

Abstract
Cytochrome ba3 is a proton-pumping heme-copper oxygen reductase from the extreme thermophile Thermus thermophilus. Despite the fact that the enzyme’s active site is buried deep within the protein, the apparent second order rate constant for the initial binding of O2 to the active-site heme has been experimentally found to be 109 M−1s−1 at 298 K, at or near the diffusion limit, and two orders of magnitude faster than for O2 binding to myoglobin. To provide quantitative and microscopic descriptions of the O2 delivery pathway and mechanism in cytochrome ba3, extensive molecular dynamics simulations of the enzyme in its membrane-embedded form have been performed, including different protocols of explicit ligand sampling (flooding) simulations with O2, implicit ligand sampling analysis and in silico mutagenesis. The results show that O2 diffuses to the active site exclusively via a Y-shaped hydrophobic tunnel with two 25-Å long membrane-accessible branches that coincide with the pathway previously suggested by the crystallographically identified xenon binding sites. The two entrances of the bifurcated tunnel of cytochrome ba3 are located within the lipid bilayer, where O2 is preferentially partitioned from the aqueous phase. The largest barrier to O2 migration within the tunnel is estimated to be only 1.5 kcal/mol, allowing O2 to reach the enzyme active site virtually impeded by one-dimensional diffusion once it reaches a tunnel entrance at the protein surface. Unlike other O2-utilizing proteins, the tunnel is “open” with no transient barriers observed due to protein dynamics. This unique low-barrier passage through the protein assures that O2 transit through the protein is never rate-limiting.
Graphical Abstract
Introduction
The heme-copper oxygen reductases (HCOs) are integral membrane enzyme complexes that catalyze the exergonic reduction of O2 to water and terminate the aerobic respiratory chains of all aerobic eukaryotes and most aerobic prokaryotic organisms.1–9 These enzymes include the mitochondrial as well as all bacterial cytochrome c oxidases. The free energy release from the reduction of O2 is used to generate a proton motive force across biological membranes which, in turn, drives ATP biosynthesis, as well as other energy-requiring processes.2,10–13 The HCOs, together with closely related NO reductases (NORs), are members of the heme-copper superfamily5,10–13 which have in common a membrane subunit (Subunit I) with a minimum of 12 transmembrane α-helical spans, which contains the bimetallic active site where O2 (or NO) binds and is reduced to water (or N2O). For the O2 reductases (HCOs), the bimetallic active site consists of a heme Fe (e.g. heme a3) and a nearby Cu atom (4–5Å distant). O2 binds to an open axial coordination site on the reduced heme Fe and is reduced by a sequence of proton-coupled electron transfer reactions. The heme-copper active site is deeply buried within the membrane subunit but must allow rapid access for each of the substrates of the reaction.
The active site of many enzymes are sequestered from the bulk solution, necessitating efficient strategies by which the substrates can be delivered and products removed. These strategies include hydrophilic clefts or pores or hydrophobic tunnels or cavities which allow the substrate to diffuse towards (or products away from) the active site.14–18 Most often the pathways are gated by protein conformational changes that may be large, as in movement of a protein domain sealing the active sites, or may be small, as in single amino acid side chain fluctuations allowing passage of a small molecule between cavities within the protein, en route to the active site.18–20
The three substrates that must access the active site of the HCOs are O2, protons and electrons. Electrons are delivered to the active site one at a time by a linear sequence of proton-coupled, step-wise electron transfer reactions between redox-active metal centers from the site where cytochrome c is oxidized (on Subunit II).21 Protons are delivered to the active site through one or two different proton-conducting channels (depending on the enzyme) that have entrances at the aqueous phase on the electrically negative side of the membrane (e.g., bacterial cytoplasm or mitochondrial matrix). Conserved polar residues as well as internal water molecules provide the continuous hydrogen-bond networks needed for the long-range (25–30Å) proton diffusion to the active site by the Grotthus mechanism.6,7,22–25 In contrast to the electron and protons “wires”, pathways for O2 diffusion into the active site have not been well-characterized. Hydrophobic tunnels in the X-ray structures of the aerobic respiratory heme-copper oxygen reductases,26–32 including cytochrome ba3 from Thermus thermophilus,28,31,33,34 have been suggested as putative O2 tunnels. Xenon (Xe), which has a similar size as O2, is often used in crystallographic studies to locate hydrophobic cavities that may provide binding sites for small diatomic gas molecules, including O2, CO, H2 or N2.35–39 X-ray crystallography has located Xe binding sites in both cytochrome ba3 35,40 and the aa3-type oxygen reductase from Rhodobacter sphaeroides,29 thereby identifying hydrophobic cavities that are within their putative O2 tunnels.
Recent molecular dynamics (MD) simulations and computational analyses of the HCO from Rhodobacter sphaeroides concluded that the putative O2 tunnel suggested by X-ray crystallography is not the primary pathway by which O2 reaches the active site.41 Two additional pathways, which are not apparent in the static X-ray structures and may be formed transiently by protein dynamics, were suggested as more likely pathways that lead O2 to the enzyme’s active site. Hence, what appears to be an O2 pathway may not function as such.
In the present study, we computationally examine O2 pathways to the active site of a different respiratory HCO, cytochrome ba3 from the hyperthermophile Thermus thermophilus. Similar to HCO from R. sphaeroides, Xe binding sites have been identified by X-ray crystallography that align within a hydrophobic tunnel suggested to act as the O2 pathway.35,40 In addition apparent second order rate constant for O2 binding to the active site from solution has been directly measured to be 1×109 M−1s−1 at 298 K,42,43 near the diffusion limit and far greater than found for O2 diffusion to internal sites within other proteins, including myoglobin (1.6×107 M−1s−1).44 The motivation of this study is to understand how O2 reaches the deeply buried active site of cytochrome ba3 so rapidly.
Previous studies have characterized mutants that place bulky residues within the putative O2 tunnel that reduce the apparent rate constant 5-fold,33 supporting the X-ray observed tunnel as a functional O2 pathway in cytochrome ba3. Short (1 ns) MD simulations concentrating on the dynamics and stability of O2 at Xe binding sites of the X-ray tunnel indicated that O2 can rapidly move between individual Xe sites within the hydrophobic tunnel.33 In the present study, extensive MD simulations (repeated 50-ns simulations with multiple copies of O2) as well as implicit ligand sampling (ILS)45 are used to characterize the energetics of O2 partitioning within the protein and the trajectories of O2 molecules starting outside the protein and reaching the active site. The all-atom MD was performed using the entire 3-subunit protein embedded in a lipid bilayer and surrounded by a bulk aqueous solution. The study demonstrates that the substrate O2 utilizes the X-ray Xe-bound hydrophobic tunnel as the pathway for reaching the active site. Furthermore, unlike other proteins, such as myoglobin or the R. sphaeroides HCO, the O2 pathway in cytochrome ba3 is static and does not seem to be affected by protein dynamics. The highest free energy barrier for O2 to diffuse through the identified O2 pathway is calculated to be 1.5 kcal/mol. Cytochrome ba3, therefore, appears to have evolved so that the transit time of O2 through the 25-Å long hydrophobic tunnel is never rate-limiting. Under 298 K, the enzyme is limited by the rate of chemical catalysis (kcat ≈102 s−1) at O2 concentration above 0.1 μM, and would be limited by the rate of O2 reaching the entrances of the O2 pathway when O2 concentration is below 0.1 μM. At physiological conditions (343–363 K) where the organism (T. thermophilus) grows, the enzyme velocity may be limited by the rate of O2 reaching the pathway’s entrances.
Materials and Methods
Construction of the membrane-embedded cytochrome ba3 model
MD simulations were performed on a membrane-embedded model of the cofactor-bound cytochrome ba3 complex constructed using the crystal structure of the enzyme from T. thermophilus HB8 strain (PDB entry 1XME)46 inserted into a hydrated and ionized patch of POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) bilayer. The crystal structure contains three subunits (I, II and IIa), 75 water molecules, heme b, active-site heme a3, three copper ions (CuB and dicopper CuA) cofactors. The membrane-embedded cytochrome ba3 was constructed using VMD.47
The cofactor-bound cytochrome ba3 complex was embedded in a patch of POPE membrane by aligning the center of transmembrane regions of its subunits I, II and IIa with the midpoint of the membrane. Lipids that overlapped the protein were removed, keeping 98 lipids in the periplasmic leaflet and 92 lipids in the cytoplasmic leaflet. The membrane-embedded cytochrome ba3 was then solvated with water. Water molecules that were in the membrane were removed, keeping 15,177 water molecules. Finally, 0.2M NaCl (26 Na+ and 33 Cl− ions) was added to neutralize and ionize the complex, resulting in a fully solvated model of approximately 85,000 atoms.
Simulation protocols
MD simulations consisted of the following steps: (1) 0.5-ns melting of lipid tails during which only the lipid tails were allowed to move in order to achieve better packing of lipids around the inserted protein; (2) 0.5-ns simulation with restraints (k =1 kcal/mol/Å2) applied to heavy atoms of the protein and cofactors (all lipid atoms and water moving) and with harmonic potentials (k =0.1 kcal/mol/Å2) applied to keep water out of the membrane; (3) 0.5-ns simulations with only backbone atoms of the protein and heavy atoms of the cofactors restrained (k =1 kcal/mol/Å2); (4) 1-ns simulation with only Cα atoms of the protein and heavy atoms of the cofactors restrained; and (5) 20-ns unrestrained relaxation. Energy minimization (1,000 steps) was performed at the beginning of Steps 1, 2 and 3 using the conjugate gradient algorithm. In order to maintain the ligation of CuB to H233, H282 and H283, the His-CuB connections were described as bonds with k =65 kcal/mol/Å2 for the bonds and k =30 kcal/mol/rad2 for the angles involving the His-CuB bond.
