

Abstract
Cigarette smoke plays a substantial role in the development of airway inflammatory diseases, including asthma and chronic rhinosinusitis (CRS). Interleukin (IL)-17A might contribute to cigarette smoke-related inflammation of the airway. This study aimed to investigate the association between cigarette smoking and IL-17A expression in the nasal tissues of patients with CRS and asthma.
We prospectively recruited 24 patients (13 smokers, 11 nonsmokers) with CRS and asthma and 6 patients with asthma but without CRS (control group) in a tertiary medical center. Nasal mucosa was obtained as part of the nasal surgery. Protein and mRNA levels of IL-17A in the nasal tissues were determined by immunostaining and real-time polymerase chain reaction.
The number of unexpected emergency clinic visits for acute asthma attacks were higher among smokers than among nonsmokers. Interleukin-17A protein and mRNA levels in the nasal tissues of smokers were greater compared to those in the nasal tissues of nonsmokers (P = 0.02 both) and control patients (P = 0.05 and 0.04, respectively).
Cigarette smoking was associated with an increase in the number of unexpected emergency clinic visits due to acute asthma attack and in the expression of IL-17A in the nasal tissues of patients with airway inflammatory diseases.
Keywords: asthma, chronic rhinosinusitis, cigarette smoking, interleukin-17A, nasal tissue
1. Introduction
The airway mucosa extends continuously from the nose to the lungs and is constantly exposed to the external environment. Inflammation of the upper and lower airway tracts may be induced by simultaneous exposure to environmental factors such as irritants, allergens, and microorganisms.[1,2] As a consequence, chronic rhinosinusitis (CRS) is often comorbid with asthma and, based on the united airways concept, the conditions are considered as 2 different manifestations of a single pathological process.[3–5]
Cigarette smoke is an inhaled pollutant, that plays a substantial role in the development of airway inflammatory diseases, including asthma and CRS.[6,7] Cigarette smoking is associated with poor resolution of respiratory disease symptoms, resistance to treatment, decline in lung function, and development of irreversible airflow obstruction in asthmatics.[8–12] In comparison with nonsmokers, smokers have been reported to exhibit poorer treatment outcomes, reduced recovery of olfactory functions, lesser improvement of quality of life, and higher risk of revision surgery after endoscopic sinus surgery (ESS) for CRS.[13–16]
A type-2 helper T (Th) cell-driven cytokine pattern has long been considered as potentially the major driver of airway inflammation in CRS and asthma.[17,18] However, interleukin (IL)-17A, a pro-inflammatory cytokine produced by Th17 cells, has also been reported to be associated with neutrophil recruitment, mucosal remodeling, and resistance to corticosteroid-based therapy in airway inflammation.[19–22]
Therefore, we hypothesized that IL-17A play a crucial role in cigarette smoke-related airway inflammation and its resistance to current therapeutic regimens. Thus, this study aimed to investigate the association between cigarette smoking and IL-17A expression in the nasal tissues of patients with CRS and asthma.
2. Methods
2.1. Study population
This prospective cohort airway disease study recruited patients with asthma who fulfilled the diagnostic criteria prescribed by the Global Initiative for Asthma (GINA) guidelines[23] and exhibited comorbidity with CRS—defined based on the criteria of the European position paper[24]—from the Thoracic and Otolaryngology departments between August 2013 and December 2014. The inclusion criteria for patients with asthma were (1) regular follow-up for at least 1 year; (2) failure of medical treatment including administration of intranasal corticosteroids spray, anti-histamines, and broad spectrum oral antibiotics for CRS, for a maximum of 3 months; and (3) treatment by ESS for CRS. Asthma had been managed according to the GINA guidelines for ≥6 months prior to surgery in order to achieve stability, allowing patients to undergo nasal surgery under general anesthesia. All participants received only intranasal corticosteroid spray and nasal saline douche for their CRS before nasal surgery.
Patients with nasal polyps as well as those with a history of previous nose surgery or major medical disorders such as diabetes, nephrotic diseases, autoimmune disorders, immunodeficiency, malignancies, and other chronic illnesses were excluded. An additional group of 6 patients with asthma but without CRS who received septomeatoplasty (SMP) for nasal obstruction were enrolled as the control group. The study was approved by the institutional review board of the Chang Gung Memorial Hospital, Taoyuan, Taiwan (IRB number: 103-7085B). All of the participants provided written informed consent.
Data regarding physical and clinical characteristics of the patients were collected. The pre-operative smoking habits of patients were also recorded in terms of presence/absence of a regular habit smoking of cigarettes or other forms of tobacco, number of cigarettes smoked per day, type of cigarettes (i.e., with filter, without filter, or both), and brands of cigarettes usually preferred. Patients were considered smokers if they reported regular or ongoing smoking in the 12 months prior to surgery.[25,26] Patients who reported never having smoked cigarettes on a regular basis were considered as nonsmokers.
