

The mRNA-interactome capture method, which utilizes UV light to cross-link proteins and mRNA in planta, was used to identify the portion of the proteome that binds RNA in etiolated Arabidopsis seedlings.
Abstract
RNA binding proteins (RBPs) control the fate and expression of a transcriptome. Despite this fundamental importance, our understanding of plant RBPs is rudimentary, being mainly derived via bioinformatic extrapolation from other kingdoms. Here, we adapted the mRNA-protein interactome capture method to investigate the RNA binding proteome in planta. From Arabidopsis thaliana etiolated seedlings, we captured more than 700 proteins, including 300 with high confidence that we have defined as the At-RBP set. Approximately 75% of these At-RBPs are bioinformatically linked with RNA biology, containing a diversity of canonical RNA binding domains (RBDs). As no prior experimental RNA binding evidence exists for the majority of these proteins, their capture now authenticates them as RBPs. Moreover, we identified protein families harboring emerging and potentially novel RBDs, including WHIRLY, LIM, ALBA, DUF1296, and YTH domain-containing proteins, the latter being homologous to animal RNA methylation readers. Other At-RBP set proteins include major signaling proteins, cytoskeleton-associated proteins, membrane transporters, and enzymes, suggesting the scope and function of RNA-protein interactions within a plant cell is much broader than previously appreciated. Therefore, our foundation data set has provided an unbiased insight into the RNA binding proteome of plants, on which future investigations into plant RBPs can be based.
INTRODUCTION
The diverse and dynamic interactions with RNA binding proteins (RBPs) govern the life of cellular RNA, including its processing, modification, cellular localization, translation, and decay (Singh et al., 2015). Even though such posttranscriptional gene regulation events are ubiquitous across all kingdoms of life, relatively little is known about RNA-protein interactions in plants and how these events impact the fate and expression of the transcriptome. Instead, much of the research on posttranscriptional gene regulation in plants has focused on the role of small RNAs (sRNAs), which has been driven by the development of next-generation sequencing methodologies, enabling the relative ease of identification of sRNAs and their targets (Ma et al., 2015). By comparison, the cohort of RBPs of a plant cell remains to be explored.
Knowledge of RBPs in plants comes mainly from targeted studies on individual proteins or from bioinformatic predictions based on sequence homology with canonical RNA binding domains (RBDs) identified in other kingdoms (Silverman et al., 2013). For instance, there are hundreds of Arabidopsis thaliana genes that encode proteins exhibiting one or more canonical RBD, such as RNA recognition motif domains (RRM; 197 proteins), K homology domains (KH; 28 proteins), cold shock domains (CSD; five proteins), DEAD-box helicase domains (nine proteins), Pumilio RNA binding repeats (PUF; 26 proteins), Like-Sm domains (LSM; 36 proteins), Zinc finger CCCH-type (C-x8-C-x5-C-x3-H; 5 proteins), and pentatricopeptide repeat proteins (PPR; ∼450 proteins) (Silverman et al., 2013). However, to date, only a few of these proteins have been functionally characterized. Examples include the RRM-containing GLYCINE-RICH RNA BINDING PROTEINS (GR-RBPs), which have been implicated in mediating responses to various stresses such as cold, salinity, and drought (Kim et al., 2007a; Kim et al., 2007b; Kwak et al., 2005; Lorković, 2009) and in regulating circadian rhythm (Nolte and Staiger, 2015). Similarly, CSD proteins and RNA helicases have been shown to be involved in abiotic stress responses (Jung et al., 2013). Proteins with known RBDs also play important roles in plant developmental processes such as flowering time (Macknight et al., 1997; Schomburg et al., 2001), floral morphogenesis (Lorković, 2009; Jung et al., 2013), embryogenesis (Tripurani et al., 2011), as well as ovule development and cell size homeostasis (Bush et al., 2015). Nevertheless, for the vast majority of bioinformatically predicted plant RBPs, there is no experimental evidence for their RNA binding activity or their molecular function. Additionally, which noncanonical RBDs exist in plants remains to be determined, awaiting a global, unbiased experimental approach that can determine the cohort of plant RBPs (Silverman et al., 2013).
Recently, a method termed “mRNA interactome capture” was developed that can identify the portion of cellular proteomes that is bound to polyadenylated RNA (Castello et al., 2012; Baltz et al., 2012). The method uses irradiation of live cells with short-wave UV light (254 nm), which, unlike formaldehyde, is known to selectively cross-link proteins in direct contact to RNA, but does not induce protein-protein cross-links (Greenberg, 1979; Dreyfuss et al., 1984; Wagenmakers et al., 1980; Pashev et al., 1991; Suchanek et al., 2005). Following cell lysis, cross-linked mRNA-protein complexes are isolated using oligo(dT) beads under stringent conditions, prior to RNase treatment and protein identification by mass spectrometry (MS). mRNA interactome capture has been performed on a range of mammalian cell lines (Castello et al., 2012; Baltz et al., 2012; Beckmann et al., 2015; Kwon et al., 2013; Liao et al., 2016), Caenorhabditis elegans (Matia-González et al., 2015), Drosophila melanogaster (Wessels et al., 2016; Sysoev et al., 2016), and Saccharomyces cerevisiae (Beckmann et al., 2015; Mitchell et al., 2013; Matia-González et al., 2015). These studies have revealed unexpectedly high numbers of diverse RBPs in eukaryotic cells, indicating that many unforeseen RNA-based regulatory mechanisms have yet to be elucidated.
Here, we detail the successful adaption of mRNA interactome capture to a living, intact plant. Using Arabidopsis etiolated seedlings as source material, we identified 300 Arabidopsis proteins as RNA binding and present another set of over 400 proteins as candidate RBPs, underscoring the prevalence of RNA binding and RBD diversity within the plant proteome. Corroborating our approach, many known RBPs were isolated, along with a multitude of bioinformatically predicted RBPs, providing the first direct experimental evidence of their in vivo RNA binding activity. Moreover, we identified potential novel plant RBDs and a diverse set of proteins not previously associated with RNA biology, including proteins involved in signaling pathways, cytoskeletal organization, and membrane transport. Our study thus reports a method of broad utility in plant research, as well as providing an experimental census of the Arabidopsis mRNA binding proteome as a unique resource for future research into RBP function in plants.
RESULTS AND DISCUSSION
Development of an mRNA Interactome Capture Protocol for Arabidopsis Seedlings
Four-day-old etiolated seedlings were chosen as source material for mRNA interactome capture (Figure 1A). To adapt the original protocol (Castello et al., 2012) for use with plant material, we increased the dosage of 254-nm UV light to three cycles of irradiation at 150 mJ/cm2 (Au et al., 2014), to establish cross-links (CLs) between proteins and RNA. This did not appreciably increase RNA degradation compared with lower UV dosages or a non-cross-linked (noCL) control sample (Supplemental Figure 1A). Snap-frozen seedlings were ground in liquid nitrogen and thawed into lysis buffer that was adapted for use in plants by supplementing with β-mercaptoethanol and polyvinylpyrrolidone 40, which increased the efficiency of RNA isolation (Supplemental Figure 1B). Lysates were further passed through a shredding column to clear the lysate and two rounds of capture on oligo(dT) beads were performed to maximize RNA recovery (Supplemental Figure 1C). Finally, proteins were released from the beads and treated with RNase. Analysis of aliquots taken before and after the oligo(dT) capture (referred to as input and eluate, respectively) by SDS-PAGE and silver staining showed purification of a distinct set of proteins in the CL eluate but not the noCL control sample (Figure 1B). Immunoblot analysis confirmed that this set of proteins contained known RBPs such as ARGONAUTE1 (AGO1) and the chloroplast RBP CP29A, whereas the non-RBP, inositol phosphatase FIERY1/SAL1 protein (Robles et al., 2010), was absent (Figure 1C). Together, this demonstrated that the modified mRNA interactome capture protocol could efficiently and selectively purify plant RBPs.
Figure 1.

mRNA Interactome Capture in Arabidopsis.
(A) Overview of the mRNA interactome capture procedure in Arabidopsis and categorization of interactome proteins.
(B) and (C) RNA-protein complexes from CL and noCL samples isolated by oligo(dT) capture were treated with RNases, separated by SDS-PAGE, and analyzed by silver staining (B) and immunoblot (C) alongside input samples. Results are representative of three independent interactome capture experiments.
(D) and (E) Annotations of At-RBPs and candidate At-RBPs compared with the input proteome according to functional characteristics (RNA biology) (D) and RBDs (E).
(F) and (G) The most significantly over- and underrepresented GO terms for molecular function in At-RBPs (F) and candidate At-RBPs (G) compared with the input proteome.
Identification of an Arabidopsis mRNA Binding Proteome
Three independent biological replicates for both CL and noCL eluate samples were prepared for analysis by quantitative MS using the SP3 (single-pot solid-phase-enhanced sample preparation) method (Hughes et al., 2014). Scatterplots comparing CL/noCL fold changes between biological replicates showed reproducibility (Supplemental Figure 1D). Together, this identified 746 proteins in the eluates, and for 333 of these, a CL/noCL ratio was determined in at least two out of three replicates. Nine proteins were enriched in the noCL sample and were not considered for further analysis, while 324 proteins were enriched in the CL sample. Of the latter, 300 proteins were enriched at a false discovery rate (FDR) <1% and were defined as the Arabidopsis RBP set (At-RBPs; Figure 1A; listed in Supplemental Data Set 1), a designation to indicate our highest confidence set of potential RBPs. The 24 proteins enriched at a higher FDR and those 413 proteins without a CL/noCL ratio (i.e., “nonquantified”) were defined as “candidate At-RBPs” (Figure 1A; listed in Supplemental Data Set 1). Although candidate At-RBPs did not pass these stringent statistical criteria, their features are nevertheless examined below as they likely contain additional RBPs that warrant further investigation. The quality of all interactome MS data was as expected; only 0.26% of peptides contained two missed trypsin cleavage sites and the minimum peptide length found was seven amino acids (Supplemental Data Set 2). MS analysis was also performed on two independent input samples, which identified 8264 proteins (termed input proteome).
Interactome Capture Enriches for Proteins Related to RNA Biology
Compared with the input proteome, both At-RBPs and candidate At-RPBs were enriched for RNA-related Gene Ontology (GO) terms and canonical RBDs (Figures 1D and 1E). Based on GO annotations, 75% of At-RBPs had prior experimentally determined or predicted links to RNA biology (Figure 1D). The remaining 25% had no known or predicted function in RNA biology and therefore represent novel RBPs in Arabidopsis. Similarly, ∼80% of the At-RBPs contained known RBDs (based on Pfam and Interpro annotations and previous mRNA interactome data sets; Castello et al., 2012; Beckmann et al., 2015; Liao et al., 2016), while the other 20% did not (Figure 1E). The candidate At-RBP grouping also showed these enrichments, albeit at a lower level; 46% of them were classified as “linked to RNA biology” and 48% of proteins had recognized RBDs (Figures 1D and 1E). Multiple molecular function GO terms were enriched among At-RBPs and candidate At-RBPs, the majority of which were associated with RNA, nucleic acid binding, or translation (Figures 1F and 1G). Similarly, Biological Process (Supplemental Figures 2A and 2B) and Cellular Component (Supplemental Figures 2C and 2D) GO terms referred to a range of processes and components typically associated with RNA, such as translation and the ribosome, as well as splicing and several types of RNA granules.
Approximately 25% of At-RBPs lack known functions in RNA biology, a proportion similar to other mRNA interactomes (Beckmann et al., 2015). However, the number of 300 proteins that qualify as At-RBPs is less than what had been observed in the two other mRNA interactomes on multicellular organisms, i.e., C. elegans (594 proteins) and Drosophila (476 proteins) (Matia-González et al., 2015; Wessels et al., 2016). This may suggest the presence of fewer RBP networks in plants compared with animals, but a lower UV cross-linking efficiency for plants due to their abundant UV-absorbing pigments is a more likely explanation, although we tried to reduce this using etiolated seedlings.
