


Abstract
Thymine DNA Glycosylase (TDG) is a base excision repair enzyme functioning in DNA repair and epigenetic regulation. TDG removes thymine from mutagenic G·T mispairs arising from deamination of 5-methylcytosine (mC), and it processes other deamination-derived lesions including uracil (U). Essential for DNA demethylation, TDG excises 5-formylcytosine and 5-carboxylcytosine, derivatives of mC generated by Tet (ten-eleven translocation) enzymes. Here, we report structural and functional studies of TDG82-308, a new construct containing 29 more N-terminal residues than TDG111-308, the construct used for previous structures of DNA-bound TDG. Crystal structures and NMR experiments demonstrate that most of these N-terminal residues are disordered, for substrate- or product-bound TDG82-308. Nevertheless, G·T substrate affinity and glycosylase activity of TDG82-308 greatly exceeds that of TDG111-308 and is equivalent to full-length TDG. We report the first high-resolution structures of TDG in an enzyme-substrate complex, for G·U bound to TDG82-308 (1.54 Å) and TDG111-308 (1.71 Å), revealing new enzyme-substrate contacts, direct and water-mediated. We also report a structure of the TDG82-308 product complex (1.70 Å). TDG82-308 forms unique enzyme–DNA interactions, supporting its value for structure-function studies. The results advance understanding of how TDG recognizes and removes modified bases from DNA, particularly those resulting from deamination.
INTRODUCTION
Thymine DNA glycosylase (TDG) is an enzyme that initiates base excision repair by removing modified forms of 5-methylcytosine (mC) that are generated by deamination or oxidation (1). TDG excises thymine from G·T mispairs, thereby protecting against C→T transition mutations that arise via deamination of mC to T (2,3). TDG is also essential for active DNA demethylation, which likely accounts for findings that its depletion in mice leads to embryonic lethality (4,5). An established pathway for active DNA demethylation includes TDG excision of 5-formylcytosine or 5-carboxylcytosine (6,7), epigenetic bases that are generated via oxidation of mC by one of three ten-eleven translocation enzymes (7–11). TDG also removes many other bases (in vitro), including uracil, 5-halogenated uracils (5FU, 5ClU, 5BrU, 5IU) and 5-hydroxymethyl-U (hmU), among others (12,13). Human TDG (410 residues) contains a central catalytic domain of about 195 residues flanked by N-terminal and C-terminal regions of roughly equivalent size that are disordered and yet important for certain functions, interactions with other proteins and regulation by post-translational modifications such as acetylation, phosphorylation and SUMO conjugation (14–21).
Here, we report structural and functional studies of TDG82-308, a new construct of human TDG comprised of residues 82–308 (of 410 total) that includes the catalytic domain and an N-terminal region that contains amino acid residues involved in regulation of TDG via protein interactions or post-translational modifications. All previous structural studies of TDG have used a construct referred to as the catalytic domain, TDGcat, comprising residues 111–308 (22–25) or SUMO-conjugated TDG, which included residues 117–332 (26). TDG82-308 contains 29 additional N-terminal residues compared to TDG111-308 (Figure 1). These include the PIP degron (residues 95–106), which mediates TDG depletion during S phase of the cell cycle via interaction with PCNA and CRL4Cdt2, a ubiquitin E3 ligase (27–29). The N-terminal region also contains three Lys residues that are subject to acetylation by CBP/p300 (14) and two Ser residues that are putative phosphorylation sites (modification sites shown in Figure 1) (30). Previous studies also indicate that some portion of the N-terminal region comprising residues 55–110, which is enriched in Arg and Lys, is required for efficient binding and catalytic processing of G·T mispairs, and for efficient binding to non-specific DNA (16,22,31). In addition, N-terminal residues of TDG mediate interactions with other proteins, including CBP/p300, Dnmt3A/B, the 9-1-1 complex and SIRT1 (14,16,17,32). Importantly, all residues of the new TDG82-308 construct are native to human TDG, because the entire poly-His purification tag is removed via protease cleavage, as described below. By contrast, TDG111-308 contains six non-native N-terminal residues that remain after cleavage of the poly-His tag (Figure 1) (22,23).
Figure 1.

Amino acid sequence for the N-terminal regions of TDG82-308 and TDG111-308 and the initial residues of the bacterial homologue MUG (E. coli). Putative sites for post-translational modification are indicated for TDG82-308, including acetylation (A) and phosphorylation (P). Also shown is the PIP degron and an α-helix that has not been observed in previous structures of E·S complexes. Shown in red for TDG111-308 are the six non-native residues that remain after removal of the poly-His tag. All residues of TDG82-308 are native to TDG.
We investigated the activity of TDG82-308, TDG111-308 and full-length TDG for acting on G·T mispairs in DNA. We find that substrate binding and catalysis for TDG82-308 is equivalent to that of full-length TDG, and much greater than that of TDG111-308. Using improved crystallization methods (33), we solved high-resolution structures of the enzyme bound to a stable G·U substrate analog, for both TDG82-308 (1.54 Å) and TDG111-308 (1.71 Å). These new structures are of much higher resolution than previously reported structures of TDG in an enzyme-substrate (E·S) complex, which were solved at 3.0 Å resolution (24,34). We also solved a high-resolution (1.70 Å) enzyme-product (E·P) complex using the new TDG82-308 construct. The structures reveal important enzyme-DNA contacts that have not previously been observed, many of which are mediated by ordered water molecules (absent from previous structures). The structures also confirm many key enzyme-substrate interactions that were suggested by the previous lower-resolution structures. Notably, several key enzyme–DNA interactions are observed only for the new construct, TDG82-308, suggesting its value for future structure-function studies. Remarkably, crystal structures and solution studies using NMR spectroscopy show that most of the 29 N-terminal residues of TDG82-308 are disordered, even when the enzyme is tightly bound to G·U or G·T substrate DNA. Together, our results advance the understanding of how TDG recognizes and excises modified bases in DNA, particularly those arising from deamination.
MATERIALS AND METHODS
Materials
Full length human TDG (410 residues) and TDG111-308 (also referred to as TDGcat) were expressed in Escherichia coli and purified essentially as previously described (23,35). A vector (pJ401) for expressing TDG82-308, a new construct containing residues Ser82-Val308 of human TDG (Figure 1) plus an N-terminal poly-His tag, was obtained from DNA 2.0 (Newark, CA, USA) and transformed into E. coli BL21(DE3) cells. TDG82-308 was expressed (at 15°C) and purified essentially as described for TDG111-308, using Ni-affinity, ion-exchange (SP sepharose) and size exclusion chromatography (23,35). The poly-His tag was removed (after Ni-affinity) using the tobacco etch virus (TEV) protease (36), which cleaves on the carboxyl end of its recognition site. As such, following TEV cleavage, all residues of TDG82-308 are native to TDG. By contrast, TDG111-308 contains six non-native N-terminal residues (GSHMAS) that remain after thrombin cleavage of the N-terminal poly-His tag (Figure 1) (22,23). The enzyme preparations were >99% pure, as judged by SDS-PAGE (Coomassie stained gel), and their concentration was determined by absorbance at 280 nm (37,38). The extinction coefficient for TDG82-308 is identical to that for TDG111-308 (23).