All patients underwent thorough nasal endoscopic examination and sinus computed tomography (CT), the findings of which were interpreted using the Lund–Kennedy endoscopy scores[27] and the Lund–Mackay CT score,[28] respectively, by 2 senior rhinologists. Pulmonary function tests including evaluation of forced vital capacity (FVC), forced expiratory volume in 1 s (FEV1), and FEV1/FVC ratio were performed prior tonasal surgery. The sino-nasal outcome test-22 (SNOT-22)[29] and asthma control test (ACT)[30] questionnaires were administered for evaluation of the sino-nasal symptoms and level of control of asthma, respectively.
2.2. Collection and processing of specimens
Tissue samples of the nasal mucosa were obtained during ESS in patients with CRS and SMP in the control subjects. The nasal mucosal specimens were rinsed in phosphate buffered saline (PBS, pH 7.6), stored at –70 °C, and later processed for immunostaining and real-time polymerase chain reaction (PCR).
2.3. Immunostaining
Paraffin sections (4 mm) of the nasal tissues were prepared for immunohistochemistry. After thorough de-waxing in xylene and rinsing in absolute alcohol, the sections were incubated in 3% H2O2 for 30 minutes to quench the endogenous peroxidase. The sections were microwaved in a citric acid buffer with 0.1% Triton for 5 minutes to enhance antigen exposure and then incubated in 0.2% normal swine serum (DAKO, CA) for 30 minutes to prevent nonspecific binding with the secondary antibody. The sections were then incubated with IL-17A antibody solution (specific) or purified rabbit IgG solution (nonspecific negative control) for 1 hour (LifeSpan BioScience, WA). Antibody labeling was visualized using the avidin–biotin complex method (LSAB 2 kit; DAKO, CA and DAB peroxidase substrate kit; Vector Laboratories, CA).
2.4. RNA extraction and reverse transcription
Frozen nasal tissue was homogenized using a homogenizer (Retsch, Haan, Germany). The homogenate was centrifuged to separate the residue and supernatant. Total RNA was extracted from the supernatant using the RNeasy mini kit (Qiagen GmbH, Strasse, Germany) according to the manufacturer's instructions. The extracted RNA was quantified using the NanoDrop system (Thermo Scientific, Barrington, IL) and stained with ethidium bromide to determine the integrity of RNA. Reverse transcription was performed using random hexamer primers using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA).
2.5. Real-time polymerase chain reaction
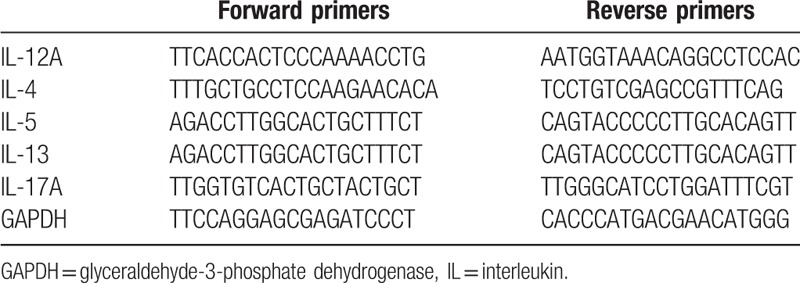
Real-time PCR was performed by the TaqMan assay using primers specific for the target IL and glyceraldehyde-3-phosphatedehydrogenase (GAPDH) genes (Table 1) using the Applied Biosystems 7500 Fast Real Time PCR System (Applied Biosystems). The amplification conditions included initial incubation at 95 °C for 10 minutes, followed by 45 cycles of 95°C for 10 seconds, 60°C for 20 seconds, and 72°C for 10 seconds, and final cooling to 40°C. Each sample was amplified in triplicates in separate tubes to permit quantification of gene expression. The mean threshold cycle (Ct) values of the test samples were normalized to those of GAPDH, and the relative mRNA levels of the target genes were analyzed with the ΔΔCt method.
Table 1.
Primer sequences specific to target genes.
2.6. Statistical analysis
The data are presented as the mean values ± standard errors and were statistically analyzed using the GraphPad Prism 5 software (GraphPad Prism Software, Inc., San Diego, CA). Comparison of categorical variables was performed using the chi-square or Fisher's exact tests, as appropriate. Comparison of continuous variables among 2 or 3 groups was performed using the Mann–Whitney U test or Kruskal–Wallis test. Correlation was determined using Spearman's correlation coefficient. Statistical significance was set at P < 0.05.