All simulations were performed using NAMD248 with a time step of 2 fs. The periodic boundary conditions (PBC) were used throughout the simulations. All bonds involving hydrogen atoms were kept rigid using the SHAKE algorithm.49 To evaluate long-range electrostatic interactions in PBC without truncation, the particle mesh Ewald (PME) method50 with a grid density of 1/Å3 was used. The cutoff for van der Waals interactions was set at 12Å. All of simulation steps except the melting of lipid tails (Step 1) were performed in a flexible cell, which allows the membrane-embedded cytochrome ba3 model to change its dimensions independently, and were performed under NPT ensemble. The temperature was maintained at 310K by Langevin dynamics51 with a damping coefficient γ of 1/ps. The Nosé-Hoover Langevin piston method51,52 with a piston period of 200 fs was used to maintain the pressure at 1 atm.
The CHARMM22 force field with ϕ/ψ corrections53,54 was used to describe the protein and heme cofactors, and CHARMM3655 was used to describe the lipids. The TIP3P model56 was used as the water model. All cofactors were treated in the reduced state. The force field parameters for the hydroxyethylfarnesyl side chain of the active-site heme cofactor (heme a3), which were not available in the CHARMM force field library, were constructed by analogy using parameterized alcohol, alkene, and alkane fragments.57 The parameters for the copper ions were those previously used by Hofacker and Schulten.58 A harmonic potential (k=400 kcal/mol/Å2) was used for the Nε-Cε bond of the His-Tyr (H233–Y237) crosslink.59
Analyses were performed mainly with VMD47 and MATLAB. The pore profile for O2 pathway was calculated using the MolAxis program,60 which was previously used to probe for tunnels in several proteins.61,62
Production runs
The final frame of wild-type cytochrome ba3 from the 20-ns relaxation simulation was used as the starting configuration for the explicit ligand sampling simulations (ELS, aka flooding simulations63,64 ) in which O2 molecules were added, for the continued 50-ns simulation in the absence of O2 (apo), and for in silico mutagenesis.
Explicit ligand sampling (ELS) simulations
Long simulations
To probe for O2 delivery pathways of cytochrome ba3 and characterize O2 delivery process, 100 O2 molecules, corresponding to 0.21M [O2] calculated with respect to the total simulation box volume of 790,000Å3, were added to the equilibrated membrane-embedded enzyme model. This O2 concentration is 161-fold higher than the saturated O2 concentration in water at ambient conditions. This high O2 concentration was introduced to maximize the sampling of O2 delivery pathways in the protein within limited timescale (tens of nanoseconds). Three independent simulation systems were designed in which O2 molecules were initially distributed in the membrane and aqueous phases at different ratios: 70:30, 100:0 and 0:100. Each system was visually inspected to ensure that the added O2 molecules do not overlap with atoms of other molecules, and was then simulated for 50 ns (Fig. 1a). As all three simulation systems showed that approximately 80 of 100 O2 molecules localized in the membrane after reaching a steady distribution (Fig. 1b), the 70:30 ratio system was repeated two more times, resulting in a total of 5×50 ns simulations. Repeated simulations are to improve statistics and provide more accurate descriptions of thermodynamic properties associated the O2 delivery. To identify O2 delivery pathways, a 3D density map representing the O2 occupancy profiles was calculated using the Volmap tool in VMD. Using equilibrated fractions (last 40 ns) of the MD trajectories, the map was projected as a free-energy (ΔGi,sol) map, which describes the energetics of O2 insertion.
Figure 1. Partitioning profiles of O2 in membrane.

a) Distributions of O2 in membrane vs. aqueous phase in the protein-free system. Top panel illustrates the diffusion of O2 molecules (shown as red balls) from the aqueous phase to the membrane. O2 is considered to be in the membrane when either oxygen atom in the molecule is in between the C22 atoms (Cα of the palmitate’s ester) of the POPE lipids in the two leaflets (−16Å ≤ z ≤ 16Å). Partitioning free-energy profiles of O2 calculated from ELS simulations and ILS indicate that more O2 molecules reside in the membrane (shaded orange) than in the aqueous phase (shaded blue). b) A representative ELS simulation system in the presence of cytochrome ba3. All systems with different initial ratio of O2 show that O2 is more distributed in the membrane than in the aqueous phase.
pi represents the probability of finding O2 at a site i calculated over the equilibrated trajectories. psol is probability of finding O2 in the bulk aqueous solution. psol = 0.00035, determined from the simulation, was used as the reference for estimating partitioning free energies of O2, as O2 appears to distribute uniformly in the aqueous phase whereas it is heterogeneously distributed in the membrane, potentially due to various degrees of hydrophobicity in different regions of the membrane (Fig. 1a). For simulations including the protein, O2 was considered to reach the active site of the enzyme when either one of its oxygen atoms was within 6Å of CuB. Their entries were confirmed by visually monitoring all delivered O2 molecules through MD trajectories and their separation from CuB. For the re-entry of O2, if O2 first moves away from CuB by more than 30Å which is already outside the protein and later diffuses into the active site, we considered it as an independent (re-entry) event.
Short simulations
Once the O2 delivery pathway was identified, the time of O2 diffusing from outside of the protein to the active site, which can relate MD simulation results to time-resolved spectroscopic experiments, can be estimated.42,43 To improve statistics, a set of 21 independent short (each 3–5 ns) simulations was performed. To reduce the computational costs, these new simulations were terminated once one O2 reached the active site, which was determined by its presence within 6Å of CuB. They were all initiated with 90 O2 in the membrane and 10 in the aqueous phase corresponding to the upper equilibrium ratio of 90:10 obtained from the 50-ns long simulations. A 0.5-ns pre-equilibration was then performed for each system to allow further randomizing of O2 molecules. A different random number seed was assigned to initialize each production run. From these short simulations, the time to observe the first O2 to reach the active site after the start of a simulation is referred to as “first entry time” or tfirst. The transit time of O2 diffusion to the entrance of the pathway (tout→ent) and the transit time of O2 diffusion from the entrance to the active site (tent→cat) can also be estimated.
Implicit ligand sampling (ILS) calculations
The implicit ligand sampling (ILS) method45 was employed as an alternative, complementary method to the ELS simulations to characterize O2 delivery pathways of cytochrome ba3. ILS is a quantitative method for characterizing gas migration pathways65–68 that might be difficult to probe with conventional MD simulations (i.e., ELS) due to limited sampling. The method is only applicable to ligands that have weak interactions with proteins. As previously described,45 ILS calculates ligand-interaction energies (Ei) at all sites of each subgrid averaged over an ensemble of protein conformations and ligand orientations, which estimate the probability of finding a ligand in a particular subgrid (pi) and then the free energy of inserting ligand into that subgrid (ΔGi).
where p0 (in vacuum)=1.
The apo trajectory of cytochrome ba3, which was simulated in the absence of O2 for 50 ns following the 20-ns relaxation simulation, was used for ILS calculations to characterize potential O2 delivery pathway. A 60×65×85Å3 grid was placed on the cytochrome ba3 complex covering portions of the membrane and aqueous phases. The grid, which defines where O2 molecules were sampled and consists of several subgrids, was 331,500Å3. Each subgrid contained 3×3×3 interaction sites. Ten orientations of O2 were sampled in all 27 interaction sites of a subgrid over 5,000 frames. The solvation free energy of O2 (ΔGsol) was used as the reference for calculating the partitioning free energy of O2 (ΔGi,sol).
We calculated ΔGsol using free-energy perturbation (FEP)69 and ILS methods over a 30×30×30Å3 ionized water box. Both methods yielded ΔGsol of 2.1 kcal/mol.
In silico site-directed mutagenesis
To examine the role of the identified O2 pathway of cytochrome ba3 in facilitating the delivery of the substrate O2, aliphatic residues along the pathway (I78, V79, A120, A149, S150 and L200) were individually mutated to either a phenylalanine or a tryptophan. These residues were characterized as lining residues by the ELS simulations and ILS calculations of the wildtype enzyme. Four single (I78F, A120F, A149F, S150W), two double (A120F/S150W and V79F/S150W) and one triple (I78F/V79F/L200F) mutants were modeled and simulated. Each mutant was energy-minimized for 1,000 steps, and equilibrated for at least 10 ns. ILS was employed to assess the effects of the mutations by exploring the free energy of O2 partitioning in the pathway. In addition, ELS simulations were performed to locate O2 delivery pathways and estimate transit times of O2 diffusion into the active site.
