

Abstract
PIN1 is a peptidyl-prolyl cis/trans isomerase that binds and catalyses isomerization of the specific motif comprising a phosphorylated serine or threonine residue preceding a proline (pSer/Thr-Pro) in proteins. PIN1 can therefore induce conformational and functional changes of its interacting proteins that are regulated by proline-directed serine/threonine phosphorylation. Through this phosphorylation-dependent prolyl isomerization, PIN1 fine-tunes the functions of key phosphoproteins (e.g., cyclin D1, survivin, β-catenin and x-protein of hepatitis B virus) that are involved in the regulation of cell cycle progression, apoptosis, proliferation and oncogenic transformation. PIN1 has been found to be over-expressed in many cancers, including human hepatocellular carcinoma (HCC). It has been shown previously that overexpression of PIN1 contributes to the development of HCC in-vitro and in xenograft mouse model. In this review, we first discussed the aberrant transcription factor expression, miRNAs dysregulation, PIN1 gene promoter polymorphisms and phosphorylation of PIN1 as potential mechanisms underlying PIN1 overexpression in cancers. Furthermore, we also examined the role of PIN1 in HCC tumourigenesis by reviewing the interactions between PIN1 and various cellular and viral proteins that are involved in β-catenin, NOTCH, and PI3K/Akt/mTOR pathways, apoptosis, angiogenesis and epithelial-mesenchymal transition. Finally, the potential of PIN1 inhibitors as an anti-cancer therapy was explored and discussed.
Keywords: Phosphorylation, Hepatocellular carcinoma, PIN1, Isomerization, Hepatocarcinogenesis
Core tip: PIN1 specifically binds and catalyses isomerization of the pSer/Thr-Pro motif of target proteins, thereby modulating their functions. Many PIN1-interacting proteins are involved in cellular transformation and maintenance of malignant phenotype, and overexpression of PIN1 is frequently found in various cancer types, including hepatocellular carcinoma (HCC). Through interacting with regulatory proteins of key signalling pathways, such as β-catenin, cyclin D1, and HBx, PIN1 drives and amplifies the oncogenic signals essential for the development of HCC. Given its diverse oncogenic functions in hepatocarcinogenesis, PIN1 represents a potential therapeutic target in the treatment of HCC.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth leading cancer in men and the ninth for women worldwide, with estimated 782000 new cases in 2012. It is one of the most common causes of cancer death, leading to 746000 deaths annually[1]. Chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) is the major risk factor[2], accounting for 92% and 43% of HCC cases in developing and developed countries, respectively. HCC is an aggressive malignant tumour associated with a poor prognosis, and only a small proportion of HCCs is detected at an early stage. Early-stage HCCs are amenable to potentially curative treatments, such as surgical resection, liver transplantation, transcatheter arterial chemoembolization (TACE) and radiofrequency ablation (RFA)[3]. Nonetheless, HCCs are frequently diagnosed at advanced-stage and patients with advanced HCCs are not candidates for curative therapies. Conventional chemotherapy is ineffective for advanced HCCs because of the development of chemo-resistance and early occurrence of metastasis[4-6]. A thorough understanding of the pathogenesis and biology of HCC provides the mechanistic basis for designing effective HCC therapies.
Protein phosphorylation is a post-translational modification that plays an important role in the regulation of signalling pathways. Through activation of multiple protein kinases, such as cyclin dependent kinases (CDKs) and mitogen activated protein kinases (MAPKs), phosphorylation of proteins in serine or threonine residues preceding proline (pSer/ Thr-Pro) motif has been linked to dysregulated cell proliferation and malignant transformation[7,8]. The identification of peptidyl-prolyl cis/trans isomerase PIN1 provides a new post-phosphorylation regulatory mechanism in cell signalling[9-12]. PIN1 is a small and highly evolutionarily conserved 18-kDa protein, and is mainly localized in the nucleus[11,13,14]. It binds specific pSer/Thr-Pro motif in certain proteins through its amino-terminal WW domain, and isomerizes the pSer/Thr-Pro peptide bonds with its carboxyl-terminal prolyl isomerase (PPIase) domain[7,11,15] (Figure 1). PIN1-catalysed isomerization induces conformational changes of its target proteins, resulting in alterations of their enzymatic activities, phosphorylation status, protein-protein interaction patterns, subcellular localization, and protein stability. Conceivably, PIN1 plays an important role in diverse cellular processes, including cell cycle progression, differentiation, apoptosis and proliferation, as well as transformation[11,16-19]. Indeed, many PIN1-interacting partners, such as β-catenin, c-Jun, cyclin D1, cyclin E, Myc, nuclear factor-kappa B (NF-κB)-p65, p53 and p73, are important in regulating cell cycle progression and cell proliferation, and are often dysregulated in cancer[17,18,20-25]. Thus, the role of PIN1 in enhancing the oncogenic potential of these proteins via phosphorylation-dependent prolyl isomerization is important during cancer development.
Figure 1.
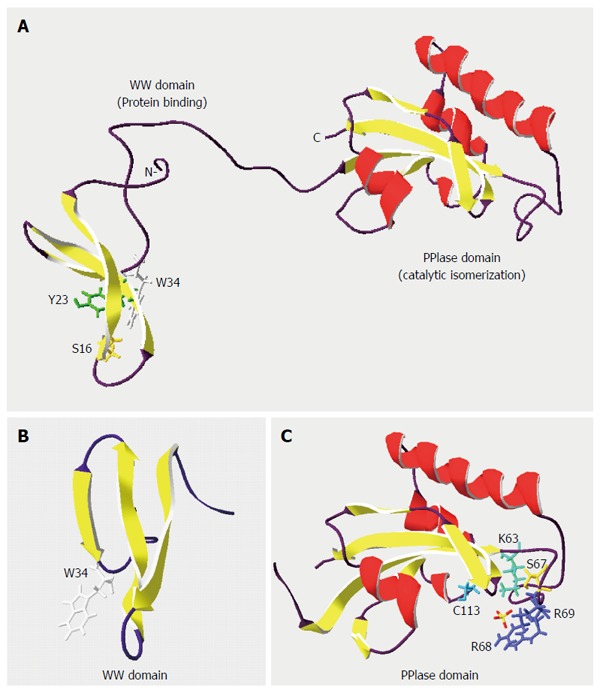
Structure of peptidyl-prolyl-isomerase PIN1 protein. Ribbon diagrams of (A) PIN1 (NCBI Structure No. 1NMV), (B) WW binding domain (NCBI Structure No. 1I8H), and (C) PPIase catalytic domain (NCBI Structure No. 1NMW) were drawn with the Swiss-Pdb Viewer[11,15,115,116]. α-helices and β-strands are denoted by coils and arrows, respectively. Residues Ser(S)16, Tyr(Y)23 and Trp(W)34 in the WW domain are critical for phospho-protein binding, while residues Lys(K)63, Ser(S)67, Arg(R)68/69 and Cys(C)113 contribute to the PPIase activity. Adapted from thesis: Identification and characterization of PIN1 binding partners, HKU 2010.
In this article, we discuss the possible mechanisms underlying dysregulated PIN1 expression in cancer, the oncogenic roles of PIN1 in hepatocarcinogenesis, and the potential of PIN1 inhibitors as anti-cancer agents.