3. Results
3.1. Physical and clinical characteristics of the study population
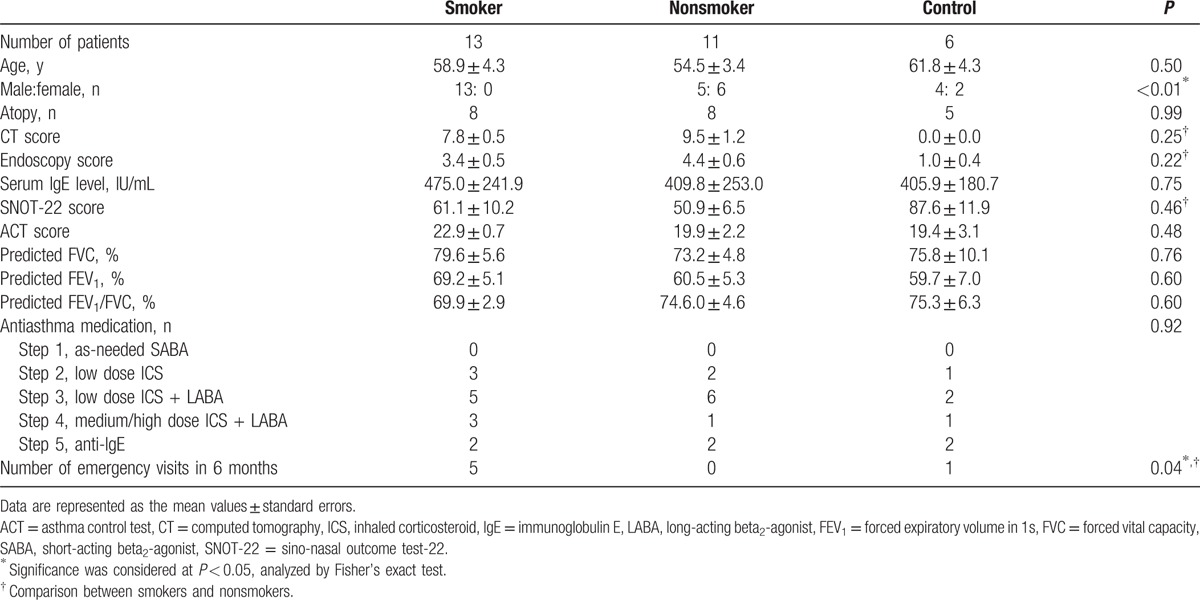
Of the 30 asthmatic patients with CRS initially enrolled in the present study, 6 with nasal polyps (CRSwNP) were excluded. Of the remaining 24 patients with CRS but without nasal polyps (CRSsNP), 13 were smokers, and 11 were nonsmokers. An additional group of 6 patients with asthma but without CRS were enrolled as control subjects. The clinical characteristics of the patients included in this study are summarized in Table 2. There were no statistically significant differences in any of the physical characteristics or medical parameters between the smoker and nonsmoker patient groups except in terms of distribution of the sexes. In addition, patients with asthma as well as smoking habits paid a greater number of emergency clinic visits for acute exacerbation of asthma than those without a smoking habit.
Table 2.
Physical and clinical characteristics of the study population.
3.2. Immunostaining for detection of IL-17A
The intensity of IL-17A immunostaining was scored based on the percentage of positively stained cells as follows: weak < 35% (scored 1; Fig. 1A), moderate, 35% to 70% (scored 3; Fig. 1B), or strong, >70% (scored 5; Fig. 1C). The immunoreactivity of IL-17A in the nasal tissues of patients with asthma as well as smoking habits was higher compared to those in the nasal tissues of nonsmokers and control subjects (Fig. 1D).
Figure 1.
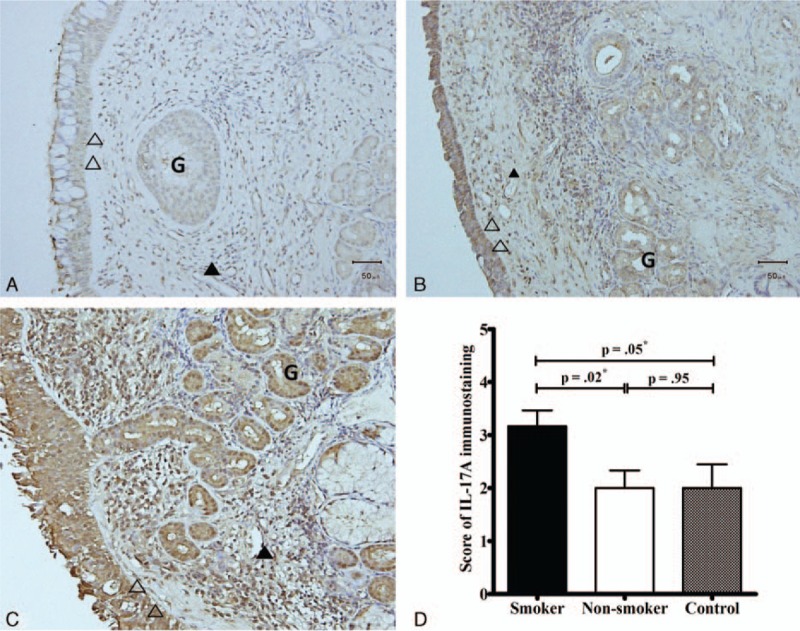
Immunohistochemistry findings of evaluation of interleukin (IL)-17Aprotein expression in nasal tissues. The intensity of immunostaining was scored as weak: <35% (A, scored 1); moderate, 35–70% (B, scored 3); or strong, >70% (C, scored 5) based on the proportion of positively stained cells. The IL-17A expression levels in nasal tissues of smokers were higher compared to those in the nasal tissues of nonsmokers and control subjects (D). ∗Significance was considered at P < 0.05, analyzed by the Mann–Whitney U test. Cells were observed at a magnification of 200×. Open head, epithelium; closed head, endothelium; arrowhead, inflammatory cells; G, mucus gland. IL = interleukin.