Next, we investigated the conservation of RBPs across the major eukaryotic kingdoms. Two hundred proteins of the At-RBP set were predicted to have orthologs in human, mouse, and/or yeast (Supplemental Figure 3A) as determined by the InParanoid database (Sonnhammer and Östlund, 2015). Of these, 64 proteins were only found in the At-RBP set (discussed below), while 136 proteins have been detected as RBPs in other mRNA interactomes including several without prior association to RNA binding (Supplemental Figure 3A and Supplemental Data Set 1). Such strong overlap strengthens the confidence that most of the At-RBPs are bona fide RBPs. Finally, 52 At-RBPs were present in interactomes of all three kingdoms (Supplemental Figure 3B). This group mainly comprises proteins involved in mRNA translation, splicing, and helicase activity (Supplemental Data Set 1), all of which are core eukaryotic RNA functions.
Biophysical Properties of Captured Proteins Are Characteristic of RBPs
Next, we examined the biophysical and amino acid sequence features of captured proteins (Figure 2). We used the properties of the input proteome as a reference for diverse proteins, whereas input proteins with the GO annotation “RNA binding” were used as a reference for expected properties of known/predicted RBPs. These two groups were compared with the properties of At-RBPs, At-RBPs without known RBDs, and candidate At-RBPs. All five groupings spanned the full range of protein sizes, with some tendency toward larger proteins among the At-RBPs with unknown RBDs (Figure 2A). Proteins within the RBP groupings range from high to low abundance with bias toward higher abundance for At-RBPs and At-RBPs with unknown RBD compared with the input proteome and proteins with the GO annotation RNA binding (Figure 2B). This tendency was also observed in previous studies (Castello et al., 2012; Liao et al., 2016) and is not surprising as At-RBPs are selected based on their CL/noCL enrichment ratios and their associated statistical significance levels (FDR values), the latter of which are dependent on sample sizes (i.e., protein abundances).
Figure 2.

Biophysical and Sequence Features of Captured Protein Sets.
(A) to (E) Density of protein length (A), density of log10 iBAQ values (as measure of protein abundance) (B), proportion of amino acid residues in disordered regions (C), isoelectric point (pI) (D), and hydrophobicity (HI) (E) were analyzed for At-RBPs (green), proteins from the input proteome with “GO RNA binding” (light purple), At-RBPs with unknown RBD (light orange), candidate At-RBP (blue), and input proteome (red). The significance of differences between RBP subsets in (A) and (C) to (E) was tested by the Kolmogorov-Smirnov test. This showed that protein size distribution (A) does not differ between all five groups. Compared with input proteome, all four subsets are significantly different in disordered region (C), isoelectric point (D), and hydrophobicity (E) (P < 0.001). Significance of differences in panel (B) was tested by a two-sided t test and found that all RBP subsets except for GO RNA binding are significantly different from the input proteome.
(F) Log2 enrichment of amino acid groups in At-RBPs (green), proteins from the input proteome annotated as “RNA binding” (light purple), At-RBPs with unknown RBD (light orange), and candidate At-RBP (blue) compared with the input proteome. The significance of enrichment/depletion was tested by two-sample test for population proportion. Amino acid groups that are significantly (P < 0.001) enriched/depleted compared with the input proteome are marked with asterisks. Number of proteins in each RBP set: input proteome n = 8246, GO RNA binding (input proteome) n = 339, At-RBPs n = 300, candidate At-RBPs n = 437, and At-RBPs (unknown RBD) n = 61.
(G) to (P) Ten most enriched amino acid motifs in At-RBPs relative to the input proteome as analyzed by the DREME software (part of the MEME suite).
Compared with the input proteome, all four groups showed significantly increased proportions of residues in intrinsically disordered regions (Figure 2C), which have been linked to protein-protein, protein-DNA, and protein-RNA interactions (Wright and Dyson, 2015; Calabretta and Richard, 2015). Furthermore, all four RBP sets exhibited significant shifts toward a more alkaline isoelectric point and a lower hydrophobicity compared with the input proteome (Figures 2D and 2E). This is also reflected in their amino acid composition. Hydrophobic and aromatic amino acids such as leucine, isoleucine, and valine, as well as amino acids with aliphatic side chains such as tryptophan and phenylalanine, which have all been shown to have low propensity to bind RNA (Jeong et al., 2003; Lejeune et al., 2005), were depleted relative to the input proteome (Figure 2F; Supplemental Figure 4B). Cysteine also showed strong depletion, which is consistent with its low propensity to bind RNA (Lejeune et al., 2005) and its depletion in intrinsically disordered regions (Theillet et al., 2013; Williams et al., 2001). By contrast, proline is enriched in all four RBP sets, in agreement with its strong enrichment in disordered regions (Theillet et al., 2013; Williams et al., 2001). Furthermore, positive and polar amino acids such as arginine, glutamine, asparagine, and histidine, which have a high propensity to bind RNA, were enriched among all RBP sets (Figure 2F; Supplemental Figure 4A). The smallest amino acid glycine, which can form a strong interaction with the nucleotide guanine (Lejeune et al., 2005), also showed strong positive enrichment (Supplemental Figure 4A). Overall, the At-RBPs showed the strongest biases in these features. At-RBPs without known RBDs and candidate At-RBPs also followed these trends albeit to a lesser extent. Taken together, these findings mirror those reported for other mRNA interactomes (Castello et al., 2012; Liao et al., 2016) and indicate that mRNA interactome capture strongly enriched for bona fide plant RBPs.
Enriched amino acid motifs among the At-RBPs mostly consist of disordered, low complexity sequences. In particular, we found many arginine-glycine (RG)-rich motifs (Figures 2H and 2K to 2M), including RGG/RG motifs, which are involved in multiple RNA-related processes (Thandapani et al., 2013). Furthermore, we found GYG and poly(Q) motifs (Figures 2G and 2P), which can promote RNA-protein granule formation (Järvelin et al., 2016). Other enriched motifs include the RGFGF (Figure 2I) and the FVGGL (and related) motifs (Figures 2J, 2N, and 2O), which are part of the RNP1 and RNP2 consensus sequences of RRM domains, respectively (Lorković and Barta, 2002).
Interactome Capture Identifies a Diverse Set of Proteins with a Range of Recognized RBDs
Next, we grouped At-RBPs based on their annotated protein domains (Figures 3A and 3B). mRNA interactome capture identified a broad array of proteins with known RBDs, covering more than 30 different known types. Of these, proteins containing RRM domains constituted the largest class (80 in the At-RBPs and 50 within candidate At-RBPs); thus, we captured the majority of the 197 bioinformatically predicted RRM domain proteins in Arabidopsis (Silverman et al., 2013). Similarly, of the 28 predicted KH domain proteins (Silverman et al., 2013), 19 have been detected in the At-RBPs (seven proteins) and candidate At-RBPs (12 proteins) (Figure 3A), indicating that the majority of RRM and KH domain proteins are expressed and bound to poly(A) RNA in the seedling. Proteins harboring diverse canonical RBDs, such as the Nuclear Transport Factor 2 (NTF2), LSM, PUF, and La domains, were all readily captured (Figure 3A). Additionally, multiple zinc-finger proteins were identified as RNA binding (Figure 3A), including those subtypes known to interact with RNA, such as zf-CCCH, zf-CCHC, and zf-C2H2 (Ciftci-Yilmaz and Mittler, 2008), as well as others such as zf-RanBP, which has been shown to interact with RNA in humans (Nguyen et al., 2011; Vandevenne et al., 2014), but not in plants. Within these zinc-finger protein classes, 10 proteins not previously associated with RNA binding have been identified (Supplemental Table 1), expanding our knowledge on zinc-finger containing RBPs.
Figure 3.

Recognized and Unknown RBDs Identified by mRNA Interactome Capture.
(A) Number of proteins harboring domains associated with RNA binding in At-RBPs (green), candidate At-RBPs (blue), or only identified in the input proteome (white).
(B) Number of proteins harboring domains not associated with RNA binding in At-RBPs (green), candidate At-RBPs (blue), or only identified in the input proteome (white).
Other canonical RBPs detected include AGO proteins that contain PAZ and PIWI domains (Figure 3A). Both AGO1 and AGO2 were detected in the At-RBP set and AGO4 in the candidate At-RBP set. This report of successful UV cross-linking of AGO proteins to mRNA in plants provides the basis for future determination of sRNA targets via methods such as immunoprecipitation followed by high-throughput sequencing (HITS-CLIP), a long established method in animal cells yet to be applied in plants (Chi et al., 2009). Lastly, we have captured many canonical RBP families that are involved in responses to various abiotic stresses including GR-RBPs, CSD proteins, tudor-SN proteins, and DEAD box RNA helicases (Figure 3A) (Kim et al., 2007b; Kwak et al., 2005; Lorković, 2009; Jung et al., 2013; Frei dit Frey et al., 2010).
Limited Capture of Mitochondrial and Chloroplastic RBPs in Etiolated Seedlings
Consistent with previous studies (Castello et al., 2012; Liao et al., 2016), we found multiple cytoplasmic ribosomal proteins (RPs) within the plant mRNA interactome (27 proteins of the large and 32 of the small ribosomal subunit; Supplemental Data Set 1). Several RPs are known to be in direct contact with mRNA (Pisarev et al., 2008) and a number of RPs also have extraribosomal functions in mRNA regulation (Warner and McIntosh, 2009). The capture of polyadenylated rRNA processing intermediates (Sikorski et al., 2015) is also likely to have contributed to the isolation of cytoplasmic RPs. By contrast, only one chloroplastic and one mitochondrial RP were captured despite a total of 46 mitochondrial and chloroplastic RPs being present in the input proteome. This stark contrast in the capture of cytoplasmic and mitochondrial/chloroplastic RPs is consistent with most mature, translatable transcripts in these organelles not harboring a poly(A) tail (Chang and Tong, 2012). Likewise, only 18 PPR proteins were detected in the interactome (six in At-RBPs and 12 in candidate At-RBPs), out of the 60 PPR proteins identified in the input proteome (Figure 3A). Again, this poor ratio of interactome/input proteome of PPR proteins is possibly explained by their mitochondrial and chloroplastic location (Colcombet et al., 2013). Although known chloroplast RBPs such as CP29A, CP31A, and CP31B were strongly enriched by interactome capture, CP29A has also been shown to interact with nuclear mRNAs (Gosai et al., 2015), so it is not certain that this RBP is being captured from the organelle. With regards to the PPR proteins, there were only 60 in the input proteome, a fraction of the 450 predicted PPR proteins in Arabidopsis (Silverman et al., 2013). Again, this strongly contrasts to other RBD classes (RRM; 160 in the input proteome of the predicted 197; Silverman et al., 2013); therefore, the lack of PPRs in general may reflect that the etioplasts have yet to differentiate. Hence, the results may be considerably different in light-grown seedlings.
Interactome Capture Provides Experimental Evidence of RNA Binding Activity for Many Predicted and Emerging Plant RBPs
Although ∼80% of the At-RBP set are annotated to contain canonical RBDs and RNA-related GO terms, this is primarily based on in silico predictions and not yet on experimental evidence. For example, although a diverse range of RRM proteins have been shown to interact with RNA in vivo (Bannikova et al., 2013; Leder et al., 2014; Xing et al., 2015; Zhang et al., 2015), the vast majority of captured RRM proteins have neither a previously demonstrated RNA binding activity nor a known biological function. Another example is the family of CTC-interacting domain (CID) proteins, which bind to the C-terminal domain of poly(A) binding proteins via their poly(A) binding protein interacting motif 2 (PAM2) (Bravo et al., 2005). CID proteins are categorized into four groups based on their other domains (Bravo et al., 2005). Until now, only CID12, which contains PAM2 and RRM domains, has been shown to interact with RNA in vitro (Hecht et al., 1997). We have identified CID12 as well as CID1, 3, 4, 7, 8, 10, and 11 in the At-RBPs and candidate At-RBPs (Figure 4A) and thus provide evidence of their RNA binding activity in vivo for the first time.
Figure 4.

mRNA Interactome Capture Provides Experimental Evidence of RNA Binding for Many Predicted Arabidopsis RBPs.
(A) Members of predicted Arabidopsis RBPs families identified as At-RBPs (green), candidate At-RBPs (blue), or only in input proteome (black). Proteins with prior experimental evidence of RNA binding are underlined; proteins not detected in the input proteome are in italic.
(B) Arabidopsis YTH domain proteins identified in input proteome, At-RBPs, candidate At-RBPs, and schematic representation of annotated domains. CPSF30 gives rise to two transcripts, a longer one containing a YTH domain and a shorter one without this domain.
