

Abstract
Background
Evolutionary histories of parasite and host populations are intimately linked such that their spatial genetic structures may be correlated. While these processes have been relatively well studied in specialist parasites and their hosts, less is known about the ecological and evolutionary consequences of relationships between generalist ectoparasites and their hosts. The aim of this study was to investigate the genetic structure and demographic history of a bat ectoparasite, Cimex adjunctus, whose host affinity is weak but the biology of the potential hosts have been well studied. This ectoparasite has been hypothesized to rely on its hosts for dispersal due to its low inherent dispersal potential. Here we describe genetic diversity and demographic history in C. adjunctus through most of its range in North America. We investigated variation at the cytochrome c oxidase 1 mitochondrial gene and nine microsatellite markers, and tested the prediction that genetic diversity in C. adjunctus is spatially structured. We also tested the prediction that demographic history in C. adjunctus is characterized by range and demographic expansion as a consequence of post-Pleistocene climate warming.
Results
We found stronger spatial structuring of genetic diversity in C. adjunctus than has been quantified in two of its hosts, but contrast in amount of variation explained by host association with different genetic markers (i.e., nuclear vs mitochondrial DNA). Also, C. adjunctus’ history is not primarily characterized by demographic and range expansion, as is the case with two of its key hosts.
Conclusions
Our study shows different patterns of genetic structure and demographic history in C. adjunctus than have been detected in two of its key hosts. Our results suggest an effect of a loose parasite-host relationship and anti-parasitism strategies on genetic structure and post-Pleistocene recovery of population size.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-016-0839-1) contains supplementary material, which is available to authorized users.
Keywords: AMOVA, Approximate Bayesian computation, Bayesian skyline plot, CO1, Genetic clustering, Isolation by distance, Phylogeography
Background
Parasites, through effects on host survival and reproduction, can modify the morphology, life history and behavior of their hosts. Parasites may also influence the dynamics of host populations thereby shaping communities [1]. Hosts in turn may also have important effects on their parasites. Many parasite species, whether endoparasites or ectoparasites, remain closely associated with their hosts through much of their life cycle [2], and often rely on their hosts for dispersal. Dispersal, in turn, influences gene flow and therefore genetic structure and diversity of a species; across a broad range of taxa, less dispersal is associated with increased spatial structure and differentiation [3]. Not surprisingly, spatial genetic structure of a parasite frequently reflects dispersal of its host. For example, population genetic structure of parasitic nematodes of cattle, sheep and white-tailed deer is explained by host movements [4]. However, relative to their hosts, parasites often show higher levels of genetic differentiation. As such, analysis of the trematode parasite (Pagioporus shawi) permitted more detailed information on population assignments in its host, the steelhead trout (Oncorhynchus mykiss) than could be obtained by examining genetic variation in the host itself [5]. In addition to dispersal, parasites and hosts may have experienced correlated demographic and range dynamics [6, 7] which will also be reflected in their population genetic structure; for instance, patterns of genetic variation among populations of the parasitic nematode Heligmosomoides polygyrus have revealed demographic and historic events affecting its host, the field mouse Apodemus sylvaticus [8]. Furthermore, differences in regional abundance of two Apodemus species likely caused differentiation of both the Apodemus host and their Heligmosomoides parasite species [9].
However, it has recently been shown that a strong link between host dispersal and parasite genetic structure is not ubiquitous, and depends on factors that include the degree of association with the host and host mobility [10]. Here, we investigated spatial genetic structure and past demography of an ectoparasite that is associated with highly mobile flying hosts, and would be considered a weak generalist based on its association with a number of different host species that are closely related to each other [10]. Our study complements a body of work on spatial genetic structure and phylogeography of various ectoparasites associated with hosts having higher mobility [11–13].
Insects in the genus Cimex (Order: Hemiptera) are temporary ectoparasites of homeothermic animals. They do not remain on their host at all times but rather remain in nests or roosts between blood meals [14]. Most Cimex species are associated exclusively with bats, while a few associate with a more diverse range of hosts [14–16]. Cimex adjunctus is a widespread ectoparasite of bats in North America, occurring from the eastern seaboard to the Rocky Mountains, and from Labrador and the Northwest Territories south to Texas [14]. It parasitizes a number of bat species, including the big brown bat (Eptesicus fuscus) and the little brown myotis (Myotis lucifugus), two species that often roost in buildings [17–19]. The generation time of C. adjunctus is unknown, but is likely similar to that of the common bed bug C. lectularius, which can range from two to 12 generations a year depending on monthly temperatures [14], and is certainly much shorter than that of its hosts.
Usinger [14] proposed that Cimex species have a very low inherent capacity for dispersal over long distances, on the scale of kilometers. He thought it unlikely that adult Cimex species disperse on their own. He therefore hypothesized that Cimex species can disperse occasionally attached to a host’s body [14]. Previous studies of genetic diversity of the big brown bat and little brown myotis in North America have reported high within-site genetic variation and generally low among-site differentiation, although there are differences between patterns at nuclear and mitochondrial markers (E. fuscus, [20, 21]; M. lucifugus, [22–25]). Overall, these studies indicate that high levels of gene flow are maintained over long distances in both bat species, while genetic structuring of mitochondrial variation suggests a higher degree of female than male philopatry. For C. adjunctus, likely only a fraction of host dispersal events result in successful parasite dispersal so gene flow may be lower in C. adjunctus relative to these two host species. Furthermore, C. adjunctus may experience frequent extirpation and recolonization events. Bartonička and Růžičková [26] identified bat bug load as a possible cause of roost-switching in bats, with numbers of bats dropping as the population of C. pipistrelli reaches a high. They also found the appearance of C. pipistrelli 21 to 56 days after the first bat visit in any given roost. Since C. adjunctus, like C. pipistrelli, does not stay on the host between blood meals, sudden host population decreases within roosts might drive local extirpation events.
Although different ectoparasite races are often associated with different host species [27–29], high gene flow among populations associated with different host species has also been documented. In Europe, Cimex pipistrelli is morphologically, but not genetically, differentiated among bat host species [30]. This suggests possible morphological plasticity, but high gene flow, among individuals associated with different host species. In North America, we might also expect gene flow among C. adjunctus populations on different host species. Many different North American bat species temporarily roost together for short intervals during the night, such as many Myotis species, including M. lucifugus, and E. fuscus [31], potentially facilitating host switching by C. adjunctus.
Much of North America was unsuitable for many bat species during the last Pleistocene glacial maximum, and both M. lucifugus and E. fuscus are hypothesized to have expanded their ranges from glacial refugia. Dixon [32] suggested that little brown myotis populations currently in Minnesota have dispersed from a single large southeastern US glacial refugium, and Neubaum et al. [21] suggested that big brown bat populations have dispersed from several eastern and western US glacial refugia into what is now Colorado. Range and demographic expansion in little brown myotis has also been proposed on the east coast of Canada [22]. We expect that the potential dependence of C. adjunctus on its host species for long-distance dispersal and colonization may have contributed to broadly congruent patterns of historical range expansion over large spatial scales.
We investigated the spatial genetic structure and phylogeography of C. adjunctus across its range in North America. Because of its comparatively shorter generation time, the likelihood that only a fraction of bat dispersal events may result in ectoparasite gene flow, and the potential for local extirpations, we predicted stronger spatial genetic structure in C. adjunctus relative to its hosts. Because of the potential for movement among host species, we also examined differentiation among populations found on different host species. Finally, based on the hypothesis that post-Pleistocene climate warming had similar effects on the demographic history of C. adjunctus as that of its hosts, we predicted genetic signatures of demographic and range expansion in C. adjunctus.
