


Abstract
DNA cleavage by Type III restriction enzymes is governed strictly by the relative arrangement of recognition sites on a DNA substrate—endonuclease activity is usually only triggered by sequences in head-to-head orientation. Tens to thousands of base pairs can separate these sites. Long distance communication over such distances could occur by either one-dimensional (1D) DNA translocation or 3D DNA looping. To distinguish between these alternatives, we analysed the activity of EcoPI and EcoP15I on DNA catenanes in which the recognition sites were either on the same or separate rings. While substrates with a pair of sites located on the same ring were cleaved efficiently, catenanes with sites on separate rings were not cleaved. These results exclude a simple 3D DNA-looping activity. To characterize the interactions further, EcoPI was incubated with plasmids carrying two recognition sites interspersed with two 21res sites for site-specific recombination by Tn21 resolvase; inhibition of recombination would indicate the formation of stable DNA loops. No inhibition was observed, even under conditions where EcoPI translocation could also occur.
INTRODUCTION
The bacterial Type III restriction endonucleases are multimeric complexes of ∼380 kDa that result from the modular assembly of proteins encoded by the mod (sequence specificity and methylation) and res (DNA and ATP hydrolysis) genes, with the stoichiometry Mod2Res2 (1–3). They recognize specific asymmetric DNA sequences, 5–6 bp in length, but subsequently cleave DNA at non-specific loci adjacent to the site (25–27 bp 3′ for EcoPI and EcoP15I) (note that the adoption of the nomenclature EcoPI which, by agreement, supercedes EcoP1I or EcoP1). DNA cleavage relies on both ATP and Mg2+ cofactors. ATP acts both as an allosteric activator of DNA binding and as a transferable source of chemical energy (1,3). The methyl donor S-adenosyl methionine (AdoMet) also influences endonuclease activity by preventing DNA cleavage between incorrect arrangements of sites and, in certain buffer conditions, by stimulating DNA binding (4).
Amino acid motifs found within the Res subunits of the Type III enzymes are characteristic of superfamily 2 (SF2) DNA helicases (3,5), implying that the ATPase activity of Type III enzymes may be coupled to polynucleotide tracking as in the helicases (3). SF2 enzymes encompass a broad range of functions and can be best described as molecular motors that couple chemical energy from nucleotide triphosphate (NTP) hydrolysis to mechanical events on nucleic acid polymers (6). SF2 motifs are also found in the HsdR subunits of the Type I restriction endonucleases (3,5), which are known to cleave DNA via a tracking/collision mechanism. The Type I enzymes remain bound tightly to their recognition sites while simultaneously translocating adjacent non-specific DNA past themselves, so extruding expanding loops of DNA (1,3,7). Motion is bi-directional (occurring from both sides of the site so that two DNA loops are produced), is highly processive (dissociation from the track or stalling events are rare) and has a rate between 100 and 500 bp/s depending on the enzyme in question [(8–10); S.McClelland, D.Dryden and M.D.Szczelkun, unpublished data]. It has been suggested that the communication between site-specific recognition and non-specific cleavage by Type III enzymes is provided by a similar, yet unidirectional, ATP-dependent DNA tracking mechanism (11,12).
The principal evidence for one-dimensional (1D) motion by Type III enzymes comes from the observation that a pair of Type III sites, in head-to-head orientation, are required for DNA cleavage (4,11,12). The sites must be oriented head-to-head—e.g. two EcoPI sequences on one 5′–3′ strand must be oriented as AGACC and GGTCT, respectively. Other arrangements are not cleaved unless relaxed reaction conditions are provided (4). ATPase activity is key to these interactions and mutations in the helicase motifs of the Type III enzyme EcoPI prevent both ATP and DNA hydrolysis (13,14). The favored explanation for these observations is that two enzyme complexes bind to the DNA, one Res2Mod2 tetramer at each site (2,4), and that both translocate directly towards each other along the linear DNA contour (12). Their subsequent collision activates DNA cleavage. Although both recognition sites are occupied, only one of the two sites is ever cleaved (4)—the top strand is cut by an Res subunit from the proximal bound enzyme while the complementary bottom strand is cut by an Res subunit donated by the second, distal enzyme (2). There therefore appears to be a surfeit of protein subunits (four Mod and four Res subunits to produce one double-strand break). The role in the reaction of each subunit has yet to be defined.