3.3. Expression of IL-4, IL-5, IL-13, and IL-17A mRNAs in nasal tissue
There were no significant differences in the mRNA levels of Th1- and various Th2-driven cytokines including IL-12A, IL-4, IL-5, and IL-13 among the smoker, nonsmoker, and control groups (Fig. 2A–D). However, the levels of IL-17AmRNA in the nasal tissues of smokers were significantly higher compared to those in the nasal tissues of nonsmokers and control subjects (P = 0.02 and 0.04, respectively, Fig. 2E).
Figure 2.
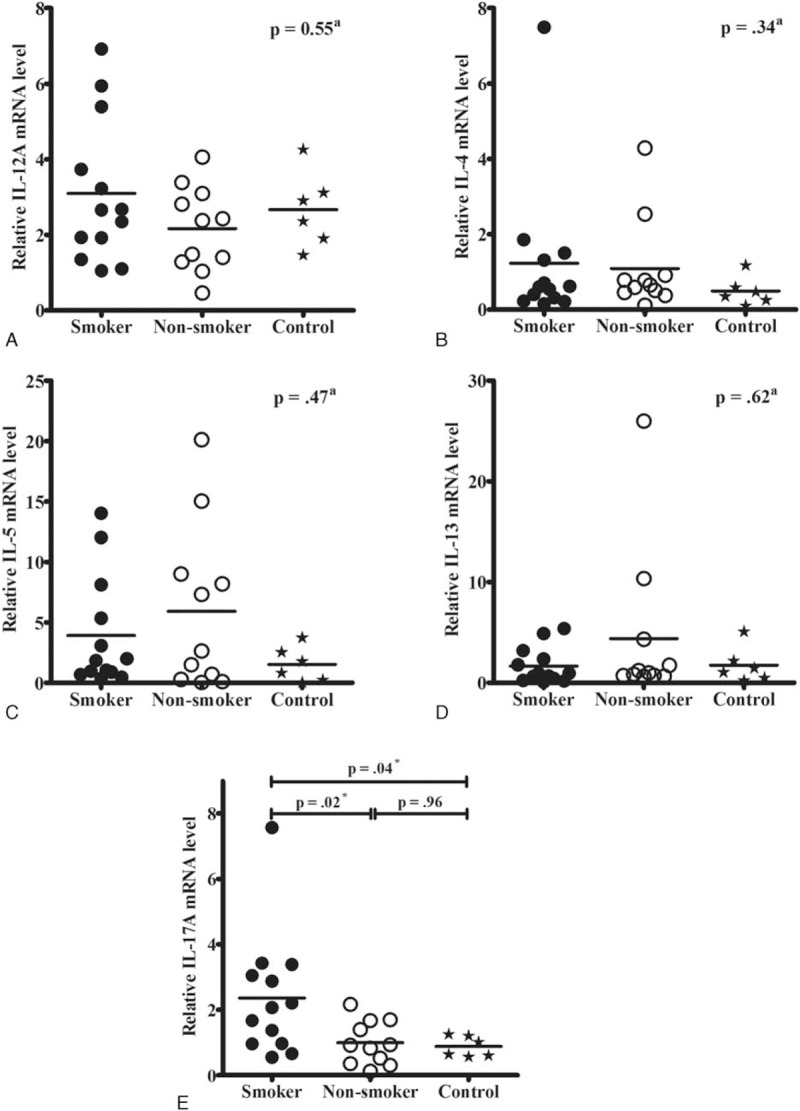
Comparison of interleukin (IL)-12A (A), IL-4 (B), IL-5 (C), IL-13 (D), and IL-17A (E) mRNA levels in nasal tissues among smokers (n = 13), nonsmokers (n = 11), and control subjects (n = 6) based on the real-time polymerase chain reaction findings. The IL-17A mRNA levels in smokers were significantly higher compared to those in nonsmokers and control subjects (E). aAnalyzed by the Kruskal–Wallis test among the 3 groups. ∗Significance was considered at P < 0.05, analyzed by the Mann–Whitney U test. IL = interleukin.