REGULATION OF PIN1 EXPRESSION AND ACTIVITY
In normal cells, PIN1 expression is usually very low and is tightly regulated by the retinoblastoma protein (Rb)-E2F pathway[26,27]. The binding between Rb and E2F proteins is controlled by the phosphorylation of Rb. Hypophosphorylated Rb binds E2F transcription factors and inhibits its transcriptional activity towards the PIN1 gene. In response to cell proliferative stimuli, CDK-cyclin complexes phosphorylate and inactivate Rb to release E2F. In turn, E2F binds to the E2F-binding sites of the PIN1 promoter and directly activates transcription of the PIN1 gene. Interestingly, PIN1 has also been found to interact with Rb and enhance its hyperphosphorylation[28,29]. Therefore, PIN1 inactivates Rb and promotes E2F target gene activation. Since dysregulation of the Rb-E2F pathway is frequently found in various cancers[30], it is speculated that abnormalities of this pathway may contribute to PIN1 overexpression in cancers. Furthermore, PIN1 interacts with phosphorylated NOTCH1 to enhance NOTCH1 transcriptional activity, which in turn, increases the transcription of PIN1, resulting in a positive feedback loop for PIN1 overexpression in cancers[31]. In addition to E2F and NOTCH1, forkhead box transcription factor FOXC1 also enhances PIN1 promoter activity, resulting in increased mRNA and protein expression of PIN1 in breast cancer cells[32].
MicroRNAs (miRNAs) are small non-coding RNA that functions as a negative regulator of gene expression by binding to the 3′UTR of target mRNA to inhibit gene expression at the post-transcriptional level[33]. Dysregulation of miRNAs expression is frequently observed in cancers[34]. In HCC, a global reduction of miRNAs expression is associated with HCC progression[35], suggesting that most of the expressed miRNAs in normal hepatocytes function as tumour suppressors. Some of these miRNAs may target the expression of PIN1 and their reduced expression may therefore result in the PIN1 overexpression observed in HCC. Currently, three miRNAs have been shown to negatively regulate PIN1 expression in cancers. miR-200b/c and miR-296-5p directly target and suppress PIN1 expression in breast cancer and prostate cancer cells, respectively[36-38]. However, no specific miRNA has been reported to target PIN1 expression in HCC.
Single nucleotide polymorphism (SNP) of the PIN1 gene promoter may also contribute to the regulation of PIN1 expression. The promoter region of PIN1 gene contains two SNPs [rs2233678 (-842G/C) and rs2233679 (-667C/T)] and one synonymous SNP [rs2233682 (Gln33GlnG>A)] in exon 2. Genotype -842CC is associated with lower PIN1 protein expression in peripheral mononuclear cells, whereas -677C/T genotype does not have significant effect on PIN1 expression[39]. Similar result was also found in squamous cell carcinoma of head and neck (SCCHN), with -842C genotype but not -677C/T associated with a lower PIN1 promoter activity and a lower risk of SCCHN[40].
In addition to the transcriptional regulation, PIN1 level is also regulated through post-translational modification. Phosphorylation at Ser65 by Polo-like kinase I (PLK1) inhibits ubiquitination of PIN1, resulting in decreased proteasomal degradation and increased protein level[41]. Moreover, multiple regulators have also been found to modulate the activity of PIN1 by post-translational modifications. Sumoylation of Lys6 on the WW domain and Lys63 on the PPIase domain suppress PIN1 protein-binding and catalytic abilities, respectively. De-sumoylation on those sites by SUMO1/sentrin specific peptidase 1 (SENP1) reverses the inhibitory function on PIN1 and increases PIN1 stability[42]. In addition, mixed-lineage kinase 3-induced phosphorylation of PIN1 at Ser138 enhances PIN1 catalytic activity[43], whereas death-associated protein kinase 1-induced phosphorylation of PIN1 at Ser71 abolishes PIN1 catalytic activity[14]. Furthermore, protein kinase A, ribosomal S6 kinase 2 and Aurora have also been shown to inhibit PIN1 function by phosphorylation at Ser16[13,44,45].
PIN1 OVEREXPRESSION IN HCC
As PIN1 regulates cellular signalling, PIN1 overexpression typically results in uncontrolled cell proliferation and tumour formation. A relationship between PIN1 and cancer was first demonstrated in breast cancer, in which PIN1 level positively correlated with tumour grade[20]. Moreover, PIN1 overexpression has been found in different cancers, including brain, breast, cervical, colon, lung and prostate[26,46].
Consistently, several studies have confirmed that PIN1 mRNA and protein are over-expressed in HCC tumours, as compared with those of the adjacent non-tumourous liver tissues[47-50]. In one of the earliest studies, PIN1 overexpression was found in more than 50% of HCC samples[47]. In addition, a study from our group has also demonstrated that PIN1 overexpression is more frequently observed in HBV-related HCCs[51]. Shinoda et al[52] has studied the association between PIN1 expression and clinicopathological characteristics in HCC. HCC tumours with higher PIN1 expression show significantly larger tumour size and higher frequency of portal vein invasion. Moreover, higher PIN1 expression is also significantly associated with poorer prognosis, lower overall survival and higher early recurrence rate (within 3 years) in patients with HCC. Thus, dysregulation of PIN1 expression is closely associated with HCC progression.
ROLES OF PIN1 IN HEPATOCARCINOGENESIS
Through catalysing isomerization of signalling molecules, PIN1 functions as a critical catalyst in many signalling pathways in cancer. The oncogenic property of PIN1 was first demonstrated in breast cancer, as PIN1 can transform breast epithelial cells through up-regulation of cyclin D1[27]. Several studies have demonstrated that PIN1 expression is positively correlated with cyclin D1 expression in various types of cancer[20,47,53-56]. A positive correlation has also been shown between PIN1 expression and centrosome amplification in breast cancer. Both in vitro and in vivo studies have demonstrated that PIN1 induces centrosome duplication, resulting in chromosomes mis-segregation and genomic instability, which in turn promote cell transformation and tumour growth in transgenic mice[57].
In hepatocytes, overexpression of PIN1 has been shown to promote malignant transformation. PIN1 overexpression in the immortalized but non-tumourigenic liver cell line MIHA leads to anchorage-independent colony formation in soft agar and tumour formation in nude mice in vivo[58]. The number of colonies in soft agar and the size of tumours in nude mice are positively correlated with PIN1 expression levels. Similarly, PIN1 depletion in human HCC cells results in the suppression of cell proliferation, reduction of colony formation in soft agar and abrogation of tumour development in nude mice. Thus, PIN1 plays a critical role in hepatocarcinogenesis and the pathways involved are further discussed in the following sections (Figure 2).
Figure 2.