(C) Meta-transcript analysis showing enrichment of m6A sites within a 100-nucleotide window upstream, and depletion of m6A sites downstream of polyadenylation sites, based on PAC sites from Wu et al. (2011) (left panel) and cleavage sites from Sherstnev et al. (2012) (right panel). Effect sizes are given as log10-transformed odds ratios (OR) and are shown by the red lines; the red shaded areas show 95% confidence intervals of the log10-transformed ORs. Statistical significance of the enrichment/depletion at the FDR = 0.001 level is denoted by the top blue bars.
(D) Arabidopsis Alba domain family members identified in input proteome, At-RBPs, candidate At-RBPs, and schematic representation of annotated domains.
Similarly, plant PUF proteins are largely only predicted to bind RNA. Of the 25 family members in Arabidopsis, only PUM2 and PUM5 have been experimentally demonstrated to bind RNA (Francischini and Quaggio, 2009; Huh et al., 2013; Huh and Paek, 2014). Here, we provide evidence that PUM1-6, all of which belong to group I with the strongest sequence similarity to the Drosophila PUF domain (Francischini and Quaggio, 2009), bind to RNA in vivo (Figure 4A). Most of the other PUF proteins were not present in the input proteome, suggesting that PUF1-6 are the major players of this family in early Arabidopsis growth and development.
Other predicted RBPs include the NTF2 protein family that contains an NTF2 domain, which is required for protein-protein interactions (Ribbeck et al., 1998; Fribourg et al., 2001), but has been shown to interact with RNA in humans (Katahira et al., 2015). In Arabidopsis, 18 proteins are predicted to contain NTF2 domains; these proteins can be classified into two groups: group I proteins, which contain both NTF2 and RRM domains; and group II proteins, which only have NTF2 domains. We have identified six out of eight group I proteins in the At-RBP set and one among the candidate At-RBPs (Figure 4A), thereby verifying their RNA binding activity.
We also captured both Arabidopsis homologs of Barentsz (BTZ; also known as METASTATIC LYMPH NODE51 [MLN51]) harboring the Btz domain (Figure 4A), which is a known RBD in animals (Bono et al., 2006). Barentsz, eIF4A3, Y14, and Mago Nashi (MAGO) form the core of the exon-junction complex (EJC). The EJC is deposited on nascent mRNA at splice junctions and functions in subsequent mRNA utilization (Nyikó et al., 2013). In addition to the Barentsz homologs, we identified eIF4A3 and Y14 in the At-RBPs and MAGO among the candidate At-RBPs (Figure 4A). In humans, direct contact with RNA has been shown for BTZ and eIF4A3 by protein-RNA co-crystal structural analysis (Bono et al., 2006). Additionally, the EJC also serves as a mark for nonsense-mediated decay (NMD) (Kim et al., 2001). Other components of NMD include UP-FRAMESHIFT proteins (UPF1-3), all three of which have been identified in the At-RBP set (Figure 4A). While the role of plant UPF1 as activator of NMD is relatively well studied, UPF2 and UPF3 are less well characterized and the molecular mechanism of NMD in plants is still poorly understood (Dai et al., 2016). Further examples of protein families that are annotated as RBPs with little experimental evidence in plants include MEI2-like proteins, La-related proteins, BRUNO-LIKE proteins, and others listed in Figure 4A. Many members of these protein families had not been shown to function as plant RBPs.
YTH Domain-Containing Proteins
Also strongly captured were YT521-B homology (YTH) domain-containing proteins. There are 13 YTH proteins in Arabidopsis (Li et al., 2014), which show distinct developmental expression patterns and responses to stress and include the 11 evolutionary conserved C-terminal domain (ECT) family proteins (Ok et al., 2005). Ten of the eleven ECT proteins were captured here, eight in the At-RBP set and two in the candidate At-RBPs (Figure 4B). The mammalian YTH proteins YTHDF1-3 and YTHDC1 were recently shown to bind the epitranscripomic mark N6-methyladenosine (m6A), impacting mRNA splicing, export, translation, or turnover (Schumann et al., 2016; Schwartz, 2016). YTH domains bind m6A in a cage of aromatic amino acids that is conserved among all Arabidopsis YTH domain proteins (Fray and Simpson, 2015). The fact that all these proteins only have the YTH domain in common (Figure 4B) makes this domain a strong candidate for an RBD also in plants.
A further YTH domain protein in the At-RBP set is the Cleavage and Polyadenylation Specificity Factor 30 (AtCPSF30), which functions as part of a larger complex in mRNA 3′ end formation (Hunt et al., 2012). Analyses with mutant Arabidopsis plants deficient in CPSF30 indicated roles in stress responses (Zhang et al., 2008), immunity, and programmed cell death (Bruggeman et al., 2014), as well as demonstrating altered mRNA 3′ end cleavage site choice in a large number of genes (Thomas et al., 2012). Interestingly, Arabidopsis expresses two CPSF30 protein variants from a single gene due to alternative mRNA 3′ end formation: a shorter form of ∼28 kD that harbors three zinc finger domains and is homologous to yeast and mammalian CPSF30 and a longer form of ∼70 kD that adds a YTH domain and is unique to plants (Hunt et al., 2012; Delaney et al., 2006). These observations, together with evidence that m6A is enriched near 3′ ends of Arabidopsis mRNAs (Luo et al., 2014; Wan et al., 2015; Bodi et al., 2012), led to recent speculation about a role of m6A in governing mRNA 3′ end formation in plants (Fray and Simpson, 2015; Chakrabarti and Hunt, 2015; Burgess et al., 2016). To formally investigate such a link, we obtained Arabidopsis data on m6A site distribution (Wan et al., 2015) and 3′ end cleavage and polyadenylation maps (polyadenylation cluster [PAC] sites from Wu et al. [2011] and cleavage site from Sherstnev et al. [2012]) and performed spatial colocation analyses. Indeed, we found an enrichment of m6A sites within a 100-nucleotide window upstream of mRNA 3′ ends (Figure 4C). No such enrichment was seen in mammals where m6A peaks were examined in a 50-nucleotide window upstream of known poly(A) cleavage sites, but no significant association was found (Meyer et al., 2012). Cleavage and polyadenylation regions in plant mRNAs consist of three signals: far upstream elements and near upstream elements (NUEs; equivalent to the mammalian AAUAA motif), as well as sequences immediately surrounding the cleavage site (Loke et al., 2005; Thomas et al., 2012). We detected maximally enriched m6A abundances at around −45 nucleotides relative to PAC and cleavage sites (Figure 4C), which lies upstream of the NUE (−13 to −30 nucleotides) at the 3′ boundary of the far upstream element region (−50 to −130 nucleotides). Similar to mammalian CPSF30, which binds to regions near the AAUAA motif (Chakrabarti and Hunt, 2015), AtCPSF30 has a role in the function of the NUE (Thomas et al., 2012), suggesting complementary roles of RNA binding through its zinc finger and YTH domains, perhaps conveying an m6A dependence for a subset of mRNA 3′ end cleavage sites that should be tested in future work.
Alba Domain-Containing Proteins
Another example is the Alba domain-containing protein family, where we identified four Alba proteins among the At-RBPs and the fifth among the candidate At-RBPs (Figure 4D), which accounts for all Alba proteins of Arabidopsis. Supporting this, an Alba protein (At1g76010) was recently found to be one of the most enriched proteins in an RNA-affinity chromatography experiment, providing evidence that Alba proteins can bind RNA in plants (Gosai et al., 2015). Currently, nothing is known about the molecular or functional role of Alba proteins in plants. Alba proteins are widely distributed in Archaea where they are a major component of chromatin and involved in transcriptional repression through binding to DNA, but are also know to interact with RNA (Bell et al., 2002; Jelinska et al., 2005; Forterre et al., 1999; Guo et al., 2003). They are structurally similar to prokaryotic translation initiation factor 3 (Aravind et al., 2003) and have been reported to play a role in translational control in multiple eukaryotes (Mani et al., 2011; Gissot et al., 2013; Mair et al., 2010), so there is precedent for proteins containing this domain to bind RNA. Interestingly, the Alba gene family of the protozoan parasite Trypanosoma brucei share a similar structure to that in Arabidopsis, where there are members encoding small proteins that only contain the Alba domain, and members encoding longer proteins with a C-terminal region rich in RGG boxes (Figure 4D; Subota et al., 2011). These motifs are known to promote RNA binding (Thandapani et al., 2013) and are often found in combination with other RBDs (Castello et al., 2012). Thus, they might function synergistically with the Alba domain to facilitate RNA-protein interactions.
WHIRLY-Domain Proteins
Another group includes WHIRLY (WHY) domain-containing proteins, which form a small family of single stranded DNA binding proteins localized to organelles where they maintain genome stability (Krause et al., 2005; Cappadocia et al., 2010; Marèchal et al., 2009). In maize (Zea mays), the chloroplast-localized WHY1 has been shown to bind to both DNA and a subset of plastid RNAs in vivo (Prikryl et al., 2008). Here, we identified all three Arabidopsis WHY proteins as At-RBPs (Figure 5A), denoting these proteins as a family of RBPs in Arabidopsis.
Figure 5.

Novel Arabidopsis RBPs.
(A) to (C) Novel families of Arabidopsis RBPs: Whirly proteins (A), DUF1296 proteins (B), and LIM proteins (C).
(D) Illustration of diverse functions of potential noncanonical RBPs in Arabidopsis.
Proteins with Potential Novel RNA Binding Activity in Plants
About one-sixth of the At-RBPs have neither recognized RBDs nor annotated roles in RNA biology. Many of these are plant specific, where they are only found in the At-RBPs but not in other mRNA interactomes, or have no identified orthologs in other kingdoms (Supplemental Figure 3A).
DUF1296-Domain Proteins
One example is a group of largely uncharacterized proteins with the Domain of Unknown Function 1296 (DUF1296). The Arabidopsis genome encodes eight proteins containing DUF1296 domains: seven kinase-related proteins of unknown function and a G-BOX TRANSCRIPTION FACTOR-INTERACTING PROTEIN1 (GIP1), which is involved in regulating the activity of transcription factors involved in plant development (Lee et al., 2014; Shaikhali, 2015). We identified three kinase-related proteins among the At-RBPs and GIP1 as well as another kinase-related protein in the candidate At-RBPs (Figure 5B). The only domain that is common to these proteins is an N-terminal DUF1296 domain (Figure 5B), suggesting that this domain might be a novel RBD in plants.
LIM-Domain and Cytoskeletal Proteins
We have identified several proteins containing the LIM domain, which consists of tandem zinc-finger structures. LIM domains are present in a wide range of eukaryotic organisms and have been shown to mediate protein-protein interactions (Kadrmas and Beckerle, 2004). We have captured four of the six Arabidopsis LIM genes in our RBP sets (Figure 5C). LIMs function in cytoskeleton organization by binding to actin filaments (Papuga et al., 2010; Ye and Xu, 2012), but have no known role in RNA binding. RNA transport along the cytoskeleton is a major mechanism of mRNA localization and requires motor proteins that move the RNA cargo in form of ribonucleoprotein (RNP) particles along the cytoskeletal tracks (Bullock, 2011; Gagnon and Mowry, 2011; Jansen, 1999). Despite some well-studied examples in Drosophila and yeast (Bullock, 2011), relatively little is known about how RNPs are connected to the motor proteins in plants. LIM proteins might carry out this connecting role in plants thereby mediating mRNA transport along actin filaments. LIM proteins are not the only cytoskeletal proteins among the At-RBPs, as we have also captured actin and tubulin (Supplemental Table 2; Figure 5D), which form microfilaments and microtubules, respectively, the major components of the cytoskeleton. Actin is also present in the nucleus where its function is less well studied (Falahzadeh et al., 2015). Interestingly, in animals, nuclear actin was found to be part of hnRNPs, proteins that are involved in mRNA processing, export, localization, and stability (Hofmann, 2009), but no such functional role is known of nuclear actin in plants.
Aquaporins
Curiously, we have also identified a subset of aquaporin proteins, which belong to the well-studied family of major intrinsic proteins (Figure 5D; Supplemental Table 2). These proteins form transmembrane channels that transport water, other small solutes, and gases (Quigley et al., 2002; Biela et al., 1999; Gaspar, 2003; Holm et al., 2005; Uehlein et al., 2003), but there are no reports of aquaporins transporting or interacting with RNA. We found the plasma membrane intrinsic proteins (PIPs) PIP2;1, PIP2;2, and PIP2;7 among the At-RBPs and PIP1;1, PIP1;2, and PIP1;3, as well as the tonoplast intrinsic protein TIP1;2, in the candidate At-RBPs. Considering the ever expanding types of substrates assigned to aquaporins (Maurel et al., 2015), it is intriguing to speculate that RNAs may also travel through aquaporins, similar to protein assisted cell-to-cell transport of RNA during virus infection (Peña and Heinlein, 2012).