The mRNA expression levels of IL-17A were not correlated with the sinus subjective SNOT-22 scores (Fig. 3A), objective CT scores for evaluation of CRS (Fig. 3B),or subjective ACT scores for asthma control (Fig. 3C); however, they were well correlated with the predicted FEV1/FVC ratio in the pulmonary function test (Fig. 3D).
Figure 3.
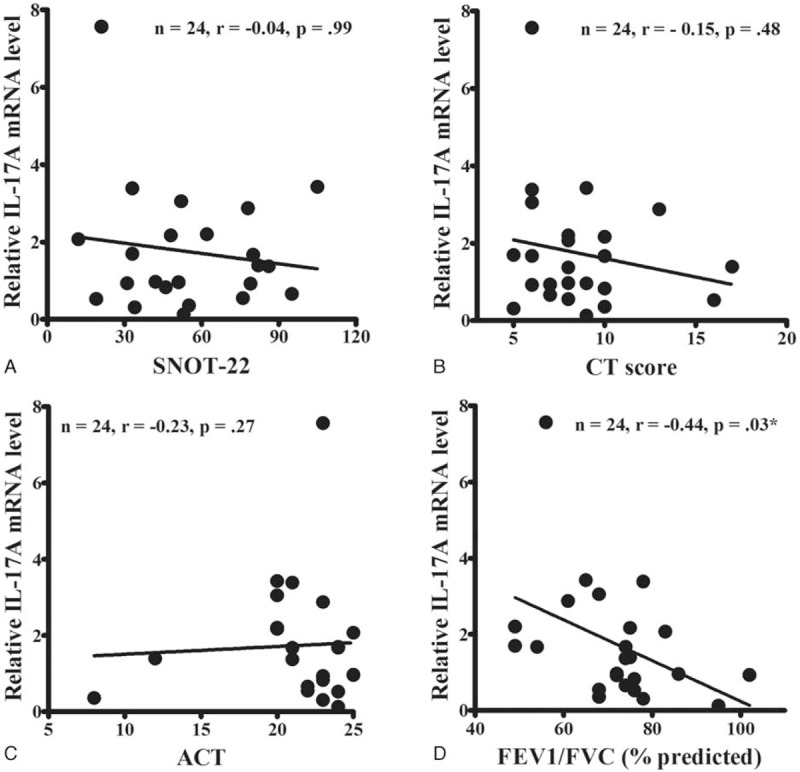
Correlation between interleukin-17A mRNA expressions levels and various airway parameters. The mRNA expression levels of IL-17A were not correlated with the sino-nasal outcome test (SNOT)-22 (A), computed tomography (CT) (B), or asthma control test (ACT) (C) scores. However, they were well correlated with the predicted ratio of forced expiratory volume in 1s (FEV1) to forced vital capacity (FVC) (D). The data were analyzed by Spearman's correlation coefficient analysis. ∗Significance was considered at P < 0.05. ACT = asthma control test, CT = computed tomography, FEV1 = forced expiratory volume in 1s, FVC = forced vital capacity, SNOT-22 = sino-nasal outcome test-22.
4. Discussion
The present study is the first to focus on the simultaneous impact of cigarette smoking on the upper and lower airway inflammation in terms of IL-17A expression. The results demonstrated increased expression of IL-17A in the nasal tissues of patients with asthma as well as smoking habits. However, the mRNA levels of Th2-related cytokines including IL-4, IL-5, and IL-13 in nasal tissues were found to be unrelated to the habit of smoking. On the other hand, patients with asthma as well as smoking habits were found to have paid a greater number of emergency visits for acute exacerbation of asthma than those with no smoking habits. These results indicate that cigarette smoking is associated with an increase in IL-17Aexpression in the airway mucosa. The increased expression of this pro-inflammatory cytokine has serious negative consequences on the Th2-cytokine-driven airway inflammation in CRS and asthma. Interleukin-17A-associated inflammation could escape treatment by corticosteroid-based medical therapy and might be responsible for a greater number of unexpected emergency hospital visits and increased healthcare needs because of acute exacerbation of asthma.