Results
We collected 160 Cimex adjunctus samples from throughout its range in North America (108 from E. fuscus, 36 from M. lucifugus and 16 from M. septentrionalis; Fig. 1; in Additional file 1: Table S1), from 45 sites (Additional file 2: Table S2). We successfully amplified a fragment of the cytochrome c oxidase 1 (CO1) gene for 154 C. adjunctus, and identified 41 haplotypes with 46 polymorphic sites (data available in Additional file 1: Table S1). We also genotyped 150 of the C. adjunctus at nine microsatellite loci (data available in Additional file 3: Table S3). We successfully obtained both mitochondrial and microsatellite data for 144 C. adjunctus (94% of the CO1 dataset and 96% of the microsatellite dataset; in Additional file 1: Table S1). For approximate Bayesian computation (ABC), which uses both types of markers, we used the overlapping dataset (144 individuals). For analyses using only microsatellite loci (genetic clustering and isolation by distance, IBD), we used the whole microsatellite dataset (150 individuals) and for analyses using only mitochondrial DNA (minimum spanning network, MSN; mismatch distribution, MD; and extended Bayesian skyline plot, EBSP), we used the complete CO1 dataset (154 individuals). Finally, we used only sites with data for at least two individuals, and for which we obtained both mitochondrial and microsatellite data, for the analysis of molecular variance (AMOVA) analysis, which resulted in a dataset of 127 individuals from 26 sites (82% of the CO1 dataset and 85% of the microsatellite dataset).
Fig. 1.
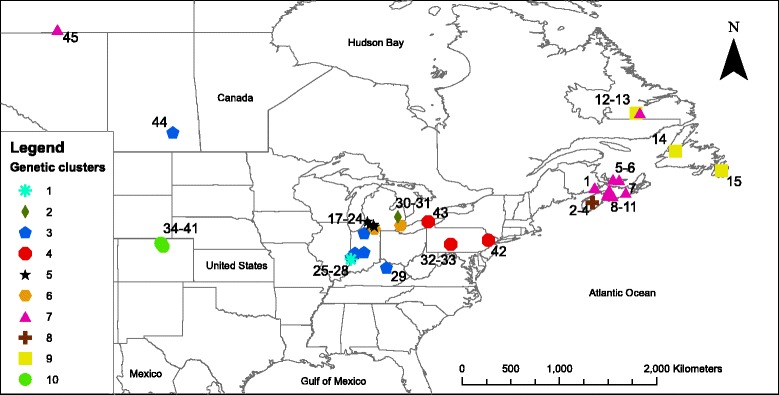
Sampling locations of Cimex adjunctus in North America. Created with ArcGIS v10.3 (ESRI, Redlands, USA). Numbers on the map correspond to site numbers in Additional file 1: Table S1. Membership to each of ten genetic clusters, defined using microsatellite data in Geneland, is shown with a unique colour and shape. Cluster numbers are given in the Legend and correspond to those in Additional file 1: Table S1
Microsatellite diversity, and Hardy-Weinberg and linkage disequilibrium
Among the nine microsatellite loci, we observed between two and 31 alleles. Across different sites and genetic clusters (identified by Geneland), average number of alleles ranged from 1.5 to 4, expected heterozygosity ranged from 0.18 to 0.62, observed heterozygosity ranged from 0.09 to 0.25, and the inbreeding coefficient varied between 0.00 and 0.77 (Table 1). Variation in genetic diversity and inbreeding coefficients did not show any obvious spatial pattern. We found three significant cases of deviation from Hardy-Weinberg equilibrium (one site at the loci Clec104 and Cle015, and another site at Clec104). Since these incidences of deviation from Hardy-Weinberg equilibrium were not systematic across loci or sites, we retained these two markers and two sites for our analyses. We did not find any evidence of significant linkage disequilibrium in any marker.
Table 1.
Genetic diversity estimates for C. adjunctus, averaged across nine microsatellite markers, for sites with five or more sampled individuals and for genetic clusters identified by Geneland (with the exception of Cluster 1, in which there was only one individual; in Additional file 1: Table S1). Site and cluster numbers correspond to those in Fig. 1 and Additional file 1: Table S1
| Site/Cluster | Average number of alleles | Expected heterozygosity | Observed heterozygosity | Inbreeding coefficient GIS |
|---|---|---|---|---|
| Site 17 | 2.000 | 0.275 | 0.278 | −0.009 |
| Site 19 | 1.889 | 0.363 | 0.093 | 0.745 |
| Site 30 | 2.778 | 0.346 | 0.201 | 0.420 |
| Site 31 | 2.111 | 0.327 | 0.254 | 0.223 |
| Site 32 | 3.000 | 0.193 | 0.193 | 0.369 |
| Site 36 | 2.444 | 0.325 | 0.224 | 0.310 |
| Site 39 | 2.556 | 0.293 | 0.241 | 0.178 |
| Site 40 | 2.222 | 0.239 | 0.145 | 0.393 |
| Site 41 | 1.778 | 0.184 | 0.160 | 0.129 |
| Cluster 2 | 3.000 | 0.349 | 0.225 | 0.431 |
| Cluster 3 | 2.556 | 0.394 | 0.246 | 0.375 |
| Cluster 4 | 3.222 | 0.332 | 0.235 | 0.291 |
| Cluster 5 | 2.778 | 0.309 | 0.186 | 0.399 |
| Cluster 6 | 2.222 | 0.327 | 0.247 | 0.245 |
| Cluster 7 | 3.889 | 0.349 | 0.225 | 0.354 |
| Cluster 8 | 3.000 | 0.615 | 0.143 | 0.768 |
| Cluster 9 | 1.667 | 0.250 | 0.194 | 0.222 |
| Cluster 10 | 3.556 | 0.301 | 0.196 | 0.349 |
Range-wide genetic structure
Genetic clustering analyses using the Geneland method revealed 10 genetic clusters (Table 2), which were generally concordant with geographic location (Fig. 1). One interesting exception was that individuals from the Northwest Territories and Saskatchewan clustered with individuals from distant regions (Clusters 3 and 7; Fig. 1). There was no association between genetic clusters identified by Geneland and any major geographic barriers that might knowingly impact dispersal. The sampling year and host species did not seem to strongly affect clustering, as individuals associated with different host species or sampled at different years were frequently assigned to the same cluster (Additional file 4: Table S4). Using K-means clustering, we obtained the lowest BIC value at K = 11, and the second lowest BIC value was at K = 10. Moreover, we observed significant IBD calculated on individual genetic relatedness values (P = 0.001, R2 = 0.19; Table 2). Conditioning for genetic structure slightly improved the fit of the IBD model (R2 = 0.21; Table 2).
Table 2.
Results of clustering and isolation-by-distance analyses of Cimex adjunctus, estimated using microsatellite markers. Most likely number of genetic clusters (K) estimated using the Geneland method, Isolation-by-distance (IBD) and IBD while correcting for population genetic structure (IBD + K) are shown
| Statistic | Value | |
|---|---|---|
| Most likely K | 10 | |
| IBD (rW) | P | 0.001* |
| R2 | 0.19 | |
| IBD (rW) + K | P (IBD) | 0.001* |
| P (K) | 0.001* | |
| R2 | 0.21 | |
*Statistically significant at α = 0.05
AMOVA results were very different between the two types of markers. For microsatellites, considerably less of the total variation was explained by among (22.8%; Table 3) than within sites (37.0%), but for mitochondrial data the variation among (48.8%) and within sites (43.7%) were similar. The proportion of genetic variation among host species was high for microsatellite data (40.2%; Table 3), but quite low for mitochondrial data (7.4%).
Table 3.
Results of analysis of molecular variance (AMOVA) on Cimex adjunctus, using mitochondrial and microsatellite data. Percentage of total variation among host species, among sample sites (population), and within sample sites are shown
| Source of variation | Mitochondrial | Microsatelite |
|---|---|---|
| Among host species | 7.4 | 40.2 |
| Among populations | 48.8 | 22.8 |
| Within populations | 43.7 | 37.0 |
Demographic history
Considering those haplotypes represented by four or more individuals, there was some degree of spatial structuring in their distribution. Specifically, distinct haplotypes were associated with the western and eastern ends of C. adjunctus’ range (Fig. 2). One interesting observation was that individuals from Northwest Territories and Saskatchewan had very similar haplotypes to individuals from the Midwest of the United States. The MSN did not show a well-defined starburst pattern (Fig. 3). Also, the MD showed multiple peaks rather than a single peak that would have indicated potential demographic expansion in the past (Fig. 4). Evolution of NEτ through time, estimated using EBSP, showed mostly constant population size with a possible gradual decrease from about 200,000 to 30,000 years ago to about half of the initial population size, followed by a small increase to the present (Fig. 5). Finally, ABC analysis gave strongest support to a scenario mimicking a decrease in effective population size of at least an order of magnitude between 10 million years ago and 10,000 years ago (Table 4; See Additional file 5: Figure S1 for pre-evaluation of prior distributions of scenarios with the observed values).