An expression vector for the R110A variant of TDG82-308 was generated via site-directed mutagenesis using the Quickchange II system (Agilent Technologies), as previously described (39); the variant enzyme was expressed and purified as described above.
Uniformly 15N-labeled TDG82-308 was produced by expression in MOPS minimal media with 99% [15N]-NH4Cl (1g/L) (C.I.L.), as previously described (40,41). Briefly, transformed BL21 (DE3) cells (Novagen) were grown overnight on an LB plate (37°C); several colonies were used to inoculate 0.2 L of LB medium and the culture was grown at 37°C to an OD600 of about 0.6. Cells were harvested, suspended in 2 l of MOPS minimal media, and grown to OD600 of 0.7. The temperature was reduced to 15°C, expression was induced with IPTG (0.4 mM) overnight (∼16 h) and 15N-labeled TDG82-308 was purified as described above.
TEV protease (S219V variant) was expressed and purified as previously described (36) using a bacterial expression vector (pRK793) obtained from Addgene (Cambridge, MA, USA).
The Oligodeoxynucleotides (ODNs) were obtained from IDT or the Keck Foundation Biotechnology Resource Laboratory at Yale University. ODNs were purified by reverse phase HPLC (33), exchanged into 0.02 M Tris-HCl pH 7.5, 0.04 M NaCl and quantified by absorbance as described (35). ODNs containing the 2′-fluoroarabino analogues of deoxyuridine or deoxythymidine, referred to as UF and TF, respectively, were synthesized at the Yale facility using phosphoramidites obtained from Glen Research (UF) or Link Technologies (TF) (39). TDG binds productively to DNA containing UF or TF but it cannot hydrolyze the N-glycosyl bond because the single-atom fluorine substitution destabilizes the chemical transition-state (38,39,42,43). The duplex included a 28mer target strand, 5′-AGCTGTCCATCGCTCAxGTACAGAGCTG, where x is T, UF or TF and its complement, 5′-CAGCTCTGTACGTGAGCGATGGACAGCT, such that the target base (x) is paired with G and located in a CpG dinucleotide context (underlined), consistent with TDG specificity (35,44). The same 28 bp DNA construct was used for glycosylase assays (x = U, T), NMR experiments (x = TF) and equilibrium binding studies (x = TF; with 3′ 6-FAM on the complementary strand).
X-ray crystallography
Samples used for crystallization contained 0.35 mM enzyme (TDG82-308 or TDG111-308) and 0.42 mM DNA in a buffer of 5 mM Tris-HCl pH 7.5, 0.13 M NaCl, 0.2 mM DTT, 0.2 mM EDTA. The enzyme–product complex was produced by incubating TDG82-308 with G·U DNA substrate for a sufficient time to ensure full conversion to product, as confirmed by HPLC (12). Crystals were grown at room temperature (∼22°C) by sitting drop vapor diffusion, using 1 μl of the TDG–DNA sample and 1 or 2 μl of mother liquor, which was 30% (w/v) PEG 4000, 0.2 M ammonium acetate, 0.1 M sodium acetate, pH 6.0. Crystals typically appeared within in a few days. Crystals were cryo-protected using mother liquor supplemented with 18% ethylene glycol and flash cooled in liquid nitrogen.
X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL beamlines 11–1) and at the Advanced Light Source (ALS beamline 8.2.2). The images were processed using XDS (45) and scaled with Aimless (46) from the CCP4 program suite (47) with the help of the autoxds script developed by Ana Gonzalez and Yingssu Tsai (http://smb.slac.stanford.edu/facilities/software/xds). For the TDG111-308 E·S complex 3 data sets were collected from separate section of a single crystal and merged to increase resolution. Resolution cutoff was determined based on CC1/2 values (48). Structures were solved by molecular replacement using Phaser (49), and a previously reported structure of DNA-bound TDG111-308 as the search model (PDBID: 4Z47). Refinement was performed using BUSTER-TNT (50) or REFMAC5 (51), and model building was performed using Coot (52). TLS refinement protocol utilized TLSMD server (53,54) as described (33). The structural figures were made with PyMOL (http://www.pymol.org).
NMR spectroscopy
15N-HSQC experiments were collected on an 800 MHz Bruker Avance III NMR spectrometer, and the data were processed and analyzed using NMRPipe and NMRDraw (55). Sample conditions are provided in the relevant figure legends.
Equilibrium binding assays
Equilibrium binding of enzyme (TDG, TDG82-308 or TDG111-308) to a G·TF substrate analog was analyzed using electrophoretic mobility shift assays (EMSAs), performed essentially as described (20), where TF is the 2′-fluoroarabino analogue of dT (described above). Samples contained a 10 nM concentration of G·TF DNA and varying concentrations of enzyme. Binding reactions were incubated at room temperature for 30 min, loaded onto a 6% native denaturing polyacrylamide gel (Invitrogen) and run at 4°C for 2 h at 50 V. Gels were imaged using a Typhoon 9400 variable mode imager (GE Healthcare) as described (56).
Glycosylase assays
Single turnover kinetics reactions were initiated by adding enzyme (TDG, TDG82-308 or TDG111-308) to G·T substrate (0.5 uM) in HEMN.1 buffer (0.02 M HEPES pH 7.5, 0.1 M NaCl, 0.2 mM EDTA, 2.5 mM MgCl2). Aliquots were removed at desired time points, quenched with 50% (v:v) 0.3 M NaOH, 0.03 M EDTA, and heated (15 min, 85°C) to quantitatively cleave the DNA backbone at TDG-generated abasic sites. The resulting DNA fragments were resolved by HPLC (35) and peak areas were used to determine fraction product. Progress curves (fraction product versus time) were fitted by non-linear regression to eq. 1:
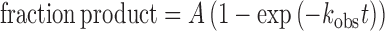 |
(1) |
where A is the amplitude, kobs is the rate constant, and t is the reaction time. Experiments were performed with saturating enzyme ([E] >> Kd; [E] > [S]) such that the observed rate constant reflects the maximal rate of product formation (kobs ≈ kmax) and is not influenced by enzyme–substrate association or by product release or product inhibition (39). Previous studies show that TDG binds G·T DNA with a Kd of 0.02 µM (38), while TDG111-308 binds G·T DNA with Kd of roughly 1.3 µM (24). Findings here indicate similar values and show that TDG82-308 binds with tighter affinity than TDG. Thus, kinetics experiments were performed with an enzyme concentration of 5 μM for TDG or TDG82-308, and 32 µM for TDG111-308. Saturating conditions were confirmed by observation of identical rate constants for experiments performed at other enzyme concentrations (not shown).