While translocation can provide a sensor of the relative orientation of a pair of sites, there are also a large number of DNA-looping enzymes that are only active with sites in a specific geometry. For instance, the resolvases from the Tn3 transposons will only function when their recognition sites are in head-to-tail orientation on supercoiled DNA (15). For many looping enzymes, the requirement for a unique array of sites is absolute and no activity is observed at other arrangements. The Type II restriction endonuclease BspMI illustrates an alternative situation where the requirement is conditional rather than fixed (16). In this case, the preference for sites in a particular orientation depends on DNA topology, overall DNA length and the number of base pairs between the sites. A further complication is that one-dimensional (1D) and 3D process are not mutually exclusive; whilse translocation by Type I enzymes has been unequivocally proven as the principal mechanism of site communication (10,17), there is also evidence that passive three-dimensional DNA looping can occur (18). The preference for a particular arrangement of Type III sites could thus be more complicated than at first glance.
Biochemical assays that can directly measure site communication by Type III enzymes have yet to be described. In their study of the ATPase and DNA cleavage activities of EcoP15I, Meisel et al. (12) used Lac repressor binding to demonstrate that a protein roadblock could inhibit DNA cleavage between a pair of sites on a multi-site linear DNA. The simplest explanation of this result is that EcoP15I must translocate along the DNA between the sites. However, we have shown that Type III reactions in the absence of AdoMet and in buffers where the monovalent cation is potassium rather than sodium [conditions used by Meisel et al. (12) in their Lac assay] can result in interactions between sites that are not in head-to-head orientation (4). This is a particular problem on multi-site substrates as used by Meisel et al. (12). It is therefore not 100% clear as to which combinations of sites participated in the generation of each cleavage product. Accordingly it is also not 100% clear what effect Lac would have on more complex, atypical interactions. In addition, the roadblock assay cannot distinguish if DNA looping plays a supporting role to translocation. To investigate fully whether the communication between Type III sites is provided by 1D translocation, 3D looping or a combination of both, we examined the activity of the EcoPI and EcoP15I enzymes on plasmid and catenane substrates that contained pairs of identical Type III sites and under buffer conditions where only head-to-head arrangements of sites should be cut. The plasmid substrates used to construct the catenanes were also used in an alternative Tn21 resolvase inhibition assay under more relaxed buffer conditions to provide a quantitative measure of any DNA looping (19–24). In all cases, the evidence points to the communication being energy-dependent 1D translocation and not thermally driven 3D looping. Furthermore, we could not find any evidence for the formation of extruded DNA loops during translocation akin to those observed with Type I enzymes.
MATERIALS AND METHODS
Proteins
Wild-type EcoPI and EcoP15I and EcoPI K918A were purified as described previously (2,4). Tn21 resolvase was obtained from S. Halford. Protein concentrations were determined from the absorption at 280 nm using extinction coefficients derived from the aromatic amino acid composition in the predicted amino acid sequences and are given in terms of the Res2Mod2 heterotetramer for EcoPI/EcoP15I and in terms of the dimer for Tn21 resolvase. All other enzymes were obtained from either New England Biolabs (MA, USA) or Promega (WI, USA) and were used as recommended (units are as specified by the manufacturer).