Signal Transduction and Transcriptional Control
Other examples of well-characterized proteins identified as At-RBPs include several plant-specific proteins involved in major signal transduction pathways of etiolated seedlings (Supplemental Table 2; Figure 5D). First, we captured ETHYLENE-INSENSITIVE2 (EIN2), which is a classic ethylene signaling protein expressed strongly in etiolated seedlings, in which ein2 mutants display the triple response of reduced apical hook formation and elongated hypocotyl and roots. Independently validating our mRNA interactome data, EIN2 has recently been shown to bind to EIN3 BINDING F-BOX1 (EBF1) and EBF2 mRNAs in the presence of ethylene, thereby promoting their translational repression and activating ethylene responsive genes (Li et al., 2015; Merchante et al., 2015). Another signaling protein among the At-RBPs is PHYTOCHROME A (PHYA), the primary red-light photoreceptor in seedlings that mediates many aspects of seedling deetiolation in response to light (Casal et al., 2014). PHYA has no previous association with RNA binding, but widespread changes in alternative splicing were observed in phyA phyB double mutants (Shikata et al., 2014) and PHYB has recently been found to regulate translation of mRNAs in the cytosol (Paik et al., 2012). Finally, the blue light receptor PHOTOTROPIN1 (PHOT1), which is required for the inhibition of hypocotyl growth during deetiolation, was also detected by interactome capture. Similar to PHYA, PHOT1 has not been ascribed an RNA binding function; however, it is required for blue-light-mediated mRNA destabilization (Folta and Kaufman, 2003). Such interactions raise the possibility that these photoreceptors regulate posttranscriptional events via RNA binding functions, or conversely, particular RNAs maybe regulating these receptors.
A number of proteins in the At-RBP set have also been implicated in transcriptional control (Figure 5A; Supplemental Table 2). First, PURA1 (PURIN-RICH ALPHA1) and AtNFXL1 are Arabidopsis homologs of known human transcription factors. PURA1 has been shown to interact with the 5′ region of many Arabidopsis genes (Tremousaygue et al., 1999) and AtNFXL1 is a NF-X1 type zinc finger protein. Additionally, MULTIPROTEIN BRIDGING FACTOR 1A, a highly conserved transcriptional coactivator (Tsuda et al., 2004), is found in the At-RBP set as well as in human, mouse, and yeast interactomes (Supplemental Data Set 1). A number of transcription factors were previously identified in mammalian mRNA interactomes (Liao et al., 2016), setting up intriguing possibilities of RNA binding being involved in the regulation of transcription.
Stress-Related Proteins
We have also captured several stress responsive proteins that have no RNA-related GO annotations, such as HEAT SHOCK PROTEIN 90.2 (HSP90.2; AT5G56030), which was identified in the At-RBPs, and HSP70.1 and HSP40, which was found in the candidate At-RBPs (Figure 5D). These proteins are highly conserved molecular chaperones involved in protein folding, stability, and activation (Wang et al., 2004). RNA binding roles are not unprecedented for HSPs as mammalian HSP70 is able to bind to the 3′ untranslated region of labile mRNAs (Henics et al., 1999) and NbHSP90 of Nicotiana benthamiana can interact with Bamboo mosaic virus genomic RNA, enhancing its replication (Huang et al., 2012). Moreover, HSP90 is involved in RNA interference in human and yeast (Wang et al., 2013) and has been captured in human, animal, and yeast mRNA interactomes (Supplemental Data Set 1). Therefore, this protein is possibly a conserved RBP, although a direct RNA binding function remains to be determined.
Another example is ANNEXIN4 (ANN4), which is a member of a multiprotein family of Ca2+-dependent membrane binding proteins (Laohavisit and Davies, 2011; Figure 5D; Supplemental Table 2). ANN4 is involved in osmotic stress and ABA signaling in a Ca2+-dependent manner (Lee et al., 2004), but has no prior association with RNA binding.
Enzymes
Finally, we captured a number of metabolic enzymes such as RAFFINOSE SYNTHASE6, which is involved in carbohydrate metabolism (Fujiki et al., 2001) (Figure 5D). Additionally, S-ADENOSYLMETHIONINE SYNTHASE4 (METK4) was captured, along with its closely associated homolog METK3 that was present in the candidate At-RBP set. DNA topoisomerase type IA, CATALASE-3, PEROXIDASE69, and the SNF1-RELATED PROTEIN KINASE 2.5 (SRK2G) were also present in the At-RBP set (Figure 5D; Supplemental Table 2). Recent mRNA interactome data in higher eukaryotes revealed the striking aspect that many enzymes bear RNA binding functions (Castello et al., 2012; Beckmann et al., 2015; Liao et al., 2016; Matia-González et al., 2015). Elucidating such interactions may lead to the discovery of novel RNA-based regulatory mechanisms.
Limitations, Challenges, and Opportunities of mRNA Interactome Capture
Our study presents a system-wide, in vivo analysis of proteins bound to mRNA in plants. We identified 300 proteins with high confidence (FDR below 1%), which gives a snapshot of the extent of RNA-protein interactions occurring in etiolated seedlings. As the biological role is unknown for the vast majority of these At-RBPs, it is clear that our knowledge of posttranscriptional gene regulation of a plant is superficial at best. There has been great variability in the capture of different families of Arabidopsis RBPs, ranging from ∼65% capture of the predicted RRM and KH domain containing proteins, to only 4% capture of the predicted 450 PPR proteins (Silverman et al., 2013). Many reasons likely underlie this variability; we have shown that strong variability exists in expression of the different RBP families in etiolated seedlings, but other reasons such as strength, stability, and regulation of RNA binding likely all contribute to the efficiency of capture. Furthermore, RBPs not bound to poly(A) RNA will not be captured by this method, where the method would require modification to capture RBPs bound to particular subsets of RNAs, such as those from organelles. Additionally, to gain a more complete picture of RNA-protein interaction in plants, multiple interactomes will need to be performed on different developmental stages and under variable environmental conditions. One immediate aim will be to determine how efficient this methodology is on tissues rich in UV-absorbing molecules, to get an idea of how widely applicable it will be regarding different plant tissues.
The 300 proteins in the At-RBP set passed stringent statistical criteria. The proteins needed a CL/noCL ratio in at least two of the three biological replicates with an FDR <1%. For determining each ratio, at least two unique peptides per protein were required, which had to be detected in both CL and noCL samples (ratio count CL/noCL of at least 2; Supplemental Data Set 1). Therefore, proteins that are only detected in the CL samples will not be in the At-RBP set, even though such proteins are being strongly enriched by interactome capture. Hence, some of the highest enriched RBPs by interactome capture maybe within the “candidate At-RBP” set.
Detection of proteins in the noCL samples implies that the ionic conditions used in the washes (including denaturing agents and chaotropic detergents) have not removed all noncovalently linked proteins or dissociated all protein-protein interactions. However, further increasing the stringency of the washing conditions of the poly(A) RNA-oligo(dT) bead samples would likely be counterproductive, first by decreasing the number of proteins detected in the noCL samples, and therefore excluding these from the At-RBP set, and second, by negatively impacting the yield of the poly(A) capture. Currently, the frequency of false-positives of mRNA interactome capture is unknown; extensive validation of RNA binding by alternative experimental means will need to be undertaken, especially for novel RBPs identified in this study that have not been previously associated with RNA.
Curiously, we found nine proteins enriched in the noCL sample, although only two of these were significantly enriched (Supplemental Data Set 1 and Supplemental Figure 1D). Both proteins were highly abundant in our sample as determined by their abundance in the input proteome (data not shown) and likely represent the background noise of the methodology. Why they are enriched in the noCL sample is hard to explain, but one possibility is that these proteins are UV light labile and have been significantly degraded in the CL sample by UV treatment.
Nevertheless, we envision that our study will help facilitate the development of other methods based on UV cross-linking such as CLIP and variations thereof, long-standing and widely utilized methodologies in animal cells (Ule et al., 2003), but rarely applied in plants (Zhang et al., 2015). Such methodologies will be important in elucidating the RNA targets of RBPs, uncovering RBP networks to give insights into the true scope of posttranscriptional gene regulation in plants.
METHODS
Plant Growth
Arabidopsis thaliana ecotype Columbia (Col-0) seeds were sown on plates containing half-strength Murashige and Skoog medium and stratified in the dark at 4°C for 48 h. Plates, wrapped in aluminum foil, were then placed into 22°C growth cabinets for 4 d.
mRNA Interactome Capture
UV Cross-Linking
For in vivo cross-linking, plates of 4-d-old etiolated seedlings were placed on ice and irradiated in a Stratalinker (Stratagene) with 254-nm UV light at 150 mJ/cm2. The irradiation was performed three times with a 1-min pause in between treatments. Seedlings were harvested immediately after irradiation and frozen in liquid N2. Seventy plates (h × w: 90 × 15 mm) were used per replicate.
Lysis and Oligo(dT) Capture
Frozen tissue was then ground into fine powder in liquid N2 and resuspended in lysis buffer (20 mM Tris HCl, pH 7.5, 500 mM LiCl, 0.5% LiDS, 1 mM EDTA, 0.02% Nonidet P-40, 5 mM DTT, 2.5% [w/v] polyvinylpyrrolidone 40, 1% [v/v] β-mercaptoethanol, and 1× EDTA-free Roche protease inhibitor). The lysate was cleared by passing it through a QIAshredder column (Qiagen), centrifuging for 2 min at 14,000 rpm. noCL seedlings were processed side-by-side as a control. Aliquots from the lysate (input) were taken for quality controls (silver stain, immunoblot) and for protein identification by MS (referred to as input proteome).
RNA-protein complexes were isolated using oligo(dT)25 magnetic beads (beads from 500 μL original bead suspension; New England Biolabs), by incubating for 1 h at 4°C on a rotator [lysate should not be frozen before oligo(dT) capture]. Beads were collected on a magnet and washed twice with 1 mL of lysis buffer, followed by two washes with 1 mL of buffer I (20 mM Tris HCl, pH 7.5, 500 mM LiCl, 0.1% LiDS, 1 mM EDTA, 0.02% Nonidet P-40, and 5 mM DTT), buffer II (20 mM Tris HCl, pH 7.5, 500 mM LiCl, 1 mM EDTA, 0.02% Nonidet P-40, and 5 mM DTT), and buffer III (20 mM Tris HCl, pH 7.5, 200 mM LiCl, 1 mM EDTA, and 5 mM DTT) for 5 min at 4°C on a rotator. RNA-protein complexes were eluted by incubating the beads with 200 μL of elution buffer (20 mM Tris HCl, pH 7.5, and 1 mM EDTA) at 50°C for 3 min. After elution, oligo(dT) beads were reactivated in lysis buffer according to the manufacturer’s recommendations, a second round of oligo(dT) capture was performed for each sample, and the two eluates were combined.
RNase Treatment and Protein Extraction
The eluate was supplemented with one-quarter volume of 5× RNase buffer (50 mM Tris HCl, pH 7.5, 750 mM NaCl, 0.25% Nonidet P-40, and 2.5 mM DTT) and treated with 0.11 µg RNase A (Sigma-Aldrich) and 0.035 units of RNase T1 (Sigma-Aldrich) at 37°C for 1 h. Proteins were extracted using trichloroacetic acid (TCA)/acetone precipitation: 3 volumes of TCA/acetone solution (13.3% [w/v] TCA and 0.07% [w/v] DTT in acetone; chilled at −20°C) were added and samples were incubated at −20°C overnight. Samples were then centrifuged at 4°C for 15 min at 14,000 rpm, supernatant was removed, and the protein pellet was washed by adding 1 mL washing solution (0.07% DTT in acetone; chilled at −20°C), vortexing and incubation at −80°C for at least 30 min. After repeating the centrifugation and wash steps, the protein pellet was dried in a SpeedVac for 2 min and resuspended in solubilization buffer (8 M urea, 0.5% SDS, 1% DTT, and 35 mM HEPES, pH 7.5). Samples were sonicated twice for 30 s, centrifuged for 10 min at 14,000 rpm, and the supernatant containing the proteins was transferred to a new tube. An aliquot (20%) was taken for protein analysis (silver staining and immunoblot) and the remainder of the eluate was used for mass spectrometry.
