

Abstract
Maladaptive epithelial repair from chronic injury is a common feature in fibrotic diseases, which in turn activates a pathogenic fibroblast response that produces excessive matrix deposition. Dysregulated microRNAs (miRs) can regulate expression of multiple genes and fundamentally alter cellular phenotypes during fibrosis. Although several miRs have been shown to be associated with lung fibrosis, the mechanisms by which miRs modulate epithelial behavior in lung fibrosis are lacking. Here, we identified miR-323a-3p to be downregulated in the epithelium of lungs with bronchiolitis obliterans syndrome (BOS) after lung transplantation, idiopathic pulmonary fibrosis (IPF), and murine bleomycin-induced fibrosis. Antagomirs for miR-323a-3p augment, and mimics suppress, murine lung fibrosis after bleomycin injury, indicating that this miR may govern profibrotic signals. We demonstrate that miR-323a-3p attenuates TGF-α and TGF-β signaling by directly targeting key adaptors in these important fibrogenic pathways. Moreover, miR-323a-3p lowers caspase-3 expression, thereby limiting programmed cell death from inducers of apoptosis and ER stress. Finally, we find that epithelial expression of miR-323a-3p modulates inhibitory crosstalk with fibroblasts. These studies demonstrate that miR-323a-3p has a central role in lung fibrosis that spans across murine and human disease, and downregulated expression by the lung epithelium releases inhibition of various profibrotic pathways to promote fibroproliferation.
miR-323a-3p downregulation by the epithelium releases inhibition on TGFA, SMAD2, and CASP3, thereby augmenting pro-fibrotic signals and promoting fibroproliferation.
Introduction
Pulmonary fibrosis is the sequela of various epithelial injuries that initiate a fibroproliferative cascade, leading to matrix deposition and ultimately organ failure (1). Under homeostatic conditions, the mucosal barrier naturally suppresses fibrosis (2–4). However, the injured epithelium induces pathways during the wound healing process that, when persistent, can surreptitiously create a fibroproliferative microenvironment that stokes the activation and proliferation of fibrogenic effector cells such as the fibroblast (5).
Multiple intertwined signaling pathways originating in the mucosal surface are involved in the early events of fibrosis (6). Central to this fibrogenic response is TGF-β activation (7). Although TGF-β has pleiotropic effects, the proximal signals driven by this cytokine are initiated in the lung epithelium (8, 9), and epithelial overexpression of TGF-β has been developed as models of lung fibrosis (10, 11). One of the major consequences of TGF-β signaling is the induction of gene transcription, particularly of profibrotic mediators (12). However, TGF-β also causes epithelial apoptosis, which partly contributes to the fibroproliferative response (10). Consistent with the concept that early maladaptive events start in the epithelium, other aberrant effects, such as excessive ER stress, apoptosis, Wnt signaling, or age-related phenomena (e.g., epigenetic modification, telomere shortening, cell senescence) also contribute to fibrosis (13–18). Because of the complicated nature of the fibrogenic signals, therapeutics likely will need to block multiple pathways. Indeed, nintedanib targets multiple tyrosine kinases and has been shown to slow the progression of idiopathic pulmonary fibrosis (IPF) (19). However, this treatment is not completely sufficient in halting pulmonary fibrosis, so further understanding of how multiple pathways are simultaneously activated could lead to novel treatments.
MicroRNAs (miRs) are cellular regulators that control protein expression primarily via suppression of mRNA translation (20). These small RNAs are roughly 22 nucleotides in length and can each control expression of hundreds of target genes and altogether regulate a third of the genome (21–23). Because of the ability to broadly regulate a large number of proteins, aberrant miR expression can fundamentally alter a cellular phenotype. For instance, deletion of miR-15a and miR-16-1 at 13q14.3 in chronic lymphocytic leukemia is procancerous through lost suppression of oncogenes such as BCL2, MCL1, CCND1, and WNT3A (24, 25). A number of miRs have also been associated with pulmonary fibrosis, but mechanisms by which dysregulated miRs affect epithelial behavior have been lacking (26).
Epithelial dysfunction is an upstream event that initiates a fibroproliferative cascade to recruit and promote expansion of effector cells, such as the fibroblast (1). Therefore, we speculated that altered miR expression within the epithelial compartment produces aberrant profibrotic signals that contribute to lung fibrosis. We found that miR-323a-3p was downregulated in the lung epithelium of patients with BOS and IPF and of mice with bleomycin-induced lung fibrosis. Suppression of miR-323a-3p augmented lung fibrosis in mice after bleomycin injury. Conversely, miR-323a-3p overexpression suppressed fibrosis. We demonstrated that miR-323a-3p controls several fibrogenic pathways as the mechanism by which it regulates lung fibrosis. Our results indicate that miR-323a-3p attenuates TGF-α and TGF-β signaling by directly targeting TGFA and SMAD2. CASP3 was also suppressed to prevent programmed cell death. Furthermore, epithelial overexpression of miR-323a-3p attenuated expression of matrix proteins by fibroblasts. Overall, this work demonstrates that miR-323a-3p is a key mediator of multiple profibrotic signals in the lung epithelium that control epithelial-mesenchymal crosstalk to regulate the development of lung fibrosis.
Results
The lung epithelium downregulates miR-323a-3p in fibrotic diseases.
Patients with lung transplants develop fibrotic airways (i.e., BOS) as part of chronic lung allograft dysfunction (27). We postulated that lung epithelial cells alter their miR expression as part of the pathogenesis of BOS after lung transplantation. Airway epithelial cells were procured by cytologic brushing during bronchoscopy from lung transplant patients, and over 700 miRs were measured using PCR arrays. We identified 20 miRs that had significantly lower expression in lung transplant patients with BOS compared with those without BOS (Table 1). Putative targets for these miRs were determined (Targetscan) followed by KEGG pathway analysis (Webgestalt), and enrichment for profibrotic signaling pathways such as MAPK, Wnt, insulin, ErbB, TGF-β, calcium, hedgehog, and apoptosis was identified (Table 2 and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/jci.insight.90301DS1) (7, 14, 28–32). These findings suggest that downregulation of the 20 miRs in BOS will augment signaling via fibrogenic pathways.
Table 1. miRs downregulated in the bronchial epithelium of patients with BOS compared with control lung transplant patientsA.

Table 2. Profibrotic signaling pathways predicted to be regulated by the 20 downregulated miRs in BOSA.