Fig. 2.
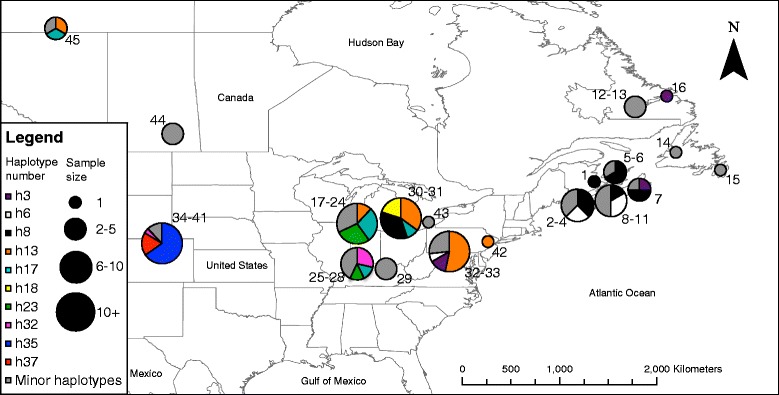
Frequencies of mitochondrial DNA haplotypes of Cimex adjunctus across its range. Data for nearby sampling sites are combined in a single pie chart. Rare haplotypes represented by fewer than four individuals in the entire data set are shown in grey. Haplotypes represented by four or more individuals in the entire dataset are identified with unique colors as indicated in the Legend, and corresponding to haplotype colors in Fig. 3. Site numbers correspond to those in Fig. 1 and in Additional file 1: Table S1. Sizes of circles indicate sample sizes
Fig. 3.
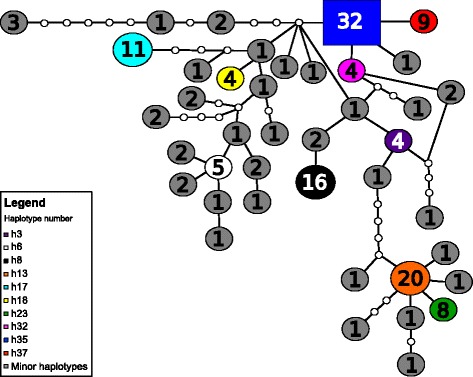
Minimum-spanning network of mitochondrial cytochrome c oxidase 1 (CO1) sequences of Cimex adjunctus. Haplotypes represented by fewer than four individuals are shown in grey. Haplotypes represented by four or more individuals are identified with unique colors as indicated in the Legend, and corresponding to those in Fig. 2. Each circle represents a unique sequence, each line segment is a mutational step, numbers are sample sizes for each unique sequence, small circles without a sample size are intermediate, unsampled haplotypes, and the square represents the putative ancestral sequence
Fig. 4.
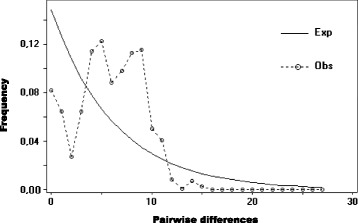
Frequency of pairwise mismatches among cytochrome c oxidase 1 (CO1) sequences of Cimex adjunctus in North America
Fig. 5.
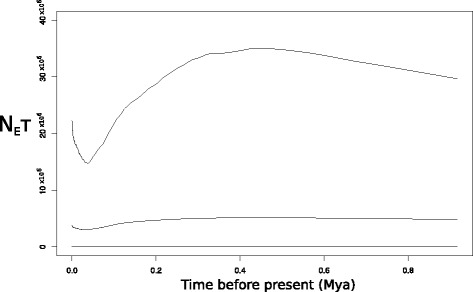
Extended Bayesian skyline plot estimated using cytochrome c oxidase 1 (CO1) data of Cimex adjunctus. Shown are the mean and 95% highest posterior density interval of the product of effective population size (NE) and generation time (τ) through time (in million years ago; Mya)
Table 4.
Results of approximate Bayesian computation analysis of effective population size (NE) history of Cimex adjunctus. Posterior probabilities of each scenario (with confidence interval in parentheses) are shown
| Scenario | Posterior probability |
|---|---|
| NE Increase | 0.297 (0.285 – 0.308) |
| NE Decrease | 0.522 (0.508 – 0.536) |
| NE Constant | 0.181 (0.167 – 0.196) |
Discussion
Range-wide genetic structure
Analyses of mitochondrial and microsatellite genetic markers supported our prediction of high range-wide genetic structure, mediated by geographic distance, in C. adjunctus, an ectoparasite of bats. Across the range of C. adjunctus, we found significant genetic structure, a large proportion of which was explained by geographic distance. Whereas IBD has not been previously investigated in most bat parasites (but see [33]), it has been investigated in two of the key hosts of C. adjunctus, the big brown bat and the little brown myotis. A relationship between genetic and geographic distance has been observed in both the big brown bat [20] and little brown myotis across a considerably smaller spatial scale [24] than examined here. Range-wide IBD has also been described for little brown myotis [25], based on population-level analyses using F ST. Thus, geographic distance explains a lot of the variation in genetic structure of C. adjunctus as it does in two of its hosts, which could potentially reflect the reliance of C. adjunctus on their hosts for dispersal.
However, the overall degree of genetic structuring appears to be higher in C. adjunctus than in its hosts. Analysis of microsatellite genotypes has revealed only two genetic clusters in both big brown bat [34] and little brown myotis [25], both at continental spatial scales, whereas our results point to ten genetic clusters in C. adjunctus. Likewise, very little genetic variation (<10% with microsatellite data, and < 20% with mitochondrial data) occurs among spatially separate sites in big brown bat [20] and in little brown myotis [22–25]. In C. adjunctus, about one third of the microsatellite variation and about one half of mitochondrial variation occur among sites (after taking out variation among host species). These observations suggest that C. adjunctus is more subdivided within its range than at least two of its hosts, and that its genetic structure does not entirely reflect the dispersal patterns of its hosts. Interestingly however, both genetic clustering and MSN results also offer some evidence of possible continent-scale long-distance movement in C. adjunctus, as reflected in the relationships among individuals from the Northwest Territories, Saskatchewan, Maritime Canada and the US Midwest. Relationships among C. adjunctus samples from these locations echo a pattern that was observed in M. lucifugus, where a set of sites in the central United States and central to north-western Canada are connected by high gene flow [25].
Spatial structuring of genetic diversity can arise when gene flow is not sufficiently high to homogenize allele frequencies throughout the study area, and across a broad range of animal species dispersal ability is correlated with both gene flow and population genetic structure [3]. This has led to the prediction that genetic structure of many parasites will reflect host dispersal and genetic structure [10]. However, the association between host dispersal and parasite genetic structure has recently been shown to be generally weak [10]. Furthermore, genetic structure in parasites is often found to be stronger than that of their host, as we have observed here for C. adjunctus. For example, a finer genetic structure was found in an endoparasitic nematode H. polygyrus than in its host, the field mouse A. sylvaticus [8]. One reason for stronger genetic structuring in parasites than their hosts could be that, for parasites using their host as a means of dispersal, not every host dispersal event will result in dispersal by the parasite. This is likely to be the case for C. adjunctus, which spends a considerable proportion of time living off of its hosts within cracks and crevices in roosting sites. First, only a small subset of dispersing bats are likely to be accompanied by C. adjunctus. Second, dispersal mortality in the parasite may be very high due to grooming behaviour of bats that can cause the parasites to fall off [35]. Additionally, parasites that have a generation time that is much shorter than that of their hosts, that are associated with more than one host species, or that are associated with highly mobile hosts typically show a much stronger genetic structure than their host, as highlighted by Mazé-Guilmo et al. [10]. All of these factors are true for C. adjunctus, and could explain the much stronger genetic structure we observed for relative to two of its key hosts.