RESULTS AND DISCUSSION
Residues 82–110 of TDG confer tight substrate binding and full glycosylase activity for G·T mspairs
To compare the activity of TDG82-308 with the smaller construct, TDG111-308 and with full-length TDG, we examined binding affinity and glycosylase (base excision) activity for a G·T substrate. This provides a stringent test for the role of N-terminal residues 82–110 (absent on TDG111-308), because substrate binding and base excision are weak for G·T mispairs relative to other TDG substrates (24,38,57). We examined the binding affinity for a G·TF analog, where TF is 2′-flouroarabino-deoxythymidine, an analog of thymidine that flips into the TDG active site but is not cleaved (34,38,39). Using EMSA, we find that G·TF binding is actually a bit tighter for TDG82-308 compared to TDG, and dramatically weaker for TDG111-308 (Figure 2A). The EMSAs indicate G·TF binds with a Kd of roughly 10 nM for TDG82-308 and about 20 nM for TDG. The result for TDG is in excellent agreement with the reported Kd of 18 nM obtained by fluorescence anisotropy (38). In sharp contrast, we find that TDG111-308 binds G·TF DNA with a Kd of over 1 µM, consistent with a previously reported Kd of 1.3 µM (24). Similarly, we find that the G·T glycosylase activity is equivalent for TDG82-308 and TDG, but is much (6-fold) lower for TDG111-308 (Figure 2B). Taken together, the results show that residues 82–110 (or some fraction thereof) are essential for proper G·T substrate binding and catalysis. While previous studies suggested that a larger construct comprising residues 56–308 (TDG56-308) is needed for full G·T glycosylase activity (22), the results here show that TDG82-308 retains full G·T activity. These results suggested that TDG82-308 could be a good construct for structural studies.
Figure 2.

Biochemical studies of full length TDG, TDG82-308 and TDG111-308. (A) Equilibrium binding of a given TDG construct to DNA (10 nM) containing a G·TF mismatch, where TF is a non-cleavable Thd analog, monitored by electrophoretic mobility shift assays (EMSA). The concentration of enzyme (nM) is indicated. For TDG111-308, the left lane marked ‘20*’ indicates binding to abasic DNA product (10 nM, 28 bp), to show the mobility of a tight 1:1 complex, given that such a complex is not clearly observed for G·TF DNA in the gel. (B) Single-turnover kinetics for excision of T from a G·T DNA substrate by the three TDG constructs, at 37°C and 22°C (TDG111-308 not stable at 37°C). Rate constants are kmax = 0.651 ± 0.044 min−1 for TDG and kmax = 0.655 ± 0.020 min−1 for TDG82-308 at 37°C, and kmax = 0.126 ± 0.008 for TDG, kmax = 0.108 ± 0.006 min−1 for TDG82-308, and kmax = 0.022 ± 0.002 min−1 for TDG111-308 at 22°C.
First high-resolution structures of enzyme-substrate complexes for TDG
Previous structures of enzyme-substrate (E·S) complexes for TDG111-308, with either G·U or G·caC, revealed important features of substrate recognition and catalysis, but were solved at moderate (3.0 Å) resolution (24,34). Moreover, the crystals used for these previous structures were obtained under conditions that give 2:1 binding (TDG:DNA), involving substantial interactions between the two TDG subunits; 2:1 binding is likely an artifact of crystallization and 1:1 binding appears to be more physiologically relevant (23,38,58). Structures of enzyme-product (E·P) complexes have been determined using crystals generated under conditions that give 1:1 binding (TDG:DNA) (25), including our recent structures solved at high resolution (up to 1.45 Å) (33). Following this approach, we solved high-resolution structures of E·S complexes, for G·UF DNA bound to TDG82-308 (1.54 Å) or TDG111-308 (1.77 Å) (Supplementary Table S1). These structures are of much higher quality than previous structures of E·S complexes, revealing new enzyme–DNA interactions. Moreover, the new structures feature hundreds of water molecules, some mediating key enzyme-DNA interactions. By contrast, no water molecules were observed in previous structures of E·S complexes, except for the putative nucleophile in the structure of TDG111-308 bound to a G·UF mispair (34). Structural comparisons using the percentile-based spread (p.b.s.) approach (59) reveal that the previous TDG111-308-G·UF structure (2.97 Å resolution) differs substantially from the new structures reported here, with a backbone p.b.s. of 0.51 Å for TDG111-308-G·UF and 0.58 Å for TDG82-308-G·UF (Supplementary Figure S1). By contrast, the two new E·S structures reported here are quite similar, with a backbone p.b.s. of 0.23 Å, even though they feature different TDG constructs and crystal cell parameters.
N-terminal residues in crystal structures of TDG82-308
While the results above show that the 29 N-terminal residues of TDG82-308 confer tight binding to G·T DNA (Figure 2), only four of these residues (107–110) are observed in the new structures, for both the E·S complex (Figure 3) and the E·P complex (not shown), indicating that the 25 N-terminal residues (82–106) are disordered. This conclusion is supported by NMR studies, as discussed below. Nevertheless, the structures reported here provide new information regarding the N-terminal residues that are observed. First, it is important to note that many more N-terminal residues are observed in structures that feature 1:1 versus 2:1 binding stoichiometry (TDG:DNA), as shown by ourselves and others (25,33). These previous studies showed that, depending on the DNA construct, TDG111-308 can bind DNA with either 1:1 or 2:1 stoichiometry (25,33). Importantly, all previous structures of enzyme–substrate complexes for TDG were obtained from crystals generated using DNA that yielded 2:1 binding (TDG:DNA), and therefore lack structural information for residues 111 to 122 (23,24,34). By contrast, the two enzyme–substrate structures reported here were generated from DNA that gives 1:1 binding and include N-terminal residues beginning at 107 for TDG82-308 and at 111 for TDG111-308. These N-terminal residues interact with other regions of the TDG catalytic domain, and feature an α-helix (α0; Glu116-Thr121) (Figure 3). Notably, residues that participate in the TDG:TDG dimer interface for the 2:1 complex (Leu143, Tyr147, Thr196 and Pro198) form contacts with the N-terminal helix (α0) and other N-terminal residues in the 1:1 complexes, which could explain why more N-terminal residues are disordered in structures featuring 2:1 versus 1:1 binding (Supplementary Figure S1).
Figure 3.

Crystal structure of the E·S complex for TDG82-308 bound to G·UF DNA (PDBID: 5HF7), solved at 1.54 Å, focusing on the N-terminal region (residues 108–122). TDG is shown in cartoon format (blue) with some residues in stick format (white with nitrogen blue and oxygen red). DNA is shown in both space and stick formats, with the dUrd-containing strand yellow and the complementary strand green. Water molecules are shown as red spheres and dashed lines represent hydrogen bonds.
The E·S and E·P structures for TDG82-308 reveal that the Arg110 side chain contacts the phosphate of the nucleotide located immediately 5′ of the flipped nucleotide (Figure 3). Mutational studies show that substrate binding is about 2-fold weaker and the maximal rate of base-excision (kmax) is 4-fold slower for R110A-relative to wild-type TDG82-308 (Supplementary Figure S2). Thus, Arg110 contributes significantly to substrate binding and glycosylase activity. The structures also reveal that the backbone N–H of Asp109 contacts a phosphate in the complementary DNA strand. Structures of TDG82-308 exhibit reasonable electron density for the N-terminal residues Lys107-Arg110 (Supplementary Figure S3). The residues that contact DNA backbone phosphates, Asp109 and Arg110, have B-factors of 49.5 Å2 and 44.5 Å2 in the E·S complex and 46.4 Å2 and 44.7 Å2 in the E·P complex, respectively. Together, the structural and biochemical findings suggest that the minimal catalytic domain of TDG should be redefined to include Asp109 and Arg110.
