L5‐05/2/15 | CRDS | 0.13 | 0.02 | 0.21 | 0.02 | −51.81 | 0.10 | −53.5 | 0.1 | 23 | 14 | 20.3 |
| L1‐09/2/15 | CRDS | 0.16 | 0.02 | 0.27 | 0.04 | −51.59 | 0.27 | −53.8 | 0.1 | 13 | 14 | 20.7 |
| L2‐09/2/15 | CRDS | 0.12 | 0.02 | 0.18 | 0.03 | −51.88 | 0.09 | −53.3 | 0.2 | 21 | 13 | 20.5 |
| L3‐10/2/15 | CRDS | 0.18 | 0.03 | 0.29 | 0.04 | −51.63 | 0.19 | −54.0 | 0.2 | 18 | 16 | 20.9 |
| L6‐18/2/15 | CRDS | 0.07 | 0.03 | 0.11 | 0.04 | −51.80 | 0.18 | −53.4 | 0.2 | 20 | 12 | 20.8 |
| L1‐22/2/15 | CRDS | 0.20 | 0.02 | 0.33 | 0.03 | −51.24 | 0.20 | −54.7 | 0.2 | 32 | 10 | 20.7 |
| L3‐04/2/15 | CRDS | 0.17 | 0.03 | 0.30 | 0.04 | −50.39 | 0.23 | −52.9 | 0.1 | 11 | 13 | 20.7 |
| L4‐02/3/15 | CRDS | 0.16 | 0.02 | 0.29 | 0.02 | −50.80 | 0.08 | −53.1 | 0.2 | 12 | 11 | 20.1 |
| L1‐03/3/15 | CRDS | 0.19 | 0.02 | 0.32 | 0.03 | −51.27 | 0.19 | −53.8 | 0.2 | 17 | 13 | 20.9 |
| L2‐05/3/15 | CRDS | 0.23 | 0.02 | 0.41 | 0.02 | −50.36 | 0.35 | −53.5 | 0.5 | 18 | 7 | 20.6 |
| L3‐06/3/15 | CRDS | 0.37 | 0.04 | 0.66 | 0.05 | −50.80 | 0.18 | −56.1 | 0.3 | 17 | 15 | 20.2 |
| L4‐08/3/15 | CRDS | 0.18 | 0.02 | 0.32 | 0.03 | −51.74 | 0.13 | −54.3 | 0.2 | 12 | 15 | 21.9 |
| L5‐09/3/15 | CRDS | 0.28 | 0.03 | 0.50 | 0.05 | −50.64 | 0.19 | −54.6 | 0.2 | 19 | 10 | 19.9 |
| L6‐11/3/15 | CRDS | 0.22 | 0.01 | 0.40 | 0.01 | −51.31 | 0.10 | −54.5 | 0.2 | 3 | 11 | 20.7 |
| L1‐12/3/15 | CRDS | 0.28 | 0.03 | 0.52 | 0.03 | −51.16 | 0.06 | −55.3 | 0.2 | 12 | 13 | 20.8 |
| L6‐13/3/15 | CRDS | 0.18 | 0.02 | 0.31 | 0.05 | −50.63 | 0.36 | −53.1 | 0.2 | 15 | 15 | 20.7 |
| L3‐22/3/15 | CRDS | 0.25 | 0.02 | 0.45 | 0.03 | −50.50 | 0.30 | −54.5 | 0.5 | 6 | 16 | 20.4 |
| L4‐23/3/15 | CRDS | 0.21 | 0.03 | 0.39 | 0.03 | −51.29 | 0.18 | −53.2 | 0.4 | 9 | 15 | 20.5 |
| L6‐26/3/15 | CRDS | 0.22 | 0.03 | 0.43 | 0.03 | −50.28 | 0.21 | −53.7 | 0.2 | −5 | 16 | 20.8 |
| L6‐27/3/15 | CRDS | 0.24 | 0.01 | 0.41 | 0.02 | −50.81 | 0.22 | −54.1 | 0.1 | 18 | 10 | 20.7 |
| L5‐30/3/15 | CRDS | 0.21 | 0.03 | 0.37 | 0.03 | −51.10 | 0.23 | −54.0 | 0.2 | 16 | 12 | 21.2 |
| L4‐01/4/15 | CRDS | 0.16 | 0.03 | 0.29 | 0.03 | −50.81 | 0.15 | −53.1 | 0.4 | 0 | 12 | 20.9 |
| L3‐02/4/15 | CRDS | 0.22 | 0.02 | 0.37 | 0.03 | −50.64 | 0.39 | −53.7 | 0.2 | 19 | 15 | 20.8 |
| L1‐20/4/15 | CRDS | 0.16 | 0.03 | 0.29 | 0.03 | −50.01 | 0.27 | −52.4 | 0.4 | 5 | 13 | 20.5 |
| L2‐21/4/15 | CRDS | 0.04 | 0.06 | 0.04 | 0.12 | −50.66 | 0.65 | −50.3 | 0.4 | 27 | 9 | 20.3 |
| L5‐21/4/15 | CRDS | 0.13 | 0.04 | 0.21 | 0.06 | −50.87 | 0.38 | −52.6 | 0.3 | 6 | 13 | 20.9 |