DNA
Unless stated otherwise, all DNA and protein manipulations were carried out using standard procedures (25). Oligodeoxyribonucleotides were supplied by Cruachem (Glasgow, Scotland) and purified using COP cartridges. The synthesis of pMDS36a and pMDS37a are described elsewhere (4). The EcoP15I sites at positions 1214 and 1423 in pUC19 (26) were point-mutated by overlapping primer PCR using the QuickChange Site-Directed protocol (Stratagene, CA, USA). The 1214 site was altered using overlapping forward and reverse primers, 5′-TTGCAAGCACCAGATTACGC-3′ and 5′-GCGTAATCTGGTGCTTGCAA-3′ (with the mutated EcoP15I site in boldface type and the point mutation underlined), to produce pUC19Δ1214. The altered base pair was close to the RNAI/II region of the pMB1 origin (27), but did not produce measurable changes in the copy number of the plasmid (data not shown). The 1423 site in pUC19Δ1214 was then altered using overlapping primers, 5′-ACTTATCGCCACTGGTAGCAG-3′ and 5′-CTGCTACCAGTGGCGATAAGT-3′ (with the mutated EcoP15I site in boldface type and the point mutation underlined), to produce pUC19Δ1214/1423. The altered locus may also form part of the RNAII fold (28), but did not produce measurable changes in the copy number of the plasmid (data not shown). The 1862 bp NdeI–HindIII restriction fragment from pMDS2a (23), which carries two head-to-tail 21res sites (note that the use of the nomenclature 21res to represent the Tn21 recombination sites to avoid confusion with the Type III res genes and Res gene products) for site-specific recombination by Tn21 resolvase, was then cloned into pUC19Δ1214/1423 linearized with NdeI and HindIII, to generate the one-site substrate pMDS32. Two-site, head-to-head substrates were created by directional cloning of the following duplex oligonucleotide into the appropriate SfiI site of pMDS32: 5′-TGGCTTCAGCAGGAAGCGCAGATACCAAATACTGTCCTTCTAGTAAC-3′ was annealed to 5′-ACTAGAAGGACAGTATTTGGTATCTGCGCTCTGCTGAAGCCAGTT-3′ (EcoP15I sequence in boldface type), and the duplex cloned into pMDS32 at the 834 site to give pMDS34a or at the 1611 site to give pMDS35a.
The plasmids were used to transform Escherichia coli HB101, the transformants grown in M9 minimal medium supplemented with 37 MBq/l [methyl-3H]thymidine, and the covalently closed circular form of the DNA purified by density gradient centrifugation in CsCl–ethidium bromide (EtBr) (29). Catenanes were prepared by treatment of supercoiled plasmid DNA with Tn21 resolvase (4). Relaxed catenane DNA was generated by extensive treatment with Wheat Germ DNA topoisomerase I (30). DNA concentrations were determined from absorbance at 260 nm, assuming that an absorbance of 1 corresponds to 50 μg/ml DNA and that the molecular weight of DNA is 6.6 × 105 Da/kb.
DNA cleavage assays
Cleavage reactions contained 10 nM plasmid or catenane, 4 mM ATP and 100 μM AdoMet in ‘NaCl buffer’ [50 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 50 mM NaCl and 1 mM DTT]. Only head-to-head sites can be cut in this buffer (4). Cleavage was initiated by the addition of 40 nM EcoPI or EcoP15I, or by 10 U of SmaI or PstI, as indicated. Reactions were carried out at 37°C and left for 1 h. The relative proportions of DNA substrates and products represent true endpoints—incubation for longer periods of time did not result in further cleavage. Cleavage was stopped by addition of 0.5 vols of STEB buffer [0.1 M Tris–HCl (pH 8.0), 0.2 M EDTA, 40% (w/v) sucrose and 0.4 mg/ml bromophenol blue]. DNA substrate and product fragments were separated by agarose gel electrophoresis. Where necessary, the relative proportion of DNA species in each lane was evaluated by scintillation counting (29). Digital images of agarose gels were captured on a Kodak Image Station 440CF and edited for presentation purposes only using a linear intensity scale.