In addition to gene flow and dispersal, genetic structure may also be influenced by genetic drift in small populations, which acts by increasing differentiation [36]. Bat-associated Cimex populations might be much smaller than populations of their hosts, although information on C. adjunctus population sizes is limited. In addition, it is possible that C. adjunctus experiences localized extirpations and recolonizations when roosts are abandoned by bats and subsequently re-occupied. The resulting founder events would further reduce effective population sizes and lead to higher genetic differentiation in C. adjunctus via genetic drift.
We also examined the proportion of genetic variance among samples of C. adjunctus associated with different host species. Interestingly, we found a sharp difference between mitochondrial DNA and microsatellite markers in this regard. Mitochondrial data suggested considerably less variation among populations associated with different host species compared to microsatellite data. At the same time, microsatellite data showed less variation among populations than did the mitochondrial data, indicating that the difference we observed with respect to host species does not reflect a generally poorer ability of the mitochondrial data to detect differentiation in C. adjunctus.
Our mitochondrial data are consistent with an earlier study on C. pipistrelli that found no genetic differentiation among individuals associated with different host species, using mitochondrial CO1 and four nuclear loci [30]. Our microsatellite results contradict these results from C. pipistrelli, although it is important to point that all nuclear loci in the study of Balvin et al. [30] showed almost no variation. Mitochondrial DNA is maternally inherited and therefore variation in it will reflect dispersal and history of the maternal lineage only. It is possible therefore that sex-biased behaviour in C. adjunctus could be the reason for our results. Male-biased dispersal among roosts could lead to the higher proportion of genetic variation among sites in mitochondrial data than in microsatellite data. On the other hand, female-biased switching of hosts within roosts could be responsible for the lower proportion of genetic variation among host species observed in the mitochondrial versus microsatellite data. Autonomous (i.e., not host-assisted) female-biased movements over short distances, such as between neighbouring apartment units, have been described in the common bed bug, C. lectularius [37]. If female C. adjunctus also move more readily at short distances within roosts, that could explain both a higher rate of host-switching among females and a lower rate of transport among roosts by their hosts (since females might spend more time off of the hosts while they engage in exploratory behaviour). However, there is currently no information available on sex-biased dispersal or host switching in C. adjunctus. Our results not only suggest sex-biased dispersal or host switching in C. adjunctus, but also highlight the need to use more than one type of marker when investigating genetic diversity in an understudied species.
The most well studied member of the genus Cimex is the human associated common bed bug, C. lectularius. Several studies have examined genetic structure in C. lectularius across a range of spatial scales [38–42]. However, most such studies focus on a considerably smaller scale than we do here, making direct comparisons of genetic structure difficult. For example, Saenz et al. [42] describe a weaker IBD pattern in C. lectularius than we observed for C. adjunctus, which could be due in part to the smaller spatial scale of their sampling (eastern USA only). On the other hand, our genetic diversity estimates for C. adjunctus were strikingly similar to those found in one study on C. lectularius [41], although we report slightly higher average numbers of alleles. In an interesting parallel, a study of C. lectularius populations associated with bats and humans found higher average numbers of alleles in the bat-associated populations than human-associated populations [38]. Another study of C. lectularius in Europe [39] found higher mitochondrial DNA variation among bat and human associated populations than we observed among populations of C. adjunctus associated with different bat species. One likely reason for this dissimilarity between C. adjunctus and C. lectularius is that the former is a weak generalist, associated with closely related species [10], while the former is a strong generalist, associated with phylogenetically very different species. Overall, sample sizes and the number of microsatellite markers used were lower in our study than in several studies of C. lectularius genetic structure [34, 40, 41], but were nonetheless appropriate given the much broader spatial and temporal scale of resolution of our analyses [5, 8, 12, 43].
Demographic history
We predicted signals of range and demographic expansion in C. adjunctus, based on the fact that there are widespread signatures of historic population expansion in many vertebrates, invertebrates and plant populations, including in the bat hosts of C. adjunctus. Such patterns are most probably attributable to postglacial climate warming [44]. However, we found that the history of this ectoparasite is marked most strongly by demographic decline, with only a weak signal of recent demographic expansion, and no clear pattern of range expansion. For example, typical starburst patterns were previously observed in the haplotypic networks of E. fuscus and M. lucifugus [21–23], indicative of range expansion. However, we found no clear starburst pattern for C. adjunctus. This is unlikely to be a result of inadequate spatial sampling since our samples cover most of the known range of this species [14].
We found evidence of population decline in the demographic history of C. adjunctus using a variety of approaches. According to EBSP results, a gradual decline might have started at around 200,000 years ago, corresponding roughly to the Illinoian glaciation, a time of likely very harsh climate for most species in North America [45]. A small demographic recovery may have started at around 30,000 years ago. Our ABC results confirmed a population decline as the most likely historical scenario. Two previous studies found signals of demographic expansion in M. lucifugus in eastern Canada [22] and Minnesota, United States [46]. A small potential increase in C. adjunctus effective population size indicated in the EBSP starting 30,000 years ago is in a similar timeframe as, but is of much smaller amplitude than, the demographic expansion found in both M. lucifugus studies. Relative to those studies, our analysis was able to span a larger amount of time, probably due to the larger spatial scale of our sampling.
Conclusions
Parasites that are mostly free-living, associate with multiple species of hosts, and have hosts that are highly mobile, such as some ectoparasites of bats, may be expected to show a genetic structure that contrasts with the dispersal patterns and genetic structure of their hosts [10]. These same factors may also lead to a difference in historic patterns of change in host and parasite ranges and population sizes. We have found exactly this pattern in C. adjunctus, an insect ectoparasite associated with a number of bat species in North America. This free-living parasite moves off the host between blood meals and could be actively removed by the host through anti-parasitism behaviour. Our results highlight that the genetic structure and demographic history of a weak generalist ectoparasite, particularly one that has a loose relationship with its hosts, can be very different from that of its hosts.
Methods
Sample collection
We collected C. adjunctus across much of its North American range. Most samples are from mist-netted host individuals of E. fuscus, M. lucifugus or M. septentrionalis. Mist net capture locations were adjacent to a known summer roost (house, barn, cabin, church, school or abandoned mine) of either of the three bat species, or within forested national, provincial, state or territorial lands (Additional file 2: Table S2). Most mist-netted bats and the C. adjunctus individuals they harboured likely came from the adjacent known roost, although it is possible that a small proportion came from different roosts in the area. Overall, between 3 and 15% of mist-netted bats harboured a parasite, depending on the location. We also sampled C. adjunctus individuals from the interior of two summer roosts. One roost was in a church attic inhabited by M. lucifugus, and one was in a house attic inhabited by E. fuscus (Additional file 2: Table S2). Because we could be certain of the roost site in these cases, we considered these two sampling locations as distinct from their adjacent mist-netting capture locations. Upon collection, we stored samples immediately in a 95% ethanol solution until further analyses. We then generated CO1 mitochondrial DNA sequence data and nine nuclear microsatellite genotype data for all individuals. All samples included in this study were confirmed as being C. adjunctus using a DNA barcoding approach [47]. We compared the CO1 sequence for each sample to known CO1 sequences for Cimex species from published sources [48].
Genetic analyses
We extracted DNA from the whole insect for all samples using the DNeasy Blood & Tissue Kit (QIAGEN, Germantown, Maryland, United States). We then amplified a 576-bp fragment of the CO1 gene from each individual using the primers: F 5’- TATGAGCAGGCATGTTAGGG and R 5’-ATAGATGTTGATAAAGAATTGGG (Designed by our group based on published sequences of Balvin et al. [48]). We used a DNAEngine PTC-200 Thermal Cycler (BIO-RAD, Hercules, California, United States) to execute the Polymerase Chain Reaction (PCR) amplification. We performed PCR in 25 μL final volume using the following recipe: 1X Taq Polymerase Buffer excluding MgCl2 (Applied Biosystems, Foster City, California, United States), 1.5 mM of MgCl2, 0.2 mM of each type of dNTP, 0.3 μM of each primer, 1 U of Taq polymerase (ABI), and 1 μL of DNA extraction product. We used the following PCR program: an initial denaturation step of 1 min at 94 °C, followed by 36 cycles of 30 s of denaturation at 94 °C, 45 s of annealing at 49 °C and 45 s of extension at 72 °C, finished by a final extension step of 5 min at 72 °C. We visualized PCR products by 1.5% agarose gel electrophoresis using SYBR Green (BIO-RAD) on a UV transluminator to check the quality and size of amplified fragments. Then, we sequenced the amplified gene fragment for every sample using Sanger sequencing with BigDye terminator chemistry (ABI) and analyzed the fragments on a 3730xl DNA Analyzer (ABI). We aligned all sequences using MEGA 6.06.