Both CRS and asthma are multifactorial diseases with heterogeneous phenotypic subgroups and diverse underlying etiologies, pathophysiologies, and therapeutic responses.[31,32] Therefore, the concept of endotypes of CRS and asthma has been proposed according to their distinct clinical features and divergent underlying molecular causes and treatment responses.[33–37] The development of endotype-specific therapies has been advocated in order to enhance the likelihood of therapeutic success especially in patients refractory to conventional therapy.[38] Recent evidence has implicated IL-17A as a key driver of disease exacerbation in patients with severe asthma. It has been reported to be associated with increased airway inflammation following viral infection during ongoing allergic airway inflammation, with viral infection being the major trigger of exacerbation of asthma.[39] Besides, IL-17A is also associated with resistance to corticosteroids in the more severe phenotypes of asthma.[22] Furthermore, a previous study on the lungs of a mouse model reported that induction of IL17A via exposure to diesel exhaust particles, a major source of traffic-related air pollution, contributes to severe asthma.[40] Another study demonstrated that exposure to polycyclic aromatic hydrocarbons (PAHs)led to the enhancement of polarization of Th17cells,[41] which is relevant to the present study because cigarette smoke is known to contain substantial concentration of PAHs. Taken together, IL-17A-mediated airway inflammation induced by cigarette smoke contributes to the acute exacerbation of asthma.
In the present study, the mRNA expression levels of IL-17A were well correlated with the predicted FEV1/FVC ratio. This finding corresponds with previously published evidence indicating the association of IL-17A with increased airway smooth muscle proliferation, migration, and airway remodeling.[42,43] Interleukin-17A has been reported to play an important role in both mucosal remodeling and nasal polyp formation in CRS.[20,44] However, the mRNA expression levels of IL-17A were not correlated with the findings of the objective and subjective evaluation of CRS in the present study. The correlation between the severity of CRS determined based on radiological findings and self-reported measures such as SNOT-22 scores has been reported to be poor, and the matter of which one of these evaluation methods best represents the severity of CRS is controversial.[45,46] As a result, future studies focusing on the adverse effects of cigarette smoke and IL-17A upregulation on the upper airway tract are required.
During inhalation of cigarette smoke, both the upper and lower airway tracts are exposed to and influenced by the associated irritants. Therefore, we investigated the impact of cigarette smoking on airway inflammation in patients with both upper and lower airway inflammatory diseases. Based on the united airways concept, CRS and asthma maybe considered as 2 different manifestations of a single pathological process.[3–5] Additionally, previous studies have shown that the inflammatory responses of nasal epithelial cells could reflect those of bronchial cells, and, therefore, nasal epithelial cells could be used as surrogates for lower airway cells.[47,48]
This study has several limitations that warrant consideration. First, we enrolled patients with asthma with or without CRS. We did not evaluate the IL-17A expression levels in patients with asthma who did not undergo nasal surgery or in subjects without asthma. Inclusion of a control group of healthy subjects with no airway diseases in the present study might have provided more conclusive results. However, the recruitment of healthy control subjects is challenging because of concerns regarding the invasive nature of biopsy of nasal mucosa. Therefore, we enrolled patients with asthma but without CRS or smoking habits as the control population instead. Second, our sample size was small. Six patients with CRSwNP were excluded because of the small number of cases and the heterogeneous pathogenesis and subtypes of nasal polyp, such as eosinophilic, neutrophilic, Th2 predominant, and Th17 predominant subclasses. Subgroup analysis of different phenotypes of CRS requires future large-scale prospective studies. This study focused on the effect of cigarette smoking and airway inflammation. Thus, we only enrolled patients with CRSsNP and asthma for analysis. Third, further longitudinal studies are required for the validation of our findings regarding the association between cigarette smoking and the increased expression of IL-17A in the airway mucosa of patients with CRS and asthma.
5. Conclusions
In the present study, cigarette smoking was found to be associated with the increased expression of IL-17A in the nasal tissues of patients with airway inflammatory diseases as well as a greater number of unexpected emergency visits due to acute exacerbation of asthma. These findings contribute towards increasing our understanding of the possible mechanisms of development of inflammatory airway diseases by exposure to environmental factors such as cigarette smoke. Future novel therapeutics that target the IL-17A-associated inflammatory response might benefit patients with asthma or CRS who are refractory to the prevalent standard treatments.
Acknowledgments
The authors thank Ms. Meng-Chieh Tsai for her assistance with data collection in this study. The authors wound also like to thank all the patients who have participated in this study.
Footnotes
Abbreviations: ACT = Asthma Control Test, CRS = chronic rhinosinusitis, CRSsNP = CRS without nasal polyps, CRSwNP = CRS with nasal polyps, CT = computed tomography, ESS = endoscopic sinus surgery, Th cell helper T cell, FEV1 = forced expiratory volume in 1s, FVC = forced vital capacity, GAPDH = glyceraldehyde-3-phosphate dehydrogenase, GINA = Global Initiative for Asthma, ICS = inhaled corticosteroid, IgE = immunoglobulin E, IL = interleukin, LABA = long-acting beta2-agonist, PAH = polycyclic aromatic hydrocarbon, PCR = polymerase chain reaction, SABA = short-acting beta2-agonist, SMP = septomeatoplasty, SNOT-22 = Sino-Nasal Outcome Test-22.