Although all of these miRs may have a role in the development of BOS, we focused on miR-323a-3p, which had an 18-fold decrease in expression in patients with BOS. In silico evaluation predicted that miR-323a-3p also targets profibrotic pathways such as TGF-β, Wnt, and MAPK signaling (Table 3 and Supplemental Table 2). Therefore, lower miR-323a-3p expression will augment profibrotic signals. We posited that downregulation of miR-323a-3p was not confined to airway fibrosis and may have more widespread effects in various types of pulmonary fibrosis. To test this idea, we procured alveolar type II epithelial cells from human lungs and found a 12.5-fold decrease in miR-323a-3p levels in the lung epithelium from IPF patients compared with control (Figure 1A, P < 0.05). We also evaluated the expression of miR-323a-3p in the murine lung epithelium after bleomycin-induced lung fibrosis. Consistent with our findings in BOS and IPF, miR-323a-3p expression also decreased in a dose-dependent manner in lung epithelial cells from fibrotic lungs after bleomycin treatment (Figure 1B, P < 0.05). These results indicate that miR-323a-3p downregulation consistently occurs across human and experimental murine lung fibrosis and may augment profibrotic pathways as a mechanism of fibroproliferation.
Table 3. Predicted signaling pathways regulated by miR-323a-3pA.

Figure 1. miR-323a-3p is downregulated by the lung epithelium in human and murine lung fibrosis.
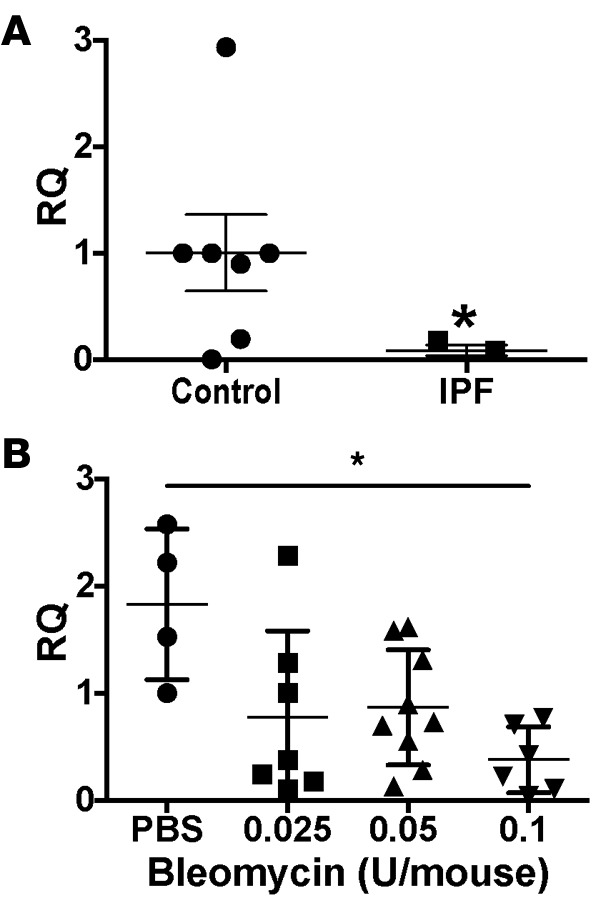
Expression of miR-323a-3p was evaluated by PCR in (A) alveolar type II cells isolated from lungs of control and idiopathic pulmonary fibrosis (IPF) patients. n = 7 control, n = 3 IPF, Student’s t test, *P < 0.05. (B) Epithelial cells were collected from bleomycin-injured lungs. n = 4 (PBS), n = 7 (0.025 U), n = 9 (0.05 U), n = 6 (0.1 U), ordinary 1-way ANOVA, *P < 0.05. RQ, relative quantification.
miR-323a-3p suppression exacerbates and overexpression attenuates lung fibrosis.
To determine if lower miR-323a-3p levels may have a causative role in fibrosis, we developed antagomirs toward miR-323a-3p and delivered them to mice after bleomycin injury (33). As predicted, miR-323a-3p antagomirs increased lung fibrosis in comparison with control antagomirs (Figure 2A). As a measurement of the fibrotic burden, we measured lung hydroxyproline levels and found higher levels in antagomir versus control conditions after bleomycin injury (Figure 2B; 72.61 ± 6.2 vs. 55.38 ± 3.7 μg/ml/lung, respectively; P < 0.0001). Notably, treatment with miR-323a-3p antagomirs without bleomycin did not induce any fibrosis, suggesting that downregulation of this miR can augment existing profibrotic signals, but alone is not sufficient to generate fibrosis. These findings demonstrate that lung fibrosis is accentuated after downregulation of miR-323a-3p.
Figure 2. miR-323a-3p antagomirs augment bleomycin-induced lung fibrosis.

(A) Representative images of H&E- and trichrome-stained lungs 21 days after bleomycin injury (0.05 U/kg) from mice treated with miR-323a-3p antagomir or a scrambled control 5, 10, and 15 days after bleomycin injury. Scale bar: 100 μm. (B) Lungs were processed for measuring hydroxyproline levels. n = 5 (PBS control and antagomir), n = 7 (control bleomycin), n = 8 (mimic bleomycin), *P < 0.05, **P < 0.005 by 2-way ANOVA and post-hoc analysis.
Next, we tested the effects of miR-323a-3p overexpression on fibrosis. In contrast with antagomirs, miR-323a-3p mimics suppressed lung fibrosis in mice after bleomycin treatment (Figure 3A). Accordingly, we found lower hydroxyproline levels and improved survival in mice given mimics compared with control (Figure 3B). These results demonstrate the ability of miR-323a-3p to suppress fibrosis after bleomycin injury.
Figure 3. miR-323a-3p mimics attenuate bleomycin-induced lung fibrosis.

(A) Representative images of H&E- and trichrome-stained lungs 21 days after bleomycin injury (0.075 U/kg) from mice treated with miR-323a-3p mimic or a scrambled control 5, 10, and 15 days after bleomycin injury. Scale bar: 100 μm. (B) Lungs were processed for measuring hydroxyproline levels. n = 9 (control), n = 11 (mimic), *P < 0.01 by Student’s t test. (C) Survival of mice treated with control or mimic after bleomycin injury, *P < 0.05.
miR-323a-3p regu lates TGF-β signaling by targeting SMAD2.
