

Abstract
Since axon damage and retinal ganglion cell (RGC) loss lead to blindness, therapies that increase RGC survival and axon regrowth have direct clinical relevance. Given that NFκB signaling is critical for neuronal survival and may regulate neurite growth, we investigated the therapeutic potential of NFκB signaling in RGC survival and axon regeneration. Although both NFκB subunits (p65 and p50) are present in RGCs, p65 exists in an inactive (unphosphorylated) state when RGCs are subjected to neurotoxic conditions. In this study, we used a phosphomimetic approach to generate DNA coding for an activated (phosphorylated) p65 (p65mut), then employed an adeno-associated virus serotype 2 (AAV2) to deliver the DNA into RGCs. We tested whether constitutive p65mut expression prevents death and facilitates neurite outgrowth in RGCs subjected to transient retinal ischemia or optic nerve crush (ONC), two models of neurotoxicity. Our data indicate that RGCs treated with AAV2-p65mut displayed a significant increase in survival compared to controls in ONC model (77±7% vs. 25±3%, P-value=0.0001). We also found protective effect of modified p65 in RGCs of ischemic retinas (55±12% vs. 35±6), but not to a statistically significant degree (P-value=0.14). We did not detect a difference in axon regeneration between experimental and control animals after ONC. These findings suggest that increased NFκB signaling in RGCs attenuates retinal damage in animal models of neurodegeneration, but insignificantly impacts axon regeneration.
Keywords: RGCs, phosphomimetic, adeno-associated virus serotype 2, transient retinal ischemia model, optic nerve crush model
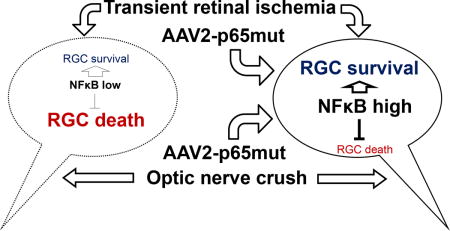
Graphical abstract
We used a phosphomimetic approach to generate DNA coding for an activated (phosphorylated) NFκB subunit p65 (p65mut), then employed an adeno-associated virus serotype 2 (AAV2) to deliver the DNA into RGCs. We found that increased activity of the NFκB in RGCs (using AAV2-p65mut) attenuates retinal damage in animal models of neurodegeneration. However, NFκB activity in RGCs has a limited impact on axon regeneration.
Introduction
Retinal ganglion cells (RGCs) are known to be susceptible to degeneration in several acute and chronic retinal diseases (Quigley, 1999; Osborne et al., 2004; Levin, 2007; Kern & Barber, 2008). RGC loss and axon damage resulting from these diseases progressively lead to blindness. Since there is currently no technology that allows us to regenerate lost RGCs, therapies to maintain the viability of damaged RGCs and mediate the regrowth of axons back to the visual cortex of the brain are sorely needed (Sernagor et al., 2001). Many different approaches have been tested (Harvey et al., 2006; Park et al., 2008; Dvoriantchikova et al., 2014c). But the levels of survival of damaged RGCs and the extent of axon regeneration observed in these studies are still limited (Park et al., 2004; Harvey et al., 2006; Park et al., 2008; Sun et al., 2011; Dvoriantchikova et al., 2014a; Dvoriantchikova et al., 2014c). Thus, new therapeutic strategies to save RGCs and restore patient vision must be identified.
NFκB signaling plays a key role in regulating inflammatory responses, cell survival, and cell proliferation (Oeckinghaus et al., 2011). Several lines of evidence suggest that NFκB signaling in neurons prevents neuronal death and regulates neurite outgrowth: 1) hippocampal pyramidal neurons of mice lacking the p50 subunit of NFκB are more susceptible to high neurotoxin levels (Yu et al., 1999); 2) reduced p65 levels in spinal cord motoneurons facilitate death of these neurons (Mincheva et al., 2011); 3) high p65 expression within cerebellar granule neurons prevents low potassium-induced apoptosis (Koulich et al., 2001), while inhibition of NFκB signaling in these cells promotes amyloid β-induced apoptosis (Kaltschmidt et al., 1999); 4) neuron-specific ablation of NFκB-dependent gene expression in the forebrain increases neurodegeneration of this tissue (Fridmacher et al., 2003); 5) the neuroprotective role of NFκB signaling has also been demonstrated in sympathetic neurons (Maggirwar et al., 1998); 6) a variety of neurotrophic factors facilitate neuronal survival and neurite outgrowth by activating NFκB (Foehr et al., 2000; Koulich et al., 2001; Gallagher et al., 2007; Gutierrez et al., 2008; Gavalda et al., 2009; Mincheva et al., 2011). Surprisingly, in contrast to many other neuronal types, RGCs fail to activate NFκB signaling under neurotoxic conditions, despite the fact that all NFκB family members are present inside them (Tezel & Yang, 2005; Dvoriantchikova & Ivanov, 2014). This absence of NFκB activity was associated with increased levels of RGC death (Dvoriantchikova & Ivanov, 2014). In vivo studies revealed NFκB activity only in the RGCs that survived optic nerve crush injury, whereas inhibition of NFκB activity accelerated the degeneration of RGCs (Choi et al., 2000). Thus, augmenting NFκB signaling in RGCs could be an efficient therapeutic strategy, serving to prevent the death of RGCs with undamaged axons in disease-mediated neurotoxicity or support survival of RGCs with damaged axons long enough to allow them to regenerate their axons under neurotoxic conditions. In this study, we tested the efficiency of NFκB signaling-based therapy in a transient retinal ischemia model and an optic nerve crush model.
Materials and Methods
Animals
All experiments were performed in compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, and the Association for Research in Vision and Ophthalmology (ARVO) statement for use of animals in ophthalmic and vision research. The protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Miami. C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Mice were housed under standard conditions of temperature and humidity, with free access to food and water and a 12-hour light/dark cycle. All animals used in our experiments were 3-month-old male mice. The mice were euthanized by CO2 inhalation.
Preparation of modified p65/Rela plasmids and AAV2 viral particles
The modified p65/Rela coding cDNA (refer to S1 Appendix) was synthesized and cloned into a pUC57 plasmid by GENEWIZ company (South Plainfield, NJ). To make the adeno-associated virus serotype-2 (AAV2), the cDNA was subcloned from the pUC57 plasmid into a pAAV-MCS plasmid (Agilent Technologies, Santa Clara, CA) that contained AAV2 inverted terminal repeats and a human growth hormone polyA signal. The pAAV-MCS plasmids were then used to produce AAV2 (1×1013 genome copies/ml) viral particles at the University of Miami Viral Vector Core.