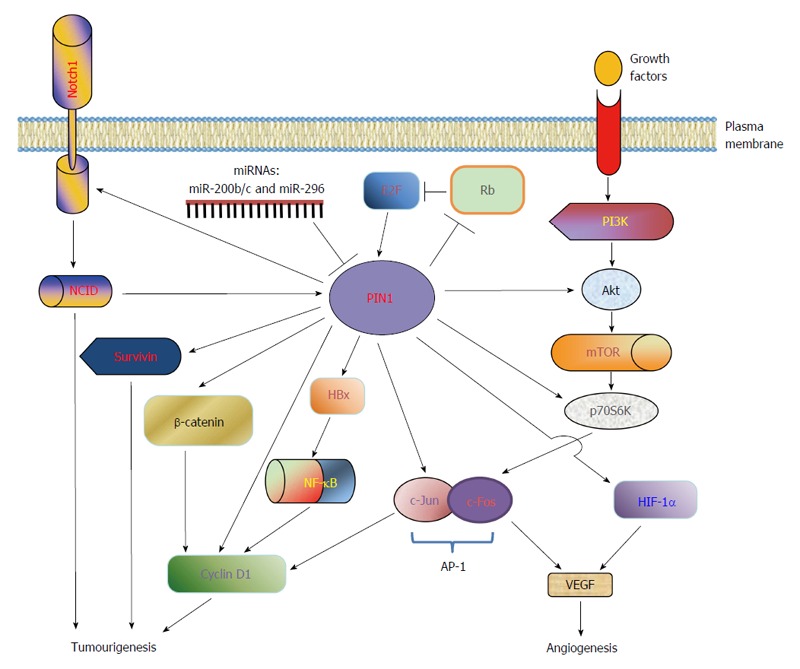
PIN1 dysregulation and targets in hepatocellular carcinoma. PIN1 functions as an amplifier to augment the oncogenic activities of key phosphoproteins involved in HCC tumourigenesis. PIN1 gene expression is up-regulated by various transcription factors, including E2F family and activated NOTCH1 intracellular domain (NCID), but is down-regulated by miRNAs (miR-200b/c and miR-296). In addition, PIN1 inactivates retinoblastoma protein (Rb), resulting in the release of E2F for activation of PIN1 expression. Through phosphorylation-dependent prolyl isomerization, PIN1 activates the β-catenin/cyclin D1 signalling pathway by induction of β-catenin transcriptional activity and stabilization of cyclin D1 protein. In parallel, PIN1 increases the transcriptional activities of c-Jun and NF-κB, leading to an increase in cyclin D1 transcription. Furthermore, PIN1 stabilizes hepatitis B virus X protein (HBx) and enhances its transactivating activity on downstream target nuclear factor-kappa B (NF-κB), which in turn increases cyclin D1 transcription. Up-regulation of cyclin D1 leads to uncontrolled cell proliferation and tumourigenesis. In addition, PIN1 increases the antiapoptotic function of survivin to inhibit apoptosis and contribute to tumourigenesis. Through interaction with Akt and ribosomal S6 kinase (p70S6K), PIN1 also activates the PI3K/Akt/mTOR pathway to promote tumourigenesis. PIN1 enhances the transcriptional activities of hypoxia-inducible factor (HIF)-1α and activator protein (AP)-1, resulting in up-regulation of angiogenic factor vascular endothelial growth factor (VEGF) and promotion of angiogenesis.
PIN1 AND β-CATENIN/CYCLIN D1 SIGNALLING PATHWAY
Dysregulation of the β-catenin signalling pathway resulting from β-catenin gene mutations contributes to HCC development. Activation of the β-catenin signalling pathway results in nuclear translocation of β-catenin protein that in turn activates the transcription of its target genes, including cyclin D1 and c-Myc[59-62]. Up-regulation of cyclin D1 and c-Myc leads to uncontrolled cell proliferation, malignant transformation and tumour development. In addition to β-catenin gene mutations, PIN1 overexpression has been shown to increase the β-catenin transcriptional activity towards its target genes, such as cyclin D1, c-Myc, PPAR-δ and fibronectin[18]. The mechanism of PIN1 regulating β-catenin transcriptional activity is dependent on the direct interaction between PIN1 and the phosphorylated Thr246-Pro motif of β-catenin. Such interaction stabilizes β-catenin by inhibiting its binding with the adenomatous polyposis coli protein for nuclear export and subsequent glycogen synthase kinase-3β-mediated degradation of β-catenin. This leads to increased nuclear accumulation and transcriptional activity of β-catenin. We have previously demonstrated that overexpression of PIN1 in HCC is associated with β-catenin accumulation[47]. More importantly, PIN1 overexpression and β-catenin gene mutations are found to be mutually exclusive events, further underscoring the role of PIN1 overexpression in causing β-catenin accumulation in HCC. Therefore, in addition to the somatic mutations of β-catenin that occur only in 20% of HCCs[63,64], PIN1 overexpression is the major mechanism leading to β-catenin accumulation in HCC.
In addition to β-catenin, PIN1 also binds other transcription factors to increase their transactivation activity towards the cyclin D1 gene. In response to activated c-Jun N-terminal kinases or oncogenic Ras, PIN1 interacts with c-Jun via its phosphorylated Ser63/73-Pro motif and increases the transcriptional activity of c-Jun on its target genes, such as cyclin D1[20]. PIN1 binds the phosphorylated Thr254-Pro motif in the p65/RelA subunit of NF-κB[23]. This interaction stabilizes NF-κB by inhibiting its interaction with its inhibitor IκB to prevent the nuclear export and subsequent ubiquitin-mediated degradation of NF-κB. Moreover, PIN1 increases NF-κB transactivation activity by promoting phosphorylation of NF-κB at Ser276 residue[52]. The PIN1-induced nuclear accumulation and activation of NF-κB result in increased cyclin D1 expression. Interestingly, PIN1 has also been shown to interact directly with cyclin D1 itself via its phosphorylated Thr286-Pro motif, resulting in protein stabilization and nuclear accumulation of cyclin D1[17].
PIN1 and HBx
Chronic infection with HBV is a major cause of HCC[65,66]. HBV is a DNA virus that facilitates malignant transformation by integration into the host genome to induce chromosome instability[67,68] and to alter the expression of cancer-related genes by insertional mutagenesis[69]. In addition, HBV modulates cell proliferation through the expression of viral proteins, in particular, the hepatitis B virus X protein (HBx)[70]. HBx is a gene transactivator that contributes to hepatocarcinogenesis through up-regulation of the proto-oncogenes such as c-myc, c-jun and NF-κB[71-73]. Studies have also shown that HBx interacts with p53 to inhibit the translocation of p53 into nucleus, resulting in the inhibition of p53-mediated cellular apoptosis and the development of liver tumour in transgenic mouse[74].
PIN1 overexpression is more frequently observed in HBV-related HCCs. Moreover, PIN1 directly binds to the phosphorylated Ser41-Pro motif in HBx[51]. This interaction results in the stabilization of HBx protein, leading to augmentation of its transactivating activity on the downstream target genes Bcl-XL, c-myc, and NF-κB[51]. Overexpression of both PIN1 and HBx leads to synergistic increase in cell proliferation in vitro and tumour growth in vivo, as compared with cells overexpressing PIN1 or HBx alone. These synergistic effects were totally dependent on the interaction between PIN1 and HBx. Neither the expression of the non-PIN1-binding HBx mutants nor PIN1 mutants that are defective for protein binding or isomerase activity cause any synergistic increase in cell proliferation and tumour growth.
PIN1 and survivin
In addition to the dysregulation of cell proliferation, the tightly regulated programmed cell death (cellular apoptosis) is frequently impaired in cancers. PIN1 has also been found to affect cellular apoptosis in breast, prostate and cervical cancer cells[56,75,76]. A recent study by our group has demonstrated an interaction between PIN1 and phosphorylated Thr34-Pro motif of survivin, an inhibitor of apoptosis protein (IAP), in HCC cells[48]. The function of survivin is to inhibit apoptosis by facilitating its interaction with hepatitis B X-interacting protein (HBXIP) and pro-caspase-9, thereby blocking caspase-9 activation[77,78]. The antiapoptotic function of survivin is critical in a number of cancers, including HCC[79]. In our study, we showed that PIN1 overexpression increases the binding between survivin and pro-caspase-9 via HBXIP, leading to suppression of caspase-9 and caspase-3-dependent apoptosis in HCC cells. Moreover, PIN1 promotes HCC tumour growth through inhibition of apoptosis and PIN1 expression is positively correlated with survivin expression in HCC tumours. Therefore, PIN1 enhances the antiapoptotic function of survivin and plays an important role in hepatocarcinogenesis through inhibition of apoptosis.