| L3‐22/4/15 | CRDS | 0.18 | 0.02 | 0.32 | 0.02 | −50.59 | 0.27 | −53.1 | 0.2 | 13 | 15 | 20.9 |
| L4‐22/4/15 | CRDS | 0.07 | 0.02 | 0.09 | 0.02 | −51.39 | 0.18 | −52.4 | 0.2 | 20 | 14 | 20.8 |
| L3‐22/4/15 | CRDS | 0.14 | 0.03 | 0.24 | 0.06 | −50.99 | 0.39 | −52.8 | 0.4 | 8 | 13 | 20.9 |
| L4‐22/4/15 | CRDS | 0.03 | 0.02 | 0.03 | 0.02 | −51.24 | 0.18 | −51.7 | 0.1 | 17 | 14 | 20.7 |
| AVG | CRDS | 0.18 | 0.31 | −51.02 | −53.4 | 15 | 20.7 | |||||
| STD | ±0.07 | ±0.13 | ±0.49 | ±1.1 | ±8 | ±0.4 | ||||||
| AVG | IRMS | 0.27 | 0.49 | ‐ | ‐ | ‐ | 8 | 20.7 | ||||
| STD | ±0.07 | ±0.14 | ±4 | ±0.2 |
Under correct operating conditions, the long‐term precision of the method was ± 0.07‰ for δ17O values, ±0.13‰ for δ18O values, and ±0.49‰ for δ2H values (n = 37, 7 injections each, ±1SD) for analyses of NEWGYP (δ17O = 0.18‰, δ18O = 0.31‰ and δ2H = −51.02‰) using CRDS (Fig. 3 and Table 2). The IRMS analysis of the same gypsum standard (n = 8, 3 or 4 analyses each) yielded mean values and reproducibility (±1SD) of 0.28 ± 0.07‰ and 0.51 ± 0.14‰ for the δ17O and δ18O values, respectively, which are within the two‐sigma error of the CRDS measurements (Fig. 3 and Table 2).
Figure 3.
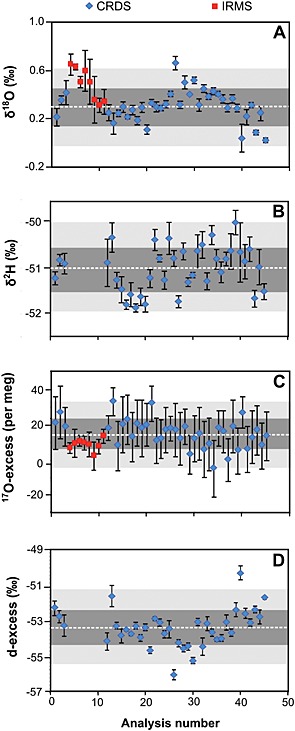
The δ18O (A), δ2H (B), 17O‐excess (C) and d‐excess (D) values of GHW from the repeated analysis of a gypsum standard (NEWGYP), extracted using the WASP and analyzed by CRDS (n = 37) and IRMS (n = 8). δ17O displayed a similar trend to δ18O. Data are displayed following the order in which the samples were extracted using the WASP. Errors bars refer to the internal error (±1‐sigma obtained from the repeated analysis of the same hydration water (7 injections for CRDS and 3–4 for IRMS). The long‐term means (dashed line) and external errors are shown for ±1‐sigma (deep‐grey shade) and ±2‐sigma (light‐grey shade).
No differences in the value of the in‐house water standard were found when the MCM was not used and no systematic drift has been observed in the δ17O, δ18O and δ2H measurements of the standard over time (Fig. 3). In principle, any long‐term drift can be monitored and corrected since a gypsum standard is extracted with each five unknown samples. Because no such drift was observed during the course of this study, there is currently no benefit in correcting the values. However, if a drift is observed in the future, a correction could be applied to improve the long‐term reproducibility of the method.