Funding: A research grant from Chang Gung Memorial Hospital (CMRPG3D0831, CMRPG3E0121, and CMRPG391991).
The authors have no conflicts of interest to disclose.
References
- [1].Bresciani M, Paradis L, Des Roches A, et al. Rhinosinusitis in severe asthma. J Allergy Clin Immunol 2001;107:73–80. [DOI] [PubMed] [Google Scholar]
- [2].Rosati MG, Peters AT. Relationships among allergic rhinitis, asthma, and chronic rhinosinusitis. Am J Rhinol Allergy 2016;30:44–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dixon AE, Kaminsky DA, Holbrook JT, et al. Allergic rhinitis and sinusitis in asthma: differential effects on symptoms and pulmonary function. Chest 2006;130:429–35. [DOI] [PubMed] [Google Scholar]
- [4].Feng CH, Miller MD, Simon RA. The united allergic airway: connections between allergic rhinitis, asthma, and chronic sinusitis. Am J Rhinol Allergy 2012;26:187–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ek A, Middelveld RJ, Bertilsson H, et al. Chronic rhinosinusitis in asthma is a negative predictor of quality of life: results from the Swedish GA(2) LEN survey. Allergy 2013;68:1314–21. [DOI] [PubMed] [Google Scholar]
- [6].Reh DD, Higgins TS, Smith TL. Impact of tobaccosmoke on chronicrhinosinusitis: a review of the literature. Int Forum Allergy Rhinol 2012;2:362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jayes L, Haslam PL, Gratziou CG, et al. SmokeHaz: systematic reviews and meta-analyses of the effects of smoking on respiratory health. Chest 2016;150:164–79. S0012-3692(16)48547-8. doi: 10.1016/j.chest.2016.03.060. [DOI] [PubMed] [Google Scholar]
- [8].Thomson NC, Chaudhuri R, Livingston E. Asthma and cigarette smoking. Eur Respir J 2004;24:822–33. [DOI] [PubMed] [Google Scholar]
- [9].Thomson NC, Spears M. The influence of smoking on the treatment response in patients with asthma. Curr Opin Allergy Clin Immunol 2005;5:57–63. [DOI] [PubMed] [Google Scholar]
- [10].Chalmers GW, Macleod KJ, Little SA, et al. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 2002;57:226–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chaudhuri R, Livingston E, McMahon AD, et al. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am J RespirCrit Care Med 2003;168:1308–11. [DOI] [PubMed] [Google Scholar]
- [12].Hancox RJ, Gray AR, Poulton R, et al. The effect of cigarette smoking on lung function in young adults with asthma. Am J Respir Crit Care Med 2016;194:276–84. [DOI] [PubMed] [Google Scholar]
- [13].Sugiyama K, Matsuda T, Kondo H, et al. Postoperative olfaction in chronic sinusitis: smokers versus nonsmokers. Ann Otol Rhinol Laryngol 2002;111:1054–8. [DOI] [PubMed] [Google Scholar]
- [14].Briggs RD, Wright ST, Cordes S, et al. Smoking in chronic rhinosinusitis: a predictor of poor long-term outcome after endoscopic sinus surgery. Laryngoscope 2004;114:126–8. [DOI] [PubMed] [Google Scholar]
- [15].Das S, Khichi SS, Perakis H, et al. Effects of smoking on quality of life following sinus surgery: 4-year follow-up. Laryngoscope 2009;119:2284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krzeski A, Galewicz A, Chmielewski R, et al. Influence of cigarette smoking on endoscopic sinus surgery long-term outcomes. Rhinology 2011;49:577–82. [DOI] [PubMed] [Google Scholar]
- [17].Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol 2015;16:45–56. [DOI] [PubMed] [Google Scholar]
- [18].Akdis CA, Bachert C, Cingi C, et al. Endotypes and phenotypes of chronic rhinosinusitis. J Allergy Clin Immunol 2013;131:1479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Newcomb DC, Peebles RS. Th17-mediated inflammation in asthma. Curr Opin Immunol 2013;25:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu Y, Zeng M, Liu Z. Th17 response and its regulation in inflammatory upper airway diseases. Clin Exp Allergy 2015;45:602–12. [DOI] [PubMed] [Google Scholar]
- [21].Cao PP, Li HB, Wang BF, et al. Distinct immunopathologic characteristics of various types of chronic rhinosinusitis in adult Chinese. J Allergy Clin Immunol 2009;124:478–84. [DOI] [PubMed] [Google Scholar]
- [22].Chambers ES, Nanzer AM, Pfeffer PE, et al. Distinct endotypes of steroid-resistant asthma characterized by IL-17Ahigh and IFN-γhigh immunophenotypes: potential benefits of calcitriol. J Allergy Clin Immunol 2015;136:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].From the Global Strategy for Asthma Management and Prevention, Global Initiative for Asthma (GINA) 2014. Available from: http://www.ginasthma.org/. [Google Scholar]