TGF-β signaling, particularly in the lung epithelium, is a central pathway involved in the development of pulmonary fibrosis (7–9). SMAD2, an important adaptor molecule in the TGF-β canonical pathway, was identified by Targetscan as a putative miR-323a-3p target. We identified 2 sequences complementary to the miR-323a-3p seed sequence in the 3′ UTR of SMAD2 (Figure 4A). To determine if miR-323a-3p regulates SMAD2 expression, we immunoblotted for SMAD2 in 16HBE14o- cells transfected with miR-323a-3p mimics and found decreased SMAD2 levels in cell lysates (Figure 4B). Next, we subcloned the SMAD2 3′ UTR distal to the luciferase ORF and determined that miR-323a-3p mimics could suppress luciferase expression (Figure 4C).
Figure 4. miR-323a-3p targets SMAD2 to suppress TGF-β signaling.

(A) miR-323a-3p binding sites in the SMAD2 3′ UTR. (B) Western blot for SMAD2 in cell lysates from 16HBE14o- cells transfected with miR-323a-3p mimics or control. A representative blot is presented from 3 individual experiments. (C) Luciferase activity of HEK cells transfected with a reporter plasmid containing a chimeric firefly luciferase with the SMAD2 3′ UTR. All data were normalized to Renilla luciferase activity. n = 3, *P < 0.001. (D and E) 16HBE14o- cells transfected with miR-323a-3p mimic or control were stimulated with TGF-β and processed for PCR for measuring levels of (D) CDH2 and (E) VIM. n = 4, *P < 0.01. Data are presented as box-and-whisker plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate 1.5 times the interquartile range from the bottom and the top of the box. Statistical analysis in this figure was performed with the Student’s t test.
Recent work has demonstrated that epithelial-mesenchymal transition does not play a role in lung fibrosis (34). However, a well-established in vitro phenomenon of TGF-β stimulation of cultured epithelial cells is the expression of mesenchymal markers (35). Therefore, we used this in vitro readout to functionally interrogate the effect of miR-323a-3p in regulating TGF-β signaling in the lung epithelium. Consistent with our other results, we found that cells transfected with miR-323a-3p mimics had lower expression of CDH2 and VIM after TGF-β stimulation in comparison with control (Figure 4, D and E). Collectively, our findings indicate that miR-323a-3p directly targets SMAD2 to blunt TGF-β signaling. Conversely, miR-323a-3p downregulation, as occurs in fibrosis, augments TGF-β signaling.
miR-323a-3p regulates TGF-α signaling by targeting TGFA.
The TGFA gene, which encodes another cytokine (TGF-α) that can mediate profibrotic effects via epithelial signaling, was also found to be a putative miR-323a-3p target via Targetscan. We identified 2 sequences in the TGFA 3′ UTR that are complementary to the miR-323a-3p seed sequence (Figure 5A). Therefore, TGFA expression was assessed in 16HBE14o- cells transfected with miR-323a-3p mimics and we found a significant decrease in mRNA (Figure 5B) and protein (Figure 5C) compared with control conditions. Moreover, miR-323a-3p suppressed luciferase expression when the TGFA 3′ UTR was subcloned distal to the luciferase ORF, confirming TGFA as a direct miR-323a-3p target (Figure 5D).
Figure 5. miR-323a-3p targets and suppresses TGFA signaling.

(A) miR-323a-3p bindings sites in the TGFA 3′ UTR. (B) qPCR, n = 6, *P < 0.0001 and (C) Western blot for TGFA from 16HBE14o- cells transfected with miR-323a-3p mimics or control. A representative blot is presented from 3 individual experiments. (D) Luciferase activity of HEK cells transfected with a reporter plasmid containing a chimeric firefly luciferase with the TGFA 3′ UTR. All data were normalized to Renilla luciferase activity. n = 3, *P < 0.05. (E) 16HBE14o- cells transfected with miR-323a-3p mimetic or control were processed for MMP9 PCR. n = 3, *P < 0.005. Data are presented as box-and-whisker plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate 1.5 times the interquartile range from the bottom and the top of the box. Statistical analysis in this figure was performed with the Student’s t test. RQ, relative quantification.
To investigate downstream TGF-α signaling, we assessed the effect of miR-323a-3p on MMP9 expression, which is induced by TGF-α in a p38-dependent manner (36, 37). Consistent with our data showing the ability of miR-323a-3p to suppress TGFA, 16HBE14o- cells had decreased levels of MMP9 expression in miR-323a-3p mimic conditions compared with control (Figure 5E). There are no sequences in the MMP9 3′ UTR that are complementary to the miR-323a-3p seed sequence, which suggests that the changes in MMP9 expression are due to the suppression of TGFA. These data confirm that miR-323a-3p can directly suppress and affect downstream TGF-α signaling.
miR-323a-3p suppresses caspase-3 to attenuate programmed cell death.
TGF-β, in addition to inducing expression of profibrotic genes, also causes epithelial apoptosis as part of the fibroproliferative mechanism (10). Therefore, we evaluated the effect of miR-323a-3p on programmed cell death. Cells treated with staurosporine, a broad protein kinase inhibitor that stimulates programmed cell death, caused less apoptosis in cells overexpressing miR-323a-3p (Figure 6A). ER stress, which has also been implicated in the pathogenesis of lung fibrosis, also causes caspase-3–mediated cell death (13, 38). Therefore, we induced the unfolded protein response by treating cells with tunicamycin, a nucleoside antibiotic that blocks N-glycosylation, and found that 16HBE14o- cells transfected with miR-323a-3p mimics had reduced apoptosis from ER stress-induced cell death (Figure 6B). We evaluated various components of the programmed cell death pathway and found that CASP3 expression was suppressed by miR-323a-3p (Figure 6, C and D). However, we could not identify any complementary miR-323a-3p binding sites in the 3′ UTR of CASP3, and subcloning of the 3′ UTR into a luciferase reporter assay did not demonstrate suppression (Figure 6E). These results indicate that miR-323a-3p downregulation augments programmed cell death in the presence of ER stress and other apoptotic stimuli by regulating CASP3 expression through indirect mechanisms.
Figure 6. miR-323a-3p attenuates apoptosis by suppressing CASP3 levels.