Gypsum samples with isotopically enriched and depleted hydration water were extracted consecutively in the same line of the WASP in order to determine the potential memory effect of the system. The samples extracted in the first run were found not to influence the δ17O, δ18O and δ2H values of the samples extracted in the second run of the apparatus. The values for the isotopically enriched gypsum (δ17O = 13.05‰, δ18O = 25.10‰ and δ2H = 34.83‰, n = 3, 7 injections each) showed analytical errors (±1SD) of ±0.08‰, ±0.16‰ and ±0.24‰ for the δ17O, δ18O and δ2H values, respectively, and the errors for the isotopically depleted gypsum (δ17O = −6.81‰, δ18O = −12.86‰ and δ2H = −150.39‰, n = 4, 7 injections each) were ±0.06‰, ±0.12‰ and ±0.53‰, respectively (Table 1). These results show that there is no measureable memory effect in the WASP system with our pumping protocol.
For natural gypsum samples, CRDS using the MCM produced values ranging from −1.00‰ to 5.61‰ for δ17O, from −1.93‰ to 10.74‰ for δ18O and from −53.36‰ to 13.21‰ for δ2H (Table 3). The in‐sample reproducibility (±1SD) of the CRDS analyzer for the analysis of natural gypsum samples was typically ±0.03‰ for δ17O values, ±0.04‰ for δ18O values, and ±0.24‰ for δ2H values, found by taking the average of seven consecutive injections for each GHW sample. These results are similar to those observed for the analysis of water standards. Regarding the long‐term reproducibility of natural samples, the replicate GHW extraction and subsequent analysis in different runs of sample Salina 1 (n = 3, 7 injections each) gave an error (±1SD) of ±0.02‰ for δ17O values, ±0.03‰ for δ18O values, and ±0.16‰ for δ2H values (Table 3). This demonstrates the long‐term precision obtainable for the hydration water of natural gypsum samples using our method.
Table 3.
Isotopic analysis (δ17O, δ18O and δ2H values) of hydration water of a gypsum standard (NEWGYP) and natural samples
| Sample | CRDS (MCM ON) | CRDS (MCM OFF) | IRMS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ17O (‰) | δ18O (‰) | δ2H (‰) | d‐excess (‰) | 17O‐excess (per meg) | δ17O (‰) | δ18O (‰) | δ2H (‰) | d‐excess (‰) | 17O‐excess (per meg) | δ17O (‰) | δ18O (‰) | 17O‐excess (per meg) | |
| BG‐10 | −1.00 ± 0.02 | −1.93 ± 0.02 | −53.36 ± 0.21 | −37.9 ± 0.2 | 20 ± 13 | −0.89 ± 0.01 | −1.78 ± 0.03 | −52.90 ± 0.14 | −38.6 ± 0.2 | 57 ± 11 | −1.01 ± 0.02 | −1.96 ± 0.04 | 25 ± 7 |
| CRI‐01 | −0.06 ± 0.02 | −0.15 ± 0.04 | −41.91 ± 0.23 | −40.7 ± 0.2 | 15 ± 7 | −0.04 ± 0.02 | −0.16 ± 0.04 | −42.3 ± 0.30 | −42.9 ± 0.3 | 54 ± 12 | 0.09 ± 0.01 | 0.12 ± 0.01 | 27 ± 5 |
| NEWGYP (n=36/8)* | 0.17 ± 0.07 | 0.30 ± 0.13 | −51.03 ± 0.48 | −53.4 ± 1.0 | 15 ± 8 | 0.12 ± 0.04 | 0.15 ± 0.05 | −51.23 ± 0.35 | −53.0 ± 0.6 | 42 ± 12 | 0.27 ± 0.07 | 0.49 ± 0.14 | 8 ± 4 |
| SBL‐2.3 | 1.55 ± 0.01 | 2.92 ± 0.03 | −27.71 ± 0.27 | −51.6 ± 0.3 | −7 ± 15 | 1.88 ± 0.02 | 3.40 ± 0.02 | −27.47 ± 0.11 | −54.7 ± 0.1 | 77 ± 13 | 1.85 ± 0.01 | 3.52 ± 0.02 | −7 ± 1 |
| SBL‐8 | 1.68 ± 0.02 | 3.13 ± 0.02 | −27.72 ± 0.07 | −53.2 ± 0.1 | 7 ± 15 | 1.74 ± 0.02 | 3.17 ± 0.02 | −28.31 ± 0.07 | −53.7 ± 0.2 | 61 ± 14 | 1.90 ± 0.01 | 3.60 ± 0.03 | 1 ± 5 |