Simulations of O2 in protein-free membrane
We also performed simulations of a protein-free membrane system in order to calculate the partitioning profiles of O2 between the membrane and aqueous phases (without any perturbation caused by the protein in the lipid structure) and to compare the results with those previously obtained from experiments and MD simulations.70–74 The protein-free membrane model was composed of 40,666 atoms (172 POPE lipids, 6,372 water molecules, 24 Na+, and 24 Cl− ions). The 0.5-ns melting of lipid tails, 5-ns relaxation and 10-ns production simulations were performed as described above. The 10-ns production trajectory was analyzed by ILS for O2 insertion. Then, two independent ELS simulations were performed starting from the 5-ns apo relaxation step: one with 100 O2 initially in the aqueous phase, and one with 100 O2 initially in the membrane. The O2 concentration calculated with respect to the total volume of 435,000Å3 was 0.38 M. A 0.1-ns equilibration at 400K followed by a gradual cooling from 400K to 310K with the total time of 0.2 ns was performed to allow rapid randomizing of O2. 50-ns production simulations were performed after the cooling. The partition coefficient of O2 was determined from the ratio of O2 in the membrane to the aqueous phase. O2 was considered to be in the membrane when either oxygen atom in the molecule resided between the C22 atoms (Cα of the palmitate’s ester) of the POPE lipids in both leaflets or −16Å ≤ z ≤ 16Å (Fig. 1a). With the estimated volume of the aqueous phase of 210,000Å3 being slightly smaller than that of the membrane, the number of O2 residing in the aqueous phase must be multiplied by a factor of 1.1 to calculate the partition coefficient.
Results
Partitioning of O2 between the membrane and aqueous phases
Before starting the ELS simulations on cytochrome ba3, we determined the partition coefficient of O2 between the membrane and aqueous phases using the protein-free membrane system (Fig. 1a). The total volume of this system, 435,000Å3, consists of water and phospholipids in a volume ratio of approximately 9:10. 100O2 molecules or 380mM [O2] were added and allowed to distribute in the system for 50 ns. Starting with different initial distributions of the O2 molecules in the membrane and aqueous phases (100:0 or 0:100) yielded the same equilibrium distribution within 10 ns, with approximately 85 O2 molecules in the membrane and 15 in the aqueous phase (Supp. Fig. 1). After normalizing to the volumes of the aqueous and membrane phases, the ratio of concentrations gives a partition coefficient of 5.5 for O2 favoring the membrane, close to the values obtained previously in a similar simulation study performed in the absence of a protein, 4.7,65 and still comparable to the experimental value of 3.9 estimated using fluorescence quenching.71 The equilibrium profile of O2 distribution within the membrane was also calculated independently using the ELS trajectories and ILS (Fig. 1a). The results are equivalent to those reported previously for simulations with just the aqueous solution and phospholipid65 and also agree with experimental data using NMR and ESR.73,74
For the membrane-embedded complex of cytochrome ba3, the total volume of the simulation system, 790,000Å3 yields an O2 concentration of 210mM (100 O2 molecules) incorporated in the system. Similar to the system, different initial distributions of the O2 molecules in the protein-free membrane and aqueous phases resulted in the same equilibrium distribution with approximately 80 O2 molecules in the membrane-protein phase and 20 in the aqueous phase (Fig. 1b). Since the volume ratio of the aqueous phase to the combined membrane-protein phase is 9:7, the equilibrium concentration of O2 in the aqueous phase has to be normalized, yielding a partition coefficient of 5.2 which is in comparable to that of the protein-free system and previous studies.65,71
O2 pathway to the active site
O2 access to the active site of cytochrome ba3 was monitored in ELS trajectories starting with 100 O2 molecules distributed between the membrane and aqueous phases, but not within the protein. Regardless of the initial O2 distribution, the first O2 reached the active site in each simulation in less than 5 ns (Fig. 2). Taking together the trajectories of all five ELS simulations (250-ns total), a total of 148 O2 molecules reached the active site, averaging to one per 1.7 ns. Of these 148 events, 26 are cases where the same O2 molecule had two or more entries, since the simulation allowed molecules to diffuse back into the solution and return. The data are summarized in Table 1. The density map of O2 within the protein was determined by combining the last 40 ns segments of the five simulations, and was normalized with respect to the average O2 density in the aqueous phase. The results are displayed as a 3D isosurface image (Fig. 3) in order to facilitate comparison to the ILS results (Fig. 4).
Figure 2. Entry of O2 into the active site observed during the ELS simulations.
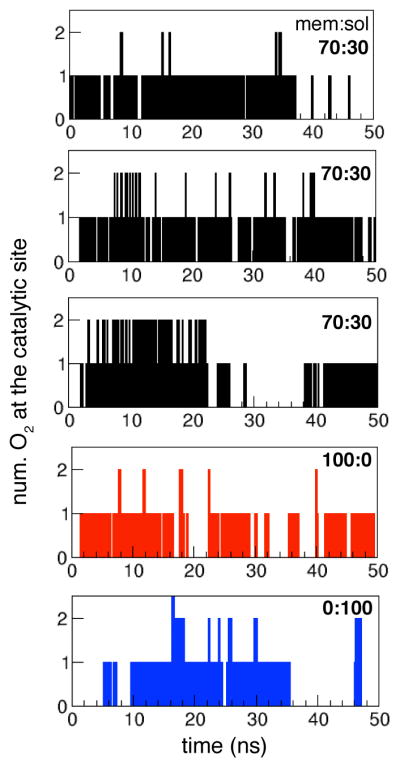
Three of the five simulations are with 70 O2 molecules initially in the membrane and 30 in the aqueous phase, one is with all 100 O2 molecules initially in the membrane, and one with all 100 O2 molecules initially in the aqueous phase. O2 is considered to be the active site when it is within 6Å of CuB.
Table 1.
O2 delivery events observed during the long ELS simulations
| System | Delivery events | Unique delivery O2 |
|---|---|---|
| 1) 70:30 | 29 | 26 |
| 2) 70:30 | 34 | 27 |
| 3) 70:30 | 28 | 23 |
| 4) 100:0 | 36 | 28 |
| 5) 0:100 | 21 | 18 |
| sum | 122 | 148 |
| average | 24.4 | 29.6 |
Systems=initial O2 mem:sol ratio. Unique delivered O2 = number of distinguishable O2 molecules (different ID numbers) found in the active site. Delivery events=number of O2 delivery events observed during the 50-ns simulations. A few O2 molecules made multiple entries to both the enzyme and active site.
Figure 3. O2 delivery pathway of cytochrome ba3 identified by the ELS simulations.

All images were taken from the same top view of the enzyme. a) Highly occupied O2 regions characterized by the 3D density map calculated using the combined last 40-ns trajectories of five independent simulations. One region, which appears in a Y-shaped form, is the pathway that has been found to deliver O2 to the active site (green-dashed box). It is connected to the membrane via two branches, which we refer to and label as Branches A and B. Red-solid isosurfaces represent O2 partitioning free energy (ΔGi,sol) of −3.5 kcal/mol. b) The O2 delivery pathway. Purple-solid and red-wireframe isosurfaces correspond to ΔGi,sol of −4.0 and −3.0 kcal/mol, respectively. c) Xe binding sites identified by X-ray crystallography.35,40 Xe atoms were detected in a hydrophobic tunnel, displayed as a yellow-transparent surface using a probe radius of 1.8Å; they are numbered according to PDB entry 3BVD.35 The O2 delivery pathway identified in the present study (b) largely overlaps with this Xe-bound tunnel.
Figure 4. Favorable O2 partitioning regions in cytochrome ba3 characterized by ILS.

a) Highly probable O2 binding regions characterized by the 3D free-energy map calculated by ILS. Red-solid and red-wireframe isosurfaces correspond to ΔGi,sol of −3.5 and −3.0 kcal/mol, respectively. b) Region in the 3D free-energy map that is equivalent to the O2 delivery pathway characterized by ELS: purple-solid and red-wireframe isosurfaces correspond to ΔGi,sol of −4.0 and −3.0 kcal/mol, respectively. c) O2 delivery pathway calculated by MolAxis60 for the crystal structure and displayed as corridors. Green corridors represent pore radii of 1.2–3.0Å. Blue corridors represent pore radii of >3.0Å. d) Pore profiles of Branches A and B of the O2 delivery pathway.
The ELS simulations reveal high-occupancy regions within the protein which form a Y-shaped O2 pathway with two openings at the protein interface with lipids, pathways leading from these openings (Branches A and B, Fig. 3a) to a common branch-point, and a single pathway leading from the branch-point to the active site. The branch-point is surrounded by residues I78, A149 and L200 (Fig. 3b). The red surfaces in Fig. 3a are isosurfaces where the free energy with respect to the aqueous phase (ΔGi,sol) is −3.5 kcal/mol. This represents favorable partitioning from the aqueous phase to these regions of about 300 folds and a 10-to-50-fold favorable partitioning of O2 with respect to the membrane phase (depending on the location within the membrane). Fig. 3b shows the isosurfaces defining ΔGi,sol =−3.0 (red-wireframe) and −4.0 kcal/mol (purple-solid), indicating higher and lower O2 affinity sites along the pathway. Examination of all O2 trajectories shows that all the O2 reaching the active site use the Y-shaped pathway shown in Figs. 3a and b. All the O2 comes from the lipid bilayer and through the hydrophobic tunnel to reach the active site (Supp. Fig. 2). Fig. 3c shows that the pathways correspond to the Xe binding sites observed by X-ray crystallography.35,40 The X-ray Xe binding sites overlap with several purple isosurfaces in Fig. 3b indicating O2 binding sites (ΔGi,sol =−4.0 kcal/mol).