Culture and transfection of the AAV-293 cell line
The AAV-293 cells were obtained from Agilent Technologies (Santa Clara, CA). The cells were grown and maintained in high-glucose DMEM containing 10% FBS and 2 mM of L-glutamine. The AAV-293 cells were split two days before transfection to obtain 70–80% confluency. To transfect the AAV-293 cells with a plasmid, we used a calcium phosphate-based protocol according to the manufacturer’s instruction manual (Agilent Technologies, Santa Clara, CA).
Isolation, culture, and viral transduction of primary retinal ganglion cell (RGC) cultures
To isolate RGCs, we used the two-step immunopanning protocol as described in our articles (Dvoriantchikova & Ivanov, 2014; Dvoriantchikova et al., 2014b; Dvoriantchikova et al., 2014c). Briefly, retinas were incubated in papain solution (16.5 U/mL; Worthington Biochemical Corp, Lakewood, NJ) for 30 minutes to obtain the cell suspension. Macrophages and endothelial cells were removed from the cell suspension by panning with the anti-macrophage antibody (Accurate Chemical, Westbury, NY). RGCs were bound to the panning plates containing CD90.2/Thy1.2 hybridoma supernatant and released by trypsin incubation. To transduce RGC cultures with AAV2, RGCs were plated in 24-well plates at a density of 50,000 cells per well. At 1–2 hours after seeding, the virus was added (1×1012 genome copies per well). RGCs were then cultured in serum-free media (Neurobasal/B27 media; Thermo Fisher Scientific, Grand Island, NY).
Immunocytochemistry
Primary RGCs and AAV-293 cells were fixed in 4% paraformaldehyde (PFA) for 30 minutes, rinsed in PBS (3 × 10 minutes) and blocked with 5% normal donkey serum with 0.15% Tween-20 in PBS at pH 7.4. Cells were then incubated with either both beta III Tubulin antibody (Tubb3, 1:250, Covance, Denver, PA) and p65 antibody (1:400; sc-372, Santa Cruz Biotechnology, Dallas, TX) or with Tubb3 alone, followed by species-specific secondary fluorescent antibodies (Thermo Fisher Scientific, Grand Island, NY). Negative controls were incubated with secondary antibodies only. The cells were imaged using an EVOS FL Auto fluorescent microscope with onstage incubation capabilities (Thermo Fisher Scientific, Grand Island, NY).
Western blot analysis
AAV-293 cells were lysed with RIPA buffer, and protein concentrations were measured using the BCA kit, according to the manufacturer’s protocol (Thermo Scientific, Brookfield, WI). An equal amount of protein samples were loaded and separated on SDS-PAGE gradient 4–12% Bis-Tris gels, then transferred to PVDF membranes (Thermo Fisher Scientific, Grand Island, NY). The PVDF membranes were blocked in 5% milk in Tris-buffered saline (TBS, pH 7.6), probed with the primary antibody against p65 (1:400; sc-372, Santa Cruz Biotechnology, Dallas, TX) overnight, washed in 0.15% Tween20 in TBS, and incubated for one hour with secondary antibody (1:10,000, Amersham Biosciences, NJ) diluted in TBS. Anti-GAPDH antibodies (1:4,000, GeneTex, Irvine, CA) were utilized to control the loading. Proteins were visualized using SuperSignal chemiluminescent substrates (Thermo Scientific, Brookfield, WI) and quantified using the FUJIFILM software.
Intravitreal injections of AAV2
Animals were first anaesthetized via intraperitoneal injection of ketamine (80 mg/kg)/xylazine (10 mg/kg). 1 μl of viral particles was then injected into the temporal part of one eye via a glass micropipette inserted just behind the ora serrata (intravitreal injection). All vector concentrations were 1–2×1013 genome copies/ml. Animals were treated with AAV2 two weeks before transient retinal ischemia or optic nerve crush.
Transient retinal ischemia model
To induce anesthesia with isoflurane (2% gas) for 45 minutes, anesthetics were administered to the breathing mice through a firmly fitting nose cone. The animals’ body temperature was maintained at 37°C with a temperature-controlled heating pad. After anesthesia, pupils were dilated with 1% tropicamide/2.5% phenylephrine hydrochloride (NutraMax Products Inc., Gloucester, MA), and corneal analgesia was achieved with one drop of 0.5% proparacaine HCl (Bausch and Lomb Pharmaceuticals, Tampa, FL). Transient retinal ischemia was induced for 45 minutes by introducing into the anterior chamber of the left eye a 33-gauge needle attached to a normal (0.9% NaCl) saline-filled reservoir. The reservoir was elevated above the animal in order to increase intraocular pressure (IOP) to 120 mmHg. The right eye was cannulated and maintained at normal IOP to serve as a normotensive control. Complete retinal ischemia, evidenced by a whitening of the anterior segment of the eye and blanching of the retinal arteries, was confirmed by microscopic examination. Erythromycin ophthalmic ointment (Fera Pharmaceuticals, Locust Valley, NY) was applied to the conjunctival sac as soon as the needle was removed.
Optic nerve crush model
After the animals were properly anesthetized via intraperitoneal injection of ketamine (80 mg/kg)/xylazine (10 mg/kg), their optic nerves were exposed intraorbitally and crushed with jeweler’s forceps (Dumont #5; tip dimension, 0.1 × 0.06 mm) for 10 seconds approximately 1 mm behind the optic disc. We then employed an anterograde labeling strategy to study levels of axon regrowth. To implement this strategy, we injected 1 μl of cholera toxin β subunit CTB-Alexa 555 (2 μg/μl, Thermo Fisher Scientific, Grand Island, NY) into the vitreous with a Hamilton syringe two days before sacrifice. After the animals were euthanized, their optic nerves were dissected, fixed in 4% PFA and cryoprotected overnight in 30% sucrose. The optic nerves were embedded in Optimal Cutting Temperature medium (OCT) and sectioned at 12 μm. Regenerating axons were recognized by CTB tracing and were counted at 500 μm from the site of injury. At least four sections were counted for each animal (5 animals per group).
Immunohistochemistry and counting of ganglion cell layer neurons
After the animals were euthanized, their eyes were enucleated and fixed in 4% PFA. Retinas were removed after one hour and cryoprotected overnight in 30% sucrose. The following day, the retinas were freeze-thawed three times, rinsed for 3 × 10 minutes in 0.1 M Tris buffer, blocked by 5% donkey serum and 0.1% Triton X-100 in 0.1 M Tris buffer for one hour, and then incubated overnight with either beta III Tubulin antibody (Tubb3, 1:250, Covance, Denver, PA) or with both Tubb3 and p65 (1:400; sc-372, Santa Cruz Biotechnology, Dallas, TX). The next day, the retinas were rinsed in 0.1 M Tris buffer (3 × 10 minutes), flat-mounted, cover-slipped, and imaged using a Leica TSL AOBS SP5 confocal microscope (Leica Microsystems, Exton, PA). To count the number of surviving RGCs, individual retinas were sampled randomly to collect a total of 20 images from four retinal quadrants using a 20X objective lens. Five images were collected from each quadrant: one from the center, two from the middle, and two from the peripheral regions. Tubb3-positive neurons (RGCs) were counted using ImageJ software. RGC survival was calculated as a percentage of the mean cell density in fellow control eyes.