Interestingly, the presence of residues 82–110 (or some fraction thereof) has a greater effect on overall protein structure than does a transition from the E·S to the E·P complex (for a given TDG construct). More specifically, the percentile-based spread (p.b.s.) (59) between the E·S and E·P complexes is 0.18 Å for TDG82-308 and 0.11 Å for TDG111-308. However, when comparing corresponding structures for TDG82-308 versus TDG111-308, the p.b.s is 0.23 Å for the E·S complexes and 0.29 Å for E·P complexes.
High-resolution structure of an E·P complex for TDG82-308
We also solved a crystal structure of the TDG82-308 E·P complex at 1.70 Å, using crystals obtained by incubating the enzyme with a G·U substrate (Supplementary Table S1). As observed for the TDG82-308 E·S complex, Arg110 contacts the DNA phosphate 5′ of the flipped nucleotide, Asp109 (backbone N-H) contacts a phosphate in the complementary strand, but electron density is not observed for residues 82–106. The new TDG82-308 E·P complex is very similar to those we reported recently for TDG111-308 (33), as indicated by the backbone p.b.s. of 0.29 Å (noted above). As observed for our high-resolution TDG111-308 product complexes (33), the TDG82-308 structure shows unambiguously that the excised base (Ura) is absent from the E·P complex. Similarly, the abasic sugar adopts a roughly even mix of the α and β anomers (not shown). Although the product complexes of TDG111-308 and TDG82-308 exhibit very similar overall structures, the DNA conformation differs somewhat, particularly for several nucleotides 5′ of the flipped site (not shown), which may reflect DNA contacts involving Asp109 and Arg110 (lacking for TDG111-308). Additional differences are also noted in relevant sections below.
NMR studies of TDG82-308 N-terminal residues
The crystal structures indicate that the 26 N-terminal residues of TDG82-308 are disordered, even when the enzyme is bound to DNA in a tight E·S or E·P complex. Nevertheless, these N-terminal residues greatly enhance G·T substrate binding for TDG82-308 relative to TDG111-308 (Figure 2). We sought to further explore these findings using NMR chemical-shift-perturbation experiments, a powerful and widely used approach for monitoring protein–ligand interactions, particularly for disordered protein regions such as those of TDG (19,60–62). We compared the backbone 1H-15N chemical shifts for disordered residues of TDG82-308, in the presence and absence of DNA containing a G·TF substrate analog (same DNA used for binding studies, Figure 2). The 15N-HSQC spectrum for DNA-free TDG82-308 reveals about 30 resonances that are likely from disordered residues, as indicated by their relatively high intensity and chemical shifts similar to that expected for random coil (Figure 4, black peaks). Notably, nearly all of these peaks are absent in the 15N-HSQC of free TDG111-308 (Supplementary Figure S4), indicating that they likely reflect disordered N-terminal residues of TDG82-308. The same NMR experiment was collected for a sample containing TDG82-308 with a saturating concentration of G·TF DNA (Figure 4, red peaks). The overlaid spectra for free and G·TF-bound TDG82-308 reveal substantial chemical shift perturbations for most of the disordered N-terminal residues, indicating a change in the conformational ensemble upon binding G·TF DNA. Together, the crystallographic and NMR results indicate that the dramatically enhanced DNA binding affinity and glycosylase activity afforded by the TDG82-308 N-terminal residues is attained without adopting an ordered structure, suggesting non-specific interactions between the numerous cationic side chains (Lys, Arg) of TDG82-308 (Figure 1) and the anionic DNA phosphates.
Figure 4.

NMR studies indicate the N-terminal residues of TDG82-308 are disordered, when the enzyme is free or bound to G·TF DNA. Shown are 15N-HSQC spectra for TDG82-308 (0.13 mM) in the absence of DNA (black peaks) and with a saturating concentration (0.20 mM) of G·TF DNA (red peaks). Samples were in 0.02 M sodium phosphate pH 6.5, 0.15 M NaCl, 0.2 mM EDTA, 0.2 mM DTT, 7% D2O. Resonances in the upper right (within dotted lines) are side chain amino groups. NMR spectra were collected at 18°C using on 800 MHz NMR spectrometer. Four resonances are equivalent for free and DNA-bound TDG82-308 (*) and likely correspond to C-terminal residues (305NMDV308), which are far removed from the DNA-binding surface and not seen in crystal structures of TDG111-308 or TDG82-308 (23,33,34), including those reported here. This assignment is supported by observation that the same four resonances appear in NMR spectra for DNA-free TDG111-308 (Supplementary Figure S3).
Interactions with the flipped dUrd nucleotide in the enzyme–substrate complex
The new E·S structures of TDG82-308 and TDG111-308 bound to a G·UF mismatch reveal detailed interactions with the flipped Ura base, some involving ordered water molecules that have not previously been observed in any structure of TDG or the related bacterial MUG enzymes (Figure 5). Notably, the E·S interactions shown in Figure 5 for TDG82-308 are also observed for TDG111-308 (not shown). The O2 of Ura forms two hydrogen bonds with TDG backbone N-H groups (Ile139, Asn140), as noted previously (34), and one of these contacts is relatively short (2.8 Å) in the new structures. Ura O2 forms a weak interaction with a water molecule, which is itself tightly bound by a backbone N–H and the Ser271 side chain. Notably, Ser271 is structurally analogous to the catalytic His residue of the related enzyme, uracil DNA glycosylase (UNG), which uses the catalytic His to contact Ura O2 directly (63). The O4 of Ura contacts a backbone amide (residue 152) of TDG and forms a close contact with a water molecule that is coordinated by the side chains of His151 and Asn191 and another ordered water molecule. The imino N3-H of Ura contacts the side chain oxygen of Asn191, confirming a previous observation (34). For uracil and its analogues (including thymine), these interactions are likely important for stabilizing the flipped conformation of the target nucleotide. Moreover, the contacts with O2 and O4 likely stabilize the anionic leaving group that is generated upon cleavage of the N-glycosyl bond (64).
Figure 5.

Interactions with the flipped Ura base in the enzyme–substrate complex of TDG82-308 bound to G·UF DNA (PDBID: 5HF7; 1.54 Å). TDG residues are in stick format (white with nitrogen blue and oxygen red), the Ura-containing DNA is yellow (with 2′-F colored cyan) and water molecules are red spheres. The 2Fo–Fc electron density map, contoured at 1.0 σ, is shown for the DNA and water molecules, but not the enzyme residues (for clarity). Dashed lines represent hydrogen bonds, with interatomic distances (Å).