PIN1 and phosphoinositide 3-kinase/serine/threonine-specific protein kinase B (Akt)/mammalian target of rapamycin pathway
In addition to the β-catenin signalling pathway, the phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is another important oncogenic pathway involved in HCC. Activation of this pathway is triggered by binding of multiple growth factors, including insulin and cytokines, to their respective cell surface receptors. This leads to the activation of PI3K and its subsequent downstream targets, including Akt, mTOR, translation initiation factor 4E-binding protein (4E-BP1) and ribosomal S6 kinase (p70S6K). Both 4E-BP1 and p70S6K regulate the translation of cell cycle regulatory proteins, and therefore, aberrant activation of the PI3K/Akt/mTOR pathway results in dysregulated cell cycle progression and tumourigenesis. It has been reported that mTOR phosphorylation is found in 15% of HCCs and total p70S6K expression is up-regulated in 45% of HCCs[80]. The occurrence of multiple serine and threonine phosphorylation events in this pathway suggests that PIN1 may be involved in and contribute to the development of HCC. Recently, PIN1 has been found to interact with the phosphorylated Thr92/450-Pro motifs of Akt, resulting in the stabilization of both total and activated Akt (phosphorylated Ser473)[81]. Moreover, PIN1 expression is positively correlated with Akt activation in various types of human tumours including breast, nasopharynx and salivary gland[81]. Therefore, the PIN1-mediated Akt stabilization may be closely associated with tumourigenesis.
In addition, Lee et al[44] revealed that PIN1 interacts with p70S6K and facilitates the insulin-induced phosphorylation of p70S6K as well as of its downstream target extracellular signal-regulated protein kinase (ERK)1/2 in human HCC cells. Through p70S6K-ERK signalling, co-expression of PIN1 and p70S6K leads to a synergistic increase in transcriptional activity of activator protein (AP)-1, as compared with expression of PIN1 or p70S6K alone[44]. The transcription factor AP-1 regulates gene expression through the formation of heterodimeric protein with other DNA binding proteins, such as c-Fos and c-Jun[82]. Activation of AP-1 regulates various cellular processes, such as cell proliferation, apoptosis and transformation, through activation of genes that encode the cell cycle regulatory proteins including cyclin D1, p53 and p21. Therefore, enforced expression of PIN1 enhances the insulin-induced neoplastic cell transformation, whereas the mTOR inhibitor rapamycin that blocks the activation of p70S6K suppresses PIN1-induced neoplastic cell transformation[44]. Hence, the PIN1-p70S6K signalling pathway enhances AP-1 activity and mediates the insulin-induced neoplastic cell transformation. Thus, PIN1 promotes hepatocarcinogenesis partly through the activation of the PI3K/Akt/mTOR pathway.
PIN1 and NOTCH1
NOTCH1 signalling pathway plays a critical role in oncogenesis through the regulation of cell proliferation, differentiation and apoptosis. Ligand (γ-secretase)-induced cleavage of the membrane-bound NOTCH1 receptor releases its intracellular domain. This intracellular domain of NOTCH1 translocates into the nucleus and then activates the expression of target genes, including oncogenes c-Myc and HES1. Hence, activation of NOTCH1 signalling has been implicated in tumourigenesis. With immunohistochemistry and immunoblotting, NOTCH1 overexpression was found in 60% of HCCs[83]. In addition, enforced expression of the activated NOTCH1 intracellular domain completely rescues the inhibitory effect of γ-secretase inhibitor (GSI) on the cell proliferation of HCC cells[84]. As mentioned above, interaction between PIN1 and NOTCH1 stimulates the NOTCH1 signalling by releasing the activated NOTCH1 intracellular domain[31]. Therefore, PIN1 overexpression increases the transcriptional activity of activated NOTCH1 intracellular domain and enhances the colony formation in vitro. Likewise, down-regulation of PIN1 decreases NOTCH1 transcriptional activity, which in turn abrogates tumour growth in vivo[31]. Taken together, both in vitro and in vivo studies demonstrate that activation of NOTCH1 signalling by PIN1 may potentially contribute to hepatocarcinogenesis.
PIN1 and angiogenesis
Angiogenesis is the formation of new blood vessels that enhances the development and progression of tumours. HCC is a highly aggressive vascular tumour that relies on active angiogenesis. Tumour angiogenesis is mainly regulated by the expression of an angiogenic factor, vascular endothelial growth factor (VEGF). VEGF functions to stimulate vascular permeability and subsequently promotes tumour angiogenesis[85,86]. Its expression is positively correlated with the angiogenic activity or tumour progression in HCCs[87-91]. Recently, PIN1 has been found to interact with hypoxia-inducible factor (HIF)-1α under hypoxic conditions[92,93]. HIF-1α is an important transcription factor for VEGF gene expression. Down-regulation of PIN1 or suppression of PIN1 activity reduces the protein stability of HIF-1α under hypoxic conditions, resulting in decreased transcriptional activity of HIF-1α, down-regulation of VEGF expression and inhibition of angiogenesis in vivo[92]. In addition, PIN1 overexpression has been found to increase the transcriptional activity of AP-1 and the protein level of VEGF in breast cancer cells[94]. The transcriptional activity of AP-1 is regulated through the formation of heterodimer with c-Fos and c-Jun[82]. Since PIN1 binds c-Fos and c-Jun, and increases their transcription activity[20,95], it also enhances the transactivation activity of AP-1 (c-Jun/c-Fos dimer), leading to increased VEGF gene transcription. Therefore, PIN1 may facilitate VEGF-mediated angiogenesis of HCCs through the regulation of AP-1 activity.
ROLES OF PIN1 IN TUMOUR INVASIVENESS
Several studies have reported that PIN1 is involved in cell motility and contributes to cancer cell invasiveness. PIN1 overexpression promotes migration of immortalized human breast epithelial cells and induces epithelial-mesenchymal transition (EMT) with up-regulation of the mesenchymal markers including N-cadherin, Zeb1 and vimentin, and down-regulation of the epithelial marker E-cadherin[37]. Moreover, the protein binding WW domain and catalytic isomerization PPIase domain of PIN1 are essential for the induction of EMT in breast cancer cells. Consistently, PIN1 silencing suppresses protein expression and promoter activity of EMT regulator SNAIL, leading to increased expression of its downstream effector E-cadherin in tamoxifen-resistant breast cancer cells[96]. In epidermal growth factor receptor (EGFR) Thr790Met mutant lung cancer tissues, PIN1 expression is also positively correlated with the mesenchymal markers vimentin and Zeb1[97]. PIN1 expression enhances the survival of EGFR-mutant lung adenocarcinoma cells with an EMT phenotype. In prostate cancer, PIN1 induces cellular migration and invasion by interacting with Smad2/3[98], while PIN1 depletion by siRNA inhibited migration, invasion and wound healing ability[75]. In addition to EMT, PIN1 also regulates focal adhesion kinase (FAK), which is a critical focal adhesion component for cell-cell interaction and migration. By binding to protein tyrosine phosphatase (PTP)-PEST, PIN1-induced isomerization enhances the interaction of PTP-PEST with FAK and dephosphorylates FAK at the Tyr397 site[99]. Dephosphorylation of FAK promotes migration, invasion and metastasis of Ras-induced transformed cells. In conclusion, PIN1 enhances cancer cell motility by controlling the EMT regulating proteins expression as well as focal adhesion protein activity.