The ILS calculations result in an energetic picture that is fully in line with ELS simulations, as shown by the free energy isosurfaces in Figs. 4a and b. Additional regions of the protein that favorably bind O2 are also revealed by both the ELS simulations and ILS calculations, but these are isolated cavities that do not constitute a pathway. Fig. 5a shows a 2D-projection of the free energy map, which strongly supports the conclusion that no pathway other than the Y-shaped pathway is available for O2 to reach the enzyme’s active site. The Y-shaped pathway has a radius throughout its length of at least 1.5Å (Figs. 4c and d), which is large enough for unhampered diffusion of O2. During the simulations, no protein fluctuations of blocked or altered the pathways, and the protein structure was not altered by the presence of O2, even at such a high concentration as 210mM (Supp. Fig. 3). The agreement of the ELS and ILS results demonstrates that the 50-ns simulations are sufficiently long for adequate sampling of O2 diffusion throughout the protein.
Figure 5. Energetic profiles of O2 along the delivery pathway.

a) A 2D projection of the free-energy map, calculated using MATLAB, describing the partitioning of O2 in the enzyme and its surroundings. The map was calculated by collapsing the normalized 3D density profiles of O2 calculated from ELS over a 50×50×30Å3 grid, which spans the enzyme region covering the entire O2 delivery pathway, heme cofactors, CuB, and surrounding lipids. Dark colors indicate high probability O2 regions. Light colors indicate O2 scarce regions. b) Estimated number of O2 binding sites along the pathway calculated by integrating O2 probability densities (pd) from the active site to the entrances of the pathway. c) 1D free-energy profiles of O2 partitioning along its delivery pathway. The coordinates of O2 that have been within 3Å of pathway’s residues are clustered into normalized O2 density profiles with respect to the aqueous phase using the distance from the active site (d) as the reaction coordinate. d=(((x − x0)2 +(y − y0)2 +(z − z0)2)1/2) - 6. (x0, y0, z0) is the position of CuB cofactor. O2 is considered to be in the active site when it has been within 6Å of CuB. BP specifies intersection point of Branches A and B, referring to as “branch-point”. ΔΔGTS–BP = +1.5 kcal/mol. The average speed of O2 diffusion along the pathway (v0) is 0.39 (0.29)Å/ps.
There are 7 to 8 binding sites of O2 along the Y-shaped pathway (Figs. 3b, 5a and 5b). These sites have ΔGi,sol values ranging from −3.5 to −4.5 kcal/mol, corresponding to dissociation constants of 0.6 to 3mM. If the concentration is 250 μM (air saturated), these data predict between 0.5 and 1 O2 present within the pathway. Under more physiologically relevant conditions, the amount of O2 present within the protein at equilibrium will be negligible. Figs. 3 and 5b provide more details relating the Xe binding sites35,40 to the regions of the protein favorable for O2 binding. The site closest to the active site coincides with Xe1 in the X-ray structure (Figs. 3b and 3c35 ). The affinity for O2 at the branch-point (BP) is most favorable and corresponds to the combination of Xe5 and Xe6 densities in the X-ray structure.35 The two O2 sites in Branch B coincide with Xe3 and Xe4. Branch A contains 3 O2 binding sites. Two are located near the entrance of Branch A (residues V79 and A120) and are not visualized by X-ray crystallography. The third O2 site in Branch A coincides with the region around Xe2 and Xe7. The 1D-projection free energy profiles of O2 in Fig. 5c show the largest free-energy barrier along the Y-shaped pathway of only 1.5 kcal/mol, located at the transition from the BP to the Xe1 site, which is very close to the active site. Other free-energy barriers are all below 1 kcal/mol.
The rate of delivery of O2 to the active site
To obtain an estimate of the rate of O2 delivery to the active site, a series of 21 short ELS simulations (3–5 ns each) were performed to determine the time required for the first O2 from the solution to reach the active site. This was defined arbitrarily as an O2 molecule getting to within 6Å of CuB. The initial distribution of O2 was 90 molecules in the membrane and 10 molecules in the aqueous phase, which is equivalent to an O2 concentration of 37mM in the aqueous phase and 185mM in the membrane. Similar to the 50-ns ELS simulations, O2 molecules were initially placed outside the protein in these short ELS simulations. In all 21 trials, O2 diffused to the active site within the first 5 ns. In 14 trials, O2 reaching the active site entered via Branch B and in the remaining 7 trials, through Branch A (Table 2). On the average, it took 1.7 ns for O2 to diffuse to the immediate vicinity of CuB, essentially the same as observed in the 50-ns ELS simulations. Using the nominal concentration in the aqueous phase (37 mM), we may estimate an apparent second order rate constant of (1/0.037M)(1/1.7×10−9s)=1.6×1010 M−1s−1. This is an “apparent” second order rate constant since the O2 molecules that reach the active site are initially within the membrane and then must pass through the protein tunnel. The apparent rate constant has been experimentally determined to be 1×109 M−1s−1 by determining the rate of formation of the initial O2 adduct with heme a3, using enzymes dissolved in detergent micelles.42 The important point is that the calculated estimate is consistent with a remarkably rapid rate of diffusion of O2 to reach the active site.
Table 2.
O2 migration times estimated from the ensemble of 21 short ELS simulations
| Branch | Sim. | tfirst (ns) | tent→cat | tBP →cat | tent→BP |
|---|---|---|---|---|---|
| A | 7 | 2.1±0.9 | 0.7±0.2 | 0.4±0.2 | 0.3±0.3 |
| B | 14 | 1.5±0.7 | 0.6±0.2 | 0.4±0.3 | 0.2±0.2 |
| total | 21 | 1.7±0.8 | 0.6±0.4 | 0.4±0.3 | 0.2±0.2 |
These results are determined from the first delivered O2 molecules in all the simulations. Sim.=number of simulations. tfirst =time taken to observe the first O2 reaching the active site after the start of a simulation. tent→cat =transit time of O2 diffusion from the entrance to the active site. tBP →cat =transit time of O2 diffusion to the BP to the active site. tent→BP =transit time of O2 from the entrances to the intersection point of Branches A and B.
Further insight is obtained by observing transit times for successful trajectories of O2 for the molecule to reach the entrance of either Branch A or B, and the transit from the pathway’s entrance(s) to CuB (Table 2). The average time for O2 (at 37mM) to get into the entrance of the pathway (either branch) is about tout→ent ≈1 ns. Once at the entrance of the pathway, the average transit time to the active site (CuB) is tent→cat ≈0.6±0.4 ns. All the successful trajectories took O2 to the BP (tent→BP ≈0.2 ns) and to CuB (tBP →cat ≈0.4 ns). The fraction of O2 molecules that started at the entrance of either Branch A or B and the went on the reach CuB is about 40 %. It should be noted that the time for O2 to diffuse to the entrance of the pathway will depend inversely on O2 concentration in the membrane, but the transit times within the O2 pathway and the fraction of molecules that reach the active site once entering the pathway will be independent of O2 concentration.
In silico mutagenesis of the O2 pathway
Seven different mutants were constructed in silico to determine their influence on the diffusion of O2 through the Y-shaped pathway (Table 3). In each of these mutants, the native residues were replaced by a bulky side chain, namely phenylalanine or tryptophan. These residues are I78, A149, A120, S150, V79 and L200, which correspond to F67, L150, H151, L112, F68 and L202 of the bovine cytochrome c oxidase or F108, V194, A153, H195, F109 and L246 of the R. sphaeroides enzyme.
Table 3.
Alterations of O2 access by in silico mutagenesis
| Mutant | Sim | tfirst | Branch | tent→cat | tBP →cat | tent→BP |
|---|---|---|---|---|---|---|
| A120F | 20 | 3.2±2.0 | B | 0.8±0.5 | 0.5±0.3 | 0.3±0.2 |
| A149F | 20 | 2.6±2.0 | A/B | 0.8±0.6 | 0.3±0.4 | 0.5±0.4 |
| S150W | 20 | 2.7±1.1 | A | 1.2±0.7 | 0.8±0.8 | 0.4±0.3 |
| A120F/S150W | 18 | 3.8±2.0 | B | 2.0±1.6 | 1.0±1.0 | 1.0±1.1 |
| V79F/S150W | 8 | 4.0±2.3 | (A)/B | 1.6±1.1 | 0.4±0.2 | 1.2±1.0 |
| I78F/V79F/L200F | 29 | 6.7±3.9 | (A)/B | 2.4±2.0 | 0.6±0.8 | 1.8±2.0 |
The results are from the short ELS simulations. Sim=number of independent simulations. Branch(es)= entrance(s) of the first observed O2 molecule entering the active site in each simulation. In the A120F/S150W mutant, O2 delivery was observed in only 8 of its 10-ns simulations. In the V79F/S150 mutant, O2 delivery was observed in only 18 of its 20-ns simulations. In the triple mutant, O2 delivery was observed in 29 of its 15-ns simulations. Of all repeated simulations, O2 entry via Branch A was found only in one simulation for V79F/S150W and triple mutants.
I78F (Fig. 6 and Supp. Fig. 4): Although I78 is at the BP, its replacement by phenylalanine has only a modest effect on the free energy barrier of O2 translocation along the pathway.