We note that the sugar pucker of the flipped 2′-F-dUrd nucleotide, C1′-exo-O4′-endo, is well defined by the electron density for E·S structures of TDG82-308 and TDG111-308 (Supplementary Figure S5). High-resolution structures of other 2′-fluoroarabino deoxynucleotides, in free DNA or flipped into a glycosylase active site, reveal either the same or a similar sugar pucker, including O4′-endo and C2′-endo (65–68). Each of these conformations is observed in high-resolution structures of B-DNA, where pyrimidines often exhibit a pucker other than C2′-endo (69). A previous structure of the E·S complex for TDG111-308-G·UF indicated a slight O4′-endo pucker, though such determination is difficult to make with confidence given relatively low resolution (34). For the MutY E·S complex, a C2′-endo pucker is observed for 2′-F-dAde and for natural dAde when these nucleotides are flipped into the active site of wild-type or mutant MutY, respectively (66,70). Thus, at least for this example, the 2′-F substituent does not appear to alter the sugar pucker of the flipped deoxynucleotide.
Notably, both of our E·S structures reveal that the flipped dUrd is partially exposed to solvent owing to a solvent-filled channel that runs along the DNA from the enzyme surface to the active site (Supplementary Figure S6). The solvent-filled channel was recently observed in high-resolution structures of product complexes for TDG111-308 (33). The new structure reveals a similar channel for the enzyme–substrate complex, and indicates that it is not occluded by the additional N-terminal residues of TDG82-308. The channel could potentially allow for escape of the excised base, perhaps involving movement of the enzyme and/or DNA. It could also be important for catalysis, given the finding that TDG excision of caC is acid catalyzed, involving a proton derived from solvent rather than a general acid of the enzyme (56).
Remarkably, the structures also reveal that the backbone carbonyl oxygen of Asn140 points directly at the N-glycosyl bond of the flipped dUrd (Figure 5) and is proximal to C1′ (3.2 Å) and to three nuclei of the Ura base (N1, 2.9 Å; C2, 2.8 Å; O2, 3.0 Å). Such an interaction could potentially serve to drive the anionic leaving group away from the cationic sugar, which could potentially suppress reformation of the N-glycosyl bond and thereby favor nucleophile addition. The Asn140 backbone oxygen could also stabilize the cationic oxacarbenium ion intermediate that likely arises upon cleavage of the C-N bond (64,71).
Coordination of the water nucleophile
The Asn140 side chain is essential for TDG activity (39,72), likely because it coordinates the nucleophilic water molecule. The previous structure of TDG111-308 bound to G·UF suggested how TDG and MUG enzymes coordinate the water nucleophile (34). However, the putative nucleophile was the sole water molecule observed in the relatively low-resolution (2.97 Å) structure, and confidence in its assignment and placement was therefore somewhat limited. By contrast, the electron density for the two structures reported here is excellent (Figure 6), defining clearly how TDG coordinates the water nucleophile and confirming our findings in the lower-resolution structure (34). The Asn140 side chain oxygen forms a short (2.7 Å) hydrogen bond to the putative nucleophilic water molecule. Loss of this interaction likely accounts, at least in part, for findings that the N140A mutation completely depletes G·T glycosylase activity and causes a huge (27 000-fold) loss in G·U activity (34). As shown in Figure 6, the nucleophile is also contacted by the backbone oxygen and side chain hydroxyl of Thr197, a residue that is also strictly conserved and important for base excision, as indicated by findings that the T197A mutation causes a 32-fold reduction in G·T glycosylase activity (34). Previous findings that the N140A mutant retains a low level of glycosylase activity for some substrates (G·U, G·5FU) (39,72) is likely explained by contacts to the nucleophile from Thr197 and perhaps other enzyme groups. Notably, the Asn140 side chain is positioned by contacts to the Thr197 hydroxyl and a backbone oxygen (195). Given the substantial homology between TDG and MUG enzymes (E. coli MUG 32% identical to human TDG), and the strict conservation of nucleophile-coordinating residues (Asn140, Thr197), this nucleophile-binding mechanism for TDG likely applies to MUG enzymes. This is significant because no putative nucleophile is observed the structure of an E·S complex for MUG bound to G·UF DNA (43).
Figure 6.

Binding of the nucleophilic water molecule in the E·S complex of TDG82-308 bound to G·UF DNA. TDG is shown in cartoon format with residues of interest in stick format (white with nitrogen blue and oxygen red), the flipped dUrd nucleotide is in yellow stick format, and the nucleophilic water molecule is a red sphere (other waters not shown, for clarity). The 2Fo–Fc map, contoured at 1.0 σ, is shown for the nucleophilic water. Dashed lines represent hydrogen bonds, with interatomic distances (Å). The distance between the nucleophilic water molecule and C1′ of the flipped dUrd nucleotide (4.0 Å) is indicated by a thin dashed line.
Our structure reveals a distance of 4.0 Å between the nucleophile and the nascent electrophile, C1′ of dUrd, in the enzyme–substrate complex (Figure 6). Previous studies of UNG indicate this distance would be reduced following cleavage of the N-glycosyl bond and migration of the electrophile (C1′ of an oxacarbenium ion intermediate) and possibly the nucleophile (73). As we noted previously (34), the proximity and relative position of the nucleophile and electrophile (C1′) observed for the E·S complex of TDG is nearly identical to that observed in a high-resolution (1.8 Å) structure of an E·S complex for the related enzyme UNG (nucleophile to C1′ distance of 3.5 Å) (63). Notably, a structure (1.9 Å resolution) of UNG bound to DNA containing an analog of the glycosyl cation intermediate reveals that the nucleophile–electrophile distance is reduced to 2.8 Å, via nucleophile migration (73).
Arg ‘plug’ residue
Previous structures of DNA-bound TDG show that the strictly conserved Arg275 side chain penetrates the DNA minor groove and fills a void generated by nucleotide flipping (23–25,34). The E·S structures here reveal direct and water-mediated contacts for Arg275 (Figure 7), most of which have not been observed in previous structures of E·S complexes. The cationic Arg275 side chain directly contacts each of the two anionic phosphates that flank the flipped nucleotide and forms an additional water-mediated contact with the 5′ phosphate. Remarkably, Arg275 also contacts the backbone oxygen of Pro198, which resides in a loop that contains key catalytic groups (Figures 6 and 7). Notably, the Arg275-Pro198 contact is also observed in our new E·P structure for TDG82-308 here, but not observed in any structures of TDG111-308, including high-resolution structures reported here and previously (33). Formation of the Arg275-Pro198 contact by TDG82-308 but not TDG111-308 might be due in part to the DNA contact provided by Arg110 (Figure 7), a residue that is absent in TDG111-308.
Figure 7.

Interactions involving the Arg275 ‘plug’ residue in the E·S complex for TDG82-308 and G·UF DNA. Arg275 penetrates the minor groove and occupies the void generated by flipping of the dUrd nucleotide. TDG is shown in cartoon format with key residues in stick format (white with nitrogen blue and oxygen red). The Ura-containing DNA is yellow and the complementary strand green; water molecules are red spheres. Dashed lines represent hydrogen bonds, with interatomic distances (Å) shown. The ‘opposing G’ is the Gua of the G·UF mispair. The contact involving Arg110, unique to TDG82-308, is also shown.