SDS-PAGE, Silver Staining, and Immunoblot
For SDS-PAGE, protein samples in 1× NuPAGE LDS sample buffer and 1× NuPAGE reducing reagent (Thermo Fisher) were loaded on NuPAGE Novex 4-12% Bis-Tris protein gels (Thermo Fisher) and electrophoresed at 130 V for 2 h in 1× MES buffer (Thermo Fisher). For silver staining, the gel was fixed in 50% methanol and 5% acetic acid for 30 min, followed by a wash with 50% ethanol and a second wash with 30% ethanol, each for 5 min. After a wash with water for 10 min, the gel was sensitized with 0.02% sodium thiosulfate for 60 s and washed three times with water for 30 s each. The gel was then placed in silver solution (6 mM silver nitrate and 0.0185% formaldehyde) for 20 min followed by three washes with water for 30 s each. The gel was developed with 2% sodium carbonate, 0.0185% formaldehyde, and 0.0004% sodium thiosulfate. The developing reaction was stopped with 5% acetic acid. All solutions were prepared freshly and all procedures were performed on a rocking platform at room temperature.
When restaining was necessary, the gel was incubated in destain solution (prepared by mixing two solutions, 30 mM potassium ferricyanide and 100 mM sodium thiosulfate, at equal volume immediately prior to use) until gels were clear again; typically this occurred within 10 min. The gel was then washed with water to remove any yellow color. The gel was then restained as described above.
For immunoblot, gels were electroblotted onto nitrocellulose membrane (GE Healthcare Life Sciences). The membrane was blocked in 5% nonfat milk in 1× PBST (1× PBS with 0.2% Tween 20) for 30 min at room temperature, followed by incubation with primary antibodies in 5% nonfat milk in 1× PBST on a rotating wheel at 4°C overnight. The membrane was then washed three times with 1× PBST, for 5 min each at room temperature. The membrane was incubated with secondary antibody in 5% nonfat milk in 1× PBST for 1 h at room temperature. Protein signals were detected using Super Signal Femto chemiluminescent reagent (Pierce), visualized on an ImageQuant LAS 4000 system (GE Healthcare Life Sciences). Primary antibodies used were anti-CP29A (kind gift from Christian Schmitz-Linneweber), anti-AGO1 (Agrisera; AS09-527), and anti-SAL1 (kind gift from Barry Pogson). An anti-rabbit HRP-conjugated antibody (AP132P; EMD Millipore) was used as secondary antibody.
MS for mRNA Interactome Samples
Sample Preparation Using SP3
For reduction and alkylation of the samples, 1 μL 200 mM DTT in 200 mM HEPES was added to the eluates followed by incubation at 56°C for 30 min. After placing the samples on ice for 2 min, 2 μL of 400 mM iodoacetamide in 200 mM HEPES (pH 8.5) was added and samples were incubated at 24°C for 30 min in the dark. Then, 2 μL of 200 mM DTT was added followed by incubation at room temperature for 5 min.
For protein cleanup and digestion, 2 μL of Sera-Mag bead mix (Thermo Scientific) was added to the eluates followed by the addition of 5 μL 5% formic acid. After ensuring that samples were acidic, acetonitrile was added to a final concentration of 50% and the samples were incubated for 8 min at room temperature. Samples were then placed on a magnetic rack and incubated for 2 min at room temperature. The supernatant was removed and samples were washed by adding 200 μL 70% ethanol and incubation for 15 s on the magnetic rack. The wash was repeated once followed by the addition of 180 μL acetonitrile and incubation on the magnetic rack for 15 s. The supernatant was removed and samples were air-dried for 30 s. Samples were then taken off the rack and digested by adding 800 ng of trypsin in 50 mM HEPES (pH 8) and incubating at 37°C for 14 h.
After the digest, beads were resuspended by pipetting and samples were placed on a magnetic rack. Dimethyl labeling was performed by adding 1 μL of formaldehyde (CH2O for light; 13CD2O for heavy) and 1 μL of sodium cyanoborohydride (NaBH3CN for light; NaBD3CN for heavy) followed by incubation on a magnetic rack for 30 min at room temperature. One microliter of the respective formaldehyde and sodium cyanoborohydride solutions were added again for more efficient labeling and samples were incubated for another 30 min at room temperature on the magnetic rack. After that, 1 μL of quench mix was added and samples were incubated for 5 min at room temperature. Samples were then taken off the magnetic rack and beads were resuspended by pipetting. Acetonitrile was added to the samples to a final percentage of 95% or higher, and samples were mixed by pipetting followed by incubation off the magnetic rack for 8 min at room temperature and further 2 min on the magnetic rack at RT. After removal of the supernatant, samples were washed by adding 180 μL of acetonitrile and incubation for 15 s on magnetic rack. The wash was repeated once, supernatant was removed and beads were air-dried for 30 s. The beads were then resuspended in 9 μL of 2% DMSO and sonicated in a water bath for 5 min. Finally, samples were placed on a magnet, and the supernatant was recovered to a new tube, acidified with 1 μL of 10% formic acid, and used for MS.
Liquid Chromatography-Tandem MS Analysis
Samples were analyzed on an LTQ-Orbitrap Velos Pro mass spectrometer (Thermo Scientific) coupled to a nanoAcquity UPLC system (Waters). Peptides were loaded onto a trapping column (nanoAcquity Symmetry C18, 5 μm, 180 μm × 20 mm) at a flow rate of 15 μL/min with solvent A (0.1% formic acid). Peptides were separated over an analytical column (nanoAcquity BEH C18, 1.7 μm, 75 μm × 200 mm) at a constant flow of 0.3 μL/min using the following gradient: 3% solvent B (acetonitrile and 0.1% formic acid) for 10 min, 7 to 25% solvent B within 210 min, 25 to 40% solvent B within 10 min, and 85% solvent B for 10 min. Peptides were introduced into the mass spectrometer using a Pico-Tip Emitter (360 μm outer diameter × 20 μm inner diameter, 10 μm tip; New Objective). MS survey scans were acquired from 300 to 1700 m/z at a nominal resolution of 30,000. The 15 most abundant peptides were isolated within a 2D window and subjected to tandem MS (MS/MS) sequencing using collision-induced dissociation in the ion trap (activation time, 10 ms; normalized collision energy, 40%). Only 2+/3+ charged ions were included for analysis. Precursors were dynamically excluded for 30 s (exclusion list size was set to 500).
MS for the Input Proteome of Etiolated Seedlings
Sample Preparation
Proteins were extracted from aliquots (500 µL) saved from the input by TCA/acetone extraction as described above, and protein pellets were resuspended in 40 μL of 50 mM Tris-HCl, pH 8, 1% SDS, and 1× protease inhibitor. Samples were digested with 2 μL (25 units/µL) benzonase for 45 min at 37°C. Samples were prepared for MS using SP3 as described above, except that the protein digestion was performed with trypsin/LysC, at 2 µg per sample at 37°C overnight.
High pH Reverse-Phase Offline Fractionation
Offline high pH reverse-phase fractionation was performed using an Agilent 1200 Infinity HPLC system equipped with a quaternary pump, degasser, variable wavelength UV detector (set to 254 nm), peltier-cooled autosampler, and fraction collector (both set at 10°C for all samples). The column was a Gemini C18 column (3 μm, 110 Å, 100 × 1.0 mm; Phenomenex) with a Gemini C18, 4 × 2.0-mm SecurityGuard (Phenomenex) cartridge as a guard column. The solvent system consisted of 20 mM ammonium formate (pH 10.0) as mobile phase (A) and 100% acetonitrile as mobile phase (B). The separation was accomplished at a mobile phase flow rate of 0.1 mL/min using the following linear gradient: 100% A for 2 min, from 100% A to 35% B in 59 min, to 85% B in a further 1 min, and held at 85% B for an additional 15 min, before returning to 100% A and reequilibration for 13 min. Thirty-two fractions were collected along with the LC separation that were subsequently pooled into 10 fractions. Pooled fractions were dried under vacuum centrifugation, reconstituted in 10 μL 0.1% formic acid, and then stored at −80°C until liquid chromatography-mass spectrometry analysis.
Liquid Chromatography-Mass Spectrometry Analysis for Input Proteome
Peptides in the pooled fractions were separated using the nanoAcquity ultra-performance liquid chromatography (UPLC) system (Waters) fitted with a trapping (nanoAcquity Symmetry C18, 5 μm, 180 μm × 20 mm) and an analytical column (nanoAcquity BEH C18, 1.7 μm, 75 μm × 200 mm). The outlet of the analytical column was coupled directly to a LTQ (linear trap quadrupole) Orbitrap Velos Pro (Thermo Fisher Scientific) using the Proxeon nanospray source. Solvent A was water and 0.1% formic acid, and solvent B was acetonitrile and 0.1% formic acid. The samples (7.5 μL out of 10 μL for input proteome analysis) were loaded with a constant flow of solvent A at 5 μL/min onto the trapping column. Trapping time was 6 min. Peptides were eluted via the analytical column with a constant flow of 0.3 μL/min. During the elution step, the percentage of solvent B increased in a linear fashion from 3 to 7% in 10 min, then increased to 25% in 100 min and finally to 40% in a further 10 min. The peptides were introduced into the mass spectrometer (Orbitrap Velos; Thermo) via a Pico-Tip Emitter (360 μm OD × 20 μm ID, 10-μm tip; New Objective), and a spray voltage of 2.2 kV was applied. The capillary temperature was set at 300°C. Full-scan MS spectra with mass range 300 to 1700 m/z were acquired in profile mode in the FT (Fourier transform) with resolution of 30,000. The filling time was set at maximum of 500 ms with limitation of 1.0 × 106 ions. The most intense ions (up to 15) from the full scan MS were selected for sequencing in the LTQ. Normalized collision energy of 40% was used, and the fragmentation was performed after accumulation of 3.0 × 104 ions or after filling time of 100 ms for each precursor ion (whichever occurred first). MS/MS data were acquired in centroid mode. Only multiply charged (2+, 3+, and 4+) precursor ions were selected for MS/MS. The dynamic exclusion list was restricted to 500 entries with maximum retention period of 30 s and relative mass window of 10 ppm. In order to improve the mass accuracy, a lock mass correction using the ion (m/z 445.12003) was applied.
Peptide Identification and Quantification
Raw data were processed using MaxQuant (version 1.4.1.2) (Cox and Mann, 2008). MS/MS spectra were searched against the UniProt Arabidopsis database (input proteome and interactome capture: version 05/06/2015 including 54,193 entries) concatenated to a database containing protein sequences of common contaminants. Enzyme specificity was set to trypsin/P, allowing a maximum of two missed cleavages. Cysteine carbamidomethylation was set as fixed modification, and methionine oxidation and protein N-terminal acetylation were used as variable modifications. For the mRNA interactome study, the required modifications for the dimethyl labeling were added as variable modification (DimethyLys0, DimethyNter0, DimethylLys8, and DimethylNter8). The minimal peptide length was set to six amino acids and a minimum of one unique peptide was required for the identification. The mass tolerances were set to 20 ppm for the first search, 6 ppm for the main search, and 0.5 D for product ion masses. FDRs for peptide and protein identification were set to 1%. Match between runs (time window 2 min) and requantify options were enabled, as well as the IBAQ function. Protein quantification was based on razor and unique peptides where at least two unique peptides needed to be detected in both CL and noCL (ratio count CL/noCL of at least 2).
Definition of mRNA Interactome Proteins
Statistical analysis for CL/noCL enrichment of protein groups quantified in at least two out of three biological replicates was performed using an empirical Bayes moderated t test within the R/Bioconductor package limma (Smyth, 2004). P values were adjusted for multiple testing using the method of Benjamini-Hochberg. The UniProt accession numbers of each protein group were converted into Arabidopsis gene IDs. Where multiple gene IDs applied, the gene ID corresponding to the majority protein ID was used. Proteins with a CL/noCL enrichment >0 at an FDR below 1% were defined as At-RBPs, whereas proteins with an FDR >1% and proteins where no CL/noCL ratio could be determined were defined as candidate At-RBPs.