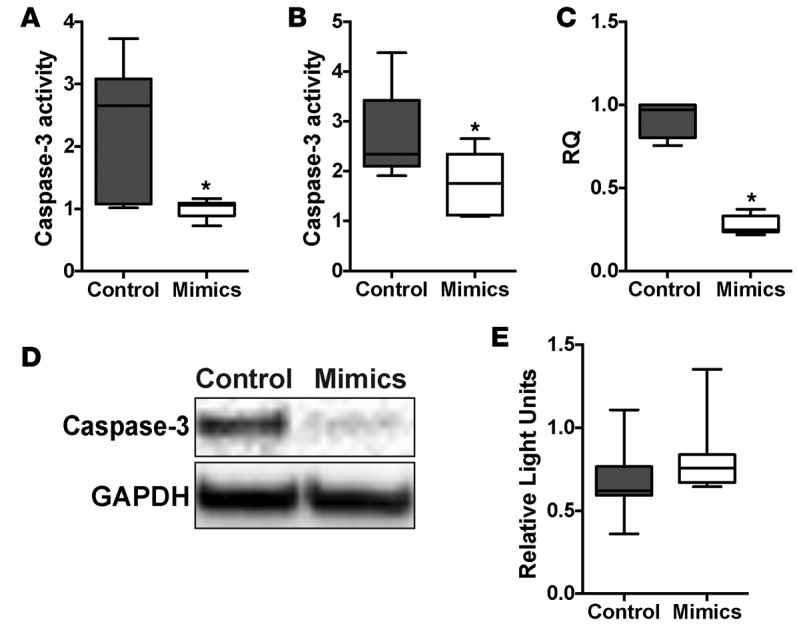
(A and B) 16HBE14o- cells transfected with miR-323a-3p mimic or control were stimulated with (A) staurosporine or (B) tunicamycin for 24 hours followed by measurement of caspase-3 activity. n = 3, *P < 0.05. (C) PCR and (D) Western blot for CASP3 in cells transfected with miR-323a-3p mimic or control. A representative blot is presented from 3 individual experiments. (E) Luciferase activity of HEK cells transfected with a reporter plasmid containing a chimeric firefly luciferase with the CASP3 3′ UTR. All data were normalized to Renilla luciferase activity. n = 3. Data are presented as box-and-whisker plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate 1.5 times the interquartile range from the bottom and the top of the box. Statistical analysis in this figure was performed with the Student’s t test. RQ, relative quantification.
Epithelial-mesenchymal cell crosstalk is regulated by miR-323a-3p.
Previous publications showed baseline suppression of fibroblast activation by the epithelium (2–4). Because miR-323a-3p can regulate several profibrotic pathways, we hypothesized that miR-323a-3p modulates the intimate interaction between epithelial and mesenchymal cells. Consistent with published findings, conditioned medium from epithelial cultures suppressed fibroblast activation with suppressed expression of α-smooth muscle actin (α-SMA) (Figure 7A), type I collagen (Figure 7B), type III collagen (Figure 7C), and fibronectin (Figure 7D) in cultured fibroblasts. More importantly, overexpression of miR-323a-3p in epithelial cells augmented these suppressive effects. Similarly, conditioned medium from epithelial cells transfected with miR-323a-3p mimics attenuated α-SMA immunostaining (Figure 7E) and F-actin formation (Figure 7F) in fibroblasts. Therefore, attenuated miR-323a-3p expression releases these inhibitory effects on fibroblasts and results in higher expression of fibroproliferative genes.
Figure 7. Epithelial suppression of fibroblasts is augmented by miR-323a-3p.

Conditioned medium from 16HBE14o- cells expressing either miR-323a-3p or control were incubated with primary human fibroblasts and compared with untreated fibroblasts. Expression of (A) α-SMA (ACTA2), (B) type I collagen (COL1A1), (C) type III collagen (COL3A1), and (D) fibronectin (FN1) was measured by PCR in fibroblasts. n = 4, *P < 0.005. (E) Immunofluorescence staining for α-SMA and (F) phalloidin in fibroblasts treated with epithelial cell–conditioned medium. Scale bar: 100 μm. Data are presented as whisker-box plots. The central horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate 1.5 times the interquartile range from the bottom and the top of the box. Statistical analysis in this figure was performed with a 1-way ANOVA. RQ, relative quantification.
Discussion
Pulmonary fibrosis is a complex disease that involves multiple fibrogenic signals causing a malignant interplay between various cells, leading to scar formation and tissue destruction (5). Here, we demonstrate that miR-323a-3p has a central role in regulating the epithelial-mesenchymal interaction during pulmonary fibrosis. Our findings indicate that miR-323a-3p regulates TGF-α, TGF-β, and apoptosis by targeting key mediators in these profibrotic pathways. Thus, the downregulation of this miR, which we found occurs within the lung epithelium in various instances of lung fibrosis, releases their inhibitory effects on TGFA, SMAD2, and CASP3 and augments fibrogenic signals to promote fibroproliferation (Figure 8). Moreover, miR-323-3p antagomirs and mimics respectively augment and suppress fibrosis after bleomycin injury. Altogether, our data demonstrate miR-323a-3p as a central mediator of the epithelial fibroproliferative response by controlling signaling through multiple profibrotic pathways.
Figure 8. Proposed model of the mechanism by which miR-323a-3p regulates fibroproliferation.