The Arg275-Pro198 contact could help to stabilize the ‘plug’ conformation of Arg275, and thereby stabilize nucleotide flipping. Moreover, Pro198 is in a loop that would block reverse flipping of the target nucleotide out of the active site and back into the DNA duplex. Together, these effects might contribute to the much tighter binding to G·T DNA observed for TDG82-308 relative to TDG111-308 (Figure 2). Because Pro198 flanks Thr197, which helps coordinate the water nucleophile, the Arg275-Pro198 contact might also serve to couple nucleotide flipping with the chemical step or help to properly position the nucleophilic water. Such a mechanism might account in part for findings that the maximal enzymatic rate (kmax) for G·T activity is 4-fold higher for TDG82-308 relative to TDG111-308 (Figure 2).
Notably, the closely related bacterial MUG enzymes have a Leu rather than an Arg residue in the corresponding ‘plug’ position (43,74), which cannot form any of the direct or water-mediated contacts observed here for TDG. MUG acts on G·U and other substrates but not on G·T mispairs. An Arg residue serves as the ‘plug’ in MBD4 and MIG enzymes (75–78), which act on G·T (and G·U) mispairs but are unrelated to TDG. These observations suggest that some of the detailed electrostatic contacts observed here for the Arg plug of TDG may be generally important for G·T glycosylase activity and may be conserved for MBD4 and MIG. Indeed, for MBD4, the Arg ‘plug’ forms similar contacts with the two phosphates flanking the flipped nucleotide as observed for TDG (75,77). No DNA-bound structures have been reported for MIG.
Interactions that may confer sequence context
TDG exhibits a strong preference for excising thymine, uracil and 5-substituted uracil analogs when these bases are flanked by a 3′ Gua and base-paired with Gua rather than Ade (35,44,79). Structures of the two G·UF complexes reported here (TDG82-308 and TDG111-308) validate the contacts to these two Gua bases that were suggested by a previous structure of TDG111-308 with G·UF solved at moderate (2.97 Å) resolution (34). In the previous structure, potential contacts to the opposing Gua from backbone oxygens were suggested, but the interatomic distances were rather long (d ≥ 3.6 Å), indicating weak interactions. By contrast, the two high-resolution E·S structures reported here reveal three clear contacts to the opposing Gua (d ≤ 3.0 Å) (Supplementary Figure S7). The backbone oxygens that contact the opposing Gua would likely present a repulsive environment to the corresponding regions of Ade (N1, C2), suggesting that specificity against canonical A·T pairs might involve repulsion of Ade as the pairing partner for Thy or other uracil analogs as the target base.
The new structures also demonstrate that the Gua located 3′ of the flipped Ura is contacted (at its N2H2) by the side chain of Gln278 and the backbone nitrogen of Ala277 (Figure 8). These contacts, which were not seen in the previous structure of the E·S complex (TDG111-308 with G·UF), offer an explanation for findings that TDG excision of Thy, and Ura analogues, is most efficient when the 3′ base is Gua, that is, a CpG (or CG) sequence context (relative activity: XpG >> XpA > XpC > XpT; where X is Thy or a Ura analog) (35). The structures also reveal that the side chain of Gln278 and potentially Lys201 can contact the second base located 3′ to the flipped Ura (Figure 8). While Lys201 does not contact the Cyt base located two nucleotides away from dUrd for TDG82-308, it does so for the TDG111-308 structure reported here (not shown). This could be significant because cytosine methylation (mC) is found in non-CG sites, with mCAC and mCAG observed most frequently (80,81). Our structures suggest TDG would not form specific interactions with a 3′ Ade (relative to the flipped nucleotide) but can potentially contact Cyt and perhaps Gua in the second position located 3′ of the flipped nucleotide.
Figure 8.

TDG contacts two bases on the 3′ side of the flipped nucleotide, including the 3′-Gua. TDG82-308 is shown in surface representation with residues that contact the 3′-Gua shown as sticks. The dUrd-containing DNA strand is yellow. For clarity, the complementary strand is not shown except for the opposing Gua (green). Water molecules are red spheres. Dashed lines represent hydrogen bonds with interatomic distances (Å).
CONCLUSION
Our results indicate that the 29 N-terminal residues of TDG82-308 confer enhanced substrate binding and faster base excision such that the activity of TDG82-308 is equivalent to that of intact TDG and much greater than TDG111-308. At the same time, crystal structures and NMR studies indicate that most (25) of these N-terminal residues are disordered, even when TDG82-308 is tightly bound to G·T or G·U substrate DNA. Thus, the enhanced biochemical activity provided by the N-terminal residues is attained without a gain in ordered structure. It will be of interest to investigate the nature of the disordered state of these N-terminal residues in future studies. One possibility might be that they participate in the search for specific sites and that this function might lead to the observed disorder through exchange between search and recognition conformations. Another possibility is that these residues, many of which are cationic (Lys, Arg), form non-specific and transient and interactions with the DNA backbone, thereby enhancing the binding affinity of TDG for specific and non-specific sites. In addition, transient DNA interactions for the N-terminal residues might enable TDG to bind DNA and allow residues 95–106, the PIP degron, to interact with PCNA and the E3 ligase CRL4Cdt2 to mediate degradation of TDG in S phase (27–29). The new high-resolution structures of TDG82-308 reported here reveal new and important enzyme–DNA contacts, some of which are not observed in corresponding structures for the smaller TDG111-308 construct. Together, our findings indicate that TDG82-308 is superior to TDG111-308 as a model for studying the structure and function of TDG. Our findings also suggest that the catalytic domain of TDG should be redefined to include Asp109 and Arg110, which contact the DNA substrate but are not found in TDG111-308.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office (DOE) by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS; including P41GM103393) and the National Center for Research Resources (NCRR; P41RR001209). The Berkeley Center for Structural Biology is supported in part by the NIH, NIGMS and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. DOE under Contract No. DE-AC02-05CH11231. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR or NIH.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited in the Protein Data Bank (http://www.rcsb.org/) with accession numbers 5HF7, 5FF8 and 5JXY.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM072711 to A.C.D. in part]; National Institutes of Health [S10-OD011969 to Support for procuring the imaging system (GE Typhoon FLA 9500)]. Funding for open access charge: National Institutes of Health (NIH) [grant GM072711 to A.C.D.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Bellacosa A., Drohat A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair (Amst) 2015;32:33–42. doi: 10.1016/j.dnarep.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neddermann P., Jiricny J. The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem. 1993;268:21218–21224. [PubMed] [Google Scholar]