Bioinformatic Analyses
GO Analyses
TAIR (version 10) ATH_GO_GOSLIM.txt .gz (version 2015-08-02) downloaded from https://www.arabidopsis.org/ was used for GO annotations. Enrichment of GOMF, GOBP, and GOCC categories was analyzed for At-RBPs and candidate At-RBPs compared with the background of proteins identified from the input proteome using fisher.test (Fisher’s exact test), and multiple testing was performed using p.adjust with the Benjamini and Hochberg method in the R package (R Core Team, 2015). GO terms with Benjamini and Hochberg P value < 0.05 were defined as enriched/depleted.
Classification of RNA Biology Status and Protein Domains
The RNA biology status and RBDs of At-RBPs and candidate At-RBPs were classified as described by Beckmann et al., (2015).
Analysis of Biophysical Properties and Sequence Features
Analyses of disordered regions, length of proteins, hydrophobicity, and amino acid composition were performed as described by Castello et al. (2012). Hydrophobicity for each amino acid residue was obtained from https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/hydrophob.html. Isoelectric point was calculated using the IsoelectricPoint module in the Biopython package (http://biopython.org/DIST/docs/api/Bio.SeqUtils.IsoelectricPoint-pysrc.html). Distribution biases for the sequence features and biophysical properties were evaluated using R packages, adk.test (Anderson-Darling k-sample test), and ks.test (two-sample Kolmogorov-Smirnov test). Protein abundance distribution plots of the different RBP sets are based on median iBAQ values from the input proteome data.
Analysis of amino acid motif enrichment in At-RBPs relative to the input proteome was performed using DREME (discriminative regular expression motif elicitation) (Bailey, 2011), which is part of the MEME suite (version 4.11.2, available from http://meme-suite.org/index.html). DREME analysis was performed using an E-value threshold of 0.1 and allowing for motif widths between 3 and 12 amino acids.
Conservation of At-RBPs
The list of predicted orthologs between plant and human, mouse, and yeast were obtained from InParanoid (version 8.0) (Sonnhammer and Östlund, 2015). The list of two-way predictions (A. thaliana-H. sapiens, A. thaliana-M. musculus, and A. thaliana-S. cerevisiae) were downloaded from http://inparanoid.sbc.su.se/download/. InParanoid uses a clustering method based on genome-wide pairwise sequence similarity matches to identify putative orthologous proteins between two species and predicts ortholog groups, where each group contains one (the highest sequence similarity matches) or more (with high pairwise similarity matches relative to the best pair) pairs, including those in-paralogs within a defined cutoff value (Sonnhammer and Östlund, 2015). We included all pairs in each group, which may include more than one combination (in-paralogs), i.e., all plant proteins that are predicted orthologs to either human, mouse. or yeast. The list of mRNA interactome proteins were obtained from Baltz et al. (2012) (HEK293, H. sapiens), Kwon et al. (2013) (mESC, M. musculus), Liao et al. (2016) (HL-1, M. musculus), and Beckmann et al. (2015) (HuH-7, H. sapiens; S. cerevisiae).
Meta-Transcript Analysis of Arabidopsis m6A and Poly(A) Sites
RNAModR (Evers et al., 2016) was used to perform a meta-transcript analysis of published m6A (Wan et al., 2015) and polyadenylation sites (PAC sites from Wu et al. [2011]; cleavage sites from Sherstnev et al. [2012]). In a first step, all reported m6A and polyadenylation sites were mapped to a custom reference transcriptome; the latter was constructed by collapsing all TAIR10-based transcript isoforms per gene and keeping the transcript isoform with the longest coding sequence and longest coding sequence-adjoining 5′/3′ untranslated regions. RNAModR then evaluates transcript-level spatial enrichment of m6A and suitable null sites relative to polyadenylation sites using multiple Fisher’s exact tests to calculate odds ratios
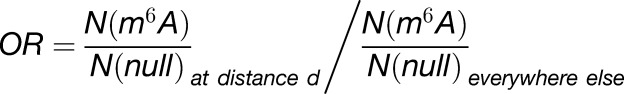 |
and test against a null hypothesis corresponding to OR = 1. Here, d corresponds to the minimum distance between an m6A or null site and all polyadenylation sites in the same transcript
The total number of m6A and null sites, respectively, at distance d is denoted by N.
Resulting P values were corrected for multiple hypotheses testing using the method of Benjamini and Hochberg (Benjamini and Hochberg, 1995). Suitable null sites were constructed within RNAModR from the genomic loci of all nonmethylated adenosines in transcripts that contain at least one m6A site.
Accession Numbers
Sequence data from this article can be found in the Arabidopsis Genome Initiative or GenBank/EMBL databases under the accession numbers listed in Supplemental Data Set 1.
Supplemental Data
Supplemental Figure 1. Optimization of the Arabidopsis mRNA interactome capture protocol.
Supplemental Figure 2. GO enrichment analysis of At-RBPs and candidate At-RBPs.
Supplemental Figure 3. Conservation of RBPs across kingdoms.
Supplemental Figure 4. Analysis of amino acid enrichment/depletion in the Arabidopsis mRNA interactome.
Supplemental Table 1. Zinc-finger proteins not associated with RNA binding identified by mRNA interactome capture.
Supplemental Table 2. Examples of noncanonical RBPs identified by mRNA interactome capture.
Supplemental Data Set 1. Arabidopsis mRNA interactome.
Supplemental Data Set 2. Peptides identified by mRNA interactome capture.
NOTE ADDED IN PROOF
During the revision of this manuscript, another article (Marondedze et al., 2016) using mRNA interactome capture on Arabidopsis was published. Here, interactome capture was performed on a variety of cell types, mainly suspension cell cultures of roots from Arabidopsis (Columbia-0) and Landsberg erecta, and to a lesser extent on 4-week-old Arabidopsis leaves. In terms of classes or families of proteins, there is not a tight overlap between the two data sets. For instance, unlike our data set, they identified many more enzymes of intermediate metabolism as potentially RNA binding but did not identify classes such as DUF1296 or WHIRLY proteins.
Marondedze, C., Thomas, L., Serrano, N.L., Lilley, K.S., and Gehring, C. (2016) The RNA-binding protein repertoire of Arabidopsis thaliana. Sci. Rep. 6: 29766.
Supplementary Material
Acknowledgments
We thank Christian Schmitz-Linneweber and Barry Pogson for kindly providing the CP29A and SAL1 antibody, respectively. We also thank the EMBL Proteomics core facility for the MS analysis and Chris Cazzonelli for constructive discussions at the initial phase of the project. This work was supported by an EMBO short-term fellowship and an International ANU PhD scholarship to M. Reichel, a John Stocker Postdoctoral Fellowship (PF14-079) from the Science and Industry Endowment Fund to Y.L., an ERC Advanced Grant (ERC-2011-ADG_20110310) to M.W.H., a grant from the National Health and Medical Research Council of Australia (#1045417) to T.P. and M.W.H., and an Australian Research Council Discovery Grant (DP110103493) to A.A.M.
AUTHOR CONTRIBUTIONS
M. Reichel, Y.L., M.W.H., T.P., and A.A.M. designed the project. M. Reichel and Y.L. performed the majority of experiments with assistance from A.-M.A. M. Rettel carried out the MS experiments and initial data analysis. C.R. performed the bioinformatic analysis of the MS data. M.E. carried out the meta-transcript analysis of m6A and poly(A) sites and the motif search. R.H. gave conceptual advice and assisted with experiments. M. Reichel, Y.L., T.P., and A.A.M. wrote the manuscript with input from all authors. All authors approved the manuscript.
Glossary
- RBP
RNA binding protein
- sRNA
small RNA
- RBD
RNA binding domain
- MS
mass spectrometry
- CL
cross-link
- noCL
non-cross-linked
- FDR
false discovery rate
- GO
Gene Ontology
- RP
ribosomal protein
- EJC
exon-junction complex
- NMD
nonsense-mediated decay
- PAC
polyadenylation cluster
- NUE
near upstream element
- RNP
ribonucleoprotein
- TCA
trichloroacetic acid
References
- Aravind L., Iyer L.M., Anantharaman V. (2003). The two faces of Alba: the evolutionary connection between proteins participating in chromatin structure and RNA metabolism. Genome Biol. 4: R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au P.C., Helliwell C., Wang M.B. (2014). Characterizing RNA-protein interaction using cross-linking and metabolite supplemented nuclear RNA-immunoprecipitation. Mol. Biol. Rep. 41: 2971–2977. [DOI] [PubMed] [Google Scholar]
- Bailey T.L. (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz A.G., et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 46: 674–690. [DOI] [PubMed] [Google Scholar]
- Bannikova O., Zywicki M., Marquez Y., Skrahina T., Kalyna M., Barta A. (2013). Identification of RNA targets for the nuclear multidomain cyclophilin atCyp59 and their effect on PPIase activity. Nucleic Acids Res. 41: 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann B.M., Horos R., Fischer B., Castello A., Eichelbaum K., Alleaume A.M., Schwarzl T., Curk T., Foehr S., Huber W., Krijgsveld J., Hentze M.W. (2015). The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 6: 10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S.D., Botting C.H., Wardleworth B.N., Jackson S.P., White M.F. (2002). The interaction of Alba, a conserved archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science 296: 148–151. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57: 289–300. [Google Scholar]
- Biela A., Grote K., Otto B., Hoth S., Hedrich R., Kaldenhoff R. (1999). The Nicotiana tabacum plasma membrane aquaporin NtAQP1 is mercury-insensitive and permeable for glycerol. Plant J. 18: 565–570. [DOI] [PubMed] [Google Scholar]
- Bodi Z., Zhong S., Mehra S., Song J., Graham N., Li H., May S., Fray R.G. (2012). Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front. Plant Sci. 3: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bono F., Ebert J., Lorentzen E., Conti E. (2006). The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126: 713–725. [DOI] [PubMed] [Google Scholar]
- Bravo J., Aguilar-Henonin L., Olmedo G., Guzmán P. (2005). Four distinct classes of proteins as interaction partners of the PABC domain of Arabidopsis thaliana poly(A)-binding proteins. Mol. Genet. Genomics 272: 651–665. [DOI] [PubMed] [Google Scholar]
- Bruggeman Q., Garmier M., de Bont L., Soubigou-Taconnat L., Mazubert C., Benhamed M., Raynaud C., Bergounioux C., Delarue M. (2014). The polyadenylation factor subunit CLEAVAGE AND POLYADENYLATION SPECIFICITY FACTOR30: a key factor of programmed cell death and a regulator of immunity in Arabidopsis. Plant Physiol. 165: 732–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock S.L. (2011). Messengers, motors and mysteries: sorting of eukaryotic mRNAs by cytoskeletal transport. Biochem. Soc. Trans. 39: 1161–1165. [DOI] [PubMed] [Google Scholar]
- Burgess A., David R., Searle I.R. (2016). Deciphering the epitranscriptome: A green perspective. J. Integr. Plant Biol. 58: 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush M.S., Crowe N., Zheng T., Doonan J.H. (2015). The RNA helicase, eIF4A-1, is required for ovule development and cell size homeostasis in Arabidopsis. Plant J. 84: 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabretta S., Richard S. (2015). Emerging roles of disordered sequences in RNA-binding proteins. Trends Biochem. Sci. 40: 662–672. [DOI] [PubMed] [Google Scholar]
- Cappadocia L., Maréchal A., Parent J.S., Lepage E., Sygusch J., Brisson N. (2010). Crystal structures of DNA-Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell 22: 1849–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal J.J., Candia A.N., Sellaro R. (2014). Light perception and signalling by phytochrome A. J. Exp. Bot. 65: 2835–2845. [DOI] [PubMed] [Google Scholar]
- Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M., Krijgsveld J., Hentze M.W. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406. [DOI] [PubMed] [Google Scholar]