| PI 6A‐13H‐2 7–8 cm | 4.46 ± 0.01 | 8.53 ± 0.02 | 3.76 ± 0.14 | −64.5 ± 0.2 | −26 ± 12 | 5.08 ± 0.04 | 9.41 ± 0.05 | 6.93 ± 0.20 | −68.4 ± 0.4 | 123 ± 18 | 4.87 ± 0.00 | 9.30 ± 0.01 | −30 ± 2 |
| PI 6C‐7H‐2 25–26 cm | 4.89 ± 0.03 | 9.30 ± 0.05 | 9.76 ± 0.26 | −64.7 ± 0.3 | −17 ± 9 | 5.32 ± 0.02 | 9.74 ± 0.02 | 11.21 ± 0.06 | −66.8 ± 0.2 | 190 ± 7 | 4.98 ± 0.02 | 9.51 ± 0.03 | −30 ± 2 |
| Salina 1 (n=3)* | 5.61 ± 0.02 | 10.74 ± 0.03 | 13.21 ± 0.16 | −72.7 ± 0.2 | −48 ± 8 | 5.74 ± 0.04 | 10.74 ± 0.04 | 13.55 ± 0.18 | −72.4 ± 0.4 | 72 ± 16 | 5.88 ± 0.03 | 11.26 ± 0.07 | −49 ± 5 |
Samples analyzed in replicate (in these cases, the analytical error refers to the external precision, taking the average of the indicated number of samples).
For most natural samples, the δ17O, δ18O and δ2H values of GHW did not differ significantly when the MCM was used or not (within the two‐sigma error) from the results obtained by IRMS (Fig. 4). However, the 17O‐excess differed considerably between CRDS and IRMS when the MCM was not used (see next section). The 17O‐excess values measured by CRDS were considerably higher than those obtained by IRMS when the MCM was turned off. The difference in 17O‐excess values with MCM On and Off reflects the degree of spectral interference caused by contamination.
Figure 4.
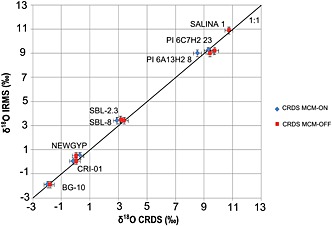
Cross‐plot of δ18O values in GHW of natural samples analyzed by CRDS/MCM compared with measurements of the same samples by IRMS. The long‐term ±2‐sigma error of the method (0.26‰ for the δ18O values from the CRDS measurements and 0.28‰ for the δ18O values from the IRMS analyses) is given for all samples. The in‐sample ±2‐sigma errors (7 injections for CRDS and 3–4 for IRMS) are smaller than the data symbols.
17O‐excess and deuterium‐excess in GHW
CRDS and IRMS produced similar 17O‐excess values for the NEWGYP standard (15±8 per meg for CRDS and 8±4 per meg for IRMS) when the MCM accessory was used with the CRDS analyzer (Table 3 and Fig. 5). However, the 17O‐excess value differed considerably (~25 per meg higher for CRDS) when the MCM was configured in WARM mode (Fig. 5).
Figure 5.
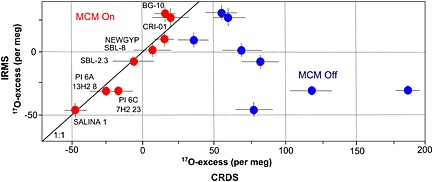
Cross‐plot of 17O‐excess in synthetic and natural gypsum analyzed by CRDS and IRMS. The analyses by CRDS that used the MCM yielded results similar to those obtained by IRMS. Error bars refer to the consecutive analyses (7 injections for CRDS and 3–4 analyses for IRMS) of the same hydration water.
Similarly, the 17O‐excess values in natural samples analyzed by CRDS and IRMS did not agree when the MCM was not used (Fig. 5 and Table 3). The 17O‐excess difference was up to ~40 per meg in the GHW from the hydrothermal gypsum sample (CRI‐01 and BG‐10), and the results from lake samples showed an even larger disagreement of up to ~200 per meg in GHW (e.g. PI 6C‐7H‐2 26 cm). For all samples, the 17O‐excess of hydration water was greater when the MCM was not powered on. By contrast, the 17O‐excess results from CRDS were similar to those achieved by IRMS when the MCM was used (Table 3), resulting in a close 1:1 relationship (Fig. 5).