A149F (Figs. 6 and 8, and Supp. Figs. 4 and 5): A149 is located at the BP. Although its replacement by phenylalanine creates a barrier, O2 can readily bypass it and still reach the active site via both Branches A and B. Short ELS simulations show that this mutation has only a modest effect on the transit time of O2 to the active site.
A120F (Figs. 6 and 8, and Supp. Figs. 4 and 5): A120 is located within Branch A and the X-ray structure of the A120F mutant has been reported.31 Although the structure of this mutant perfectly aligns with the wild-type structure, the introduced phenylalanine at this position occupies the Xe2–Xe7 sites, as shown both in the X-ray structure31 and in the simulation (Fig. 6), and reduces O2 diffusion through branch A. However, since Branch B is still fully functional, the net effect on the rate of O2 diffusion to the active site is modest.
S150W (Figs. 6 and 8, and Supp. Figs. 4 and 5): S150 is at the entrance of Branch B. A tryptophan at this position creates a significant barrier to O2 diffusion and reduces the radius of the pathway to below 1.2Å. However, Branch A is still fully functional, and the time required for O2 to reach the active site is only modestly influenced.
A120F/S150W double mutant (Figs. 7 and 8, and Supp. Figs. 5 and 6): Ten simulations (5–10 ns each) were performed for this mutant, and O2 was observed to reach the active site in eight of them. This mutant increases the barrier to O2 diffusion through both Branches A and B, but the net result is only to double the average time required for the first O2 to reach the active site during the simulations (Table 3).
V79F/S150W double mutant (Figs. 7 and 8, and Supp. Figs. 5 and 6): V79 is located at the entrance of Branch A, near A120. This double mutant also increases the barrier against O2 diffusion through both Branches A and B. However, in 18 of 20 simulations (5–10 ns each), O2 reached the active site and the time for the first O2 was only increased by a factor of 2.
I78F/V79F/L200F triple mutant (Figs. 7 and 8, and Supp. Figs. 5 and 6): L200 is located near S150 at the entrance of Branch B. This triple mutant increases barriers to O2 diffusion at the BP as well as the entrances of each branch. A total of 30 simulations ranging from 5 to 15 ns were performed and in 29 of these O2 reached the active site. The time required for the first O2 to reach the active site is about 4-fold greater than in the wild type (6.7 ns vs. 1.7 ns).
Figure 6. Alterations of the O2 delivery pathway by in silico mutagenesis of pathway-lining residues to bulky substitutions.

Left panels highlight predicted O2 binding regions calculated by ILS performed on the 10-ns of apo trajectories. The pink transparent surface indicates regions with ΔGi,sol of −2.5 kcal/mol for O2 binding. Right panels show the O2 delivery pathway calculated from the equilibrated structure (at t = 10 ns) of each mutant using MolAxis.60 Red corridors represent pore radii of <1.2Å. Green corridors represent pore radii of 1.2–3.0Å. Blue corridors represent pore radii of >3. 0Å.
Figure 8. Migration profiles of O2 along the delivery pathway in the wild-type and mutants of cytochrome ba3 explicitly determined from multiple short ELS simulations.

tent→cat is the transit time of O2 from the entrances of the pathway to the active site. It is directly measured from the first delivered O2 molecule in each simulation. Histograms in each plot represent tent→cat distribution of a mutant. The shaded curved behind the bar histogram represents tent→cat distribution of the wild-type enzyme, providing a direct comparison with each mutant. tent→cat for the wild-type is 0.6±0.4 ns.
Figure 7. Alterations of the O2 delivery pathway upon in silico blockages of pathway’s entrances.

Double (V79F/S150W and A120F/S150W) and triple (I78F/V79F/L200F) mutants have been introduced to hinder the O2 delivery pathway. Descriptions of left and right panels are the same as in Fig 6.
Hence, the expectation for these mutations is that the effects on the transit of O2 through the protein will be modest. We note that the length of the simulations performed here may not be sufficient to detect potentially large conformational changes, beyond the observed local constrictions of the O2 pathway described above. However, we also note that the crystal structures of different types of cytochrome oxidases in which the regions studied here by in silico mutagenesis show variations in their amino acid compositions, have indicated no global structural differences. Furthermore, a crystal structure of one of the mutants studied here (A120F)31 perfectly aligns with the wild-type structure. Based on these, we do not expect significant global structural changes in the enzyme due to the introduced mutations.
Discussion
This study is motivated by the report of the remarkably fast rate of O2 diffusion into the deeply buried active site of cytochrome ba3 for T. thermophilus The experimentally determined second order rate constant at 298K for the formation of the initial complex of O2 with the active-site heme a3 is k1 =1×109 M−1s−1.42,43 This is ten times faster than the rate constant for the equivalent first step in the reaction of the mitochondrial cytochrome c oxidase, k1 =1×108 M−1s−1,42,43 100 times faster than O2 binding to myoglobin, k1 =1.6×107 M−1s−1,75 and 100 times larger than O2 diffusing into the diiron active site of toluene/oxylene monooxygenase hydroxylase, k1 =2.1×105 M−1s−1.76 The rate constant for CO or O2 to reach the buried active site of NiFe hydrogenase has been measured to be 6.3×107 M−1s−1,77 also considerably less than cytochrome ba3.
The first experiments to measure the diffusion of O2 within proteins determined the rate constant for the collisional quenching by O2 of the fluorescence of tryptophan within small soluble proteins.78,79 The second order rate constants for this were found to be diffusion-limited, in the range of 2–8×109 M−1s−1. Residues internally buried within the proteins that are not accessible to O2 based on the static X-ray structures were readily quenched by collisional contact with O2. It was estimated that O2 could diffuse through the protein at rates that are 20–50% of the diffusion rate in water,79 providing one of the first demonstrations of protein dynamics, necessary to allow O2 to penetrate the protein. Although these data suggest that there may be no need for specific pathways to facilitate the diffusion of O2 or other small, hydrophobic gases within proteins, considerable experimental and computational evidence indicates that such pathways have evolved for enzymes that use small gas molecules as substrates, e.g., nitrogenase,80 methane monooxygenase,81,82 laccase,83,84 NiFe hydrogenase,77,85 FeFe hydrogenase,36,86 D-amino acid oxidase,67 copper amine oxidase, 87 myoglobin,45,75,88–90 hemoglobin,91 CO dehydrogenase/acetyl-CoA synthase,92 and heme-copper oxygen reductases, including cytochrome ba3.33–35,40–43,58,93–95 There are also several examples of enzymes with multiple active sites that have molecular tunnels for delivering the product generated at one site to a second distant site where it is consumed as a substrate.15
In many cases, the gas channels include hydrophobic cavities that are identified by locating Xe binding sites using X-ray crystallography.36 An important caveat is that the Xe binding sites identified crystallographically may not be functional and need not be involved in O2 diffusion within the protein. For example, photosystem II from Thermosynechococcus elongates 96 has 25 Xe binding sites scattered through the membrane domain of the protein, but they are clearly not involved in O2 diffusion away from the oxygen evolving cluster. Myoglobin has 4 Xe binding sites, which may also bind O2, but it remains unclear which, if any, of these sites are on the primary pathway for O2 to reach the heme Fe.75,90
Protein conformational changes are often crucial either in the transient formation of the hydrophobic cavities or in opening the passage connecting adjacent hydrophobic cavities, thus allowing passage of O2. These are dynamic tunnels. An example of this is the pathway for CO2 to reach the buried metal cluster where it is reduced to CO in the CO dehydrogenase/acetyl-CoA synthase.92 Dynamic tunnels have also been proposed for FeFe hydrogenase,36,86 the Scarpharca dimeric hemoglobin,91 laccase,84 and for the HCO from R. sphaeroides.41
A recent MD study in the cytochrome c oxidase from R. sphaeroides 41 concluded that the X-ray inferred tunnel is not an important route for O2 to reach the active site and are in contrast with the finding reported here on cytochrome ba3. The HCO from R. sphaeroides is an A-family heme-copper oxygen reductase, and crystallographic studies previously identified a putative O2 pathway with two Xe binding sites.29 This tunnel is roughly in the same location as the tunnel observed here in cytochrome ba3, which is a B-family HCO. The functionality of the X-ray inferred tunnel in two different A-family heme-copper oxygen reductases has been tested experimentally by site-directed mutations.94,97,98 However, these mutations are too close to the active site to allow straightforward interpretation. Using computational techniques similar to those employed in the present study, three putative O2 pathways were suggested in the R. sphaeroides HCO,41 one corresponding to that inferred by the X-ray studies. A detailed examination of the structures of the A-family HCOs33 as well as the MD simulations41 have revealed a constriction in the X-ray inferred tunnel that would substantially impede the passage of O2 molecules to the active site. MD simulations did not observe any delivery event of O2 to the active site over 100-ns periods.41 Nevertheless, free-energy profiles estimated from data generated by ILS calculations suggested two probable alternative pathways for O2 diffusion from the solvent to the active site.41 Since the free-energy barriers of the two pathways were estimated to be 5 kcal/mol lower than the X-ray inferred tunnel, it was concluded that these alternative pathways, not observed in the static X-ray structures, are the preferred routes used in the R. sphaeroides HCO.41
In contrast, the inferred pathway from X-ray data for O2 diffusion to the active site of cytochrome ba3 is supported by both previous experimental data and by MD simulations, which only concentrated on the dynamics and stability of O2 binding at individual X-ray Xe sites,33,34 as well as the present study. The tunnel in this B-family heme-copper oxygen reductase does not have the constriction observed in the A-family enzymes.33,35 The constriction in the A-family HCOs is due to bulky residues W172 and F282 (numbering for the R. sphaeroides enzyme) or residues W126 and F238 (numbering for the bovine enzyme), whereas the equivalent residues in the T. thermophilus enzyme are Y133 and T231. The examination of the Y133W mutant of cytochrome ba3 showed that the second order rate constant for O2 to diffuse to the active site is slowed down 5-fold to k1 =2×108 M−1s−1.33 Short (1 ns) MD simulations showed that there is effectively no barrier within this tunnel for O2 diffusion.34
The present study employs long ELS MD simulations (50 ns) with a model that places the entire 3-subunit cytochrome ba3 in a lipid bilayer surrounded by an aqueous solution. By initially placing O2 molecules outside the protein, this allows not only substrate O2 to unequivocally search for its delivery pathways, but also to address the role of protein dynamics in modulating the X-ray inferred tunnel and to forming other alternative pathways. A sufficient number of trajectories were obtained for O2, starting outside the protein and reaching the active site, to estimate a free energy map of O2 within the protein based on occupancy. In addition, ILS calculations were performed to obtain an independent free energy map of O2 in the protein. The results of both the ELS and ILS calculations are in full agreement with each other as well as previous experimental and computational studies.33,34 The most important points are summarized below.