- Chakrabarti M., Hunt A.G. (2015). CPSF30 at the interface of alternative polyadenylation and cellular signaling in plants. Biomolecules 5: 1151–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.H., Tong L. (2012). Mitochondrial poly(A) polymerase and polyadenylation. Biochim. Biophys. Acta 1819: 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi S.W., Zang J.B., Mele A., Darnell R.B. (2009). Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciftci-Yilmaz S., Mittler R. (2008). The zinc finger network of plants. Cell. Mol. Life Sci. 65: 1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colcombet J., Lopez-Obando M., Heurtevin L., Bernard C., Martin K., Berthomé R., Lurin C. (2013). Systematic study of subcellular localization of Arabidopsis PPR proteins confirms a massive targeting to organelles. RNA Biol. 10: 1557–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dai Y., Li W., An L. (2016). NMD mechanism and the functions of Upf proteins in plant. Plant Cell Rep. 35: 5–15. [DOI] [PubMed] [Google Scholar]
- Delaney K.J., Xu R., Zhang J., Li Q.Q., Yun K.Y., Falcone D.L., Hunt A.G. (2006). Calmodulin interacts with and regulates the RNA-binding activity of an Arabidopsis polyadenylation factor subunit. Plant Physiol. 140: 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G., Adam S.A., Choi Y.D. (1984). Physical change in cytoplasmic messenger ribonucleoproteins in cells treated with inhibitors of mRNA transcription. Mol. Cell. Biol. 4: 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers M., Shafik A., Schumann U., Preiss T. (2016). RNAModR: Functional analysis of mRNA modifications in R. bioRxiv, http://dx.doi.org/10.1101/080051. [Google Scholar]
- Falahzadeh K., Banaei-Esfahani A., Shahhoseini M. (2015). The potential roles of actin in the nucleus. Cell J. 17: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folta K.M., Kaufman L.S. (2003). Phototropin 1 is required for high-fluence blue-light-mediated mRNA destabilization. Plant Mol. Biol. 51: 609–618. [DOI] [PubMed] [Google Scholar]
- Forterre P., Confalonieri F., Knapp S. (1999). Identification of the gene encoding archeal-specific DNA-binding proteins of the Sac10b family. Mol. Microbiol. 32: 669–670. [DOI] [PubMed] [Google Scholar]
- Francischini C.W., Quaggio R.B. (2009). Molecular characterization of Arabidopsis thaliana PUF proteins--binding specificity and target candidates. FEBS J. 276: 5456–5470. [DOI] [PubMed] [Google Scholar]
- Fray R.G., Simpson G.G. (2015). The Arabidopsis epitranscriptome. Curr. Opin. Plant Biol. 27: 17–21. [DOI] [PubMed] [Google Scholar]
- Frei dit Frey N., Muller P., Jammes F., Kizis D., Leung J., Perrot-Rechenmann C., Bianchi M.W. (2010). The RNA binding protein Tudor-SN is essential for stress tolerance and stabilizes levels of stress-responsive mRNAs encoding secreted proteins in Arabidopsis. Plant Cell 22: 1575–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribourg S., Braun I.C., Izaurralde E., Conti E. (2001). Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol. Cell 8: 645–656. [DOI] [PubMed] [Google Scholar]
- Fujiki Y., Yoshikawa Y., Sato T., Inada N., Ito M., Nishida I., Watanabe A. (2001). Dark-inducible genes from Arabidopsis thaliana are associated with leaf senescence and repressed by sugars. Physiol. Plant. 111: 345–352. [DOI] [PubMed] [Google Scholar]
- Gagnon J.A., Mowry K.L. (2011). Molecular motors: directing traffic during RNA localization. Crit. Rev. Biochem. Mol. Biol. 46: 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar M., Bousser A., Sissoëff I., Roche O., Hoarau J., Mahé A. (2003). Cloning and characterization of ZmPIP1–5b, an aquaporin transporting water and urea. Plant Sci. 165: 21–31. [Google Scholar]
- Gissot M., Walker R., Delhaye S., Alayi T.D., Huot L., Hot D., Callebaut I., Schaeffer-Reiss C., Dorsselaer A.V., Tomavo S. (2013). Toxoplasma gondii Alba proteins are involved in translational control of gene expression. J. Mol. Biol. 425: 1287–1301. [DOI] [PubMed] [Google Scholar]
- Gosai S.J., Foley S.W., Wang D., Silverman I.M., Selamoglu N., Nelson A.D., Beilstein M.A., Daldal F., Deal R.B., Gregory B.D. (2015). Global analysis of the RNA-protein interaction and RNA secondary structure landscapes of the Arabidopsis nucleus. Mol. Cell 57: 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg J.R. (1979). Ultraviolet light-induced crosslinking of mRNA to proteins. Nucleic Acids Res. 6: 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R., Xue H., Huang L. (2003). Ssh10b, a conserved thermophilic archaeal protein, binds RNA in vivo. Mol. Microbiol. 50: 1605–1615. [DOI] [PubMed] [Google Scholar]
- Hecht V., Stiefel V., Delseny M., Gallois P. (1997). A new Arabidopsis nucleic-acid-binding protein gene is highly expressed in dividing cells during development. Plant Mol. Biol. 34: 119–124. [DOI] [PubMed] [Google Scholar]
- Henics T., Nagy E., Oh H.J., Csermely P., von Gabain A., Subjeck J.R. (1999). Mammalian Hsp70 and Hsp110 proteins bind to RNA motifs involved in mRNA stability. J. Biol. Chem. 274: 17318–17324. [DOI] [PubMed] [Google Scholar]
- Hofmann W.A. (2009). Cell and molecular biology of nuclear actin. Int. Rev. Cell Mol. Biol. 273: 219–263. [DOI] [PubMed] [Google Scholar]
- Holm L.M., Jahn T.P., Møller A.L., Schjoerring J.K., Ferri D., Klaerke D.A., Zeuthen T. (2005). NH3 and NH4+ permeability in aquaporin-expressing Xenopus oocytes. Pflugers Arch. 450: 415–428. [DOI] [PubMed] [Google Scholar]
- Huang Y.W., Hu C.C., Liou M.R., Chang B.Y., Tsai C.H., Meng M., Lin N.S., Hsu Y.H. (2012). Hsp90 interacts specifically with viral RNA and differentially regulates replication initiation of Bamboo mosaic virus and associated satellite RNA. PLoS Pathog. 8: e1002726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C.S., Foehr S., Garfield D.A., Furlong E.E., Steinmetz L.M., Krijgsveld J. (2014). Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10: 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh S.U., Paek K.H. (2014). APUM5, encoding a Pumilio RNA binding protein, negatively regulates abiotic stress responsive gene expression. BMC Plant Biol. 14: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh S.U., Kim M.J., Paek K.H. (2013). Arabidopsis Pumilio protein APUM5 suppresses Cucumber mosaic virus infection via direct binding of viral RNAs. Proc. Natl. Acad. Sci. USA 110: 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A.G., Xing D., Li Q.Q. (2012). Plant polyadenylation factors: conservation and variety in the polyadenylation complex in plants. BMC Genomics 13: 641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R.P. (1999). RNA-cytoskeletal associations. FASEB J. 13: 455–466. [PubMed] [Google Scholar]
- Järvelin A.I., Noerenberg M., Davis I., Castello A. (2016). The new (dis)order in RNA regulation. Cell Commun. Signal. 14: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinska C., Conroy M.J., Craven C.J., Hounslow A.M., Bullough P.A., Waltho J.P., Taylor G.L., White M.F. (2005). Obligate heterodimerization of the archaeal Alba2 protein with Alba1 provides a mechanism for control of DNA packaging. Structure 13: 963–971. [DOI] [PubMed] [Google Scholar]
- Jeong E., Kim H., Lee S.W., Han K. (2003). Discovering the interaction propensities of amino acids and nucleotides from protein-RNA complexes. Mol. Cells 16: 161–167. [PubMed] [Google Scholar]
- Jung H.J., Park S.J., Kang H. (2013). Regulation of RNA metabolism in plant development and stress responses. J. Plant Biol. 56: 123–129. [Google Scholar]
- Kadrmas J.L., Beckerle M.C. (2004). The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 5: 920–931. [DOI] [PubMed] [Google Scholar]
- Katahira J., Dimitrova L., Imai Y., Hurt E. (2015). NTF2-like domain of Tap plays a critical role in cargo mRNA recognition and export. Nucleic Acids Res. 43: 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim V.N., Kataoka N., Dreyfuss G. (2001). Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon-exon junction complex. Science 293: 1832–1836. [DOI] [PubMed] [Google Scholar]
- Kim Y.O., Pan S., Jung C.H., Kang H. (2007a). A zinc finger-containing glycine-rich RNA-binding protein, atRZ-1a, has a negative impact on seed germination and seedling growth of Arabidopsis thaliana under salt or drought stress conditions. Plant Cell Physiol. 48: 1170–1181. [DOI] [PubMed] [Google Scholar]
- Kim J.Y., Park S.J., Jang B., Jung C.H., Ahn S.J., Goh C.H., Cho K., Han O., Kang H. (2007b). Functional characterization of a glycine-rich RNA-binding protein 2 in Arabidopsis thaliana under abiotic stress conditions. Plant J. 50: 439–451. [DOI] [PubMed] [Google Scholar]
- Krause K., Kilbienski I., Mulisch M., Rödiger A., Schäfer A., Krupinska K. (2005). DNA-binding proteins of the Whirly family in Arabidopsis thaliana are targeted to the organelles. FEBS Lett. 579: 3707–3712. [DOI] [PubMed] [Google Scholar]
- Kwak K.J., Kim Y.O., Kang H. (2005). Characterization of transgenic Arabidopsis plants overexpressing GR-RBP4 under high salinity, dehydration, or cold stress. J. Exp. Bot. 56: 3007–3016. [DOI] [PubMed] [Google Scholar]
- Kwon S.C., Yi H., Eichelbaum K., Föhr S., Fischer B., You K.T., Castello A., Krijgsveld J., Hentze M.W., Kim V.N. (2013). The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 20: 1122–1130. [DOI] [PubMed] [Google Scholar]
- Laohavisit A., Davies J.M. (2011). Annexins. New Phytol. 189: 40–53. [DOI] [PubMed] [Google Scholar]
- Leder V., Lummer M., Tegeler K., Humpert F., Lewinski M., Schüttpelz M., Staiger D. (2014). Mutational definition of binding requirements of an hnRNP-like protein in Arabidopsis using fluorescence correlation spectroscopy. Biochem. Biophys. Res. Commun. 453: 69–74. [DOI] [PubMed] [Google Scholar]
- Lee H.W., Park J.H., Park M.Y., Kim J. (2014). GIP1 may act as a coactivator that enhances transcriptional activity of LBD18 in Arabidopsis. J. Plant Physiol. 171: 14–18. [DOI] [PubMed] [Google Scholar]
- Lee S., Lee E.J., Yang E.J., Lee J.E., Park A.R., Song W.H., Park O.K. (2004). Proteomic identification of annexins, calcium-dependent membrane binding proteins that mediate osmotic stress and abscisic acid signal transduction in Arabidopsis. Plant Cell 16: 1378–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune D., Delsaux N., Charloteaux B., Thomas A., Brasseur R. (2005). Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins 61: 258–271. [DOI] [PubMed] [Google Scholar]
- Li D., Zhang H., Hong Y., Huang L., Li X., Zhang Y., Ouyang Z., Song F. (2014). Genome-wide identification, biochemical characterization, and expression analyses of the YTH domain-containing RNA-binding protein family in Arabidopsis and rice. Plant Mol. Biol. Report. 32: 1169–1186. [Google Scholar]
- Li W., Ma M., Feng Y., Li H., Wang Y., Ma Y., Li M., An F., Guo H. (2015). EIN2-directed translational regulation of ethylene signaling in Arabidopsis. Cell 163: 670–683. [DOI] [PubMed] [Google Scholar]
- Liao Y., et al. (2016). The cardiomyocyte RNA-binding proteome: links to intermediary metabolism and heart disease. Cell Reports 16: 1456–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke J.C., Stahlberg E.A., Strenski D.G., Haas B.J., Wood P.C., Li Q.Q. (2005). Compilation of mRNA polyadenylation signals in Arabidopsis revealed a new signal element and potential secondary structures. Plant Physiol. 138: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorković Z.J. (2009). Role of plant RNA-binding proteins in development, stress response and genome organization. Trends Plant Sci. 14: 229–236. [DOI] [PubMed] [Google Scholar]
- Lorković Z.J., Barta A. (2002). Genome analysis: RNA recognition motif (RRM) and K homology (KH) domain RNA-binding proteins from the flowering plant Arabidopsis thaliana. Nucleic Acids Res. 30: 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G.-Z., MacQueen A., Zheng G., Duan H., Dore L.C., Lu Z., Liu J., Chen K., Jia G., Bergelson J., He C. (2014). Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 5: 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Tang Z., Qin J., Meng Y. (2015). The use of high-throughput sequencing methods for plant microRNA research. RNA Biol. 12: 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macknight R., Bancroft I., Page T., Lister C., Schmidt R., Love K., Westphal L., Murphy G., Sherson S., Cobbett C., Dean C. (1997). FCA, a gene controlling flowering time in Arabidopsis, encodes a protein containing RNA-binding domains. Cell 89: 737–745. [DOI] [PubMed] [Google Scholar]