PIN1 AS A NEW DRUG TARGET FOR HCC TREATMENT
The oncogenic role of PIN1 makes it an attractive target for the development of anticancer drugs. Most of the PIN1 inhibitors developed are small molecules that inhibit its isomerase activity by binding to its catalytic active site[100] (Table 1). The first described PIN1 inhibitor is Juglone, which irreversibly inhibits the PIN1 PPIase activity[101]. Juglone was found to suppress tumourigenicity in human cancer cells by inducing apoptosis[102] and reducing number and size of colonies in human HCC cells in soft agar assay[44]. However, in addition to its PIN1-inhibitory activity, Juglone also directly inhibits RNA polymerase II[103], rendering it not suitable for clinical treatment of human cancers. Tatara et al[104] and Uchida et al[105] have also screened for additional PIN1 PPIase inhibitors using a chemical compound library. PiB and dipentamethylene thiuram monosulfide (DTM) were identified to exhibit specific inhibitory activity toward PPIase through binding to the active site of PIN1. Both PiB and DTM inhibit proliferation of colon carcinoma HCT116 cells by delaying or blocking cell cycle progression. Moreover, the same concentration of PiB inhibits proliferation of wild-type mouse embryonic fibroblasts (MEFs), but not Pin1-null MEFs, suggesting that PiB is more specific in suppression of cell proliferation through inhibition of PIN1. Recently, all-trans retinoic acid was also found to inhibit PIN1 activity and to suppress cell proliferation in breast cancer and acute promyelocytic leukaemia[106]. However, the specificity of those PIN1 inhibitors remains a concern as some of them also possess parvulin-type PPIases inhibitory activity[100]. In addition to directly inhibiting PIN1 activity, miRNA-mediated gene silencing may also be used to knock-down PIN1 expression in cancer cells. The first miRNA mimic MRX34 has already been tested in phase I clinical trial for HCC[107]. PIN1 silencing by miR-200b/c or miR-296-5p may provide a new approach for HCC treatment.
Table 1.
Potential PIN1 inhibitors for cancer treatment
| Drug | Details | Status |
| Juglone | First PIN1 inhibitor | Preclinical |
| Irreversibly inhibits PIN1 PPIase activity | ||
| PiB | Specifically inhibits PIN1 PPIase activity | Preclinical |
| Inhibits colon cancer cell proliferation | ||
| Dipentamethylene thiuram monosulfide | Specifically inhibits PIN1 PPIase activity | Preclinical |
| Inhibits colon cancer cell proliferation | ||
| All-trans retinoic acid | Binds PIN1 and inhibits its activity | FDA approved for treatment of APL |
| Inhibits breast cancer and APL cell proliferation | ||
| miRNAs | Bind to the 3’UTR of PIN1 mRNA | Preclinical |
| miR-200b/c | Suppress PIN1 expression in breast cancer and prostate cancer cells | |
| miR-296-5p | ||
| Sorafenib | Multi-kinase inhibitor targeting Raf/Mek/Erk signalling pathway and tyrosine receptors | FDA approved for treatment of HCC |
| Inhibits angiogenesis and growth of HCC tumours in vivo | ||
| Inhibits phosphorylation of PIN1-interacting proteins (Mcl-1 and p70S6K) | ||
| Improves overall survival and increases time to progression in HCC patients | ||
| Bortezomib | Proteasome inhibitor | FDA approved for treatment of multiple myeloma |
| Suppresses expression of PIN1 and its transcription factor E2F | ||
| Inhibits HCC cell proliferation in vitro |
APL: Acute promyelocytic leukaemia; FDA: Food and Drug Administration; HCC: Hepatocellular carcinoma; PPIase: Peptidyl-prolyl isomerase.
The application of PIN1 inhibitors or miRNAs in the treatment of human cancers may be challenged by the fact that PIN1 is also expressed in normal cells for the regulation of cell division. PIN1 interacts with p53 to enhance protein stability and transactivation activity of p53 to promote cellular apoptosis in response to DNA damage[16,24,108]. Depletion of PIN1 has also been reported to induce transformation and tumourigenesis of MEFs through stabilization of oncogenic proteins Myc and cyclin E[21,22]. Therefore, it remains uncertain whether PIN1 inhibitors would have any adverse effect on normal tissues. To minimize the detrimental effects of PIN1 inhibitors on normal cells, a targeted delivery system may be employed to ensure specific drug delivery to HCC cells. More importantly, preclinical or clinical studies are necessary to examine the safety and effectiveness of the PIN1 inhibitors in cancer treatment.
Sorafenib is the only approved small molecular targeting agent for the treatment of advanced stage HCC. It is a multi-kinase inhibitor that blocks the Raf/Mek/Erk signalling pathway and inhibits several receptor tyrosine kinases, including VEGF receptor 2 and 3. In clinical studies, patients who received sorafenib had longer overall survival and time to progression[109,110]. Recently, in vitro and in vivo studies have provided further evidence of the efficacy of sorafenib in suppressing the growth of HCC cells[111,112]. Through inhibition of Raf/Mek/Erk signalling, sorafenib suppresses proliferation and enhances apoptosis of HCC cells. Moreover, sorafenib inhibits the angiogenesis and growth of patient-derived HCC tumour xenografts in mice. Sorafenib has also been shown to block the Erk-mediated phosphorylation of Mcl-1, which is required for interaction with PIN1 and subsequent protein stabilization[76]. Indeed, sorafenib also inhibits the phosphorylation of another PIN1 target, p70S6K, which induces cell transformation through enforced PIN1 expression. Therefore, inhibition of the phosphorylation of oncogenic PIN1 interacting partners may be an effective treatment strategy for PIN1-overexpressing tumours. Furthermore, proteasome inhibitor bortezomib (BZB) has been shown to suppress HCC cell growth through down-regulation of PIN1 and its transcription factor E2F[113]. As a single agent, however, the benefit of BZB in advanced HCC patients is limited[114]. Combination of BZB with other agents as treatment for HCC should be further evaluated.
CONCLUSION
HCC is an aggressive cancer associated with a poor prognosis. Conventional treatment options available for advanced HCC patients are very limited. Identification of important molecular targets in HCC may lead to the development of a new therapeutic approach. Dysregulation of PIN1 expression is associated with HCC and its expression is positively correlated with tumour size. Through phosphorylation-dependent prolyl isomerization, PIN1 functions as an amplifier to augment the oncogenic activities of its interacting proteins in hepatocarcinogenesis. The diverse oncogenic effects exerted by PIN1 overexpression in HCC render PIN1 as an attractive therapeutic target for treatment. In fact, several studies have already demonstrated that inhibiting PIN1 activity results in the suppression of cell growth and tumour development. Further studies and clinical trials are required to examine the safety and efficacy of PIN1 inhibition in the treatment of HCC.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C, C, C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: All authors declare no conflict of interest related to this publication.