Statistical analysis
To reduce variability, we selected founders for all colonies from littermates. This design was insensitive to individual variations and allowed the decreasing of animal group size without lowering the statistical power of the analysis. We performed group-size estimation with two means to evaluate the number of animals needed to detect the true difference between group 1 (AAV2-p65mut treated animals) and group 2 (AAV2-GFP treated animals) means, which is equal to (or greater than) 20%. In our calculations we assumed that standard deviation in group 1 is 12%, standard deviation in group 2 is 6%, type I error (level of significance) is 0.05 (5%), and type II error (statistical power) is 0.8 (80%). Therefore, if the true difference in the two group means is (or greater than) 20%, we will need 5 mice in group 1 and 5 mice in group 2 to be able to reject the null hypothesis that there is no difference between group 1 and group 2 with the probability (power) 0.8 (80%). The Type I error probability associated with this test of this null hypothesis is 0.05 (5%). Statistical analysis was performed with the Student t-test. P values less than or equal to 0.05 were considered statistically significant.
Results
Genetic modifications of p65 on serine residues 276 and 536 do not affect the expression or stability of the modified protein
To augment NFκB signaling in RGCs, we sought to increase activity of the NFκB subunit p65/Rela. The NFκB transcription factor is comprised of two subunits, p65 and p50/Nfkb1, which assemble as heterodimers (Oeckinghaus & Ghosh, 2009; Oeckinghaus et al., 2011). Unlike its p50 counterpart, the p65 subunit must be activated by phosphorylation; this activation step is necessary for NFκB signaling to occur (Oeckinghaus & Ghosh, 2009; Oeckinghaus et al., 2011). Our previously published results and other evidence indicate that while both p65 and p50 are present in RGCs, RGCs exposed to neurotoxic conditions contain inactive (unphosphorylated) forms of p65 (Tezel & Yang, 2005; Dvoriantchikova & Ivanov, 2014). We therefore reasoned that NFκB signaling in RGCs could be augmented by simply increasing the expression of phosphorylated (activated) p65. Several studies have demonstrated that phosphorylation of p65’s serine residue 276 (Ser276) is critical for NFκB-mediated pro-survival activity (Dong et al., 2008; Arun et al., 2009; Nihira et al., 2010; Wang et al., 2011). While none of these studies were conducted in RGCs, the preponderance of evidence in other tissues convinced us that a strategy targeting Ser276 was a worthwhile gamble. It was shown that changing Ser276 to aspartic acid (S276D) mimics p65 phosphorylation (activation) and significantly increases expression of many NFκB target genes; this approach was appropriately termed a “phosphomimetic” strategy (Dong et al., 2010). Since NFκB signaling can promote neuronal survival, and since p50 (the other half of the NFκB heterodimer) is present in RGCs, we hypothesized that constitutive expression of a mutant form of p65 (with the Ser276D substitution) would enable NFκB signaling, thereby promoting RGC survival after exposure to neurotoxic conditions. In addition, we also noted that enhanced NFκB signaling promotes neurite growth from sensory neurons, but inhibits neurite growth from sympathetic neurons (Gutierrez et al., 2008; Gavalda et al., 2009). This effect was mediated by phosphorylation of p65 on Ser536 (Gutierrez et al., 2008; Gavalda et al., 2009). It was also shown that a serine-to-alanine substitution on Ser536 (S536A) prevents phosphorylation at this site while preserving p65’s transcriptional activity (Gutierrez et al., 2008; Gavalda et al., 2009). Although phosphorylation of p65 on Ser536 is unlikely (given the absence of NFκB activation in wild type RGCs exposed to neurotoxic conditions), we opted nevertheless to employ a S536A substitution in order to prevent any possibility of p65 phosphorylation on Ser536. Since a serine-to-alanine substitution at residue 536 (S536A) can prevent the undesirable phosphorylation of p65 at this site and since changing Ser276 to aspartic acid (S276D) can mimic p65 phosphorylation (activation), we suggested that constitutive expression of a p65 S276DS536A double mutant (p65mut) in RGCs could prevent RGC death and possibly facilitate neurite outgrowth in animal models of retinal disease. To this end, we created an adeno-associated virus serotype 2 (AAV2) vector bearing a constitutively active p65 S276DS536A double mutant (AAV2- p65mut) (Fig. 1A). Prior to preparing viral particles, we tested for expression of p65mut by transiently transfecting AAV-293 cells with an AAV2-p65mut vector and subsequently analyzing them by immunocytochemistry (Fig. 1B) and western blot analysis (Fig. 1C). We found increased expression of p65mut in these cells, indicating that the two p65 modifications employed in our study do not affect p65 expression or stability.
Figure 1.
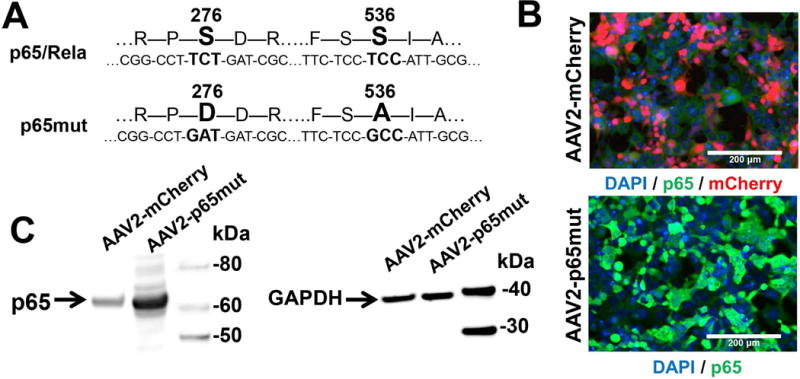
The genetically modified p65/Rela subunit of NFκB is highly expressed in AAV-293 cells. A) An AAV2 vector was used in the cloning of modified p65. The p65mut DNA was synthesized by Genewiz company (USA). B) AAV-293 cells were transfected with an AAV2-p65mut plasmid and followed by immunocytochemistry; these cells revealed high expression of modified p65 (p65mut—green). Control cells transfected with AAV2-mCherry were used as controls (mCherry—red). To identify p65mut proteins, we used an anti-p65 antibody. DAPI (blue) was used to identify cell nuclei. C) We confirmed quantitatively high p65mut expression in experimental AAV-293 by Western blot analysis using lysates of experimental and control cells. P65 band intensity normalized to GAPDH (housekeeping gene) was 3.9±0.5 (P-value=0.025) times higher in lysates of AAV-293 cells transfected with an AAV2-p65mut plasmid than in lysates of AAV-293 cells transfected with an AAV2- mCherry plasmid.