In previous work on the isotopic composition of GHW in lake samples measured by CRDS, Hodell et al.6 analyzed their spectra using Picarro's ChemCorrect software that identifies irregularities caused by traces of hydrocarbons.27 These authors found that none of their samples showed any signs of spectroscopic interference affecting the δ18O or δ2H values, probably because the hydrocarbons and fatty acids present in the gypsum tend to be of longer mean chain length, as suggested in earlier work.28 In our study, we analyzed some similar lake gypsum samples, which showed the largest discrepancies when the MCM was not used (PI 6A‐13H‐2 8 cm, PI 6C‐7H‐2 26 cm). Our results strongly indicate that contaminant gases released from natural gypsum samples (VOCs, H2S, etc.) during dehydration cause spectral interferences at the wavenumber used to determine 1H2 17O by CRDS (7193 cm−1),16 but not in a meaningful way to the 1H2 18O and 2H1H16O signals; thus, although the 17O‐excess is affected by these contaminants, the δ18O and δ2H values are not.
Use of the micro‐combustion module (MCM) removes impurities that affect the 17O‐excess determination. As stated above, the differences in 17O‐excess in natural samples in our MCM On/Off experiments are greater in the case of gypsum samples generated in organic‐ and microbe‐rich environments (e.g. lake sediments). This, as expected, points to higher concentration of contaminants in these types of material and suggests that the cause of the spectral interferences could be H2S or organic compounds released by natural gypsum samples during dehydration. Further evidence for the presence of impurities in GHW is given by a strong smell of H2S in water after extraction, especially in water from lacustrine gypsum samples.
Our results reveal that the use of the MCM accessory is crucial for accurate determination of the 17O‐excess in GHW by CRDS. In earlier work, this device was found to be necessary for isotopic analysis of water samples with high concentrations of organic compounds, such as water extracted from the xylem of plants that usually contains alcohols that spectroscopically interfere with the CRDS analysis.18, 19 To date, the efficiency of the MCM has been demonstrated for the removal of alcohols and other organic compounds from water.19 However, catalytic oxidation of other substances also occurs, such as H2S, VOCs and long‐chain compounds, and this is important for the analysis of GHW samples. Although this accessory removes most impurities, we suspect that organic‐rich gypsum samples (e.g. those from lake sediments) may require additional pre‐treatment to remove organic compounds (H2O2, sodium hypochlorite, etc.). However, any pre‐treatment procedure will require testing to ensure that it does not alter the δ18O and δ2H values of the hydration water.
Using the MCM, the long‐term precision (±1SD) of our method for 17O‐excess determination by CRDS of the analytical‐grade gypsum standard (NEWGYP) was ±8 per meg (n = 37) (Table 2). In addition, no systematic drift in the 17O‐excess of the standard has been observed with time (Fig. 3). Remarkably, this reproducibility is better than the typical in‐sample precision obtained from seven consecutive injections (±13 per meg) observed in both water standards and GHW. Similar precision (typically ±15 per meg) is also obtained by randomly choosing the 17O‐excess of injections from NEWGYP samples, extracted on different days and analyzed in different CRDS analyzer runs. This is a consequence of the minimized importance of drift, memory effect and potential isotopic fractionation occurring during the 17O‐excess measurements, compared with the measurement of the δ17O and δ18O values. As demonstrated by Barkan and Luz,17 and later by Schoenemann et al.,26 the errors in the δ17O and δ18O values are covarying. This means that any isotopic fractionation during the analytical procedure may affect the individual δ17O and δ18O values, but does not change significantly the relative difference between the δ17O and δ18O values (17O‐excess).
Likewise, the repeated extraction and measurement (n = 3, 7 injections each) of one natural gypsum sample (Salina 1) by CRDS using the MCM produced an analytical error (±1SD) of ±7 per meg. This indicates that the long‐term 17O‐excess precision of our method using CRDS is similar for synthetic and natural gypsum when the MCM is powered on. The long‐term precision is also similar to that obtained for the analysis of water standards, as observed from the repeated measurement of our internal water standard (SPIT, ±8 per meg, n = 23) in different runs over the period of this study. No measurable memory effects were detected in our experiments with ENR‐gyp and DPL‐gyp for the 17O‐excess (Table 1).
The repeated extraction of our NEWGYP standard (n = 8) and subsequent analysis by IRMS produced a typical in‐sample precision of ±6 per meg (3 or 4 consecutive analyses of each hydration water sample) and an external precision of ±4 per meg, which is similar to the typical precision achieved for water standards using CoF3 fluorination IRMS17, 26 and better than the precision obtained by CRDS (±8 per meg).