Peer-review started: August 24, 2016
First decision: September 12, 2016
Article in press: October 30, 2016
P- Reviewer: Grassi G, Kuboki S, Sun BC, Tomizawa M S- Editor: Gong ZM L- Editor: Filipodia E- Editor: Wang CH
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13–20. doi: 10.1111/j.1749-6632.2002.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 3.Poon RT, Fan ST, Tsang FH, Wong J. Locoregional therapies for hepatocellular carcinoma: a critical review from the surgeon’s perspective. Ann Surg. 2002;235:466–486. doi: 10.1097/00000658-200204000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow AK, Ng L, Lam CS, Wong SK, Wan TM, Cheng NS, Yau TC, Poon RT, Pang RW. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One. 2013;8:e78675. doi: 10.1371/journal.pone.0078675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gish RG, Porta C, Lazar L, Ruff P, Feld R, Croitoru A, Feun L, Jeziorski K, Leighton J, Gallo J, et al. Phase III randomized controlled trial comparing the survival of patients with unresectable hepatocellular carcinoma treated with nolatrexed or doxorubicin. J Clin Oncol. 2007;25:3069–3075. doi: 10.1200/JCO.2006.08.4046. [DOI] [PubMed] [Google Scholar]
- 6.Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, Koh J, Mo FK, Yu SC, Chan AT, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97:1532–1538. doi: 10.1093/jnci/dji315. [DOI] [PubMed] [Google Scholar]
- 7.Lu KP, Liou YC, Zhou XZ. Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 2002;12:164–172. doi: 10.1016/s0962-8924(02)02253-5. [DOI] [PubMed] [Google Scholar]
- 8.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 9.Lu KP. Phosphorylation-dependent prolyl isomerization: a novel cell cycle regulatory mechanism. Prog Cell Cycle Res. 2000;4:83–96. doi: 10.1007/978-1-4615-4253-7_8. [DOI] [PubMed] [Google Scholar]
- 10.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 11.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 12.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, et al. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 13.Lu PJ, Zhou XZ, Liou YC, Noel JP, Lu KP. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J Biol Chem. 2002;277:2381–2384. doi: 10.1074/jbc.C100228200. [DOI] [PubMed] [Google Scholar]
- 14.Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RH, Zhou XZ, et al. Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol Cell. 2011;42:147–159. doi: 10.1016/j.molcel.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–853. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 17.Liou YC, Ryo A, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchida T, Hunter T, Lu KP. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci USA. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- 19.Bernis C, Vigneron S, Burgess A, Labbé JC, Fesquet D, Castro A, Lorca T. Pin1 stabilizes Emi1 during G2 phase by preventing its association with SCF(betatrcp) EMBO Rep. 2007;8:91–98. doi: 10.1038/sj.embor.7400853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeh ES, Lew BO, Means AR. The loss of PIN1 deregulates cyclin E and sensitizes mouse embryo fibroblasts to genomic instability. J Biol Chem. 2006;281:241–251. doi: 10.1074/jbc.M505770200. [DOI] [PubMed] [Google Scholar]
- 22.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 23.Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 24.Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853–857. doi: 10.1038/nature01120. [DOI] [PubMed] [Google Scholar]
- 25.Mantovani F, Piazza S, Gostissa M, Strano S, Zacchi P, Mantovani R, Blandino G, Del Sal G. Pin1 links the activities of c-Abl and p300 in regulating p73 function. Mol Cell. 2004;14:625–636. doi: 10.1016/j.molcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rizzolio F, Lucchetti C, Caligiuri I, Marchesi I, Caputo M, Klein-Szanto AJ, Bagella L, Castronovo M, Giordano A. Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with Pin1. Cell Death Differ. 2012;19:1152–1161. doi: 10.1038/cdd.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tong Y, Ying H, Liu R, Li L, Bergholz J, Xiao ZX. Pin1 inhibits PP2A-mediated Rb dephosphorylation in regulation of cell cycle and S-phase DNA damage. Cell Death Dis. 2015;6:e1640. doi: 10.1038/cddis.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 31.Rustighi A, Tiberi L, Soldano A, Napoli M, Nuciforo P, Rosato A, Kaplan F, Capobianco A, Pece S, Di Fiore PP, et al. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol. 2009;11:133–142. doi: 10.1038/ncb1822. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Ray PS, Sim MS, Zhou XZ, Lu KP, Lee AV, Lin X, Bagaria SP, Giuliano AE, Cui X. FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-κB signaling. Oncogene. 2012;31:4798–4802. doi: 10.1038/onc.2011.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289:3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 34.Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314. doi: 10.1146/annurev-pathol-012513-104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong CM, Wong CC, Lee JM, Fan DN, Au SL, Ng IO. Sequential alterations of microRNA expression in hepatocellular carcinoma development and venous metastasis. Hepatology. 2012;55:1453–1461. doi: 10.1002/hep.25512. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Zhang B, Gao J, Wang X, Liu Z. Regulation of the microRNA 200b (miRNA-200b) by transcriptional regulators PEA3 and ELK-1 protein affects expression of Pin1 protein to control anoikis. J Biol Chem. 2013;288:32742–32752. doi: 10.1074/jbc.M113.478016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo ML, Gong C, Chen CH, Lee DY, Hu H, Huang P, Yao Y, Guo W, Reinhardt F, Wulf G, et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 2014;74:3603–3616. doi: 10.1158/0008-5472.CAN-13-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee KH, Lin FC, Hsu TI, Lin JT, Guo JH, Tsai CH, Lee YC, Lee YC, Chen CL, Hsiao M, et al. MicroRNA-296-5p (miR-296-5p) functions as a tumor suppressor in prostate cancer by directly targeting Pin1. Biochim Biophys Acta. 2014;1843:2055–2066. doi: 10.1016/j.bbamcr.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici M, Arosio B, Crovella S. PIN1 promoter polymorphisms are associated with Alzheimer’s disease. Neurobiol Aging. 2007;28:69–74. doi: 10.1016/j.neurobiolaging.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Lu J, Hu Z, Wei S, Wang LE, Liu Z, El-Naggar AK, Sturgis EM, Wei Q. A novel functional variant (-842G& gt; C) in the PIN1 promoter contributes to decreased risk of squamous cell carcinoma of the head and neck by diminishing the promoter activity. Carcinogenesis. 2009;30:1717–1721. doi: 10.1093/carcin/bgp171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eckerdt F, Yuan J, Saxena K, Martin B, Kappel S, Lindenau C, Kramer A, Naumann S, Daum S, Fischer G, et al. Polo-like kinase 1-mediated phosphorylation stabilizes Pin1 by inhibiting its ubiquitination in human cells. J Biol Chem. 2005;280:36575–36583. doi: 10.1074/jbc.M504548200. [DOI] [PubMed] [Google Scholar]
- 42.Chen CH, Chang CC, Lee TH, Luo M, Huang P, Liao PH, Wei S, Li FA, Chen RH, Zhou XZ, et al. SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013;73:3951–3962. doi: 10.1158/0008-5472.CAN-12-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rangasamy V, Mishra R, Sondarva G, Das S, Lee TH, Bakowska JC, Tzivion G, Malter JS, Rana B, Lu KP, et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc Natl Acad Sci USA. 2012;109:8149–8154. doi: 10.1073/pnas.1200804109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee NY, Choi HK, Shim JH, Kang KW, Dong Z, Choi HS. The prolyl isomerase Pin1 interacts with a ribosomal protein S6 kinase to enhance insulin-induced AP-1 activity and cellular transformation. Carcinogenesis. 2009;30:671–681. doi: 10.1093/carcin/bgp027. [DOI] [PubMed] [Google Scholar]