Tn21 resolvase recombination assays
For the recombination time courses, 10 nM pMDS36a was pre-incubated for 15 min at 20°C in 300 μl of ‘KGlu buffer’ [50 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 20 mM K-glutamate and 1 mM DTT], with or without EcoPI or EcoPI K918A (concentrations shown in Figure 3). This K-based buffer can support promiscuous DNA cleavage (4). Where indicated, 100 μM AdoMet and/or 4 mM NTP (ATP, ATPγS or AMP-PNP) were also included. A zero-time point aliquot (20 μl) was taken and then Tn21 resolvase added to a final concentration of 240 nM. At timed intervals thereafter, aliquots (20 μl) were removed from the reactions and mixed immediately with 3 μl of 42 μg/ml EtBr, in order to stop any further recombination. EcoRV was then added (10 U) and the samples incubated at 37°C for further 1 h. EcoRV generated characteristic fragments from the intact (unrecombined) plasmid and catenated product that were separated by electrophoresis through agarose. To evaluate the relative amounts of each fragment, the gels were stained for 1 h after electrophoresis in TAEE [40 mM Tris-acetate (pH 8.0), 1 mM EDTA and 0.5 mg/ml EtBr], destained for 20 min in TAE [40 mM Tris-acetate (pH 8.0) and 1 mM EDTA] and the resultant DNA fluorescence recorded digitally using a Kodak Image Station 440CF. Substrate (∼3.7 kb) and product (∼3.1 kb) bands that fell within a linear range of fluorescence were quantified from unedited images—the intensity values were then corrected for the DNA size differences. The percentage recombination at each time point during the reaction was calculated as described previously (24): the cited values include a correction factor for nicked DNA in the plasmid preparations (typically 5–10%) that cannot be recombined by resolvase. The kinetics of the recombination was fitted to the appropriate double-exponential function (24) by non-linear least-squares regression in GraFit 5 (Erithacus Software, Staines, UK).
Figure 3.
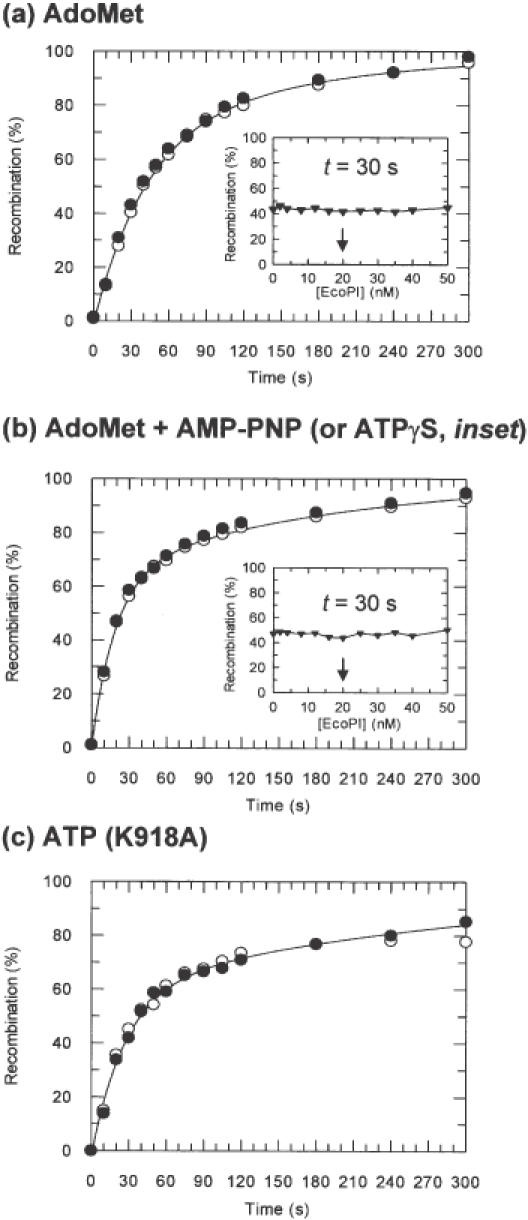
Tn21 resolvase recombination as a function of EcoPI binding. For kinetic reactions, 10 nM pMDS36a (20 nM Type III sites) was pre-incubated either with (black circles) or without (white circles) EcoPI (see Materials and Methods and text for further details). For titration experiments (black triangles), 10 nM pMDS36a was pre-incubated with EcoPI at the concentrations indicated (see Materials and Methods and text for further details). Curves represent least squares best fits of the data in the absence of EcoPI to a double exponential function (24). (A) Recombination time course in the presence of AdoMet and absence of NTP. Where added, EcoPI was at 20 nM. (a, inset) Extent of recombination after 30 s in the presence of AdoMet and absence of NTP following pre-incubation with EcoPI at the concentrations indicated. The arrow indicates where the Res2Mod2:site ratio is 1:1. (b) Recombination time course in the presence of AdoMet and AMP-PNP. Where added, EcoPI was at 20 nM. (b, inset) Extent of recombination after 30 s in the presence of AdoMet and ATPγS following pre-incubation with EcoPI at the concentrations indicated. The arrow indicates where the Res2Mod2:site ratio is 1:1. (c) Recombination time course in the presence of ATP alone. Where added, EcoPI K918A was at 40 nM.