A distinct advantage of CRDS over IRMS is the possibility of simultaneously determining 2H/1H, along with 17O/16O and 18O/16O, on the same hydration water sample. This enables the calculation of both d‐excess and 17O‐excess in GHW. The long‐term d‐excess of the NEWGYP standard (n = 37) determined using CRDS was −53.4 ± 1.1‰ (Fig. 3 and Table 2). This reproducibility is comparable with the long‐term precision observed from the repeated measurement of our SPIT water standard (±0.9‰, n = 23). No systematic drift in the d‐excess of the NEWGYP standard has been observed with time (Fig. 4). As for the natural gypsum samples, the d‐excess in GHW analyzed by CRDS using the MCM ranged from −39.7‰ to −71.9‰. As expected, the values are positively correlated with those of the 17O‐excess across the dataset.11
Distillation of brines and isotopic analysis by CRDS
The isotopic values of the untreated and distilled standards agree within the internal error of the CRDS water analyzer (Table 4), suggesting that there was complete water vapour recovery in the cryogenic traps and no isotopic fractionation during distillation in the WASP. The reproducibility (±1SD) for two repeated analyses (7 injections each) of the distilled brines was better than ±0.04‰ and ±0.06‰ for the δ17O and δ18O values, respectively, and better than ±0.27‰ for the δ2H values (Table 4), which is similar to the long‐term precision of the CRDS analyzer for the analysis of water standards. When comparing the untreated and the distilled standards, the derived d‐excess and 17O‐excess values differed by less than 0.3‰ and 10 per meg, respectively. This demonstrates that the WASP can be used for the distillation of saline solutions for isotopic analysis by CRDS, with no contamination of the analyzer by salt deposition. However, earlier investigations found that isotopic fractionation may occur during distillation of highly saline brines.29 The degree of fractionation depends on the nature and the molar concentration of salts, and can be corrected using 'salt effect' coefficients as long as the activity of the major elements (Ca2+, Mg2+, K+) is known (i.e.30). Additional corrections may need to be applied to obtain accurate isotopic values after distillation and CRDS analysis of brines.
Table 4.
Isotopic composition of water samples distilled using the WASP and analysed by CRDS
| Sample | δ17O (‰) | 1σ (‰) | δ18O (‰) | 1σ (‰) | δ2H (‰) | 1σ (‰) | d‐excess (‰) | 1σ (‰) | 17O‐excess (per meg) | 1σ (per meg) |
|---|---|---|---|---|---|---|---|---|---|---|
| JRW | −9.99 | 0.01 | −18.85 | 0.02 | −146.01 | 0.19 | 5.6 | 0.2 | −4 | 13 |
| JRW‐WASP | −10.02 | 0.03 | −18.90 | 0.05 | −146.19 | 0.20 | 5.9 | 0.2 | 2 | 13 |
| BOTTY | −3.95 | 0.02 | −7.52 | 0.02 | −50.18 | 0.21 | 10.6 | 0.2 | 20 | 13 |
| BOTTY‐WASP | −3.96 | 0.02 | −7.54 | 0.03 | −50.22 | 0.11 | 10.7 | 0.3 | 24 | 10 |
| SPIT | −0.07 | 0.02 | −0.13 | 0.03 | 0.16 | 0.14 | 1.5 | 0.3 | −6 | 11 |
| SPIT‐WASP | −0.08 | 0.02 | −0.17 | 0.02 | −0.17 | 0.20 | 1.4 | 0.3 | 6 | 13 |
| ENR | 5.64 | 0.02 | 10.76 | 0.01 | 40.23 | 0.10 | −46.4 | 0.1 | −36 | 18 |
| ENR‐WASP | 5.55 | 0.02 | 10.59 | 0.02 | 39.58 | 0.12 | −45.6 | 0.1 | −37 | 12 |
| DEPO‐03A | 3.03 | 0.03 | 5.81 | 0.02 | 21.34 | 0.16 | −25.3 | 0.2 | −43 | 21 |
| DEPO‐03B | 3.03 | 0.02 | 5.80 | 0.02 | 21.33 | 0.08 | −25.3 | 0.2 | −40 | 17 |
| DEPO‐06A | 4.15 | 0.01 | 7.95 | 0.01 | 34.02 | 0.18 | −29.9 | 0.2 | −47 | 10 |
| DEPO‐06B | 4.20 | 0.03 | 8.03 | 0.03 | 34.40 | 0.11 | −30.1 | 0.2 | −36 | 16 |
Conclusions
The method described enables the determination of the isotopic composition (δ17O, δ18O, and δ2H values) of gypsum hydration water (GHW) using the newest generation of CRDS analyzer. Our procedure presents several analytical advantages over earlier methods, including better long‐term precision, higher sample throughput, reduced sample size and less memory effect between consecutive samples.