An adeno-associated virus type serotype 2 (AAV2) efficiently delivers modified p65 directly and exclusively to RGCs
We demonstrated previously that therapy based on indiscriminately increasing NFκB signaling in all cell types of the ganglion cell layer (RGCs, astrocytes, microglia) is ineffective and even dangerous (Dvoriantchikova et al., 2009; Barakat et al., 2012; Brambilla et al., 2012; Dvoriantchikova & Ivanov, 2014; Dvoriantchikova et al., 2014c). Thus, precise delivery of p65mut directly and exclusively into RGCs was critical to the success of this project. Since many studies have shown AAV2 to be an ideal mechanism for gene delivery into RGCs we therefore selected the AAV2 vector for delivery p65mut into RGCs (refer to above). We began by testing p65mut expression in RGCs in vitro. To this end, primary RGCs isolated from retinas were transduced using AAV2-p65mut viral particles. An “empty” AAV2-MCS vector was used as a control. Our data indicated that p65mut was highly expressed in these RGCs, predominantly in their nuclei (Fig. 2A). To demonstrate the efficiency and specificity of RGC transduction with AAV2-p65mut in vivo, AAV2-p65mut or AAV2-MCS (control) viral particles were injected intravitreally into the eyes of anesthetized mice. Two weeks later, the treated and control retinas were collected, flatmounted, and analyzed by immunohistochemistry with antibodies against p65 and Tubulin-βIII (Tubb3; an RGC marker). We found that p65mut was highly expressed in many cells, and was always co-localized with Tubb3 (Fig. 2B). And like the RGCs in vitro, p65mut expression was largely confined to the cells’ nuclei. Meanwhile, no p65 expression was detected in control retinas (Fig. 2B). We therefore demonstrated that the AAV2 vector efficiently and specifically delivers the modified p65 into RGCs of adult animals.
Figure 2.
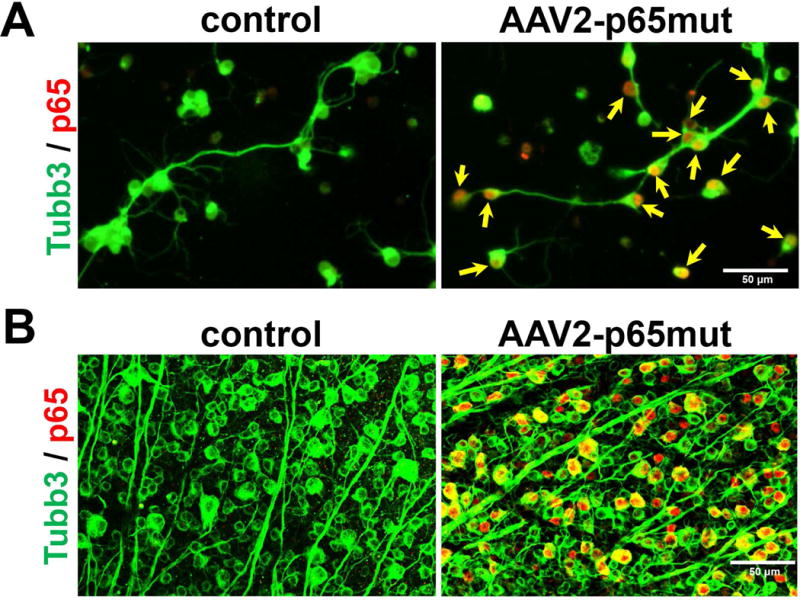
RGCs transduced with AAV2-p65mut highly express modified p65 (p65mut), mostly in the cell nuclei. A) To test the expression of p65mut in RGCs in vitro, we transduced primary RGCs isolated from wild type mouse retinas with AAV2-p65mut viral particles or with “empty” AAV2-MCS viral particles (controls). Experimental and control RGCs were fixed ten days after transduction and immunolabeled for Tubulin-βIII (Tubb3; green) and p65 (red). The arrows (yellow) indicate the cell nuclei. B) AAV2 efficiently and exclusively transduces RGCs in the ganglion cell layer of adult animals to mediate high p65mut expression, predominantly in nuclei of these cells.
Constitutively active p65mut facilitates survival of damaged RGCs in a transient retinal ischemia model and an optic nerve crush model
We employed two models—transient retinal ischemia and optic nerve crush—to evaluate the influence of constitutive p65mut expression on RGC survival. NFκB signaling has been shown to prevent both apoptosis and necroptosis (programmed necrosis) in neurons subjected to neurotoxic conditions (Oberst & Green, 2011; Vanden Berghe et al., 2014). The two models enabled us to separately evaluate p65’s role in the contexts of apoptosis and necroptosis. In the transient retinal ischemia model, most injured RGCs die via necroptosis (Fujita et al., 2009; Dvoriantchikova et al., 2010; Rosenbaum et al., 2010; Dvoriantchikova et al., 2014a). After optic nerve crush, wherein the axons of RGCs (but not the cell bodies themselves) are destroyed, most RGCs die by apoptosis (Berkelaar et al., 1994; Bien et al., 1999; Magharious et al., 2011). Thus, to study p65mut effects, AAV2-p65mut viral particles were injected intravitreally into the left eyes of anesthetized mice at least two weeks before ischemia or optic nerve crush. Control animals were transduced with AAV2-GFP at this same time. The animals’ right eyes were used as untreated controls. Retinas were collected either seven days after ischemia or two weeks after optic nerve crush, then stained for the RGC marker Tubulin-βIII (Tubb3) (Figs. 3A and 4A). By counting the number of Tubb3-positive cells, we determined that the numbers of viable RGCs were significantly higher in the AAV2-p65mut treated retinas compared to controls (Figs. 3B and 4B). However, individual analysis of the transient ischemia model detected no statistically significant difference in RGC survival between treated retinas and controls (55±12% vs. 35±6%, P-value=0.14, n=5). Meanwhile, in the optic nerve crush model, AAV2-p65mut treated RGCs did survive at a significantly higher rate than control RGCs (77±7% vs. 25±3%, P-value=0.0001, n=5). Importantly, though RGCs treated with AAV2-p65mut survived at significantly higher rates overall, they did not always look healthy. Many of the surviving RGCs were reduced in size, and displayed degenerate, disorganized axons (Figs. 3B, 3C, 4B and 4C). This effect was particularly evident in RGCs from the optic nerve crush model (Figs. 4B, 4C and S2 Appendix). Given the degenerate, disorganized axons observed in many of the surviving RGCs, we hypothesized that p65mut plays an insignificant role in promoting axon regeneration. To test this hypothesis, a group of animals were treated with AAV2-p65mut two weeks before optic nerve crush. Their control cohorts were treated with AAV2-GFP at this same time. Two weeks after optic nerve crush, axon regrowth in experimental and control retinas was measured by examining axonal fibers labeled with cholera toxin β (CTB, an anterograde tracer) in the optic nerve sections across the lesion site (Fig. 5A). We detected no significant difference in axon regeneration between experimental and control animals after optic nerve crush (Fig. 5B; 3.91±0.96 [AAV2-p65mut] vs. 2.83±0.93 [control], P-value=0.45, n=5), even though a significant number of the surviving RGCs expressed p65mut (Fig. 5C). Thus, our data suggest that increased expression of p65mut efficiently reduces apoptosis in RGCs subjected to neurotoxic conditions. Increased p65mut expression also appears to reduce necroptosis in these neurons, but not to a statistically significant degree. Less encouragingly, augmenting p65mut expression alone fails to promote RGC axonal regeneration or normalize RGC metabolism (as the shrunken, sickly morphologies of the surviving RGCs suggest).