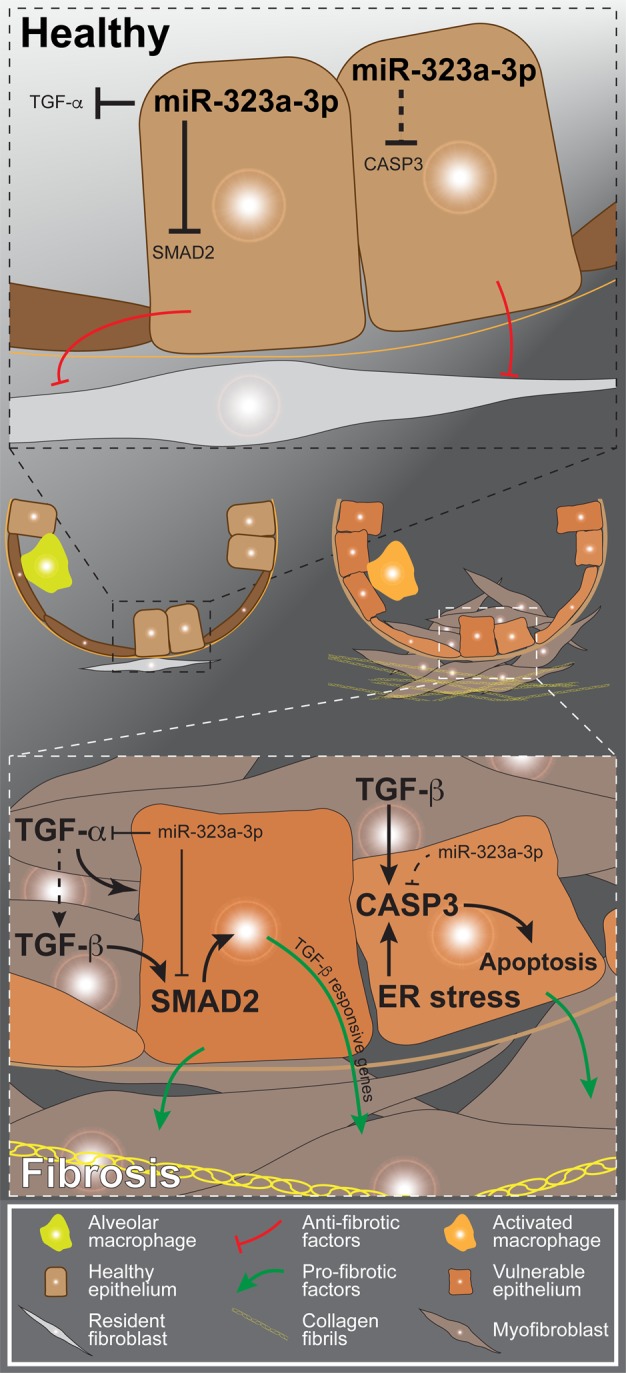
In the healthy epithelium, miR-323a-3p suppresses TGF-α, SMAD2, and caspase-3 to limit signaling through the respective pathways. Moreover, the epithelium naturally suppresses resident fibroblasts in a quiescent state under homeostatic conditions. In fibrosis, chronic injury downregulates miR-323a-3p expression in the lung epithelium, thereby releasing the suppression over multiple intertwined profibrotic signals such as apoptosis and TGF-α and TGF-β signaling. The augmented fibrogenic signals within the damaged epithelium induce it to secrete factors that promote fibroblast activation and fibroproliferation.
TGF-β signaling by the lung epithelium is a key pathway that is critical in the development of lung fibrosis (7–11). We determined that miR-323a-3p targets SMAD2 to functionally alter downstream TGF-β signaling events. A recent study by Wang et al. also found that miR-323a-3p targets SMAD2 and SMAD3 (39). Although we validated the effects on SMAD2, we could not replicate miR-323a-3p suppression of SMAD3 (unpublished data). Similar to TGF-β, TGF-α signaling in the lung epithelium has also been implicated in pulmonary fibrosis (32, 40, 41). We confirmed TGF-α as another direct target of miR-323a-3p that is suppressed with subsequent attenuation of downstream events. Coincidentally, TGF-α mediates part of its profibrotic effects via αvβ6 integrin activation of TGF-β (41). Thus, by controlling multiple steps in an interdigitated signaling network, miR-323a-3p can exert a more profound impact on profibrotic signals than by regulating any individual pathway.
Another consequence of TGF-β stimulation of the lung epithelium is the induction of apoptosis (10). Certainly, the association between lung epithelial apoptosis and IPF has been well established (42, 43). Moreover, murine models of pulmonary fibrosis demonstrate epithelial apoptosis as an early event after injury by bleomycin or TGF-β overexpression (10, 43). Suppression of apoptosis after epithelial induction of TGF-β also attenuates lung fibrosis, suggesting that TGF-β–mediated programmed cell death promotes fibroproliferation (10). We found that miR-323a-3p suppresses CASP3 mRNA and protein levels and prevents programmed cell death. However, CASP3 is not a direct target of miR-323a-3p, and we speculate that miR-323a-3p is regulating 1 or more transcription factors that induce CASP3 mRNA expression. Altogether, our findings demonstrate that miR-323a-3p limits the fibrotic effects of TGF-β by attenuating apoptotic cell death in addition to suppressing TGF-β activation (via TGFA) and signaling (via SMAD2).
In recent years, ER stress has been identified as contributing to the development of pulmonary fibrosis (44). Indeed, ER stress markers are found in the lung epithelium of patients with IPF (38, 45). Moreover, ER stress induction within the lung epithelium augments fibrosis after bleomycin injury (13). Notably, ER stress induces apoptotic cell death and is likely a mechanism contributing to the development of lung fibrosis. Here, we show that CASP3 suppression by miR-323a-3p attenuates tunicamycin-induced apoptosis. Thus, miR-323a-3p exerts another level of regulation over fibrogenic processes by limiting the apoptotic death secondary to ER stress.
The prevailing concept of pulmonary fibrosis is that persistent epithelial damage and the associated wound healing signals becomes maladaptive, resulting in fibroblast activation and matrix deposition (1). Consistent with this model, suppression of miR-323a-3p augments and overexpression attenuates wound closure of a monolayer of cells (39). These findings imply that miR-323a-3p downregulation is necessary for the wound healing process. Although it is unknown how miR-323a-3p regulates wound healing, a recent high-throughout sequencing coupled with crosslinking immunoprecipitation (HITS-CLIP) study offers some clues by identifying miR-323a-3p binding to the mRNA of intracellular signaling and transport proteins (e.g., MAP4K4, DCTN4) and secreted factors and matrix proteins (e.g., STC1, LAMC1) that can participate in cell migration (analysis of the raw data was done with Tarbase) (46–51). Although downregulated miR-323a-3p promotes re-epithelialization, our data indicate that it also releases inhibition of several interlinked profibrotic pathways. Therefore, injury may disrupt epithelial-mesenchymal interactions as a normal response to wound healing but chronically cause collateral damage by tipping the balance to excessive matrix deposition and destruction of the tissue architecture. In support of this maladaptive response, we demonstrate that miR-323a-3p can regulate epithelial-fibroblast crosstalk by promoting epithelial suppression of fibroblast differentiation and expression of matrix proteins. The epithelium releases multiple stimulatory and inhibitory factors to affect fibroblast activation and survival (52). In all likelihood, miR-323a-3p is manipulating important aspects of the epithelial-mesenchymal crosstalk either via direct targets or indirectly through its regulation of fibrogenic signals in the epithelium (i.e., TGF-α, TGF-β, apoptosis).