We also genotyped all individuals at nine microsatellite loci originally designed for Cimex lectularius (Cle002, Cle003, Cle013, Cle015, from Fountain et al. [40], and Clec21, Clec48, Clec15, Clec104 and BB28B, from Booth et al. [42]; in Additional file 6: Table S5). We used a DNAEngine PTC-200 Thermal Cycler (BIO-RAD) to execute PCR amplification. For markers from Fountain et al. [41], we performed PCR using the following recipe: 1X Taq Polymerase Buffer excluding MgCl2 (ABI), 2.175 mM of MgCl2, 0.216 mM of each type of dNTP, 0.25 to 1.2 μM (Additional file 6: Table S5) of each primer, 1 U of Taq polymerase (ABI), 2 μL of DNA extraction product, in total volume of 12 μL. For markers from Fountain et al. [40], we used the following thermal cycling: an initial denaturation step of 15 min at 95 °C, followed by 11 cycles of 30 s of denaturation at 94 °C, 1 min and 30 s of annealing (initially at 65 °C and reduced 1 °C at every cycle) and 1 min of extension at 72 °C, followed by 26 cycles of 30 s of denaturation at 94 °C, 1 min and 30 s of annealing at 55 °C and 1 min of extension at 72 °C, finished by a final extension step of 10 min at 72 °C. For markers from Booth et al. [41], we used the following thermal cycling: an initial denaturation step of 3 min at 95 °C, followed by 35 cycles of 30 s of denaturation at 95 °C, 30 s of annealing at 59 to 61 °C (Additional file 6: Table S5) and 30 s of extension at 72 °C, and a final extension step of 5 min at 72 °C. We amplified each locus individually. PCR products were visualized by 1.5% agarose gel electrophoresis using SYBR Green (BIO-RAD) on a UV transluminator to check the quality and size of amplified fragments. We then sized products on a 3730xl DNA Analyzer (ABI). We called all microsatellite genotypes for each species using GeneMapper Software 4.0 (ABI), and we checked all calls manually.
Statistical analyses
Microsatellite diversity, and Hardy-Weinberg and linkage disequilibrium
For sites with data for at least five sampled C. adjunctus individuals, and for genetic clusters (see next section), we calculated average number of alleles, expected and observed heterozygosity, and inbreeding coefficient. For microsatellite loci, we tested for Hardy-Weinberg and linkage disequilibrium within each site with data for at least two sampled C. adjunctus individuals, using Genepop 4.2. For each type of test, we corrected for multiple tests using Bonferroni correction, with a threshold α of 0.05.
Range-wide genetic structure
We tested our prediction of range-wide genetic structure and an effect of geographic distance in C. adjunctus using genetic clustering, tests of isolation-by-distance (IBD), and an analysis of molecular variance (AMOVA). We conducted a Bayesian clustering analysis using Geneland 4.0.5, which takes into account geographic coordinates of individual samples. We used 100,000 iterations, thinned every 100th iteration, and a post-process burn-in of 200 (of the 1000 left after thinning), for K values between 1 and 20. We executed 10 runs, and kept the one with the highest posterior mean density, after burn-in. We attempted to identify the population to which each individual was assigned the most often, defined here as the population where the majority of Markov Chain Monte Carlo (MCMC) chains converged for any given individual. We also conducted a K-Means clustering analysis using GenoDive 2.0 on allele frequencies, for K values between 1 and 20, and using 50,000 simulation steps, to validate results obtained with the Geneland method. We used Bayesian Information Criterion (BIC) values to determine the most likely K value.
We conducted an individual-level analysis of IBD, using the estimate of genetic relatedness, rW [49], calculated with SpaGeDi 1.5. We calculated 1 – rW for each pairwise relationship, in order to obtain genetic distances. We calculated geographic distance (in km) between sample sites, corrected for sphericity of the earth, using the ‘rdist.earth’ function from the ‘fields’ package [50] in R v3.1.3 (R Development Core Team, Vienna, Austria). We then fit pairwise genetic distance to geographic distance using Multiple Regression on distance Matrices (MRM), in the ‘MRM’ function from the ‘ecodist’ package in R v3.1.3 [51], which uses a Mantel test derived linear regression model. We assessed significance through a permutation procedure (9999 replicates). An assumption of the rW relatedness index, and most other relatedness indices, is that individuals are in a large random mating population without population structure [52]. In an attempt to correct for the population structure present in our dataset, we subsequently conditioned IBD models for genetic clustering. For each pair of individuals assigned to the same population in clustering analyses, we assigned a value of 0, and for each pair of individuals assigned to different populations, we assigned a value of 1. We then tested the effect of geographic distance, together with genetic clustering, on genetic distance in an MRM model.
For all sites with at least two sampled individuals, we used AMOVA to examine the proportion of genetic variation among sites, and among individuals associated with different host species. AMOVA was executed in GenoDive 2.0 for microsatellite data, and Arlequin 3.5 for mitochondrial data.
Demographic history
We tested the prediction that C. adjunctus would show signals of demographic and range expansion, similar to some of its bat hosts, with a suite of methods for investigating demographic history using either mitochondrial data alone, or both mitochondrial and microsatellite data. First, we produced a minimum-spanning network of mitochondrial haplotypes (MSN) using TCS 1.21, with a 95% connection limit. MSNs can indicate past range expansions if they show starburst like patterns [53, 54]. We expected to find such evidence pointing towards range expansion in C. adjunctus.
We executed a Mismatch Distribution (MD) analysis with DNASP 5.1. The purpose of this analysis is to compare the distribution of the frequency of each number of pairwise mitochondrial sequence mismatches in the dataset to the expected distributions under demographic expansion or constant population size through time. A unimodal peak at a non-zero number of pairwise mismatches is associated with demographic expansion, which we expected to observe, whereas more than one non-zero number of pairwise mismatches is usually associated with a constant population size through time [55].
Then, we constructed an Extended Bayesian Skyline Plot (EBSP) using mitochondrial data in BEAST 1.8.4. We used a linear EBSP model, and random local clock, which reportedly performs better than strict and relaxed clocks for most situations using intraspecific data [56, 57]. In trial runs, we found the HKY substitution model [58] to be the best-fitting model, as has also been shown for Triatoma infestans [59], a species in a genus closely related to Cimex. We used the gamma sites model to account for heterogeneity of substitution rate among individual loci. We used the default value of 10,000,000 Markov Chain Monte Carlo (MCMC) chains, logging every 1000 chains. We set the substitution rate to 0.575%/Ma, or half of 1.15%/Ma, which is the standard Arthropod mitochondrial pairwise substitution rate as reported by [60]. All other parameters were kept at default value. EBSPs allow one to visualize effective population size (NE) multiplied by generation time (τ) since some time in the past. In the case of highly structured populations, Heller et al. [61] suggested that a pooled sampling scheme, where several individuals are taken from about ten populations, was ideal to avoid a confounding effect of population structure, as opposed to all samples taken from the same population or one sample taken for each of a large number of populations. The sampling scheme used in our analysis fits well with the described pooled scheme. We expected to see an increase in effective population size over time, corresponding with a post-Pleistocene climate warming timeline.