- 3.Neddermann P., Gallinari P., Lettieri T., Schmid D., Truong O., Hsuan J.J., Wiebauer K., Jiricny J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J. Biol. Chem. 1996;271:12767–12774. doi: 10.1074/jbc.271.22.12767. [DOI] [PubMed] [Google Scholar]
- 4.Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Le Coz M., Devarajan K., Wessels A., Soprano D., et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortazar D., Kunz C., Selfridge J., Lettieri T., Saito Y., Macdougall E., Wirz A., Schuermann D., Jacobs A.L., Siegrist F., et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 6.Maiti A., Drohat A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., et al. Tet-mediated formation of 5-Carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfaffeneder T., Hackner B., Truss M., Munzel M., Muller M., Deiml C.A., Hagemeier C., Carell T. The discovery of 5-Formylcytosine in embryonic stem cell DNA. Angew Chem. Int. Ed. Engl. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 10.Song C.X., Szulwach K.E., Dai Q., Fu Y., Mao S.Q., Lin L., Street C., Li Y., Poidevin M., Wu H., et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen L., Wu H., Diep D., Yamaguchi S., D'Alessio A.C., Fung H.L., Zhang K., Zhang Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett M.T., Rodgers M.T., Hebert A.S., Ruslander L.E., Eisele L., Drohat A.C. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J. Am. Chem. Soc. 2006;128:12510–12519. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortazar D., Kunz C., Saito Y., Steinacher R., Schar P. The enigmatic thymine DNA glycosylase. DNA Repair (Amst) 2007;6:489–504. doi: 10.1016/j.dnarep.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Tini M., Benecke A., Um S.J., Torchia J., Evans R.M., Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 15.Mohan R.D., Rao A., Gagliardi J., Tini M. SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol. Cell. Biol. 2007;27:229–243. doi: 10.1128/MCB.00323-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan X., Madabushi A., Chang D.Y., Fitzgerald M., Shi G., Drohat A.C., Lu A.L. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates DNA repair enzyme TDG glycosylase. Nucleic Acids Res. 2007;35:6207–6218. doi: 10.1093/nar/gkm678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madabushi A., Hwang B.J., Jin J., Lu A.L. Histone deacetylase SIRT1 modulates and deacetylates DNA base excision repair enzyme thymine DNA glycosylase. Biochem J. 2013;456:89–98. doi: 10.1042/BJ20130670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardeland U., Steinacher R., Jiricny J., Schar P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002;21:1456–1464. doi: 10.1093/emboj/21.6.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smet-Nocca C., Wieruszeski J.M., Chaar V., Leroy A., Benecke A. The thymine-DNA glycosylase regulatory domain: Residual structure and DNA binding. Biochemistry. 2008;47:6519–6530. doi: 10.1021/bi7022283. [DOI] [PubMed] [Google Scholar]
- 20.Coey C.T., Fitzgerald M.E., Maiti A., Reiter K.H., Guzzo C.M., Matunis M.J., Drohat A.C. E2-mediated small ubiquitin-like modifier (SUMO) modification of thymine DNA glycosylase is efficient but not selective for the enzyme-product complex. J. Biol. Chem. 2014;289:15810–15819. doi: 10.1074/jbc.M114.572081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLaughlin D., Coey C.T., Yang W.C., Drohat A.C., Matunis M.J. Characterizing requirements for SUMO modification and binding on base excision repair activity of thymine DNA glycosylase in vivo. J. Biol. Chem. 2016;291:9014–9024. doi: 10.1074/jbc.M115.706325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinacher R., Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr. Biol. 2005;15:616–623. doi: 10.1016/j.cub.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 23.Maiti A., Morgan M.T., Pozharski E., Drohat A.C. Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8890–8895. doi: 10.1073/pnas.0711061105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G.L., Luo C., Jiang H., He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012;8:328–330. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashimoto H., Hong S., Bhagwat A.S., Zhang X., Cheng X. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012;40:10203–10214. doi: 10.1093/nar/gks845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baba D., Maita N., Jee J.-G., Uchimura Y., Saitoh H., Sugasawa K., Hanaoka F., Tochio H., Hiroaki H., Shirakawa M. Crystal structure of thymine DNA glycosylase conjugated to SUMO-1. Nature. 2005;435:979–982. doi: 10.1038/nature03634. [DOI] [PubMed] [Google Scholar]
- 27.Slenn T.J., Morris B., Havens C.G., Freeman R.M., Jr, Takahashi T.S., Walter J.C. Thymine DNA glycosylase is a CRL4Cdt2 substrate. J. Biol. Chem. 2014;289:23043–23055. doi: 10.1074/jbc.M114.574194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata E., Dar A., Dutta A. CRL4Cdt2 E3 ubiquitin ligase and PCNA cooperate to degrade thymine DNA glycosylase in S-phase. J. Biol. Chem. 2014;289:23056–23064. doi: 10.1074/jbc.M114.574210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardeland U., Kunz C., Focke F., Szadkowski M., Schar P. Cell cycle regulation as a mechanism for functional separation of the apparently redundant uracil DNA glycosylases TDG and UNG2. Nucleic Acids Res. 2007;35:3859–3867. doi: 10.1093/nar/gkm337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohan R.D., Litchfield D.W., Torchia J., Tini M. Opposing regulatory roles of phosphorylation and acetylation in DNA mispair processing by thymine DNA glycosylase. Nucleic Acids Res. 2009;38:1135–1148. doi: 10.1093/nar/gkp1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallinari P., Jiricny J. A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature. 1996;383:735–738. doi: 10.1038/383735a0. [DOI] [PubMed] [Google Scholar]
- 32.Li Y.Q., Zhou P.Z., Zheng X.D., Walsh C.P., Xu G.L. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malik S.S., Coey C.T., Varney K.M., Pozharski E., Drohat A.C. Thymine DNA glycosylase exhibits negligible affinity for nucleobases that it removes from DNA. Nucleic Acids Res. 2015;43:9541–9552. doi: 10.1093/nar/gkv890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maiti A., Noon M.S., Mackerell A.D., Jr, Pozharski E., Drohat A.C. Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8091–8096. doi: 10.1073/pnas.1201010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan M.T., Bennett M.T., Drohat A.C. Excision of 5-halogenated uracils by human thymine DNA glycosylase: Robust activity for DNA contexts other than CpG. J. Biol. Chem. 2007;282:27578–27586. doi: 10.1074/jbc.M704253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tropea J.E., Cherry S., Waugh D.S. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol. Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 37.Gill S.C., von Hippel P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 38.Morgan M.T., Maiti A., Fitzgerald M.E., Drohat A.C. Stoichiometry and affinity for thymine DNA glycosylase binding to specific and nonspecific DNA. Nucleic Acids Res. 2011;39:2319–2329. doi: 10.1093/nar/gkq1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maiti A., Morgan M.T., Drohat A.C. Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase. J. Biol. Chem. 2009;284:36680–36688. doi: 10.1074/jbc.M109.062356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manvilla B.A., Varney K.M., Drohat A.C. Chemical shift assignments for human apurinic/apyrimidinic endonuclease 1. Biomol. NMR Assign. 2009;4:5–8. doi: 10.1007/s12104-009-9196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amburgey J.C., Abildgaard F., Starich M.R., Shah S., Hilt D.C., Weber D.J. 1H, 13C and 15N NMR assignments and solution secondary structure of rat Apo-S100 beta. J. Biomol. NMR. 1995;6:171–179. doi: 10.1007/BF00211781. [DOI] [PubMed] [Google Scholar]