Our study adds to the growing list of miRs found to participate in lung fibrosis (26). We recently reported that culturing of epithelial cells (and likely other cell types) alter the expression of a substantial number of miRs in comparison with freshly isolated samples (53). Moreover, murine models of lung fibrosis, such as bleomycin, may not fully replicate human disease (54). Therefore, the relevance to human disease of the miRs identified in anything other than noncultured, human disease samples needs to be further substantiated. Notably, if we focus on in vivo human studies, the majority of dysregulated miRs were identified by evaluating whole-lung explants (55–64). However, the whole lung has multiple cell types (e.g., epithelium, fibroblast, macrophage) that participated in fibroproliferation, and only a limited number of studies confirm the in vivo cellular compartment with dysregulated miR expression (61–67). In our study, we specifically procured and evaluated a purified population of epithelial cells. Furthermore, the downregulation of miR-323a-3p was not only found in the epithelium of lung transplant patients with BOS but also stood true in IPF and the murine bleomycin injury model.
Interest in using miRs as therapeutics for pulmonary fibrosis has been growing (68). The advantage of miR-based therapy is that multiple targets can be simultaneously suppressed. Accordingly, miR-323a-3p is a candidate that can be included in such an approach owing to its ability to suppress TGFA, SMAD2, and CASP3. Indeed, we found that intranasal delivery of miR-323a-3p mimics attenuated lung fibrosis. A limitation of our approach is that exogenously delivered oligonucleotide may also target nonepithelial cells. However, epithelial cells are the first barrier against environmental insults, and so the epithelium would be presented with the highest concentration of intranasally delivered RNAs and be the predominant cellular compartment targeted by this treatment. Studies are ongoing to develop nanoparticle strategies to deliver small RNAs specifically to the lung epithelium.
Concurrent suppression of several pathways by miRs is a double-edged sword, as numerous off-target effects would be anticipated that might cause toxic side effects. Delivery of lower levels of miRs will likely reduce off-target effects but may not have enough impact on inhibiting fibrogenic signals. Future strategies may also need to focus on using multiple miRs at lower doses that have overlapping effects on targets within several key fibrogenic pathways to suppress fibrosis while maintaining minimal off-target side effects. Alternatively, identifying miR clusters involved in fibrosis that are all controlled by the same regulatory mechanisms could lead to methods to pharmacologically manipulate expression of fibrogenic miRs as a therapeutic for pulmonary fibrosis. One candidate is the miR-17-92 cluster, which suppresses TGF-β signaling and lung fibrosis and can be epigenetically regulated (64, 69). Interestingly, several of the miRs that we identified (miR-323a-3p, -665, -668, and -758) in our initial studies in BOS samples are also within a tight cluster on chromosome 14, suggesting coregulation of their expression.
In summary, we have shown that miR-323a-3p is downregulated in the epithelium of human and experimental murine lung fibrosis. In addition, we demonstrated that epithelial expression of miR-323a-3p simultaneously regulates multiple profibrotic signaling (TGF-, TGF-β, and apoptosis) to moderate epithelial-mesenchymal crosstalk. Furthermore, miR-323a-3p overexpression suppressed, and its antagonism augmented, bleomycin-induced lung fibrosis. Much work is still needed to understand the regulatory mechanisms that stimulate downregulation of miR-323a-3p in lung injury and fibrosis as well as to further identify targets within profibrotic pathways. However, the fact that miR-323a-3p can simultaneously target multiple fibrogenic signals not only defines a mechanism by which downregulation promotes fibroproliferation, but also increases the possibility of further developing it as an antifibrotic therapeutic.
Methods
Isolation of human lung epithelium and miR analysis.
Bronchial epithelia from lung transplant patients were collected by airway brushing during bronchoscopy, and miR expression was profiled using PCR arrays (Exiqon) as previously described (53, 70). False discovery rate (FDR) analysis was used to determine significant differential expression of miRs between patients with BOS versus controls (71). Putative targets for differentially expressed miRs were determined using TargetScan (http://www.targetscan.org) (23). Functional enrichment analysis of target genes whose expression may be modulated by the differentially expressed miRs was performed using Webgestalt (http://www.webgestalt.org/) (72).
Alveolar type II (ATII) cells were dissociated from lungs explanted from IPF patients during transplantation and nondiseased donor lungs rejected for transplant. ATII cells were purified from single-cell suspensions by FACS using the following markers: CD326+ (Epcam; epithelial), CD45– (hematopoietic), CD31– (endothelial), and HTII-280+ (ATII).
Bleomycin injury.
Eight-week-old C57BL/6J mice (The Jackson Laboratories) were intubated, and bleomycin (0.5–0.75 U/kg of body weight) was intratracheally instilled one time. On days 5, 10, and 15 after bleomycin treatment, miR-323a-3p mimics, antagomirs, or scrambled control (50 μg in 50 μl nuclease-free ddH2O; Sigma-Aldrich) was intranasally instilled into anesthetized mice. Mice were euthanized on day 21 after injury, and lungs were harvested for histologic evaluation and hydroxyproline measurement using standard protocols (73).
Cell culture and treatments.
Primary fibroblasts were isolated from human lung explants as previously described (74). All cells (16HBE14o-, HEK293, and lung fibroblasts) were cultured in DMEM with 10% FBS (Hyclone) in 5% CO2 at 37°C. Mimics, antagomirs, or scrambled control for miR-323a-3p (10 nM, Life Technologies) were transfected into 16HBE14o- cells using Lipofectamine RNAiMAX reagent according to the manufacturer’s protocol (ThermoFisher Scientific). To investigate TGF-β signaling, 16HBE14o- cells were treated for 24 hours with TGF-β1 (5 ng/ml, PeproTech). To evaluate the effect of miR-323a-3p on apoptosis, cells were treated with either staurosporine (0.5 μM, Sigma-Aldrich) or tunicamycin (5 μg/ml, Sigma-Aldrich) for 24 hours and then processed for caspase-3 activity measurements (Biovision) as previously described (75).
For epithelial-fibroblast crosstalk experiments, we collected conditioned medium from 16HBE14o- cells transfected with miR-323a-3p mimic or control cultured for 24 hours. Subsequently, fibroblasts were cultured with the conditioned medium for 24 hours prior to downstream evaluation.
Protein assays.
Western blot analysis was performed using primary antibodies for SMAD2 (D43B4; catalog 5339), CASP3 (8G10; catalog 9665), TGFA (catalog 3715), and GAPDH (D16H11, catalog 5174) from Cell Signaling Technology and β-actin (catalog sc-1616) from Santa Cruz Biotechnology. Blots were visualized using the ChemiDoc MP imaging system and analyzed with Image Lab software (Bio-Rad).
Primary human lung fibroblasts were cultured on chamber slides (Lab-Tek) and processed for immunofluorescence. Fixed cells were stained with anti–α-SMA antibody (1A4, catalog A2547) from Sigma-Aldrich or phalloidin-Alexa-488 (Cytoskeleton). The images were taken with a Zeiss Observer Z1 fluorescence microscope using Plan Apo 5×/0.16 and Plan Apo 20×/0.8 objectives.