Figure 3.
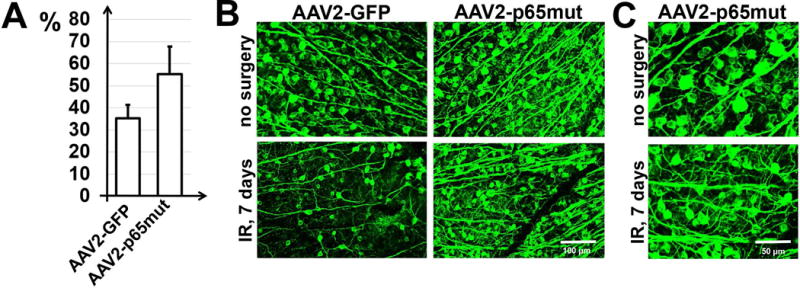
Constitutively active NFκB promotes RGC survival after ischemic injury. A) RGC survival in ischemic retinas transduced with either AAV2-p65mut or AAV2-GFP was calculated as a percentage of the mean cell density in the retinas of fellow control (no surgery) eyes of the same animals (n = 5 mice per group). To identify RGCs, flatmounted retinas were immunostained for the RGC marker Tubb3 (green). B) Representative confocal images of Tubb3-labeled GCLs (green) in flatmounted retinas were taken 7 days after reperfusion (IR). C) Higher magnification images of flatmounted retinas of control (no surgery) and ischemic eyes of AAV2-p65mut-treated animals allow for evaluation of the morphology of somata and axons of surviving RGCs.
Figure 4.
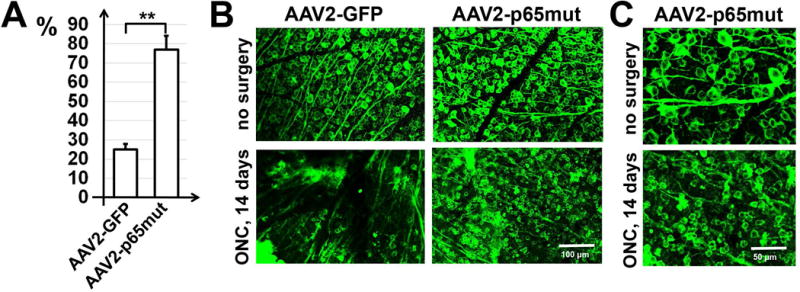
High p65mut expression efficiently prevents death of RGCs after optic nerve crush. A) RGCs of adult animals were transduced with either AAV2-p65mut or AAV2-GFP. Retinas of these animals were collected two weeks after optic nerve crush. The percentage of RGC survival was determined by bilateral comparison for individual mice (**P < 0.01, n = 5 mice per group). B) Typical confocal images of Tubb3-labeled RGCs in flatmounted retinas were collected two weeks after optic nerve crush. C) Higher magnification images of flatmounted retinas of experimental and control (no surgery) eyes of AAV2-p65mut-treated animals collected two weeks after optic nerve crush allow for evaluation of the morphology of somata and axons of surviving RGCs.
Figure 5.
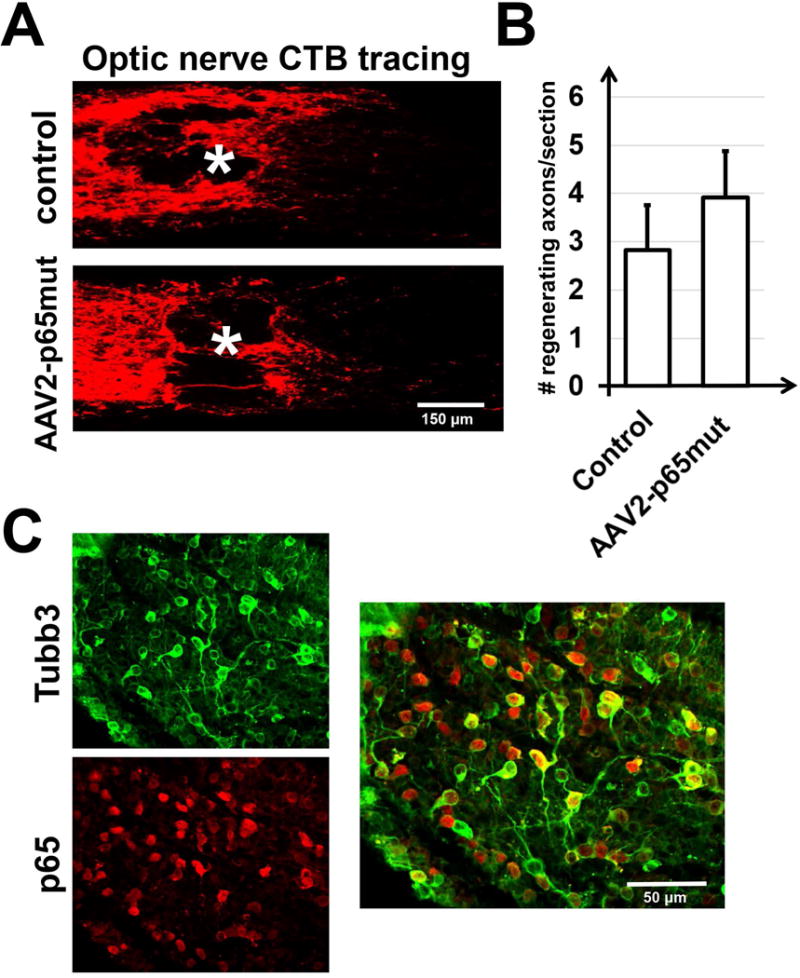
Increased p65mut expression in adult RGCs cannot alone promote axonal regeneration after optic nerve crush. A) Images of the optic nerve sections showing cholera toxin β (CTB)-labelled axons two weeks after optic nerve crush indicate no difference between animals transduced with AAV2-p65mut and those transduced with an AAV2-GFP. Asterisks label lesion sites. B) Regenerating axons identified by CTB tracing were counted at 500 μm from the site of injury; at least four sections were counted (n=5 mice per group). C) Representative confocal images of the AAV2-p65mut transduced retina of an animal whose optic nerve was injured. RGCs that highly expressed p65mut did not efficiently regrow axons, but efficiently survived for a long time with damaged axons (Tubb3—green; p65—red).