Simultaneous 17O‐excess and d‐excess values can be obtained by this method in GHW and brines. This can provide additional information about the conditions under which the gypsum formed and subsequently interacted with other fluids after deposition. Although the present procedure has been initially tested for the analysis of gypsum, other hydrated minerals could also be extracted. This opens up a broad field of possible future applications using this technique.
Supporting information
Supporting info item
Acknowledgements
The authors are grateful to Mr Vicente Suarez and Mr Francisco Márquez (Grupo Salins Company) for providing access to the Cabo de Gata Salinas. Carmen Guirado carried out the sampling in the Salinas. Professors José‐María Calaforra and Paolo Forti supplied the speleothems samples. Comments and advice from Professor Eric Steig and two anonymous reviewers are warmly acknowledged. This research was supported by the ERC WIHM Project (#339694) to DAH.
Gázquez, F. , Mather, I. , Rolfe, J. , Evans, N. P. , Herwartz, D. , Staubwasser, M. and Hodell, D. A. (2015) Simultaneous analysis of 17O/16O, 18O/16O and 2H/1H of gypsum hydration water by cavity ring‐down laser spectroscopy. Rapid Commun. Mass Spectrom., 29: 1997–2006. doi: 10.1002/rcm.7312.
The copyright line for this article was changed on 23 November 2016 after original online publication.
References
- 1. Fontes J. C., Letolle R., Nesteroff W. D., Ryan W. B. F.. Oxygen, carbon, sulfur, and hydrogen stable isotopes in carbonate and sulfate mineral phases of neogene evaporites, sediments, and in interstitial waters. Initial Rep. Deep Sea Drill. Proj. 1973, 13, 788. [Google Scholar]
- 2. Matsuyaba O., Sakai O. H.. Oxygen and hydrogen isotopic study on the water of crystallization of gypsum from the Kuroko type mineralization. Geochem. J. 1973, 7, 153. [Google Scholar]
- 3. Halas S., Krouse H. R.. Isotopic abundances of water of crystallization of gypsum from the Miocene evaporites formation, Carpathian Foredeep, Poland. Geochim. Cosmochim. Acta 1982, 46, 293. [Google Scholar]
- 4. Khademi H., Mermut A. R., Krouse H. R.. Isotopic composition of gypsum hydration water in selected landforms from central Iran. Chem. Geol. 1997, 138, 245. [Google Scholar]
- 5. Buck B. J., Van Hoesen J. G.. Assessing the applicability of isotopic analysis of pedogenic gypsum as a paleoclimate indicator, Southern New Mexico. J. Arid Environ. 2005, 60, 99. [Google Scholar]
- 6. Hodell D., Turchyn A. V., Wiseman C. J., Escobar J., Curtis J. H., Brenner M., Gilli A., Mueller A. D., Anselmetti F., Aritzegui D., Brown E.. Late Glacial temperature and precipitation changes in the lowland Neotropics by tandem measurement of δ18O in biogenic carbonate and gypsum hydration water. Geochim. Cosmochim. Acta 2012, 77, 352. [Google Scholar]
- 7. Gázquez F., Calaforra J. M., Stoll H., Sanna L., Forti P., Lauritzen S. E., Delgado A., Martínez‐Frías J.. Isotope and trace element evolution of the Naica aquifer (Chihuahua,Mexico) over the past 60,000 yr revealed by speleothems. Quaternary Res. 2013, 80, 510. [Google Scholar]
- 8. Evans N., Turchyn A. V., Gázquez F., Bontognali T. R. R., Chapman H. J., Hodell D. A.. Geochemical evidence for meteoric water and precession control of basin hydrology during gypsum‐marl deposition of the Messinian Yesares Member, Sorbas Basin (SE Spain). Earth Planet. Sci. Lett. 2015. DOI: 10.1016/j.epsl.2015.07.071. [Google Scholar]
- 9. Dansgaard W.. Stable isotopes in precipitation. Tellus 1964, 16, 436. [Google Scholar]
- 10. Barkan E., Luz B.. Diffusivity fractionations of H2 16O/H2 17O and H2 16O/H2 18O in air and their implications for isotope hydrology. Rapid Commun. Mass Spectrom. 2007, 21, 2999. [DOI] [PubMed] [Google Scholar]