To determine the effect of varying the endonuclease concentration on Tn21 resolvase recombination, EcoPI (0–50 nM) and pMDS36a (10 nM) were pre-incubated at 20°C in 20 μl of KGlu buffer supplemented with cofactors as indicated. After 15 min, Tn21 resolvase was added to each reaction to a final concentration of 240 nM. The reactions were allowed to proceed at 20°C for a further 30 s before quenching with EtBr. Recombination was analysed as above.
RESULTS
Activity of EcoPI and EcoP15I on plasmid and catenane substrates
To provide a distinction between 1D and 3D pathways for long-range communications between DNA sites we utilized catenane substrates containing two interlinked DNA rings (31). Where two interacting sites are placed onto separate rings of a catenane, the topological interlinking of the rings holds the sites in proximity to each other despite their being in trans. Since this tethering facilitates the juxtaposition of the sites in 3D space, a reaction that occurs by a 3D process, such as simple DNA looping, will occur with equal efficiency on a catenane as on a two-site plasmid where both sites are held in cis. Alternatively, where communication occurs by a 1D process that follows closely the DNA contour, the reaction will fail despite the tethering of the rings. This strategy has been applied to other restriction enzymes suspected of interacting simultaneously with multiple sites. For the Type I enzyme EcoR124I, non-specific cleavage of a catenane with a single recognition site was constrained to the ring carrying the site (17). This result is most readily accommodated by a 1D translocation process. In contrast, for the Type II enzymes SfiI (23), NgoMIV and NaeI (32), as well as many others (S. Halford, personal communication), catenanes with sites in each ring were cleaved faster than separate single-site DNA substrates and almost as rapidly as the parental two-site substrates. Communication in these cases could only have occurred by looping through 3D space.
In this study, catenanes were generated from plasmids containing two head-to-tail 21res sites for site-specific recombination by the Tn21 resolvase [(15), Figure 1]. This enzyme converts ∼98% of a 21res-containing plasmid into a [−2] catenane that can be purified from unreacted nicked substrate using standard techniques (24). The plasmids also contained two copies of the asymmetric target sites for the related Type III restriction enzymes EcoPI or EcoP15I in head-to-head orientation. The adjoining non-specific DNA was identical from 10 bp 5′ of the sites to 30 bp 3′ of the sites [sequences were as used in (4)]. The Type III and 21res sites in the parental plasmids were either: (i) interspersed (pMDS34a and pMDS36a), such that the resulting daughter catenanes carried one Type III site on each ring (34acat and 36acat); or (ii) arranged in series (pMDS35a and pMDS37a), such that the resulting daughter catenanes carried both Type III sites on the larger of the two interlinked rings (35acat and 37acat). This latter series of catenanes allowed us to check if the DNA topology of the linked rings interfered with the Type III reaction independently of any communication event. To further check the effect of topology, some substrates were relaxed with DNA topoisomerase I (Materials and Methods). In a [−2] catenane, the topological link is predominantly located at the apex of each superhelix as opposed to the interwound segments (33). As a consequence of the restricted polymer dynamics, looping interactions between sites on separate rings may be curtailed despite the physical proximity of the circles. By relaxing the DNA, the catenanes could explore more conformations and the probability of site juxtaposition will increase (30).