Discussion
RGCs transfer visual information directly to the visual cortex of the brain, and are susceptible to degeneration in several acute and chronic retinal diseases (Quigley, 1999; Osborne et al., 2004; Levin, 2007; Kern & Barber, 2008). RGC death and/or axon damage (resulting from glaucoma, diabetic retinopathy, optic neuritis, ischemic optic neuropathy, traumatic optic neuropathy, etc.) progressively lead to blindness (Quigley, 1999; Osborne et al., 2004; Levin, 2007; Kern & Barber, 2008). Thus, therapies to maintain the viability of damaged RGCs and mediate the regrowth of their axons are sorely needed. In this study we investigated the therapeutic potential of NFκB signaling in RGC survival and axon regeneration by increasing the expression of activated (modified) NFκB transcription factor subunit p65/Rela. We found that activated (modified) p65 (p65mut) promotes survival of RGCs exposed to either retinal ischemia or optic nerve crush. However, p65mut expression alone is not sufficient to promote axon regeneration or normalize RGC homeostasis after injury (as evidenced by the shrunken, unhealthy appearance of many surviving RGCs).
Transcription factor NFκB, a heterodimer comprised of the subunits p65 (Rela) and p50 (Nfkb1), has been shown to promote cell survival by inhibiting apoptosis and programmed necrosis (necroptosis) (Oeckinghaus & Ghosh, 2009; Oberst & Green, 2011; Oeckinghaus et al., 2011; Vanden Berghe et al., 2014). At the same time, NFκB can also induce the expression of a number of genes whose products mediate pro-inflammatory toxicity (Tak & Firestein, 2001; Lawrence, 2009; Oeckinghaus & Ghosh, 2009; Oeckinghaus et al., 2011). It is possible that pro-survival and pro-inflammatory NFκB activity go hand-in-hand, enabling a cell to survive in an inflammatory milieu that it instigates. Both the pro-survival and pro-inflammatory effects of NFκB signaling depend on the phosphorylation of certain residues (one or many) on the p65 subunit (Tak & Firestein, 2001; Dong et al., 2008; Arun et al., 2009; Lawrence, 2009; Oeckinghaus & Ghosh, 2009; Dong et al., 2010; Nihira et al., 2010; Oberst & Green, 2011; Oeckinghaus et al., 2011; Wang et al., 2011; Vanden Berghe et al., 2014). The specific residue or combinations of residues that get phosphorylated variably influence (along with additional transcription factors/cofactors) expression of NFκB-dependent genes (Tak & Firestein, 2001; Dong et al., 2008; Arun et al., 2009; Lawrence, 2009; Oeckinghaus & Ghosh, 2009; Dong et al., 2010; Nihira et al., 2010; Oberst & Green, 2011; Oeckinghaus et al., 2011; Wang et al., 2011; Vanden Berghe et al., 2014). Thus, the repertoire of NFκB-dependent gene expression may differ in different cell types, and may produce vastly different consequences for the tissue at large. For example, our previously published data indicate that NFκB signaling in glial cells facilitates damage in the retina, optic nerve, and spinal cord (Brambilla et al., 2009; Dvoriantchikova et al., 2009; Barakat et al., 2012; Brambilla et al., 2012). Meanwhile, reduced NFκB activity in glial cells diminishes damage and promotes neuronal survival (Brambilla et al., 2009; Dvoriantchikova et al., 2009; Barakat et al., 2012; Brambilla et al., 2012). The protective effect of reduced glial NFκB signaling was associated with reduced inflammation in the tissue (Brambilla et al., 2009; Dvoriantchikova et al., 2009; Barakat et al., 2012; Brambilla et al., 2012). By contrast, a plethora of evidence points to the neuroprotective role of NFκB signaling in neurons (Maggirwar et al., 1998; Kaltschmidt et al., 1999; Yu et al., 1999; Koulich et al., 2001; Fridmacher et al., 2003; Mattson & Meffert, 2006; Mincheva et al., 2011). In light of these findings, it is reasonable to imagine that targeted augmentation of NFκB signaling in neurons (but not glia) could form the basis for efficient new therapies to treat neurodegenerative diseases, including those involving retinal neurons. But precision would be paramount, as accidentally increasing NFκB signaling in glial cells would achieve the opposite of the desired effects. To achieve such precision in our study, we used an AAV2 vector to deliver a modified, constitutively active NFκB subunit (p65mut) into RGCs. Corroborating previously published data, our results indicate that AAV2 delivers modified p65 directly and exclusively into RGCs.
Unlike many other CNS neurons, RGCs fail to activate NFκB signaling under neurotoxic conditions. This failure of NFκB signaling occurs despite the fact that all members of the NFκB family, including p65 and p50, are present inside RGCs (Dvoriantchikova & Ivanov, 2014). We explained this discrepancy by noting that IκBα, a protein that prevents translocation of NFκB into nucleus, is highly expressed and stable in RGCs (Dvoriantchikova & Ivanov, 2014). It’s no surprise, then, that we could not find any studies describing p65 modification sites that promote NFκB signaling in RGCs. Forced to act somewhat blindly, we decided to work with p65’s serine residue 276 (Ser276), which has been established as critical for NFκB pro-survival activity in several other tissues (Dong et al., 2008; Arun et al., 2009; Nihira et al., 2010; Wang et al., 2011). We took advantage of a phosphomimetic approach and substituted Ser276 with an aspartic acid (S276D), a change that has been shown to mimic p65 phosphorylation (activation) and significantly increase expression of many NFκB target genes (Dong et al., 2010). To test the effect of constitutively active NFκB signaling on the survival of RGCs after exposure to neurotoxic conditions, we used two different models: transient retinal ischemia (to study RGCs undergoing necroptosis) and optic nerve crush (to study RGCs undergoing apoptosis) (Berkelaar et al., 1994; Bien et al., 1999; Fujita et al., 2009; Dvoriantchikova et al., 2010; Rosenbaum et al., 2010; Magharious et al., 2011; Dvoriantchikova et al., 2014a). We found that the modified p65 (p65mut) was highly expressed in transduced RGCs of adult animals, predominantly in nuclei of these neurons. We also found that constitutive expression of p65mut reduces RGC death in both animal models, but inhibits RGC apoptosis more efficiently than RGC necroptosis. However, most of the surviving p65mut-expressing RGCs appear shrunken and sickly, suggesting that Ser276 phosphorylation on p65 is not sufficient to restore homeostasis after injury. We can assume that additional p65 modifications and/or additional transcription factors/cofactors (which may be absent in RGCs) may be required to achieve normalization of RGC homeostasis post-injury.