- 45.Lee YC, Que J, Chen YC, Lin JT, Liou YC, Liao PC, Liu YP, Lee KH, Lin LC, Hsiao M, et al. Pin1 acts as a negative regulator of the G2/M transition by interacting with the Aurora-A-Bora complex. J Cell Sci. 2013;126:4862–4872. doi: 10.1242/jcs.121368. [DOI] [PubMed] [Google Scholar]
- 46.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 47.Pang R, Yuen J, Yuen MF, Lai CL, Lee TK, Man K, Poon RT, Fan ST, Wong CM, Ng IO, et al. PIN1 overexpression and beta-catenin gene mutations are distinct oncogenic events in human hepatocellular carcinoma. Oncogene. 2004;23:4182–4186. doi: 10.1038/sj.onc.1207493. [DOI] [PubMed] [Google Scholar]
- 48.Cheng CW, Chow AK, Pang R, Fok EW, Kwong YL, Tse E. PIN1 inhibits apoptosis in hepatocellular carcinoma through modulation of the antiapoptotic function of survivin. Am J Pathol. 2013;182:765–775. doi: 10.1016/j.ajpath.2012.11.034. [DOI] [PubMed] [Google Scholar]
- 49.Ao R, Zhang DR, Du YQ, Wang Y. Expression and significance of Pin1, β-catenin and cyclin D1 in hepatocellular carcinoma. Mol Med Rep. 2014;10:1893–1898. doi: 10.3892/mmr.2014.2456. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, Li L, Zhang Y, Yang H, Wei Y, Zhang L, Liu X, Yu L. Interaction of Pin1 with Nek6 and characterization of their expression correlation in Chinese hepatocellular carcinoma patients. Biochem Biophys Res Commun. 2006;341:1059–1065. doi: 10.1016/j.bbrc.2005.12.228. [DOI] [PubMed] [Google Scholar]
- 51.Pang R, Lee TK, Poon RT, Fan ST, Wong KB, Kwong YL, Tse E. Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology. 2007;132:1088–1103. doi: 10.1053/j.gastro.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 52.Shinoda K, Kuboki S, Shimizu H, Ohtsuka M, Kato A, Yoshitomi H, Furukawa K, Miyazaki M. Pin1 facilitates NF-κB activation and promotes tumour progression in human hepatocellular carcinoma. Br J Cancer. 2015;113:1323–1331. doi: 10.1038/bjc.2015.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou CX, Gao Y. Aberrant expression of beta-catenin, Pin1 and cylin D1 in salivary adenoid cystic carcinoma: relation to tumor proliferation and metastasis. Oncol Rep. 2006;16:505–511. [PubMed] [Google Scholar]
- 54.Nakashima M, Meirmanov S, Naruke Y, Kondo H, Saenko V, Rogounovitch T, Shimizu-Yoshida Y, Takamura N, Namba H, Ito M, et al. Cyclin D1 overexpression in thyroid tumours from a radio-contaminated area and its correlation with Pin1 and aberrant beta-catenin expression. J Pathol. 2004;202:446–455. doi: 10.1002/path.1534. [DOI] [PubMed] [Google Scholar]
- 55.Miyashita H, Mori S, Motegi K, Fukumoto M, Uchida T. Pin1 is overexpressed in oral squamous cell carcinoma and its levels correlate with cyclin D1 overexpression. Oncol Rep. 2003;10:455–461. [PubMed] [Google Scholar]
- 56.Li H, Wang S, Zhu T, Zhou J, Xu Q, Lu Y, Ma D. Pin1 contributes to cervical tumorigenesis by regulating cyclin D1 expression. Oncol Rep. 2006;16:491–496. [PubMed] [Google Scholar]
- 57.Suizu F, Ryo A, Wulf G, Lim J, Lu KP. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol Cell Biol. 2006;26:1463–1479. doi: 10.1128/MCB.26.4.1463-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pang RW, Lee TK, Man K, Poon RT, Fan ST, Kwong YL, Tse E. PIN1 expression contributes to hepatic carcinogenesis. J Pathol. 2006;210:19–25. doi: 10.1002/path.2024. [DOI] [PubMed] [Google Scholar]
- 59.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 60.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 62.Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol. 1999;155:703–710. doi: 10.1016/s0002-9440(10)65168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pineau P, Marchio A, Battiston C, Cordina E, Russo A, Terris B, Qin LX, Turlin B, Tang ZY, Mazzaferro V, et al. Chromosome instability in human hepatocellular carcinoma depends on p53 status and aflatoxin exposure. Mutat Res. 2008;653:6–13. doi: 10.1016/j.mrgentox.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 64.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson WS. The role of hepatitis B virus in the development of primary hepatocellular carcinoma: Part I. J Gastroenterol Hepatol. 1992;7:622–638. doi: 10.1111/j.1440-1746.1992.tb01497.x. [DOI] [PubMed] [Google Scholar]
- 66.Bréchot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004;127:S56–S61. doi: 10.1053/j.gastro.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 67.Brechot C, Pourcel C, Louise A, Rain B, Tiollais P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533–535. doi: 10.1038/286533a0. [DOI] [PubMed] [Google Scholar]
- 68.Shafritz DA, Shouval D, Sherman HI, Hadziyannis SJ, Kew MC. Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens. N Engl J Med. 1981;305:1067–1073. doi: 10.1056/NEJM198110293051807. [DOI] [PubMed] [Google Scholar]
- 69.Paterlini-Bréchot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Bréchot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–3916. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 70.Feitelson MA, Duan LX. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am J Pathol. 1997;150:1141–1157. [PMC free article] [PubMed] [Google Scholar]
- 71.Balsano C, Avantaggiati ML, Natoli G, De Marzio E, Will H, Perricaudet M, Levrero M. Full-length and truncated versions of the hepatitis B virus (HBV) X protein (pX) transactivate the cmyc protooncogene at the transcriptional level. Biochem Biophys Res Commun. 1991;176:985–992. [Google Scholar]
- 72.Chirillo P, Falco M, Puri PL, Artini M, Balsano C, Levrero M, Natoli G. Hepatitis B virus pX activates NF-kappa B-dependent transcription through a Raf-independent pathway. J Virol. 1996;70:641–646. doi: 10.1128/jvi.70.1.641-646.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Twu JS, Lai MY, Chen DS, Robinson WS. Activation of protooncogene c-jun by the X protein of hepatitis B virus. Virology. 1993;192:346–350. doi: 10.1006/viro.1993.1041. [DOI] [PubMed] [Google Scholar]
- 74.Ueda H, Ullrich SJ, Gangemi JD, Kappel CA, Ngo L, Feitelson MA, Jay G. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat Genet. 1995;9:41–47. doi: 10.1038/ng0195-41. [DOI] [PubMed] [Google Scholar]
- 75.Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem K, Kubota Y, Lu KP, Aoki I. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin Cancer Res. 2005;11:7523–7531. doi: 10.1158/1078-0432.CCR-05-0457. [DOI] [PubMed] [Google Scholar]
- 76.Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, Chang CJ, Yang Y, Lai CC, Lee DF, et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008;68:6109–6117. doi: 10.1158/0008-5472.CAN-08-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci USA. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, Reed JC. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–2740. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ito T, Shiraki K, Sugimoto K, Yamanaka T, Fujikawa K, Ito M, Takase K, Moriyama M, Kawano H, Hayashida M, et al. Survivin promotes cell proliferation in human hepatocellular carcinoma. Hepatology. 2000;31:1080–1085. doi: 10.1053/he.2000.6496. [DOI] [PubMed] [Google Scholar]
- 80.Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin Cancer Res. 2004;10:8421–8425. doi: 10.1158/1078-0432.CCR-04-0941. [DOI] [PubMed] [Google Scholar]
- 81.Liao Y, Wei Y, Zhou X, Yang JY, Dai C, Chen YJ, Agarwal NK, Sarbassov D, Shi D, Yu D, et al. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene. 2009;28:2436–2445. doi: 10.1038/onc.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 83.Giovannini C, Gramantieri L, Chieco P, Minguzzi M, Lago F, Pianetti S, Ramazzotti E, Marcu KB, Bolondi L. Selective ablation of Notch3 in HCC enhances doxorubicin’s death promoting effect by a p53 dependent mechanism. J Hepatol. 2009;50:969–979. doi: 10.1016/j.jhep.2008.12.032. [DOI] [PubMed] [Google Scholar]