The equilibrium distribution of O2 between the membrane and aqueous phases, as well as distribution of O2 within the membrane in the computational model are consistent with previous experimental and computational data.73,74
The protein contains about 7 binding sites for O2 corresponding generally to the Xe binding sites in the X-ray structure. At 298 K, each site has a dissociation constant for O2 of about 1mM with respect to the concentration in aqueous solution. At 250 μM O2, at equilibrium there will be an average of about 1 O2 molecule present in the protein, distributed among the sites within the pathway. At 10 μM O2, more realistic physiologically, the occupancy of each site is less than 1% (at 298 K). Hence, these binding sites do not act as a storage reservoir for O2.
The average time for O2 to reach an entrance of its delivery pathway is about 1 ns, starting with a concentration of 37mM in the aqueous phase (MD simulation condition). The first O2 molecule to reach the pathway entrance in each simulation always originates in the lipid bilayer. Based on the concentration of O2 in solution, the apparent second order rate constant to reach the pathway entrance is about 1.6×1010 M−1s−1. This compares favorably with the measured second order rate constant for the collisional quenching by O2 of pyrene dissolved in the lipid bilayer of 1.2×1010 M−1s−1.99 When the dissolved O2 concentration in the aqueous phase is 250 μM (air saturated water at 298 K), the rate for an O2 molecule to reach the pathway entrance can be estimated to be (2.5×10−4 M)(1.6×1010 M−1s−1)=4×106 s−1, or about one O2 per 250 ns.
The O2 binding sites right next to the pathway entrances have binding free energies of about −1.5 to −2 kcal/mol with respect to the O2 pool within the membrane, recalling that O2 in the lipid is favored by about −2 kcal/mol with respect to O2 in solution. This is equivalent to an association constant of about 20 for O2 binding from the lipid bilayer. Since the simulated second order rate constant to reach these sites from the bilayer is 1.6×1010 M−1s−1, the off rate constant can be estimated to be k−1 =(1.6×1010/20)=8×108 s−1. The transit time from the pathway entrance to the active site is 0.6 ns, so the first order rate constant to transit through the pathway is k2 =(1/6×10−10)=1.7×109 s−1. These rough estimates indicate that about half the molecules that get into the pathway make it all the way to the active site. At no point do protein conformational changes introduce any significant free energy barrier to the diffusion of O2 within the delivery pathway.
-
We can model the steady state kinetics of collision with the active site as a two-step process: i) a reversible step to enter the pathway from the lipid, followed by ii) an irreversible step to reach the active site. The O2 that reaches the active site is assumed in this model to be consumed with 100% efficiency.The observed rate for O2 reaching the active site in this model is given byFor any reasonable value of [O2], it is reasonable to assume [O2]k1 ≪ k−1, k2. Then
assuming k1 =1.6×1010 M−1s−1, k−1 =8×108 s−1 and k2 =1.7×1010 s−1. The second order rate constant for O2 to reach the active site is predicted to be about 1.5×1010 M−1s−1, which is about 15-fold larger than the measured rate constant of 1×109 M−1s−1.42 The experimental measure of the second order rate constant includes the process of forming the adduct with heme a3 at the active site, and this could be rate-limiting, explaining why the value is smaller than the rate of reaching the vicinity of the active site which is computed. In any event, the MD simulations demonstrate that diffusion through the body of the protein is not rate limiting.
In order for the rate of transit through the protein to be rate limiting, it would be necessary to slow the transit time to about 100 ns or k2 =107s−1. This would mean adding about 3 kcal/mol to the highest energy barrier (to 4.5 kcal/mol), and would result in a second order rate constant of about 108 M−1s−1, which is what is measured for the bovine cytochrome c oxidase and is the result of adding a constriction to the pathway in cytochrome ba3.
Using the measured rate constant, 1×1010 M−1s−1, and assuming that the maximum rate of catalysis is kcat ≈102 s−1 O2 reduced /sec, the enzyme will be limited by the dissolved concentration of O2 at concentrations of less than about 10−7M (0.1 μM) at 298 K. At concentrations of O2 greater than 0.1 μM, the chemistry of catalysis is rate limiting.
In the native enzyme, the highest free energy barrier is about 1.5 kcal/mol. Mutations in the pathway that slow the rate of O2 diffusion through the protein must present a free energy barrier of at least 4 kcal/mol, which would lower the second order rate constant to about 108 M−1s−1, which is what the Y133W must accomplish in cytochrome ba3.33
If the second order rate constant for O2 to reach the active site were reduced to 107 M−1s−1, similar to the value for myoglobin, the consequence would be that the concentration of O2 would limit the turnover rate of the enzyme at 10 μM instead of 0.1 μM. This is in a range that is physiologically significant, and provides a plausible rationale for why the respiratory HCOs have evolved to allow very fast access to the enzyme’s active site. The unusual hydrophobic tunnel present in cytochrome ba3 with a largest free energy barrier of 1.5 kcal/mol suggests that this enzyme has evolved for maximum turnover even at concentrations of its O2 substrate down to 0.1 μM. It is noted that this enzyme from T. thermophilus normally functions at 75°C or 348 K, so conclusions about physiological relevance involve extrapolations from the simulations and experiments done at or near 25°C (298 K).
In summary, a combination of ELS MD simulations and ILS thermodynamic calculations provide a consistent explanation for the rapid kinetics of O2 to access the active site of cytochrome ba3: the protein has a hydrophobic, static tunnel that provides an unencumbered 25-Å pathway from the lipid bilayer to the active site, allowing the enzyme to operate at maximal velocity at O2 concentrations as low as 0.1 μM at 298 K.
Acknowledgments
The authors acknowledge supercomputing resources at National Science Foundation Supercomputing Centers (XSEDE MCA06N060) and funding from the National Institutes of Health (NIH P41-GM104601 and U01-GM111251 to E.T., and R01-HL16101 to R.B.G.). P.M. gratefully acknowledges NIH support as a trainee of the Molecular Biophysics Training Program (5T32-GM008276).