- 42.Scharer O.D., Kawate T., Gallinari P., Jiricny J., Verdine G.L. Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4878–4883. doi: 10.1073/pnas.94.10.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrett T.E., Scharer O.D., Savva R., Brown T., Jiricny J., Verdine G.L., Pearl L.H. Crystal structure of a thwarted mismatch glycosylase DNA repair complex. EMBO J. 1999;18:6599–6609. doi: 10.1093/emboj/18.23.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waters T.R., Swann P.F. Kinetics of the action of thymine DNA glycosylase. J. Biol. Chem. 1998;273:20007–20014. doi: 10.1074/jbc.273.32.20007. [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCoy A.J., Grosse-Kunstleve R.W., Storoni L.C., Read R.J. Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 50.Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Rovers i.P., Sharff A., Smart O.S., Vonrhein C., et al. Cambridge: Global Phasing Ltd; 2011. [Google Scholar]
- 51.Winn M.D., Isupov M.N., Murshudov G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P., Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Painter J., Merritt E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 54.Painter J., Merritt E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006;39:109–111. [Google Scholar]
- 55.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 56.Maiti A., Michelson A.Z., Armwood C.J., Lee J.K., Drohat A.C. Divergent mechanisms for enzymatic excision of 5-formylcytosine and 5-carboxylcytosine from DNA. J. Am. Chem. Soc. 2013;135:15813–15822. doi: 10.1021/ja406444x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maiti A., Drohat A.C. Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and processing of G.T mispairs. DNA Repair (Amst) 2011;10:545–553. doi: 10.1016/j.dnarep.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buechner C.N., Maiti A., Drohat A.C., Tessmer I. Lesion search and recognition by thymine DNA glycosylase revealed by single molecule imaging. Nucleic Acids Res. 2015;43:2716–2729. doi: 10.1093/nar/gkv139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pozharski E. Percentile-based spread: a more accurate way to compare crystallographic models. Acta Crystallogr. D Biol. Crystallogr. 2010;66:970–978. doi: 10.1107/S0907444910027927. [DOI] [PubMed] [Google Scholar]
- 60.Williamson M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Joshi P., Vendruscolo M. Druggability of Intrinsically Disordered Proteins. Adv. Exp. Med. Biol. 2015;870:383–400. doi: 10.1007/978-3-319-20164-1_13. [DOI] [PubMed] [Google Scholar]
- 62.Rezaei-Ghaleh N., Blackledge M., Zweckstetter M. Intrinsically disordered proteins: from sequence and conformational properties toward drug discovery. Chembiochem. 2012;13:930–950. doi: 10.1002/cbic.201200093. [DOI] [PubMed] [Google Scholar]
- 63.Parikh S.S., Walcher G., Jones G.D., Slupphaug G., Krokan H.E., Blackburn G.M., Tainer J.A. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Drohat A.C., Maiti A. Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem. 2014;12:8367–8378. doi: 10.1039/c4ob01063a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berger I., Tereshko V., Ikeda H., Marquez V., Egli M. Crystal structures of B-DNA with incorporated 2′-deoxy-2′-fluoro- arabino-furanosyl thymines: implications of conformational preorganization for duplex stability. Nucleic Acids Res. 1998;26:2473–2480. doi: 10.1093/nar/26.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee S., Verdine G.L. Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18497–18502. doi: 10.1073/pnas.0902908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bowman B.R., Lee S.M., Wang S.Y., Verdine G.L. Structure of the E-coli DNA glycosylase AlkA bound to the ends of duplex DNA: A system for the structure determination of lesion-containing DNA. Structure. 2008;16:1166–1174. doi: 10.1016/j.str.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee S., Bowman B.R., Ueno Y., Wang S., Verdine G.L. Synthesis and structure of duplex DNA containing the genotoxic nucleobase lesion N7-methylguanine. J. Am. Chem. Soc. 2008;130:11570–11571. doi: 10.1021/ja8025328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shui X., McFail-Isom L., Hu G.G., Williams L.D. The B-DNA dodecamer at high resolution reveals a spine of water on sodium. Biochemistry. 1998;37:8341–8355. doi: 10.1021/bi973073c. [DOI] [PubMed] [Google Scholar]
- 70.Fromme J.C., Banerjee A., Huang S.J., Verdine G.L. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 71.Berti P.J., McCann J.A. Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem. Rev. 2006;106:506–555. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 72.Hardeland U., Bentele M., Jiricny J., Schar P. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem. 2000;275:33449–33456. doi: 10.1074/jbc.M005095200. [DOI] [PubMed] [Google Scholar]
- 73.Bianchet M.A., Seiple L.A., Jiang Y.L., Ichikawa Y., Amzel L.M., Stivers J.T. Electrostatic guidance of glycosyl cation migration along the reaction coordinate of uracil DNA glycosylase. Biochemistry. 2003;42:12455–12460. doi: 10.1021/bi035372+. [DOI] [PubMed] [Google Scholar]
- 74.Barrett T.E., Savva R., Panayotou G., Barlow T., Brown T., Jiricny J., Pearl L.H. Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell. 1998;92:117–129. doi: 10.1016/s0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- 75.Manvilla B.A., Maiti A., Begley M.C., Toth E.A., Drohat A.C. Crystal structure of human methyl-binding domain IV glycosylase bound to abasic DNA. J. Mol. Biol. 2012;420:164–175. doi: 10.1016/j.jmb.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hashimoto H., Zhang X., Cheng X. Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012;40:8276–8284. doi: 10.1093/nar/gks628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morera S., Grin I., Vigouroux A., Couve S., Henriot V., Saparbaev M., Ishchenko A.A. Biochemical and structural characterization of the glycosylase domain of MBD4 bound to thymine and 5-hydroxymethyuracil-containing DNA. Nucleic Acids Res. 2012;40:9917–9926. doi: 10.1093/nar/gks714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mol C.D., Arvai A.S., Begley T.J., Cunningham R.P., Tainer J.A. Structure and activity of a thermostable thymine-DNA glycosylase: evidence for base twisting to remove mismatched normal DNA bases. J. Mol. Biol. 2002;315:373–384. doi: 10.1006/jmbi.2001.5264. [DOI] [PubMed] [Google Scholar]
- 79.Sibghat U., Gallinari P., Xu Y.Z., Goodman M.F., Bloom L.B., Jiricny J., Day R.S., 3rd Base analog and neighboring base effects on substrate specificity of recombinant human G:T mismatch-specific thymine DNA-glycosylase. Biochemistry. 1996;35:12926–12932. doi: 10.1021/bi961022u. [DOI] [PubMed] [Google Scholar]
- 80.Guo J.U., Su Y., Shin J.H., Shin J., Li H., Xie B., Zhong C., Hu S., Le T., Fan G., et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kinde B., Gabel H.W., Gilbert C.S., Griffith E.C., Greenberg M.E. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. U.S.A. 2015;112:6800–6806. doi: 10.1073/pnas.1411269112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.