- 11. Luz B., Barkan E.. Variations of 17O/16O and 18O/16O in meteoric waters. Geochim. Cosmochim. Acta 2010, 74, 6276. [Google Scholar]
- 12. Gonfiantini R., Fontes J. C.. Oxygen isotopic fractionation in the water of crystallization of gypsum. Nature 1963, 200, 644. [Google Scholar]
- 13. Fontes H. C., Gonfiantini R.. Fractionnement isotopique de l'hydrogène dans l'eau de cristallistion du gypse. Cr. Acad. Sci. d. Nat. 1967, 265, 4. [Google Scholar]
- 14. Sofer Z.. Isotopic composition of hydration water in gypsum. Geochim. Cosmochim. Acta 1978, 42, 1141. [Google Scholar]
- 15. Palacio S., Azorin J., Montserrat‐Marti G., Ferrio J. P.. The crystallization water of gypsum rocks is a relevant water source for plants. Nat. Commun. 2014, 5, 4660. [DOI] [PubMed] [Google Scholar]
- 16. Steig E. J., Gkinis V., Schauer A. J., Schoenemann S. W., Samek K., Hoffnagle J., Dennis K. J., Tan S. M.. Calibrated high‐precision 17O‐excess measurements using laser‐current tuned cavity ring‐down spectroscopy. Atmos. Meas. Tech. 2014, 7, 2421. [Google Scholar]
- 17. Barkan E., Luz B.. High precision measurements of 17O/16O and 18O/16O ratios in H2O. Rapid Commun. Mass Spectrom. 2005, 19, 3737. [DOI] [PubMed] [Google Scholar]
- 18. Brand W. A., Geilmann H., Crosson E. R., Rella C. W.. Cavity ring‐down spectroscopy versus high‐temperature conversion isotope ratio mass spectrometry; a case study on δ2H and δ18O of pure water samples and alcohol/water mixtures. Rapid Commun. Mass Spectrom. 2009, 23, 1879. [DOI] [PubMed] [Google Scholar]
- 19. Saad N., Hsiao G., Chapellet‐Volpini L., Vu D.. Two‐pronged approach to overcome spectroscopically interfering organic compounds with isotopic water analysis. Geophys. Res. Abstracts 2013, 15, EGU2013–13296. [Google Scholar]
- 20. Gázquez F., Calaforra J. M.. The Gypsum Karst of Sorbas, Betic Chain, in Landscapes and Landforms of Spain, (Eds: Gutiérrez F., Gutiérrez M.). Springer, 2014, p. 127. [Google Scholar]
- 21. García‐Guinea J., Morales S., Delgado A., Recio C., Calaforra J. M.. Formation of gigantic gypsum crystals. J. Geol. Soc. 2002, 159, 347. [Google Scholar]
- 22. Playá E., Recio C., Mitchell J.. Extraction of gypsum hydration water for oxygen isotopic analysis by the guanidine hydrochloride reaction method. Chem. Geol. 2005, 217, 89. [Google Scholar]
- 23. McConnell J. D. C., Astill D. M., Hall P. L.. The pressure dependence of the dehydration of gypsum to bassanite. Mineral. Mag. 1987, 51, 453. [Google Scholar]
- 24. Conley R. F., Bundy W. M.. Mechanism of gypsification. Geochim. Cosmochim. Acta 1958, 15, 57. [Google Scholar]
- 25. Lin Y., Clayton R. N., Groening M.. Calibration of δ17O and δ18O of international measurement standards – VSMOW, VSMOW2, SLAP, and SLAP2. Rapid Commun. Mass Spectrom. 2010, 24, 773. [DOI] [PubMed] [Google Scholar]
- 26. Schoenemann S. W., Schauer A. J., Steig E. J.. Measurement of SLAP and GISP δ17O and proposed VSMOW‐SLAP normalization for 17O excess. Rapid Commun. Mass Spectrom. 2013, 27, 582. [DOI] [PubMed] [Google Scholar]
- 27. West A. G., Goldsmith G. R., Matimati I., Dawson T. E.. Spectral analysis software improves confidence in plant and soil water stable isotope analyses performed by isotope ratio infrared spectroscopy (IRIS). Rapid Commun. Mass Spectrom. 2011, 25, 2268. [DOI] [PubMed] [Google Scholar]
- 28. Barcelona M. J., Atwoods D. K.. Gypsum organic interactions in the marine environment: sorption of fatty acids and hydrocarbons. Geochim. Cosmochim. Acta 1979, 43, 47. [Google Scholar]
- 29. Horita J.. Analytical aspects of stable isotopes in brines. Chem. Geol. 1989, 79, 107. [Google Scholar]
- 30. Sofer Z., Gat J. R.. Activities and concentrations of oxygen‐18 in concentrated aqueous salt solutions: analytical and geophysical implications. Earth Planet. Sci. Lett. 1972, 15, 232. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item