- Mair G.R., Lasonder E., Garver L.S., Franke-Fayard B.M., Carret C.K., Wiegant J.C., Dirks R.W., Dimopoulos G., Janse C.J., Waters A.P. (2010). Universal features of post-transcriptional gene regulation are critical for Plasmodium zygote development. PLoS Pathog. 6: e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani J., Güttinger A., Schimanski B., Heller M., Acosta-Serrano A., Pescher P., Späth G., Roditi I. (2011). Alba-domain proteins of Trypanosoma brucei are cytoplasmic RNA-binding proteins that interact with the translation machinery. PLoS One 6: e22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A., Parent J.S., Véronneau-Lafortune F., Joyeux A., Lang B.F., Brisson N. (2009). Whirly proteins maintain plastid genome stability in Arabidopsis. Proc. Natl. Acad. Sci. USA 106: 14693–14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matia-González A.M., Laing E.E., Gerber A.P. (2015). Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 22: 1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurel C., Boursiac Y., Luu D.T., Santoni V., Shahzad Z., Verdoucq L. (2015). Aquaporins in plants. Physiol. Rev. 95: 1321–1358. [DOI] [PubMed] [Google Scholar]
- Merchante C., Brumos J., Yun J., Hu Q., Spencer K.R., Enríquez P., Binder B.M., Heber S., Stepanova A.N., Alonso J.M. (2015). Gene-specific translation regulation mediated by the hormone-signaling molecule EIN2. Cell 163: 684–697. [DOI] [PubMed] [Google Scholar]
- Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S.F., Jain S., She M., Parker R. (2013). Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 20: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C.D., Mansfield R.E., Leung W., Vaz P.M., Loughlin F.E., Grant R.P., Mackay J.P. (2011). Characterization of a family of RanBP2-type zinc fingers that can recognize single-stranded RNA. J. Mol. Biol. 407: 273–283. [DOI] [PubMed] [Google Scholar]
- Nolte C., Staiger D. (2015). RNA around the clock - regulation at the RNA level in biological timing. Front. Plant Sci. 6: 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyikó T., Kerényi F., Szabadkai L., Benkovics A.H., Major P., Sonkoly B., Mérai Z., Barta E., Niemiec E., Kufel J., Silhavy D. (2013). Plant nonsense-mediated mRNA decay is controlled by different autoregulatory circuits and can be induced by an EJC-like complex. Nucleic Acids Res. 41: 6715–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ok S.H., Jeong H.J., Bae J.M., Shin J.S., Luan S., Kim K.N. (2005). Novel CIPK1-associated proteins in Arabidopsis contain an evolutionarily conserved C-terminal region that mediates nuclear localization. Plant Physiol. 139: 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik I., Yang S., Choi G. (2012). Phytochrome regulates translation of mRNA in the cytosol. Proc. Natl. Acad. Sci. USA 109: 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papuga J., Hoffmann C., Dieterle M., Moes D., Moreau F., Tholl S., Steinmetz A., Thomas C. (2010). Arabidopsis LIM proteins: a family of actin bundlers with distinct expression patterns and modes of regulation. Plant Cell 22: 3034–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashev I.G., Dimitrov S.I., Angelov D. (1991). Crosslinking proteins to nucleic acids by ultraviolet laser irradiation. Trends Biochem. Sci. 16: 323–326. [DOI] [PubMed] [Google Scholar]
- Peña E.J., Heinlein M. (2012). RNA transport during TMV cell-to-cell movement. Front. Plant Sci. 3: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev A.V., Kolupaeva V.G., Yusupov M.M., Hellen C.U., Pestova T.V. (2008). Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J. 27: 1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prikryl J., Watkins K.P., Friso G., van Wijk K.J., Barkan A. (2008). A member of the Whirly family is a multifunctional RNA- and DNA-binding protein that is essential for chloroplast biogenesis. Nucleic Acids Res. 36: 5152–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley F., Rosenberg J.M., Shachar-Hill Y., Bohnert H.J. (2002). From genome to function: the Arabidopsis aquaporins. Genome Biol. 3: research0001.0001–research0001.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015). R: A Language and Environment for Statistical Computing. (Vienna, Austria: R Foundation for Statistical Computing).
- Ribbeck K., Lipowsky G., Kent H.M., Stewart M., Görlich D. (1998). NTF2 mediates nuclear import of Ran. EMBO J. 17: 6587–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles P., Fleury D., Candela H., Cnops G., Alonso-Peral M.M., Anami S., Falcone A., Caldana C., Willmitzer L., Ponce M.R., Van Lijsebettens M., Micol J.L. (2010). The RON1/FRY1/SAL1 gene is required for leaf morphogenesis and venation patterning in Arabidopsis. Plant Physiol. 152: 1357–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg F.M., Patton D.A., Meinke D.W., Amasino R.M. (2001). FPA, a gene involved in floral induction in Arabidopsis, encodes a protein containing RNA-recognition motifs. Plant Cell 13: 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann U., Shafik A., Preiss T. (2016). METTL3 Gains R/W access to the epitranscriptome. Mol. Cell 62: 323–324. [DOI] [PubMed] [Google Scholar]
- Schwartz S. (2016). Cracking the epitranscriptome. RNA 22: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikhali J. (2015). GIP1 protein is a novel cofactor that regulates DNA-binding affinity of redox-regulated members of bZIP transcription factors involved in the early stages of Arabidopsis development. Protoplasma 252: 867–883. [DOI] [PubMed] [Google Scholar]
- Sherstnev A., Duc C., Cole C., Zacharaki V., Hornyik C., Ozsolak F., Milos P.M., Barton G.J., Simpson G.G. (2012). Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 19: 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata H., Hanada K., Ushijima T., Nakashima M., Suzuki Y., Matsushita T. (2014). Phytochrome controls alternative splicing to mediate light responses in Arabidopsis. Proc. Natl. Acad. Sci. USA 111: 18781–18786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski P.J., Zuber H., Philippe L., Sement F.M., Canaday J., Kufel J., Gagliardi D., Lange H. (2015). Distinct 18S rRNA precursors are targets of the exosome complex, the exoribonuclease RRP6L2 and the terminal nucleotidyltransferase TRL in Arabidopsis thaliana. Plant J. 83: 991–1004. [DOI] [PubMed] [Google Scholar]
- Silverman I.M., Li F., Gregory B.D. (2013). Genomic era analyses of RNA secondary structure and RNA-binding proteins reveal their significance to post-transcriptional regulation in plants. Plant Sci. 205-206: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G., Pratt G., Yeo G.W., Moore M.J. (2015). The clothes make the mRNA: past and present trends in mRNP fashion. Annu. Rev. Biochem. 84: 325–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G.K. (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3: Article3. [DOI] [PubMed] [Google Scholar]
- Sonnhammer E.L., Östlund G. (2015). InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 43: D234–D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subota I., Rotureau B., Blisnick T., Ngwabyt S., Durand-Dubief M., Engstler M., Bastin P. (2011). ALBA proteins are stage regulated during trypanosome development in the tsetse fly and participate in differentiation. Mol. Biol. Cell 22: 4205–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchanek M., Radzikowska A., Thiele C. (2005). Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat. Methods 2: 261–267. [DOI] [PubMed] [Google Scholar]
- Sysoev V.O., Fischer B., Frese C.K., Gupta I., Krijgsveld J., Hentze M.W., Castello A., Ephrussi A. (2016). Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in Drosophila. Nat. Commun. 7: 12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandapani P., O’Connor T.R., Bailey T.L., Richard S. (2013). Defining the RGG/RG motif. Mol. Cell 50: 613–623. [DOI] [PubMed] [Google Scholar]
- Theillet F.-X., Kalmar L., Tompa P., Han K.-H., Selenko P., Dunker A.K., Daughdrill G.W., Uversky V.N. (2013). The alphabet of intrinsic disorder. Intrinsically Disord. Proteins 1: e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.E., Wu X., Liu M., Gaffney B., Ji G., Li Q.Q., Hunt A.G. (2012). Genome-wide control of polyadenylation site choice by CPSF30 in Arabidopsis. Plant Cell 24: 4376–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremousaygue D., Manevski A., Bardet C., Lescure N., Lescure B. (1999). Plant interstitial telomere motifs participate in the control of gene expression in root meristems. Plant J. 20: 553–561. [DOI] [PubMed] [Google Scholar]
- Tripurani S.K., Nakaminami K., Thompson K.B., Crowell S.V., Guy C.L., Karlson D.T. (2011). Spatial and temporal expression of cold-responsive DEAD-box RNA helicases reveals their functional roles during embryogenesis in Arabidopsis thaliana. Plant Mol. Biol. Report. 29: 761–768. [Google Scholar]
- Tsuda K., Tsuji T., Hirose S., Yamazaki K. (2004). Three Arabidopsis MBF1 homologs with distinct expression profiles play roles as transcriptional co-activators. Plant Cell Physiol. 45: 225–231. [DOI] [PubMed] [Google Scholar]
- Uehlein N., Lovisolo C., Siefritz F., Kaldenhoff R. (2003). The tobacco aquaporin NtAQP1 is a membrane CO2 pore with physiological functions. Nature 425: 734–737. [DOI] [PubMed] [Google Scholar]
- Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B. (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215. [DOI] [PubMed] [Google Scholar]
- Vandevenne M., O’Connell M.R., Helder S., Shepherd N.E., Matthews J.M., Kwan A.H., Segal D.J., Mackay J.P. (2014). Engineering specificity changes on a RanBP2 zinc finger that binds single-stranded RNA. Angew. Chem. Int. Ed. Engl. 53: 7848–7852. [DOI] [PubMed] [Google Scholar]
- Wagenmakers A.J., Reinders R.J., van Venrooij W.J. (1980). Cross-linking of mRNA to proteins by irradiation of intact cells with ultraviolet light. Eur. J. Biochem. 112: 323–330. [DOI] [PubMed] [Google Scholar]
- Wan Y., Tang K., Zhang D., Xie S., Zhu X., Wang Z., Lang Z. (2015). Transcriptome-wide high-throughput deep m(6)A-seq reveals unique differential m(6)A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 16: 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Vinocur B., Shoseyov O., Altman A. (2004). Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 9: 244–252. [DOI] [PubMed] [Google Scholar]
- Wang Y., Mercier R., Hobman T.C., LaPointe P. (2013). Regulation of RNA interference by Hsp90 is an evolutionarily conserved process. Biochim. Biophys. Acta 1833: 2673–2681. [DOI] [PubMed] [Google Scholar]
- Warner J.R., McIntosh K.B. (2009). How common are extraribosomal functions of ribosomal proteins? Mol. Cell 34: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels H.H., Imami K., Baltz A.G., Kolinski M., Beldovskaya A., Selbach M., Small S., Ohler U., Landthaler M. (2016). The mRNA-bound proteome of the early fly embryo. Genome Res. 26: 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R.M., Obradovi, Z., Mathura, V., Braun, W., Garner, E.C., Young, J., Takayama, S., Brown, C.J., Dunker, A.K. (2001). The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomp. 2001: 89–100. [DOI] [PubMed] [Google Scholar]
- Wright P.E., Dyson H.J. (2015). Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Liu M., Downie B., Liang C., Ji G., Li Q.Q., Hunt A.G. (2011). Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 108: 12533–12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D., Wang Y., Hamilton M., Ben-Hur A., Reddy A.S. (2015). Transcriptome-wide identification of RNA targets of Arabidopsis SERINE/ARGININE-RICH45 uncovers the unexpected roles of this RNA binding protein in RNA processing. Plant Cell 27: 3294–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Xu M. (2012). Actin bundler PLIM2s are involved in the regulation of pollen development and tube growth in Arabidopsis. J. Plant Physiol. 169: 516–522. [DOI] [PubMed] [Google Scholar]
- Zhang J., Addepalli B., Yun K.-Y., Hunt A.G., Xu R., Rao S., Li Q.Q., Falcone D.L. (2008). A polyadenylation factor subunit implicated in regulating oxidative signaling in Arabidopsis thaliana. PLoS One 3: e2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Gu L., Hou Y., Wang L., Deng X., Hang R., Chen D., Zhang X., Zhang Y., Liu C., Cao X. (2015). Integrative genome-wide analysis reveals HLP1, a novel RNA-binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Res. 25: 864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.