- 84.Suwanjunee S, Wongchana W, Palaga T. Inhibition of gamma-secretase affects proliferation of leukemia and hepatoma cell lines through Notch signaling. Anticancer Drugs. 2008;19:477–486. doi: 10.1097/CAD.0b013e3282fc6cdd. [DOI] [PubMed] [Google Scholar]
- 85.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 86.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 87.Mise M, Arii S, Higashituji H, Furutani M, Niwano M, Harada T, Ishigami S, Toda Y, Nakayama H, Fukumoto M, et al. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology. 1996;23:455–464. doi: 10.1053/jhep.1996.v23.pm0008617424. [DOI] [PubMed] [Google Scholar]
- 88.Yamaguchi R, Yano H, Iemura A, Ogasawara S, Haramaki M, Kojiro M. Expression of vascular endothelial growth factor in human hepatocellular carcinoma. Hepatology. 1998;28:68–77. doi: 10.1002/hep.510280111. [DOI] [PubMed] [Google Scholar]
- 89.Torimura T, Sata M, Ueno T, Kin M, Tsuji R, Suzaku K, Hashimoto O, Sugawara H, Tanikawa K. Increased expression of vascular endothelial growth factor is associated with tumor progression in hepatocellular carcinoma. Hum Pathol. 1998;29:986–991. doi: 10.1016/s0046-8177(98)90205-2. [DOI] [PubMed] [Google Scholar]
- 90.Moon WS, Rhyu KH, Kang MJ, Lee DG, Yu HC, Yeum JH, Koh GY, Tarnawski AS. Overexpression of VEGF and angiopoietin 2: a key to high vascularity of hepatocellular carcinoma? Mod Pathol. 2003;16:552–557. doi: 10.1097/01.MP.0000071841.17900.69. [DOI] [PubMed] [Google Scholar]
- 91.Poon RT, Ng IO, Lau C, Zhu LX, Yu WC, Lo CM, Fan ST, Wong J. Serum vascular endothelial growth factor predicts venous invasion in hepatocellular carcinoma: a prospective study. Ann Surg. 2001;233:227–235. doi: 10.1097/00000658-200102000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han HJ, Kwon N, Choi MA, Jung KO, Piao JY, Ngo HK, Kim SJ, Kim DH, Chung JK, Cha YN, et al. Peptidyl Prolyl Isomerase PIN1 Directly Binds to and Stabilizes Hypoxia-Inducible Factor-1α. PLoS One. 2016;11:e0147038. doi: 10.1371/journal.pone.0147038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jalouli M, Déry MA, Lafleur VN, Lamalice L, Zhou XZ, Lu KP, Richard DE. The prolyl isomerase Pin1 regulates hypoxia-inducible transcription factor (HIF) activity. Cell Signal. 2014;26:1649–1656. doi: 10.1016/j.cellsig.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 94.Kim MR, Choi HS, Heo TH, Hwang SW, Kang KW. Induction of vascular endothelial growth factor by peptidyl-prolyl isomerase Pin1 in breast cancer cells. Biochem Biophys Res Commun. 2008;369:547–553. doi: 10.1016/j.bbrc.2008.02.045. [DOI] [PubMed] [Google Scholar]
- 95.Monje P, Hernández-Losa J, Lyons RJ, Castellone MD, Gutkind JS. Regulation of the transcriptional activity of c-Fos by ERK. A novel role for the prolyl isomerase PIN1. J Biol Chem. 2005;280:35081–35084. doi: 10.1074/jbc.C500353200. [DOI] [PubMed] [Google Scholar]
- 96.Kim MR, Choi HK, Cho KB, Kim HS, Kang KW. Involvement of Pin1 induction in epithelial-mesenchymal transition of tamoxifen-resistant breast cancer cells. Cancer Sci. 2009;100:1834–1841. doi: 10.1111/j.1349-7006.2009.01260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sakuma Y, Nishikiori H, Hirai S, Yamaguchi M, Yamada G, Watanabe A, Hasegawa T, Kojima T, Niki T, Takahashi H. Prolyl isomerase Pin1 promotes survival in EGFR-mutant lung adenocarcinoma cells with an epithelial-mesenchymal transition phenotype. Lab Invest. 2016;96:391–398. doi: 10.1038/labinvest.2015.155. [DOI] [PubMed] [Google Scholar]
- 98.Matsuura I, Chiang KN, Lai CY, He D, Wang G, Ramkumar R, Uchida T, Ryo A, Lu K, Liu F. Pin1 promotes transforming growth factor-beta-induced migration and invasion. J Biol Chem. 2010;285:1754–1764. doi: 10.1074/jbc.M109.063826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X, Zhou XZ, Aldape K, Cobb MH, Xie K, et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell. 2009;35:11–25. doi: 10.1016/j.molcel.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou XZ, Lu KP. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat Rev Cancer. 2016;16:463–478. doi: 10.1038/nrc.2016.49. [DOI] [PubMed] [Google Scholar]
- 101.Hennig L, Christner C, Kipping M, Schelbert B, Rücknagel KP, Grabley S, Küllertz G, Fischer G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 1998;37:5953–5960. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- 102.Rippmann JF, Hobbie S, Daiber C, Guilliard B, Bauer M, Birk J, Nar H, Garin-Chesa P, Rettig WJ, Schnapp A. Phosphorylation-dependent proline isomerization catalyzed by Pin1 is essential for tumor cell survival and entry into mitosis. Cell Growth Differ. 2000;11:409–416. [PubMed] [Google Scholar]
- 103.Chao SH, Greenleaf AL, Price DH. Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res. 2001;29:767–773. doi: 10.1093/nar/29.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tatara Y, Lin YC, Bamba Y, Mori T, Uchida T. Dipentamethylene thiuram monosulfide is a novel inhibitor of Pin1. Biochem Biophys Res Commun. 2009;384:394–398. doi: 10.1016/j.bbrc.2009.04.144. [DOI] [PubMed] [Google Scholar]
- 105.Uchida T, Takamiya M, Takahashi M, Miyashita H, Ikeda H, Terada T, Matsuo Y, Shirouzu M, Yokoyama S, Fujimori F, et al. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem Biol. 2003;10:15–24. doi: 10.1016/s1074-5521(02)00310-1. [DOI] [PubMed] [Google Scholar]
- 106.Wei S, Kozono S, Kats L, Nechama M, Li W, Guarnerio J, Luo M, You MH, Yao Y, Kondo A, et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med. 2015;21:457–466. doi: 10.1038/nm.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bouchie A. First microRNA mimic enters clinic. Nat Biotechnol. 2013;31:577. doi: 10.1038/nbt0713-577. [DOI] [PubMed] [Google Scholar]
- 108.Wulf GM, Liou YC, Ryo A, Lee SW, Lu KP. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J Biol Chem. 2002;277:47976–47979. doi: 10.1074/jbc.C200538200. [DOI] [PubMed] [Google Scholar]
- 109.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Raoul J, Zeuzem S, Santoro A, Shan M, Moscovici M, Voliotis D, et al. Randomized phase III trial of sorafenib versus placebo in patients with advanced hepatocellular carcinoma (HCC). Proceedings of the ASCO Annual Meeting Proceedings. J Clin Oncol. 2007;25:LBA1. [Google Scholar]
- 110.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 111.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 112.Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Thng CH, Chow P, Ong HS, Chung A, Soo KC. Sorafenib and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J Cell Mol Med. 2009;13:2673–2683. doi: 10.1111/j.1582-4934.2009.00692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Farra R, Dapas B, Baiz D, Tonon F, Chiaretti S, Del Sal G, Rustighi A, Elvassore N, Pozzato G, Grassi M, et al. Impairment of the Pin1/E2F1 axis in the anti-proliferative effect of bortezomib in hepatocellular carcinoma cells. Biochimie. 2015;112:85–95. doi: 10.1016/j.biochi.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 114.Kim GP, Mahoney MR, Szydlo D, Mok TS, Marshke R, Holen K, Picus J, Boyer M, Pitot HC, Rubin J, et al. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma. Invest New Drugs. 2012;30:387–394. doi: 10.1007/s10637-010-9532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bayer E, Goettsch S, Mueller JW, Griewel B, Guiberman E, Mayr LM, Bayer P. Structural analysis of the mitotic regulator hPin1 in solution: insights into domain architecture and substrate binding. J Biol Chem. 2003;278:26183–26193. doi: 10.1074/jbc.M300721200. [DOI] [PubMed] [Google Scholar]
- 116.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]