Finally, we executed approximate Bayesian computations (ABC) on both mitochondrial and microsatellite data, using DIYABC 2.1.0. ABCs allow one to compare posterior probabilities of different demographic scenarios [62]. As per the method of [63], we input three scenarios in the analyses (for population sizes NA > N1 > NB), mimicking an increase in effective population size from N1 to NA at time t, a decrease in population size from N1 to NB at time t, and finally constancy in population size at N1. Boundaries for NA, NB, N1 and t priors are available in Table 5. We set the potential time for the population size change event between 10,000 and 10 million years ago, to encompass a broad period of major climatic changes in the northern hemisphere [64]. We set the upper boundary of effective population after an increase (NA) to 10 times the initial upper boundary of effective population size (N1), to limit our analysis to population size increases of at least an order of magnitude. Similarly, we set the lower boundary of effective population size after a decrease (NB) to 1/10 of the initial lower boundary of effective population size (N1) to limit our analysis to population size decreases of at least an order of magnitude. If no change in population size of at least an order of magnitude occurred in the analysis timeframe, or if both a population size decrease and increase of similar magnitude occurred, then the scenario of constant population size would be most likely. We conducted a series of initial trial runs to determine the effective population size parameters for ABC analysis where we could achieve convergence between priors and observed values (Table 5). As an example, our final effective population size parameter values are large in comparison with those in a study on the invasive ladybird Harmonia axyridis [65]. Trial runs also indicated the best fit was achieved when we used a mutation rate per site between 10−7 and 10−9, with a mean at 10−8, for both mitochondrial and microsatellite markers, and the Stepwise Mutation Model (setting coefficient P at 0) for microsatellite markers. The estimated mutation rate for microsatellites is on the low end for such markers, but consistent with the relatively low variability observed for markers originally developed in a different species (i.e., ascertainment bias; [66]). We used “Mean number of alleles” and “Mean genic diversity” as summary statistics for microsatellite loci, and “Number of haplotypes”, “Mean of pairwise differences” and “Private segregating sites” for the mitochondrial locus. We computed 3,000,000 simulated datasets to compare with the observed dataset. First, we pre-evaluated the fit of observed values to prior distributions of scenarios, using a Principal Component Analysis implemented with the software. In a graph of the first two principal components, a good prior assessment is reflected in the observed values being approximately in the centre of the prior values for all three scenarios. Second, we calculated posterior probabilities for all three scenarios using a logarithmic regression, to determine which scenario is the most likely given the data. We expected strong support for a scenario mimicking a demographic expansion.
Table 5.
Parameter values used in the approximate Bayesian computation analysis of demographic history of Cimex adjunctus. The set lower and upper boundaries of the three effective population size parameters are shown: N1 is the effective population size before population size change, NA is the effective population size after demographic expansion, and NB is the effective population size after demographic decline. The time period over which a population size change potentially occurred is t (in years)
| Parameter | Lower boundary | Upper boundary |
|---|---|---|
| NA | 500,000 | 50,000,000 |
| NB | 50,000 | 5,000,000 |
| N1 | 500,000 | 5,000,000 |
| t | 10,000 | 10,000,000 |
Acknowledgements
We would like to thank Alejandra Ceballos, Cal Butchkoscki, Christy Humphrey, David Elliot, Derek Morningstar, Dylan Baloun, Elizabeth Warburton, Jenna Siu, John Whitaker, Ken Luzynski, Kristin Jonasson, Laura Kaupas, Lee Johnson, Lesley Hale, Lisa Winhold, Lucas Greville, Lynne Burns, Mark Brigham, Paul Faure, Quinn Webber, Rob Valdizon, Robert Barclay, Roger Pearce, Sara McCarthy, Shelby Bohn, Stephanie Erickson, Toby Thorne and Tony Parr for their help collecting all samples for the study. We thank André Lachance for comments on an earlier draft of the manuscript, and for guidance with data analysis and interpretation.
Funding
Natural Sciences and Engineering Research Council of Canada (NSERC) and Western University funded BT during his PhD degree. NSERC and Western Michigan University funded this project.
Availability of data and material
All nucleotide sequences can be found on NCBI’s Genbank under accession numbers KU534896 to KU534936, and all microsatellite data are available as a supplementary file (Additional file 3: Table S3).
Authors’ contributions
BT’s work involved conception of the study, collection of some samples, execution of genetic and statistical analyses and writing the first draft. NK coordinated the study, supervised the collection and interpretation of genetic data and revised the writing. BF contributed to the collection of data, coordinated the study, supervised the interpretation of data and revised the writing. MJV and HGB contributed with most of the sample collection, and helped in the interpretation of data and revision of the writing. All authors read and approved the final manuscript.
Authors’ information
BT is broadly interested in phylogeography and population genetics of animals. He also has an interest in parasite and host interactions. He began this project as part of his doctoral dissertation, jointly supervised by NK and BF at the University of Western Ontario. MJV and HGB are collaborators on the project.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
We captured and released all bats according to the requirements of the University of Western Ontario’s Animal Care Committee and by our Wildlife Scientific Collector’s Authorization supplied by the Ontario Ministry of Natural Resources to Benoit Talbot and to Derek Morningstar, for all bats caught in Ontario, Canada. Also, we captured and released all bats according to the requirements of the Saint Mary's University Animal Care Committee and our permits issued by Nova Scotia, Prince Edward Island, Newfoundland and Labrador and New Brunswick government agencies to Hugh Broders, for all bats caught in the maritime provinces of Canada. Dr. Mark Brigham received permission from the University of Regina President’s Committee on Animal Care to catch the bats from which they took parasite samples in Saskatchewan, Canada. Finally, Laura Kaupas received permission from the University of Calgary’s Life and Environmental Sciences Animal Care Committee, and a Northwest Territories Government Wildlife Research Permit, to catch the bats from which they took parasite samples in Northwest Territories, Canada.
Additional files
List of Cimex adjunctus specimens included in analyses. Site refers to each unique sampling location in the study. Host species refers to the bat species from which the samples were collected, or which was inhabiting the roost from which the samples were collected (EPFU : Eptesicus fuscus, MYLU : Myotis lucifugus, MYSE : Myotis septentrionalis). Individuals with the same haplotype number share the same mitochondrial CO1 haplotype sequence (we could not obtain CO1 information for six individuals). We provide the Genbank accession number for each unique haplotype the first time it appears in the table. The genetic cluster (identified by Geneland based on microsatellite data) to which each specimen was assigned is also given (we could not obtain microsatellite data for 10 individuals). (XLSX 51 kb)
Details of sampling locations where we collected Cimex adjunctus specimens used in this study. Site refers to each unique sampling location and correspond to those in Additional file 1: Table S1. Sample size refers to the number of specimens collected at each site. Capture method refers to the way C. adjunctus samples were collected: either from the body of a bat that was captured outside a roost with a mist net or a harp trap (Bat capture), or from inside of the roost itself (Roost visit). Site characteristics refers either to the type of roost (house, barn, cabin, church or abandoned mine) in, or next to which, we collected samples, or to the forested land jurisdiction where collection was performed. (XLSX 37 kb)