- [24].Fokkens WJ, Lund VJ, Mullol J, et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology 2012;50:1–2. [DOI] [PubMed] [Google Scholar]
- [25].Das S, Khichi SS, Perakis H, et al. Effects of smoking on quality of life following sinus surgery: 4-year follow-up. Laryngoscope 2009;119:2284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Teo KK, Ounpuu S, Hawken S, et al. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: a case-control study. Lancet 2006;368:647–58. [DOI] [PubMed] [Google Scholar]
- [27].Lund VJ, Kennedy DW. Staging for rhinosinusitis. Otolaryngol Head Neck Surg 1997;117:S35–40. [DOI] [PubMed] [Google Scholar]
- [28].Lund VJ, Mackay IS. Staging in rhinosinusitus. Rhinology 1993;31:183–4. [PubMed] [Google Scholar]
- [29].Hopkins C, Gillett S, Slack R, et al. Psychometric validity of the 22-item Sinonasal Outcome Test. Clin Otolaryngol 2009;34:447–54. [DOI] [PubMed] [Google Scholar]
- [30].Nathan RA, Sorkness CA, Kosinski M, et al. Development of the asthma control test: a survey for assessing asthma control. J Allergy Clin Immunol 2004;113:59–65. [DOI] [PubMed] [Google Scholar]
- [31].Van Crombruggen K, Zhang N, Gevaert P, et al. Pathogenesis of chronic rhinosinusitis: inflammation. J Allergy Clin Immunol 2011;128:728–32. [DOI] [PubMed] [Google Scholar]
- [32].Tan BK, Schleimer RP, Kern RC. Perspectives on the etiology of chronic rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg 2010;18:21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008;372:1107–19. [DOI] [PubMed] [Google Scholar]
- [34].Lotvall J, Akdis CA, Bacharier LB, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 2011;127:355–60. [DOI] [PubMed] [Google Scholar]
- [35].Agache I, Akdis C, Jutel M, et al. Untangling asthma phenotypes and endotypes. Allergy 2012;67:835–46. [DOI] [PubMed] [Google Scholar]
- [36].Tomassen P, Vandeplas G, Van Zele T, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol 2016;137:1449–56. [DOI] [PubMed] [Google Scholar]
- [37].Akdis CA, Bachert C, Cingi C, et al. Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol 2013;131:1479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Choy DF, Hart KM, Borthwick LA, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 2015;7:301ra129. [DOI] [PubMed] [Google Scholar]
- [39].Newcomb DC, Boswell MG, Reiss S, et al. IL-17A inhibits airway reactivity induced by RSV infection during allergic airway inflammation. Thorax 2013;68:717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brandt EB, Kovacic MB, Lee GB, et al. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J Allergy Clin Immunol 2013;132:1194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].van Voorhis M, Knopp S, Julliard W, et al. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS One 2013;11:e82545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chang Y, Al Alwan L, Risse PA, et al. Th17 cytokines induce human airway smooth muscle cell migration. J Allergy Clin Immunol 2011;127:1046–53. [DOI] [PubMed] [Google Scholar]
- [43].Chang Y, Al Alwan L, Risse PA, et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J 2012;26:5152–60. [DOI] [PubMed] [Google Scholar]
- [44].Wen W, Liu W, Zhang L, et al. Increased neutrophilia in nasal polyps reduces the response to oral corticosteroid therapy. J Allergy Clin Immunol 2012;129:1522–8. (e5). [DOI] [PubMed] [Google Scholar]
- [45].Zheng Y, Zhao Y, Lv D, et al. Correlation between computed tomography staging and quality of life instruments in patients with chronic rhinosinusitis. Am J Rhinol Allergy 2010;24:e41–5. [DOI] [PubMed] [Google Scholar]
- [46].Huang CC, Wang CH, Fu CH, et al. The link between chronic rhinosinusitis and asthma: a questionnaire-based study. Medicine (Baltimore) 2016;95:e4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McDougall CM, Blaylock MG, Douglas JG, et al. Nasal epithelial cells as surrogates for bronchial epithelial cells in airway inflammation studies. Am J Respir Cell Mol Biol 2008;39:560–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Comer DM, Elborn JS, Ennis M. Comparison of nasal and bronchial epithelial cells obtained from patients with COPD. PLoS One 2012;7:e32924. [DOI] [PMC free article] [PubMed] [Google Scholar]