Luciferase reporter assay.
CASP3 3′ UTR dual luciferase construct (pEZX-MT01 plasmid) was purchased from GeneCopoeia. For SMAD2 and TGFA, the respective 3′ UTRs were subcloned into the reporter plasmid, and the CASP3 3′ UTR was replaced by cutting with EcoRI and XhoI restriction endonucleases (New England Biolabs). Luciferase reporter constructs were cotransfected with miR-323a-3p mimics or control into HEK293 cells, and cells were processed for luciferase activity using the Luc-Pair Luciferase Assay Kit following the manufacturer’s protocol (GeneCopoeia).
PCR.
RNA was isolated using TRIzol (Thermo Fisher Scientific) or an miRNeasy Mini Kit (Qiagen). Quantitative PCR (qPCR) for miR-323a-3p and SNORD38B as a housekeeping control was performed by using PCR primers and the miRCURY LNA Universal RT Kit from Exiqon. qPCR for mRNA of the designated genes and 18S as a control was performed using primers purchased from IDT and SYBR green master mix (ThermoFisher Scientific) on the ViiA 7 Real-Time PCR System (ThermoFisher Scientific).
Statistics.
Unless otherwise indicated, 2-tailed Student’s t test was used for 2 groups, and 1-way ANOVA was used for multiple groups. Results are presented as the mean ± SEM, and P < 0.05 was considered significant.
Study approval.
The human studies were approved by the Institutional Review Board at the University of Washington and Cedars-Sinai Medical Center, and all patient samples were collected after receiving signed informed consent. The animal experiments were approved by the Institutional Animal Care and use Committee at Cedars-Sinai Medical Center.
Author contributions
LG and PC designed the research studies. LG, DH, TP, RB, YH, and JA conducted experiments. LG, SG, JE, and PC acquired the data. LG, SG, and PC analyzed the data. PH, RK, MK, DJ, AK, TM, BS, PN, and CH provided reagents. LG and PC wrote the manuscript.
Supplementary Material
Acknowledgments
We appreciate the help from Ellen McCown and Sharon Kelso in enrolling patients in this study. This work was funded by the NIH grants HL120947 and HL103868, the Samuel Oschin Comprehensive Cancer Institute, the Institute of Translational Health Sciences, and the Cystic Fibrosis Foundation (all to P.C.). Additional support was also provided by the Cystic Fibrosis Research Development Program funded through the NIH-NIDDK (P30-DK089507).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:JCI Insight. 2016;1(20):e90301. doi:10.1172/jci.insight.90301.
Contributor Information
Lingyin Ge, Email: lingyin.ge@cshs.org.
Tanyalak Parimon, Email: tanyalak.parimon@cshs.org.
Rena Brauer, Email: Rena.Brauer@cshs.org.
Ying Huang, Email: ying.huang@cshs.org.
Jenieke Allen, Email: jenieke.allen@cshs.org.
Dianhua Jiang, Email: Dianhua.Jiang@cshs.org.
Takako Mizuno, Email: takako.mizuno@cshs.org.
Barry R. Stripp, Email: barry.stripp@cshs.org.
Paul W. Noble, Email: paul.noble@cshs.org.
Cory M. Hogaboam, Email: Cory.Hogaboam@cshs.org.
Peter Chen, Email: peter.chen@cshs.org.
References
- 1.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122(8):2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan T, Mason RJ, Westcott JY, Shannon JM. Rat alveolar type II cells inhibit lung fibroblast proliferation in vitro. Am J Respir Cell Mol Biol. 2001;25(3):353–361. doi: 10.1165/ajrcmb.25.3.4004. [DOI] [PubMed] [Google Scholar]
- 3.Portnoy J, et al. Alveolar type II cells inhibit fibroblast proliferation: role of IL-1alpha. Am J Physiol Lung Cell Mol Physiol. 2006;290(2):L307–L316. doi: 10.1152/ajplung.00102.2005. [DOI] [PubMed] [Google Scholar]
- 4.Moore BB, et al. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am J Physiol Lung Cell Mol Physiol. 2003;284(2):L342–L349. doi: 10.1152/ajplung.00168.2002. [DOI] [PubMed] [Google Scholar]
- 5.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208(7):1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 7.Munger JS, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–328. doi: 10.1016/S0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 8.Degryse AL, et al. TGFβ signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L887–L897. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M, et al. Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest. 2011;121(1):277–287. doi: 10.1172/JCI42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee CG, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200(3):377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18(7):816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 13.Lawson WE, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci U S A. 2011;108(26):10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Königshoff M, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119(4):772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stuart BD, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. 2014;2(7):557–565. doi: 10.1016/S2213-2600(14)70124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsakiri KD, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104(18):7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. 2014;124(11):4673–4677. doi: 10.1172/JCI74368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. 2006;3(4):350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richeldi L, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 20.Eulalio A, Huntzinger E, Izaurralde E. Getting to the root of miRNA-mediated gene silencing. Cell. 2008;132(1):9–14. doi: 10.1016/j.cell.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455(7209):58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 23.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 24.Calin GA, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010;17(2):215–220. doi: 10.1038/cdd.2009.69. [DOI] [PubMed] [Google Scholar]
- 26.Rajasekaran S, Rajaguru P, Sudhakar Gandhi PS. MicroRNAs as potential targets for progressive pulmonary fibrosis. Front Pharmacol. 2015;6:254. doi: 10.3389/fphar.2015.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weigt SS, DerHovanessian A, Wallace WD, Lynch JP, Belperio JA. Bronchiolitis obliterans syndrome: the Achilles’ heel of lung transplantation. Semin Respir Crit Care Med. 2013;34(3):336–351. doi: 10.1055/s-0033-1348467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol. 2012;46(3):380–388. doi: 10.1165/rcmb.2011-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahaman SO, et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest. 2014;124(12):5225–5238. doi: 10.1172/JCI75331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, et al. Hedgehog signaling in neonatal and adult lung. Am J Respir Cell Mol Biol. 2013;48(6):703–710. doi: 10.1165/rcmb.2012-0347OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung CF, Rohani MG, Lee SS, Chen P, Schnapp LM. Role of IGF-1 pathway in lung fibroblast activation. Respir Res. 2013;14:102. doi: 10.1186/1465-9921-14-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardie WD, et al. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1217–L1225. doi: 10.1152/ajplung.00020.2008. [DOI] [PubMed] [Google Scholar]
- 33.et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J Allergy Clin Immunol. doi: 10.1016/j.jaci.2016.04.038. [published online ahead of print June 10, 2016]. [DOI] [PubMed] [Google Scholar]