Genotypic data at 9 microsatellite markers for 150 Cimex adjunctus individuals (we could not obtain microsatellite data for 10 individuals). The first three digits in each string describe the length (in base pairs) of the first allele, and the last three describe the length of the second allele. Missing data is identified with “000000”. (XLSX 59 kb)
Host species and sampling year from which C. adjunctus samples assigned to each genetic cluster were collected. (XLSX 41 kb)
First and second principal components (percentage of explained variation in parentheses) of simulated values under each putative demographic scenario for C. adjunctus, as determined by approximate Bayesian computation (ABC), and comparison with observed values. (PDF 2969 kb)
Sequence, primer concentration ([Primer]), annealing temperature (T°), total number of alleles, and size range information for nine microsatellite markers previously designed for Cimex lectularius (by 1: Fountain et al. [41] and 2: Booth et al. [42]), used here on Cimex adjunctus samples. For each marker, we also show the average number of alleles (NA), expected heterozygosity (HE), observed heterozygosity (HO) and inbreeding coefficient (GIS) across all sites with five or more sampled individuals. (XLSX 51 kb)
References
- 1.Poulin R. The functional importance of parasites in animal communities: many roles at many levels? Int J Parasitol. 1999;29:903–14. doi: 10.1016/S0020-7519(99)00045-4. [DOI] [PubMed] [Google Scholar]
- 2.Marshall AG. The ecology of ectoparasitic insects. London: Academic; 1981. [Google Scholar]
- 3.Bohonak AJ. Dispersal, gene flow, and population structure. Q Rev Biol. 1999;74:21–45. doi: 10.1086/392950. [DOI] [PubMed] [Google Scholar]
- 4.Blouin MS, Yowell CA, Courtney CH, Dame JB. Host movement and the genetic structure of populations of parasitic nematodes. Genetics. 1995;141:1007–14. doi: 10.1093/genetics/141.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Criscione CD, Cooper B, Blouin MS. Parasite genotypes identify source populations of migratory fish more accurately than fish genotypes. Ecology. 2006;87:823–8. doi: 10.1890/0012-9658(2006)87[823:PGISPO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 6.Anderson RM, Gordon DM. Processes influencing the distribution of parasite numbers within host populations with special emphasis on parasite-induced host mortalities. Parasitology. 1982;85:373–98. doi: 10.1017/S0031182000055347. [DOI] [PubMed] [Google Scholar]
- 7.Thrall PH, Burdon JJ. Host-pathogen dynamics in a metapopulation context: the ecological and evolutionary consequences of being spatial. J Ecol. 1997;85:743–53. doi: 10.2307/2960598. [DOI] [Google Scholar]
- 8.Nieberding C, Morand S, Libois R, Michaux JR. A parasite reveals cryptic phylogeographic history of its host. Proc R Soc B Biol Sci. 2004;271:2559–68. doi: 10.1098/rspb.2004.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nieberding CM, Durette-Desset M-C, Vanderpoorten A, Casanova JC, Ribas A, Deffontaine V, et al. Geography and host biogeography matter for understanding the phylogeography of a parasite. Mol Phylogenet Evol. 2008;47:538–54. doi: 10.1016/j.ympev.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 10.Mazé-Guilmo E, Blanchet S, McCoy KD, Loot G. Host dispersal as the driver of parasite genetic structure: a paradigm lost? Ecol Lett. 2016;19:336–47. doi: 10.1111/ele.12564. [DOI] [PubMed] [Google Scholar]
- 11.McCoy KD, Boulinier T, Tirard C, Michalakis Y. Host-dependant genetic structure of parasite populations: differential dispersal of seabird tick host races. Evolution. 2003;57:288–96. doi: 10.1111/j.0014-3820.2003.tb00263.x. [DOI] [PubMed] [Google Scholar]
- 12.van der Mescht L, Matthee S, Matthee CA. Comparative phylogeography between two generalist flea species reveal a complex interaction between parasite life history and host vicariance: parasite-host association matters. BMC Evol Biol. 2015;15:105. doi: 10.1186/s12862-015-0389-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelbrecht A, Matthee S, du Toit N, Matthee CA. Limited dispersal in an ectoparasitic mite, Laelaps giganteus, contributes to significant phylogeographic congruence with the rodent host, Rhabdomys. Mol Ecol. 2016;25:1006–21. doi: 10.1111/mec.13523. [DOI] [PubMed] [Google Scholar]
- 14.Usinger RL. Monograph of Cimicidae (Hemiptera, Heteroptera) Annapolis: Entomological Society of America; 1966. [Google Scholar]
- 15.Goddard J. Bed bugs (Cimex lectularius) and clinical consequences of their bites. J Am Med Assoc. 2009;301:1358–66. doi: 10.1001/jama.2009.405. [DOI] [PubMed] [Google Scholar]
- 16.Criado PR, Belda Junior W, Criado RFJ, Vasconcelos e Silva E, Vasconcellos C. Bedbugs (Cimicidae infestation): the worldwide renaissance of an old partner of human kind. Braz. J Infect Dis. 2011;15:74–80. doi: 10.1016/s1413-8670(11)70144-1. [DOI] [PubMed] [Google Scholar]
- 17.Furlonger CL, Dewar HJ, Fenton MB. Habitat use by foraging insectivorous bats. Can J Zool. 1987;65:284–8. doi: 10.1139/z87-044. [DOI] [Google Scholar]
- 18.Ellison LE, O’Shea TJ, Neubaum DJ, Bowen RA. Factors influencing movement probabilities of big brown bats (Eptesicus fuscus) in buildings. Ecol Appl. 2007;17:620–7. doi: 10.1890/06-0315. [DOI] [PubMed] [Google Scholar]
- 19.Pearce RD, O’Shea TJ. Ectoparasites in an urban population of big brown bats (Eptesicus fuscus) in Colorado. J Parasitol. 2007;93:518–30. doi: 10.1645/GE-973R.1. [DOI] [PubMed] [Google Scholar]
- 20.Vonhof MJ, Strobeck C, Fenton MB. Genetic variation and population structure in big brown bats (Eptesicus fuscus): is female dispersal important? J Mammal. 2008;89:1411–9. doi: 10.1644/08-MAMM-S-062.1. [DOI] [Google Scholar]
- 21.Turmelle AS, Kunz TH, Sorenson MD. A tale of two genomes: contrasting patterns of phylogeographic structure in a widely distributed bat. Mol Ecol. 2011;20:357–75. doi: 10.1111/j.1365-294X.2010.04947.x. [DOI] [PubMed] [Google Scholar]
- 22.Burns LE, Frasier TR, Broders HG. Genetic connectivity among swarming sites in the wide ranging and recently declining little brown bat (Myotis lucifugus) Ecol Evol. 2014;4:4130–49. doi: 10.1002/ece3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLeod BA, Burns LE, Frasier TR, Broders HG. Effect of oceanic straits on gene flow in the recently endangered little brown bat (Myotis lucifugus) in maritime Canada: implications for the spread of white-nose syndrome. Can J Zool. 2015;93:427–37. doi: 10.1139/cjz-2014-0262. [DOI] [Google Scholar]
- 24.Johnson LNL, McLeod BA, Burns LE, Arseneault K, Frasier TR, Broders HG. Population genetic structure within and among seasonal site types in the little brown bat (Myotis lucifugus) and the northern long-eared bat (M. septentrionalis) PLoS One. 2015;10:e0126309. doi: 10.1371/journal.pone.0126309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vonhof MJ, Russell AL, Miller-Butterworth CM. Range-wide genetic analysis of little brown bat (Myotis lucifugus) populations: estimating the risk of spread of White-Nose Syndrome. PLoS One. 2015;10:e0128713. doi: 10.1371/journal.pone.0128713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartonička T, Růžičková L. Recolonization of bat roost by bat bugs (Cimex pipistrelli): could parasite load be a cause of bat roost switching? Parasitol Res. 2013;112:1615–22. doi: 10.1007/s00436-013-3316-4. [DOI] [PubMed] [Google Scholar]
- 27.Tomisawa R, Akimoto S. Host range and host preference of a flea weevil, Orchestes hustachei, parasitizing aphid galls. Entomol Sci. 2004;7:21–30. doi: 10.1111/j.1479-8298.2003.00038.x. [DOI] [Google Scholar]
- 28.McCoy KD, Chapuis E, Tirard C, Boulinier T, Michalakis Y, Bohec CL, et al. Recurrent evolution of host-specialized races in a globally distributed parasite. Proc R Soc B Biol Sci. 2005;272:2389–95. doi: 10.1098/rspb.2005.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dick CW, Patterson BD. Against all odds: explaining high host specificity in dispersal-prone parasites. Int J Parasitol. 2007;37:871–6. doi: 10.1016/j.ijpara.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Balvín O, Vilímová J, Kratochvíl L. Batbugs (Cimex pipistrelli group, Heteroptera: Cimicidae) are morphologically, but not genetically differentiated among bat hosts. J Zool Syst Evol Res. 2013;51:287–95. doi: 10.1111/jse.12000. [DOI] [Google Scholar]