Abbreviations
- HCO
heme-copper oxygen reductase
- ba3
cytochrome ba3 oxidase
- ELS
explicit ligand sampling
- ILS
implicit ligand sampling
Footnotes
Supporting Information Available
Supp. Fig. 1–7 This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Wikström M. Active site intermediates in the reactions of O2 by cytochrome oxidase, and their derivatives. Biochim Biophys Acta – Bioener. 2012;1817:468–475. doi: 10.1016/j.bbabio.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Wikström M, Verkhovsky MI. Mechanism and energetics for proton translocation by the respiratory heme-copper oxidases. Biochim Biophys Acta – Bioener. 2007;1767:1200–1214. doi: 10.1016/j.bbabio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Hosler JP, Ferguson-Miller S, Mills DA. Energy transduction: proton transfer through the respiratory complexes. Annu Rev Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaila VR, Verkhovsky MI, Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110:7062–7082. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 5.Sousa FL, Alves RJ, Ribeiro MA, Pereira-Leal JB, Teixeira M, Pereira MM. The superfamily of heme-copper oxygen reductases: Types and evolutionary considerations. Biochim Biophys Acta – Bioener. 2012;1817:629–637. doi: 10.1016/j.bbabio.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Brzezinski P, Gennis RB. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson-Miller S, Hiser C, Liu J. Gating and regulation of the cytochrome c oxidase proton pump. Biochim Biophys Acta – Bioener. 2012;1817:489–494. doi: 10.1016/j.bbabio.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belevich L, Bloch DA, Belevich N, Wikström M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci USA. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belevich L, Verkhovsky MI, Wikström M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 10.Sharma V, Wikström M. A structural and functional perspective on the evolution of the heme-copper oxidases. FEBS Lett. 2014;588:3787–3792. doi: 10.1016/j.febslet.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 11.Hemp J, Gennis RB. Diversity of the Heme-Copper superfamily in Archaea: Insights from Genomics and Structural Modeling. Results Probl Cell Differ. 2008;45:1–31. doi: 10.1007/400_2007_046. [DOI] [PubMed] [Google Scholar]
- 12.Brochier-Armanet C, Talla E, Gribaldo S. The multiple evolutionary histories of dioxygen reductases: Implications for the origin and evolution of aerobic respiration. Mol Biol Evol. 2009;26:285–297. doi: 10.1093/molbev/msn246. [DOI] [PubMed] [Google Scholar]
- 13.Ducluzeau AL, Schoepp-Cothenet B, van Lis R, Baymann F, Russell MJ, Nitschke W. The evolution of respiratory O2/NO reductases: an out-of-the-phylogenetic-box perspective. J R Soc Interface. 2014;11:20140196. doi: 10.1098/rsif.2014.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Arch Phys Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 15.Raushel FM, Thoden JB, Holden HM. Enzymes with molecular tunnels. Acc Chem Res. 2003;36:539–548. doi: 10.1021/ar020047k. [DOI] [PubMed] [Google Scholar]
- 16.Thoden JB, Huang X, Raushel FM, Holden HM. Carbamoyl-phosphate synthethase. Creation of an escape route for ammonia. J Biol Chem. 2002;277:39722–39727. doi: 10.1074/jbc.M206915200. [DOI] [PubMed] [Google Scholar]
- 17.Huang HX, McCammon A. The gates of ion channels and enzymes. Trends Biochem Sci. 2010;35:179–185. doi: 10.1016/j.tibs.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gora A, Brezovsky J, Damborsky J. Gates of enzymes. Chem Rev. 2013;113:5871–5923. doi: 10.1021/cr300384w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaikh SA, Li J, Enkavi G, Wen PC, Huang Z, Tajkhorshid E. Visualizing functional motions of membrane transporters with molecular dynamics simulations. Biochemistry. 2013;52:569–587. doi: 10.1021/bi301086x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson-Miller S, Babcock G. Heme/Copper terminal oxidases. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 22.Sharpe MA, Qin L, Ferguson-Miller S. From Biophysical and Structural Aspects of Bioenergetics. In: Wikström M, editor. Methods in Molecular Biology. RSC Publishing; Cambridge, UK: 2005. pp. 26–54. [Google Scholar]
- 23.Han H, Hemp J, Pace LA, Ouyang H, Ganesan K, Roh JH, Daldal F, Blanke SR, Gennis RB. Adaptation of aerobic respiration to low O2 environments. Proc Natl Acad Sci USA. 2011;108:14109–14114. doi: 10.1073/pnas.1018958108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agmon N. The Grotthuss mechanism. Chem Phys Lett. 1995;244:456–462. [Google Scholar]
- 25.Yamashita T, Voth GA. Insights into the mechanism of proton transport in cytochrome c oxidase. J Am Chem Soc. 2011;134:1147–1152. doi: 10.1021/ja209176e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8Å Resolution of Cytochrome C Oxidase From Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 27.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinnzawha-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Structures of Metal Sites of Oxidized Bovine Heart Cytochrome C Oxidase at 2.8Å. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 28.Soulimane T, Buse G, Bourenkov GP, Bartunik HD, Huber R, Than ME. Structure and mechanism of the abberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Svensson-Ek M, Abramson J, Larsson G, Törnroth S, Brzezinski P, Iwata S. The X-ray Crystal Structures of Wild-type and EQ(I–286) Mutant Cytochrome c Oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 30.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiefenbrunn T, Liu W, Chen Y, Katritch V, Stout CD, Fee JA, Cherezov V. High Resolution Structure of the ba3 Cytochrome c Oxidase from Thermus thermophilus in a Lipidic Environment. PLoS One. 2011;6:e22348. doi: 10.1371/journal.pone.0022348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyons JA, Aragao D, Slattery O, Pisliakov AV, Soulimane T, Caffrey M. Structural insights into electron transfer in caa3-type cytochrome oxidase. Nature. 2012;487:514–518. doi: 10.1038/nature11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald W, Funatogawa C, Li Y, Szundi I, Fee YCJA, Stout CD, Einarsdóttir O. Ligand access to the active site in Thermus thermophilus ba3 and bovine heart aa3 cytochrome oxidases. Biochemistry. 2013;52:640–652. doi: 10.1021/bi301358a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald W, Funatogawa C, Li Y, Chen Y, Szundi I, Fee JA, Stout CD, Einarsdóttir O. Conserved Glycine 232 in the Ligand Channel of ba3 cytochrome oxidase of Thermus thermophilus. Biochemistry. 2014;53:4467–4475. doi: 10.1021/bi500289h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luna VM, Chen Y, Fee JA, Stout CD. Crystallographic stuides of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry. 2008;47:4657–4665. doi: 10.1021/bi800045y. [DOI] [PubMed] [Google Scholar]
- 36.Lautier T, Ezanno P, Baffert C, Fourmond V, Cournac L, Fontecilla-Camps JC, Soucaille P, Bertrand P, Meyinal-Salles I, Léger C. The quest for a functional substrate access tunnel in FeFe hydrogenase. Faraday Discuss. 2011;148:385–407. doi: 10.1039/c004099c. [DOI] [PubMed] [Google Scholar]
- 37.Doukov TI, Blasiak LC, Servalli J, Ragsdale SW, Drennan CL. Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Biochemistry. 2008;47:3474–3483. doi: 10.1021/bi702386t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick MS, Lippard SJ. Analysis of substrate access to active sites in bacterial multicomponent monooxygenase hydroxylase: X-ray crystal structure of Xenon-pressurized phenol hydroxylase from Pseudomonas sp. OX1. Biochemistry. 2011;50:11058–11069. doi: 10.1021/bi201248b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brunori M, Gibson QH. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2001;2:676–679. doi: 10.1093/embo-reports/kve159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luna VM, Fee JA, Deniz AA, Stout CD. Mobility of Xe Atoms within the Oxygen Diffusion Channel of Cytochrome ba3 oxidase. Biochemistry. 2012;51:4669–4676. doi: 10.1021/bi3003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliveira AS, Damas JM, Baptista A, Soares CM. Exploring O2 diffusion in A-type cytochrome c oxidases: molecular dynamics simulations uncover two alternative channels towards the binuclear site. PLoS Comput Biol. 2011;10:e1004010. doi: 10.1371/journal.pcbi.1004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szundi I, Funatogawa C, Fee JA, Soulimane T, Einarsdóttir O. CO impedes superfast O2 binding in ba3 cytochrome oxidase from Thermus thermophilus. Proc Natl Acad Sci USA. 2010;107:21010–21015. doi: 10.1073/pnas.1008603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Einarsdóttir O, Funatogawa C, Soulimane T, Szundi I. Kinetic studies of the reactions of O2 and NO with reduced Thermus thermophilus ba3 and bovine aa3 using photolabile carriers. Biochim Biophys Acta – Bioener. 2012;1817:672–679. doi: 10.1016/j.bbabio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olson JS, Phillips GN., Jr Myoglobin discriminates between O2, NO, and CO by electrostatic interactions with the bound ligand. J Biol Inorg Chem. 1997;2:544–552. [Google Scholar]
- 45.Cohen J, Arkhipov A, Braun R, Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys J. 2006;91:1844–1857. doi: 10.1529/biophysj.106.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunsicker-Wang L, Pacoma R, Chen Y, Fee J, Stout C. A novel cry-oprotection scheme for enhancing the diffraction of crystals of recombinants cytochrome ba3 oxidase from Thermus thermophilus. Acta Cryst D. 2005;61:340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. VMD – Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 48.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 50.Darden T, York D, Pedersen LG. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 51.Martyna GJ, Tobias DJ, Klein ML. Constant Pressure Molecular Dynamics Algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- 52.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- 53.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 54.MacKerell AD, Jr, Feig M, Brooks CL., III Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J Comp Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 55.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 57.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell AD., Jr CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J Comp Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hofacker I, Schulten K. Oxygen and Proton Pathways in Cytochrome c Oxidase. Proteins: Struct, Func, Gen. 1998;30:100–107. [PubMed] [Google Scholar]