- 34.Rock JR, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y, et al. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol. 2009;184(2):309–322. doi: 10.1083/jcb.200806067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meissner M, et al. AP1-dependent repression of TGFα-mediated MMP9 upregulation by PPARδ agonists in keratinocytes. Exp Dermatol. 2011;20(5):425–429. doi: 10.1111/j.1600-0625.2011.01253.x. [DOI] [PubMed] [Google Scholar]
- 37.Ringshausen I, et al. Constitutive activation of the MAPkinase p38 is critical for MMP-9 production and survival of B-CLL cells on bone marrow stromal cells. Leukemia. 2004;18(12):1964–1970. doi: 10.1038/sj.leu.2403544. [DOI] [PubMed] [Google Scholar]
- 38.Lawson WE, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, et al. MicroRNA-323-3p inhibits cell invasion and metastasis in pancreatic ductal adenocarcinoma via direct suppression of SMAD2 and SMAD3. Oncotarget. 2016;7(12):14912–14924. doi: 10.18632/oncotarget.7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L741–L749. doi: 10.1152/ajplung.00208.2003. [DOI] [PubMed] [Google Scholar]
- 41.Madala SK, et al. Inhibition of the αvβ6 integrin leads to limited alteration of TGF-α-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2014;306(8):L726–L735. doi: 10.1152/ajplung.00357.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barbas-Filho JV, Ferreira MA, Sesso A, Kairalla RA, Carvalho CR, Capelozzi VL. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP) J Clin Pathol. 2001;54(2):132–138. doi: 10.1136/jcp.54.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol. 1998;275(6 Pt 1):L1192–L1199. doi: 10.1152/ajplung.1998.275.6.L1192. [DOI] [PubMed] [Google Scholar]
- 44.Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta. 2013;1832(7):940–947. doi: 10.1016/j.bbadis.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korfei M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balakrishnan I, et al. Genome-wide analysis of miRNA-mRNA interactions in marrow stromal cells. Stem Cells. 2014;32(3):662–673. doi: 10.1002/stem.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vlachos IS, et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015;43(Database issue):D153–D159. doi: 10.1093/nar/gku1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collins CS, et al. A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase. Proc Natl Acad Sci U S A. 2006;103(10):3775–3780. doi: 10.1073/pnas.0600040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- 50.Yeung BH, Wong CK. Stanniocalcin-1 regulates re-epithelialization in human keratinocytes. PLoS One. 2011;6(11):e27094. doi: 10.1371/journal.pone.0027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishikawa R, et al. Tumor-suppressive microRNA-29s inhibit cancer cell migration and invasion via targeting LAMC1 in prostate cancer. Int J Oncol. 2014;45(1):401–410. doi: 10.3892/ijo.2014.2437. [DOI] [PubMed] [Google Scholar]
- 52.Sakai N, Tager AM. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim Biophys Acta. 2013;1832(7):911–921. doi: 10.1016/j.bbadis.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen P, Edelman JD, Gharib SA. Comparative evaluation of miRNA expression between in vitro and in vivo airway epithelium demonstrates widespread differences. Am J Pathol. 2013;183(5):1405–1410. doi: 10.1016/j.ajpath.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.B Moore B, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol. 2013;49(2):167–179. doi: 10.1165/rcmb.2013-0094TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li S, et al. miR-130b-3p modulates epithelial-mesenchymal crosstalk in lung fibrosis by targeting IGF-1. PLoS One. 2016;11(3):e0150418. doi: 10.1371/journal.pone.0150418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milosevic J, et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2012;47(6):879–887. doi: 10.1165/rcmb.2011-0377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang H, et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis. 2014;5:e1238. doi: 10.1038/cddis.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Z, et al. MicroRNA-144 dysregulates the transforming growth factor-β signaling cascade and contributes to the development of bronchiolitis obliterans syndrome after human lung transplantation. J Heart Lung Transplant. 2015;34(9):1154–1162. doi: 10.1016/j.healun.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ji X, et al. The anti-fibrotic effects and mechanisms of microRNA-486-5p in pulmonary fibrosis. Sci Rep. 2015;5:14131. doi: 10.1038/srep14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Christmann RB, et al. miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res Ther. 2016;18(1):155. doi: 10.1186/s13075-016-1054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu G, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207(8):1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pandit KV, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182(2):220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das S, et al. MicroRNA-326 regulates profibrotic functions of transforming growth factor-β in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2014;50(5):882–892. doi: 10.1165/rcmb.2013-0195OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dakhlallah D, et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187(4):397–405. doi: 10.1164/rccm.201205-0888OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bodempudi V, et al. miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2014;307(4):L283–L294. doi: 10.1152/ajplung.00069.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Disayabutr S, et al. miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11(6):e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su S, et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun. 2015;6:8523. doi: 10.1038/ncomms9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montgomery RL, et al. MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol Med. 2014;6(10):1347–1356. doi: 10.15252/emmm.201303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, et al. HDAC3-dependent epigenetic pathway controls lung alveolar epithelial cell remodeling and spreading via miR-17-92 and TGF-β signaling regulation. Dev Cell. 2016;36(3):303–315. doi: 10.1016/j.devcel.2015.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gharib SA, Edelman JD, Ge L, Chen P. Acute cellular rejection elicits distinct microRNA signatures in airway epithelium of lung transplant patients. Transplant Direct. 2015;1(10) doi: 10.1097/TXD.0000000000000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41(Web Server issue):W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lovgren AK, et al. β-Arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci Transl Med. 2011;3(74):74ra23. doi: 10.1126/scitranslmed.3001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pierce EM, et al. Therapeutic targeting of CC ligand 21 or CC chemokine receptor 7 abrogates pulmonary fibrosis induced by the adoptive transfer of human pulmonary fibroblasts to immunodeficient mice. Am J Pathol. 2007;170(4):1152–1164. doi: 10.2353/ajpath.2007.060649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brauer R, et al. Syndecan-1 attenuates lung injury during influenza infection by potentiating c-met signaling to suppress epithelial apoptosis. Am J Respir Crit Care Med. 2016;194(3):333–344. doi: 10.1164/rccm.201509-1878OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.