In addition to the aforementioned pro-survival effects in neurons, some evidence suggests that NFκB signaling may regulate neurite outgrowth and axon regeneration. Gallagher et al. demonstrated that ciliary neurotrophic factor (CNTF) facilitates neurite outgrowth in nodose ganglion neurons by activating NFκB in these cells (Gallagher et al., 2007). Similarly, nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) were shown to promote neurite outgrowth in sympathetic neurons, nodose ganglion neurons, and pyramidal neurons, in part by increasing NFκB activity (Sole et al., 2004; Gutierrez et al., 2005). At least two other studies, however, indicated that NFκB signaling can also inhibit neurite outgrowth (Gutierrez et al., 2008; Gavalda et al., 2009). Importantly, the ability to activate or inhibit neurite outgrowth in nodose ganglion and superior cervical ganglion (SCG) neurons depends on the phosphorylation status of p65’s serine reside 536 (Ser536): phosphorylation of Ser536 inhibits NFκB-dependent neurite outgrowth, while de-phosphorylation of Ser536 facilitates it (Gutierrez et al., 2008; Gavalda et al., 2009). Thus, the phosphorylation status of Ser536 on p65 may function as a switch between activation and inhibition of neurite outgrowth in some classes of neurons. Although the absence of NFκB signaling in RGCs implies an unphosphorylated status for Ser536, we nevertheless decided to prevent any accidental Ser536 phosphorylation, thereby optimizing the potential for RGC neurite outgrowth. To this end, we modified a p65 protein with a serine-to-alanine substitution at residue 536, preventing phosphorylation at this site while preserving the transcriptional activity of p65 (Gutierrez et al., 2008). We found that high expression of p65mut (our modified p65 protein with a “phosphorylated” Ser276 and “unphosphorylated” Ser536) does not significantly affect neurite outgrowth in RGCs. Thus, modification of different or additional p65 sites or the presence of additional transcription factors/cofactors may be required to promote neurite outgrowth and axon regeneration in damaged adult RGCs.
In conclusion, our data indicate that constitutively active NFκB signaling promotes survival of damaged RGCs. These pro-survival effects depend on the phosphorylation status of specific residues on NFκB’s p65 subunit. However, the p65 modifications studied here were not sufficient to facilitate neurite outgrowth and axon regeneration, nor to restore homeostasis in adult RGCs exposed to neurotoxic insults. More studies are needed to identify additional NFκB modification sites, as well as additional transcription factors/cofactors, required to simultaneously promote survival, normal homeostasis, and neurite outgrowth/axon regeneration in damaged adult RGCs.
Supplementary Material
S1 Appendix. The nucleotide and amino acid sequences of modified p65/Rela (p65mut)
S2 Appendix. Representative confocal images of beta-III Tubulin (Tubb3)-labeled ganglion cell layers (green) in flat-mounted retinas acquired at the center, middle, and peripheral regions in control (no surgery) and ONC retinas 14 days after surgery.
Acknowledgments
This work was supported in part by National Eye Institute/National Institutes of Health (NIH) Grant R01 EY022348 (DI), Center Core Grant P30 EY014801 to the University of Miami Department of Ophthalmology, Dr. Nasser Al-Rashid Orbital Vision Research Center Grant (DP), GemCon Family Foundation Research Grant (DP), NIH/NEI R01 EY022961 (KKP), the Ziegler Foundation (KKP), the Pew Charitable Trust (KKP), the Craig H. Neilsen Foundation (KKP), and the Buoniconti Fund (KKP). We thank the Analytic Imaging Facility (Gabriel Gaidosh) at the Bascom Palmer Eye Institute (BPEI) and the Viral Vector Core at the Miami Project to Cure Paralysis.
Abbreviations
- RGCs
Retinal ganglion cells
- NFκB
Nuclear Factor kappa B
- AAV2
adeno-associated virus serotype 2
- ONC
optic nerve crush model
- MCS
multiple cloning site
- p65mut
p65 S276DS536A double mutant
- Tubb3
beta III Tubulin
- PFA
paraformaldehyde
- TBS
Tris-buffered saline
- FBS
fetal bovine serum
- RIPA buffer
radioimmunoprecipitation assay buffer
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- PVDF membrane
polyvinylidene difluoride membrane
References
- Arun P, Brown MS, Ehsanian R, Chen Z, Van Waes C. Nuclear NF-kappaB p65 phosphorylation at serine 276 by protein kinase A contributes to the malignant phenotype of head and neck cancer. Clin Cancer Res. 2009;15:5974–5984. doi: 10.1158/1078-0432.CCR-09-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat DJ, Dvoriantchikova G, Ivanov D, Shestopalov VI. Astroglial NF-kappaB mediates oxidative stress by regulation of NADPH oxidase in a model of retinal ischemia reperfusion injury. J Neurochem. 2012;120:586–597. doi: 10.1111/j.1471-4159.2011.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci. 1994;14:4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien A, Seidenbecher CI, Bockers TM, Sabel BA, Kreutz MR. Apoptotic versus necrotic characteristics of retinal ganglion cell death after partial optic nerve injury. J Neurotrauma. 1999;16:153–163. doi: 10.1089/neu.1999.16.153. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, Shestopalov VI. Transgenic inhibition of astroglial NF-kappaB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J Neuroinflammation. 2012;9:213. doi: 10.1186/1742-2094-9-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. 2009;182:2628–2640. doi: 10.4049/jimmunol.0802954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Kim JA, Kim DH, Chun MH, Gwag BJ, Yoon SK, Joo CK. Failure to activate NF-kappaB promotes apoptosis of retinal ganglion cells following optic nerve transection. Brain Res. 2000;883:60–68. doi: 10.1016/s0006-8993(00)02886-9. [DOI] [PubMed] [Google Scholar]