- 59.Hemp J, Christian C, Barquera B, Gennis RB, Martinez TJ. Helix switching of a key active-site residue in the cytochrome cbb3 oxidases. Biochemistry. 2005;44:10766–10775. doi: 10.1021/bi050464f. [DOI] [PubMed] [Google Scholar]
- 60.Yaffe E, Fishelovitch D, Wolfson HJ, Halperin D, Nussinov R. MolAxis: efficient and accurate identification of channels in macromolecules. PROTEINS: Structure, Function, and Bioinformatics. 2008;73:72–86. doi: 10.1002/prot.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E. Characterizing the Membrane-Bound State of Cytochrome P450 3A4: Structure, Depth of Insertion, and Orientation. J Am Chem Soc. 2013;135:8542–8551. doi: 10.1021/ja4003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong G, Pachter R. Inhibition of Biocatalysis in [Fe-Fe] Hydrogenase by Oxygen: Molecular Dynamics and Density Functional Theory Calculations. ACS Chem Biol. 2012;7:1268–1275. doi: 10.1021/cb3001149. [DOI] [PubMed] [Google Scholar]
- 63.Brannigan G, LeBard DN, Hénin J, Eckenhoff RG, Klein ML. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc Natl Acad Sci USA. 2010;107:14122–14127. doi: 10.1073/pnas.1008534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murali S, Wallner B, Trudell JR, Bertaccini E, Lindahl E. Microsecond simulations indicate that ethanol binds between subunits and could stabilize an open-state model of a glycine receptor. Biophys J. 2011;100:1642–1650. doi: 10.1016/j.bpj.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Cohen J, Boron WF, Schulten K, Tajkhorshid E. Exploring Gas Permeability of Cellular Membranes and Membrane Channels with Molecular Dynamics. J Struct Biol. 2007;157:534–544. doi: 10.1016/j.jsb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 66.Saam J, Ivanov I, Walther M, Holzhutter H, Kuhn H. Molecular dioxygen enters the active site of 12/15 lipoxygenase via dynamic oxygen access channels. Proc Natl Acad Sci USA. 2007;104:13319–13324. doi: 10.1073/pnas.0702401104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saam J, Rosini E, Molla G, Schulten K, Pollegioni L, Ghisla S. O2-reactivity of flavoproteins: Dynamic access of dioxygen to the active site and role of a H+ relay system in D-amino acid oxidase. J Biol Chem. 2010;285:24439–24446. doi: 10.1074/jbc.M110.131193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Tajkhorshid E. Nitric oxide conduction by the brain aquaporin AQP4. Proteins: Struct, Func, Bioinf. 2010;78:661–670. doi: 10.1002/prot.22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frenkel D, Smit B. Understanding Molecular Simulation From Algorithms to Applications. Academic Press; California: 2002. [Google Scholar]
- 70.Fischkoff S, Vanderkooi J. Oxygen diffusion in biological and artificial membranes determined by the Flurochrome Pyrene. J Gen Physiol. 1975;65:663–676. doi: 10.1085/jgp.65.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moller M, Botti H, Batthyany C, Rubbo H, Radi R, Denicola A. Direct measurement of nitric oxide and oxygen partitioning into liposomes and low density lipoprotein. J Biol Chem. 2005;280:8850–8854. doi: 10.1074/jbc.M413699200. [DOI] [PubMed] [Google Scholar]
- 72.Dzikovski BG, Livshits VA, Marsh D. Oxygen permeation profile in lipid membrane: comparison with transmembrane polarity profile. Biophys J. 2003;85:1005–1012. doi: 10.1016/S0006-3495(03)74539-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marsh D, Dzikovski BG, Livshits VA. Oxygen profiles in membrane. Biophys J. 2006;90:L49–L51. doi: 10.1529/biophysj.106.081042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al-Abdul-Wahid MS, Evanics F, Prosser RS. Dioxygen transmembrane distributions and partitioning thermodynamics in lipid bilayers and micelles. Biochemistry. 2011;50:3975–3983. doi: 10.1021/bi200168n. [DOI] [PubMed] [Google Scholar]
- 75.Scott EE, Gibson QH, Olson JS. Mapping the Pathways for O2 Entry Into and Exit from Myoglobin. J Biol Chem. 2001;276:5177–5188. doi: 10.1074/jbc.M008282200. [DOI] [PubMed] [Google Scholar]
- 76.Song WJ, Gucinski G, Sazinsky MH, Lippard SJ. Tracking a defined route for O2 migration in a dioxygen-activating diiron enzyme. Proc Natl Acad Sci USA. 2011;108:14795–14800. doi: 10.1073/pnas.1106514108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liebgott PP, et al. Relating diffusion along the substrate tunnel and oxygen sensitivity in hydrogenase. Nat Chem Biol. 2010;6:63–70. doi: 10.1038/nchembio.276. [DOI] [PubMed] [Google Scholar]
- 78.Lakowicz JR, Weber G. Quenching of Protein Fluorescence by Oxygen. Detection of Structural Fluctuations in Proteins on the Nanosecond Time Scale. Biochemistry. 1973;12:4171–4179. doi: 10.1021/bi00745a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lakowicz JR, Weber G. Quenching of Fluorescence by Oxygen: A Probe for Structural Fluctuations in Macromolecules. Biochemistry. 1973;12:4161–4170. doi: 10.1021/bi00745a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith D, Danyal K, Raugei S, Seefeldt LC. Substrate channel in nitrogenase revealed by a molecular dynamics approach. Biochemistry. 2014;53:2278–2285. doi: 10.1021/bi401313j. [DOI] [PubMed] [Google Scholar]
- 81.Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- 82.Lee SJ, McCormick MS, Lippard SJ, Cho US. Control of substrate access to the active site in methane monooxygenase. Nature. 2013;494:380–384. doi: 10.1038/nature11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kallio JP, Rouvinen J, Kruus K, Hakulinen N. Probing the dioxygen route in Melanocarpus albomyces laccase with pressurized xenon gas. Biochemistry. 2011;50:4396–4398. doi: 10.1021/bi200486b. [DOI] [PubMed] [Google Scholar]
- 84.Damas JM, Baptista A, Soares CM. The pathway for O2 diffusion inside CotA Laccase and possible implications on the multicopper oxidases family. J Chem Theor Comp. 2014;10:3525–3531. doi: 10.1021/ct500196e. [DOI] [PubMed] [Google Scholar]
- 85.Wang PH, Best RB, Blumberger J. Multiscale Simulation Reveals Multiple Pathways for H2 and O2 transport in a [NiFe]-Hydrogenase. J Am Chem Soc. 2011;133:3548–3556. doi: 10.1021/ja109712q. [DOI] [PubMed] [Google Scholar]
- 86.Cohen J, Kim K, Posewitz M, Ghirardi ML, Schulten K, Seibert M, King P. Molecular dynamics and experimental investigation of H2 and O2 Diffusion in [Fe]-hydrogenase. Biochem Soc Trans. 2005;33:80–82. doi: 10.1042/BST0330080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson BJ, Cohen J, Welford RW, Pearson AR, Schulten K, Klinman JP, Wilmot CM. Exploring Molecular Oxygen Pathways in Hanseluna Polymorpha Copper-Containing Amine Oxidase. J Biol Chem. 2007;282:17767–17776. doi: 10.1074/jbc.M701308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen J, Schulten K. O2 migration pathways are not conserved across proteins of a similar fold. Biophys J. 2007;93:3591–3600. doi: 10.1529/biophysj.107.108712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Olson JS, Soman J, Jr, GNP Ligand pathways in myoglobin: a review of Trp cavity mutations. IUBMB Life. 2007;59:552–562. doi: 10.1080/15216540701230495. [DOI] [PubMed] [Google Scholar]
- 90.Jiang Y, Kirmizialtin S, Sanchez IC. Dynamic void distribution in myoglobin and five mutants. Sci Rep. 2014;4:1–9. doi: 10.1038/srep04011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Knapp JE, Pahl R, Cohen J, Nichols JC, Schulten K, Gibson QH, Šrajer V, WER Ligand migration and cavities within Scapharca dimeric HbI: Studies by time-resolved crystallography, Xe binding, and computational analysis. Structure. 2009;17:1494–1504. doi: 10.1016/j.str.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang PH, Bruschi M, Gioia LD, Blumberger J. Uncovering a Dynamically Formed Substrate Access Tunnel in Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase. J Am Chem Soc. 2013;135:9493–9502. doi: 10.1021/ja403110s. [DOI] [PubMed] [Google Scholar]
- 93.Porrini M, Daskalakis V, Farantos SC. Exploring the topography of free energy surfaces and kinetics of cytochrome c oxidases interacting with small ligands. RCS Advances. 2012;2:5828–5836. [Google Scholar]
- 94.Riistama S, Puustinen A, García-Horsman A, Iwata S, Michel H, Wikström M. Channelling of dioxygen into the respiratory enzyme. Biochim Biophys Acta. 1996;1275:1–4. doi: 10.1016/0005-2728(96)00040-0. [DOI] [PubMed] [Google Scholar]
- 95.Einarsdóttir O, McDonald W, Funatogawa C, Szundi I, Woodruff WH, Dyer RB. The pathway of O2 to the active site in heme-copper oxidases. Biochim Biophys Acta – Bioener. 2015;1847:109–118. doi: 10.1016/j.bbabio.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murray JW, Maghlaoui K, Kargul J, Sugiura M, Barber J. Analysis of xenon binding to photosystem II by X-ray crystallography. Photosyn Res. 2008;98:523–527. doi: 10.1007/s11120-008-9366-2. [DOI] [PubMed] [Google Scholar]
- 97.Salomonsson L, Lee A, Gennis RB, Brzezinski P. A single-amino-acid lid renders a gas-tight compartment within a membrane-bound transporter. Proc Natl Acad Sci USA. 2004;101:11617–11621. doi: 10.1073/pnas.0402242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Riistama S, Puustinen A, Verkhovsky MI, Morgan JE, Wikström M. Binding of O2 and Its Reduction Are Both Retarded by Replacement of Valine 279 by Isoleucine in Cytochrome c Oxidase from Paracoccus denitrificans. Biochemistry. 2000;39:6365–6372. doi: 10.1021/bi000123w. [DOI] [PubMed] [Google Scholar]
- 99.Denicola A, Souza JM, Radi R, Lissi E. Nitric oxide diffusion in membranes determined by fluorescence quenching. Arch Biochem Biophys. 1996;328:208–212. doi: 10.1006/abbi.1996.0162. [DOI] [PubMed] [Google Scholar]