- 31.Adam MD, Hayes JP. Use of bridges as night roosts by bats in the Oregon Coast Range. J Mammal. 2000;81:402–7. doi: 10.1644/1545-1542(2000)081<0402:UOBANR>2.0.CO;2. [DOI] [Google Scholar]
- 32.Dixon MD. Post-Pleistocene range expansion of the recently imperiled eastern little brown bat (Myotis lucifugus lucifugus) from a single southern refugium. Ecol Evol. 2011;1:191–200. doi: 10.1002/ece3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olival KJ, Dick CW, Simmons NB, Morales J, Melnick DJ, Dittmar K, et al. Lack of population genetic structure and host specificity in the bat fly, Cyclopodia horsfieldi, across species of Pteropus bats in Southeast Asia. Parasit Vectors. 2013;6:231–48. doi: 10.1186/1756-3305-6-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nadin-Davis SA, Feng Y, Mousse D, Wandeler AI, Aris-Brosou S. Spatial and temporal dynamics of rabies virus variants in big brown bat populations across Canada: footprints of an emerging zoonosis. Mol Ecol. 2010;19:2120–36. doi: 10.1111/j.1365-294X.2010.04630.x. [DOI] [PubMed] [Google Scholar]
- 35.ter Hofstede HM, Fenton MB. Relationships between roost preferences, ectoparasite density, and grooming behaviour of neotropical bats. J Zool. 2005;266:333–40. doi: 10.1017/S095283690500693X. [DOI] [Google Scholar]
- 36.Levy F, Neal CL. Spatial and temporal genetic structure in chloroplast and allozyme markers in Phacelia dubia implicate genetic drift. Heredity. 1999;82:422–31. doi: 10.1038/sj.hdy.6885000. [DOI] [PubMed] [Google Scholar]
- 37.Cooper R, Wang C, Singh N. Mark-release-recapture reveals extensive movement of bed bugs (Cimex lectularius L.) within and between apartments. PLoS One. 2015;10:e0136462. doi: 10.1371/journal.pone.0136462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Booth W, Balvín O, Vargo EL, Vilímová J, Schal C. Host association drives genetic divergence in the bed bug, Cimex lectularius. Mol Ecol. 2015;24:980–92. doi: 10.1111/mec.13086. [DOI] [PubMed] [Google Scholar]
- 39.Balvín O, Munclinger P, Kratochvíl L, Vilímová J. Mitochondrial DNA and morphology show independent evolutionary histories of bedbug Cimex lectularius (Heteroptera: Cimicidae) on bats and humans. Parasitol Res. 2012;111:457–69. doi: 10.1007/s00436-012-2862-5. [DOI] [PubMed] [Google Scholar]
- 40.Fountain T, Duvaux L, Horsburgh G, Reinhardt K, Butlin RK. Human-facilitated metapopulation dynamics in an emerging pest species, Cimex lectularius. Mol Ecol. 2014;23:1071–84. doi: 10.1111/mec.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth W, Saenz VL, Santangelo RG, Wang C, Schal C, Vargo EL. Molecular markers reveal infestation dynamics of the bed bug (Hemiptera: Cimicidae) within apartment buildings. J Med Entomol. 2012;49:535–46. doi: 10.1603/ME11256. [DOI] [PubMed] [Google Scholar]
- 42.Saenz VL, Booth W, Schal C, Vargo EL. Genetic analysis of bed bug populations reveals small propagule size within individual infestations but high genetic diversity across infestations from the eastern United States. J Med Entomol. 2012;49:865–75. doi: 10.1603/ME11202. [DOI] [PubMed] [Google Scholar]
- 43.James PMA, Coltman DW, Murray BW, Hamelin RC, Sperling FAH. Spatial genetic structure of a symbiotic beetle-fungal system: toward multi-taxa integrated landscape genetics. PLoS One. 2011;6:e25359. doi: 10.1371/journal.pone.0025359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grant WS. Problems and cautions with sequence Mismatch Analysis and Bayesian Skyline Plots to infer historical demography. J Hered. 2015;106:333–46. doi: 10.1093/jhered/esv020. [DOI] [PubMed] [Google Scholar]
- 45.Swenson NG, Howard DJ. Clustering of contact zones, hybrid zones, and phylogeographic breaks in North America. Am Nat. 2005;166:581–91. doi: 10.1086/491688. [DOI] [PubMed] [Google Scholar]
- 46.Dixon MD. Population genetic structure and natal philopatry in the widespread North American bat Myotis lucifugus. J Mammal. 2011;92:1343–51. doi: 10.1644/10-MAMM-A-426.1. [DOI] [Google Scholar]
- 47.Hebert PDN, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc R Soc B Biol Sci. 2003;270:313–21. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balvín O, Roth S, Vilímová J. Molecular evidence places the swallow bug genus Oeciacus Stål within the bat and bed bug genus Cimex Linnaeus (Heteroptera: Cimicidae): The genus Oeciacus within the genus Cimex. Syst Entomol. 2015;40:652–65. doi: 10.1111/syen.12127. [DOI] [Google Scholar]
- 49.Wang J. An estimator for pairwise relatedness using molecular markers. Genetics. 2002;160:1203–15. doi: 10.1093/genetics/160.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fields Development Team . Fields: tools for spatial data. Boulder: National Center for Atmospheric Research; 2006. [Google Scholar]
- 51.Goslee SC, Urban DL. The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 2007;22:1–19.
- 52.Wang J. Unbiased relatedness estimation in structured populations. Genetics. 2011;187:887–901. doi: 10.1534/genetics.110.124438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan M-L, Wei D-D, Zhang K, Gao Y-Z, Liu Y-H, Wang B-J, et al. Genetic diversity and population structure of Panonychus citri (Acari: Tetranychidae), in China based on mitochondrial COI gene sequences. J Econ Entomol. 2010;103:2204–13. doi: 10.1603/EC09392. [DOI] [PubMed] [Google Scholar]
- 54.Pulgarín-R PC, Burg TM. Genetic signals of demographic expansion in downy woodpecker (Picoides pubescens) after the last North American glacial maximum. PLoS One. 2012;7:e40412. doi: 10.1371/journal.pone.0040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pereira L, Dupanloup I, Rosser ZH, Jobling MA, Barbujani G. Y-chromosome mismatch distributions in Europe. Mol Biol Evol. 2001;18:1259–71. doi: 10.1093/oxfordjournals.molbev.a003911. [DOI] [PubMed] [Google Scholar]
- 56.Drummond AJ, Suchard MA. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010;8:114. doi: 10.1186/1741-7007-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown RP, Yang Z. Rate variation and estimation of divergence times using strict and relaxed clocks. BMC Evol Biol. 2011;11:271. doi: 10.1186/1471-2148-11-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 59.Bargues MD, Klisiowicz DR, Panzera F, Noireau F, Marcilla A, Perez R, et al. Origin and phylogeography of the Chagas disease main vector Triatoma infestans based on nuclear rDNA sequences and genome size. Infect Genet Evol. 2006;6:46–62. doi: 10.1016/j.meegid.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 60.Brower AV. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci. 1994;91:6491–5. doi: 10.1073/pnas.91.14.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heller R, Chikhi L, Siegismund HR. The confounding effect of population structure on Bayesian Skyline Plot inferences of demographic history. PLoS One. 2013;8:e62992. doi: 10.1371/journal.pone.0062992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bertorelle G, Benazzo A, Mona S. ABC as a flexible framework to estimate demography over space and time: some cons, many pros. Mol Ecol. 2010;19:2609–25. doi: 10.1111/j.1365-294X.2010.04690.x. [DOI] [PubMed] [Google Scholar]
- 63.Chakraborty D, Sinha A, Ramakrishnan U. Mixed fortunes: ancient expansion and recent decline in population size of a subtropical montane primate, the Arunachal macaque Macaca munzala. PLoS One. 2014;9:e97061. doi: 10.1371/journal.pone.0097061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zachos J. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science. 2001;292:686–93. doi: 10.1126/science.1059412. [DOI] [PubMed] [Google Scholar]
- 65.Lombaert E, Guillemaud T, Thomas CE, Lawson Handley LJ, Li J, Wang S, et al. Inferring the origin of populations introduced from a genetically structured native range by approximate Bayesian computation: case study of the invasive ladybird Harmonia axyridis. Mol Ecol. 2011;20:4654–70. doi: 10.1111/j.1365-294X.2011.05322.x. [DOI] [PubMed] [Google Scholar]
- 66.Li B, Kimmel M. Factors influencing ascertainment bias of microsatellite allele sizes: impact on estimates of mutation rates. Genetics. 2013;195:563–72. doi: 10.1534/genetics.113.154161. [DOI] [PMC free article] [PubMed] [Google Scholar]