- Dong J, Jimi E, Zeiss C, Hayden MS, Ghosh S. Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev. 2010;24:1709–1717. doi: 10.1101/gad.1958410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 2008;22:1159–1173. doi: 10.1101/gad.1657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat D, Brambilla R, Agudelo C, Hernandez E, Bethea JR, Shestopalov VI, Ivanov D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur J Neurosci. 2009;30:175–185. doi: 10.1111/j.1460-9568.2009.06814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Liposome-delivered ATP effectively protects the retina against ischemia-reperfusion injury. Mol Vis. 2010;16:2882–2890. [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Degterev A, Ivanov D. Retinal ganglion cell (RGC) programmed necrosis contributes to ischemia-reperfusion-induced retinal damage. Exp Eye Res. 2014a;123:1–7. doi: 10.1016/j.exer.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Ivanov D. Tumor necrosis factor-alpha mediates activation of NF-kappaB and JNK signaling cascades in retinal ganglion cells and astrocytes in opposite ways. Eur J Neurosci. 2014;40:3171–3178. doi: 10.1111/ejn.12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Santos AR, Danek D, Dvoriantchikova X, Ivanov D. The TIR-domain-containing adapter inducing interferon-beta-dependent signaling cascade plays a crucial role in ischemia-reperfusion-induced retinal injury, whereas the contribution of the myeloid differentiation primary response 88-dependent signaling cascade is not as pivotal. Eur J Neurosci. 2014b;40:2502–2512. doi: 10.1111/ejn.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Santos AR, Saeed AM, Dvoriantchikova X, Ivanov D. Putative role of protein kinase C in neurotoxic inflammation mediated by extracellular heat shock protein 70 after ischemia-reperfusion. J Neuroinflammation. 2014c;11:81. doi: 10.1186/1742-2094-11-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehr ED, Lin X, O’Mahony A, Geleziunas R, Bradshaw RA, Greene WC. NF-kappa B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci. 2000;20:7556–7563. doi: 10.1523/JNEUROSCI.20-20-07556.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, Pfeiffer J, Kaltschmidt C, Israel A, Memet S. Forebrain-specific neuronal inhibition of nuclear factor-kappaB activity leads to loss of neuroprotection. J Neurosci. 2003;23:9403–9408. doi: 10.1523/JNEUROSCI.23-28-09403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita R, Ueda M, Fujiwara K, Ueda H. Prothymosin-alpha plays a defensive role in retinal ischemia through necrosis and apoptosis inhibition. Cell Death Differ. 2009;16:349–358. doi: 10.1038/cdd.2008.159. [DOI] [PubMed] [Google Scholar]
- Gallagher D, Gutierrez H, Gavalda N, O’Keeffe G, Hay R, Davies AM. Nuclear factor-kappaB activation via tyrosine phosphorylation of inhibitor kappaB-alpha is crucial for ciliary neurotrophic factor-promoted neurite growth from developing neurons. J Neurosci. 2007;27:9664–9669. doi: 10.1523/JNEUROSCI.0608-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavalda N, Gutierrez H, Davies AM. Developmental switch in NF-kappaB signalling required for neurite growth. Development. 2009;136:3405–3412. doi: 10.1242/dev.035295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez H, Hale VA, Dolcet X, Davies A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development. 2005;132:1713–1726. doi: 10.1242/dev.01702. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, O’Keeffe GW, Gavalda N, Gallagher D, Davies AM. Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J Neurosci. 2008;28:8246–8256. doi: 10.1523/JNEUROSCI.1941-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AR, Hu Y, Leaver SG, Mellough CB, Park K, Verhaagen J, Plant GW, Cui Q. Gene therapy and transplantation in CNS repair: the visual system. Prog Retin Eye Res. 2006;25:449–489. doi: 10.1016/j.preteyeres.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:9409–9414. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–4408. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulich E, Nguyen T, Johnson K, Giardina C, D’Mello S. NF-kappaB is involved in the survival of cerebellar granule neurons: association of IkappaBbeta [correction of Ikappabeta] phosphorylation with cell survival. J Neurochem. 2001;76:1188–1198. doi: 10.1046/j.1471-4159.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin LA. Axonal loss and neuroprotection in optic neuropathies. Can J Ophthalmol. 2007;42:403–408. [PubMed] [Google Scholar]
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magharious M, D’Onofrio PM, Hollander A, Zhu P, Chen J, Koeberle PD. Quantitative iTRAQ analysis of retinal ganglion cell degeneration after optic nerve crush. J Proteome Res. 2011;10:3344–3362. doi: 10.1021/pr2004055. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Mincheva S, Garcera A, Gou-Fabregas M, Encinas M, Dolcet X, Soler RM. The canonical nuclear factor-kappaB pathway regulates cell survival in a developmental model of spinal cord motoneurons. J Neurosci. 2011;31:6493–6503. doi: 10.1523/JNEUROSCI.0206-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihira K, Ando Y, Yamaguchi T, Kagami Y, Miki Y, Yoshida K. Pim-1 controls NF-kappaB signalling by stabilizing RelA/p65. Cell Death Differ. 2010;17:689–698. doi: 10.1038/cdd.2009.174. [DOI] [PubMed] [Google Scholar]
- Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol. 2011;12:757–763. doi: 10.1038/nrm3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Park K, Luo JM, Hisheh S, Harvey AR, Cui Q. Cellular mechanisms associated with spontaneous and ciliary neurotrophic factor-cAMP-induced survival and axonal regeneration of adult retinal ganglion cells. J Neurosci. 2004;24:10806–10815. doi: 10.1523/JNEUROSCI.3532-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA. Neuronal death in glaucoma. Prog Retin Eye Res. 1999;18:39–57. doi: 10.1016/s1350-9462(98)00014-7. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sernagor E, Eglen SJ, Wong RO. Development of retinal ganglion cell structure and function. Prog Retin Eye Res. 2001;20:139–174. doi: 10.1016/s1350-9462(00)00024-0. [DOI] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kapp B signaling. J Cell Biol. 2004;167:479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, He Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Yang X. Comparative gene array analysis of TNF-alpha-induced MAPK and NF-kappaB signaling pathways between retinal ganglion cells and glial cells. Exp Eye Res. 2005;81:207–217. doi: 10.1016/j.exer.2005.01.022. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mo X, Piper MG, Wang H, Parinandi NL, Guttridge D, Marsh CB. M-CSF induces monocyte survival by activating NF-kappaB p65 phosphorylation at Ser276 via protein kinase C. PLoS One. 2011;6:e28081. doi: 10.1371/journal.pone.0028081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Appendix. The nucleotide and amino acid sequences of modified p65/Rela (p65mut)
S2 Appendix. Representative confocal images of beta-III Tubulin (Tubb3)-labeled ganglion cell layers (green) in flat-mounted retinas acquired at the center, middle, and peripheral regions in control (no surgery) and ONC retinas 14 days after surgery.