Figure 1.
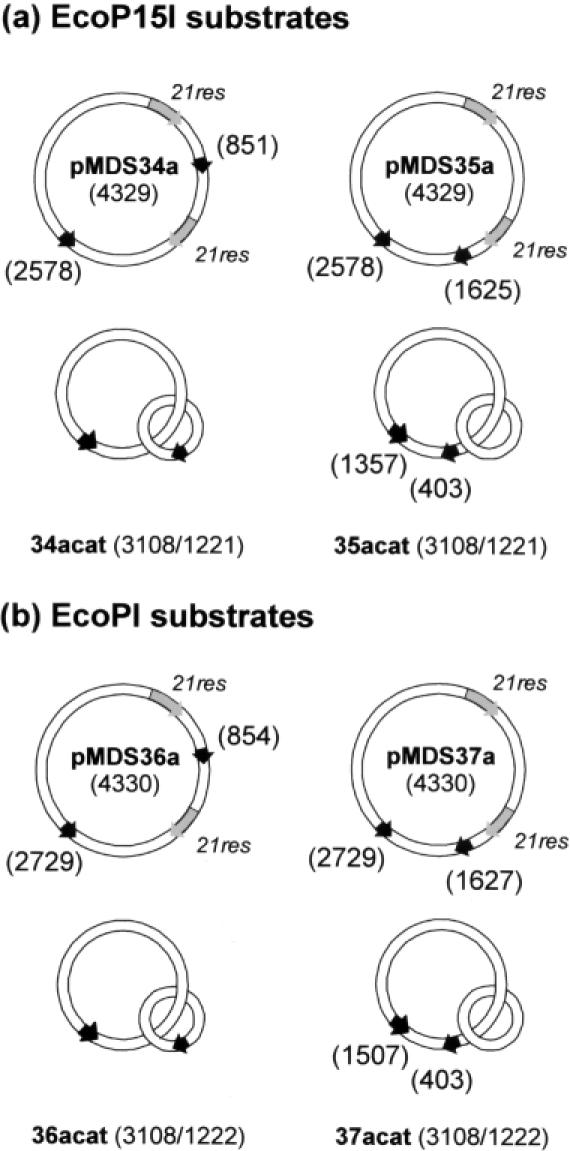
Plasmid and catenane substrates for EcoP15I and EcoPI. Recognition sites for either EcoP15I (a) or EcoPI (b) are indicated by black arrows. The locations of the Type III site are defined according to (4). Recombination sites for Tn21 resolvase (21res) are indicated by grey arrows. Daughter catenanes are illustrated directly below their corresponding parental plasmid: i.e. recombination of pMDS34a generates 34acat, etc. The numbers in parentheses correspond to total size of the plasmid, or, for the catenanes, to the sizes of the large and small rings, respectively.
Cleavage of the plasmid and catenane substrates (Figure 1) was analysed at a fixed time point and at fixed concentrations of EcoPI or EcoP15I (Figure 2). Buffer conditions were chosen to avoid DNA cleavage at site arrangements other than pairs of head-to-head sites (4). All components except the endonucleases were pre-incubated at 37°C. After 2 min, enzyme was added, DNA cleavage allowed to proceed for 1 h, the reactions stopped by the addition of EDTA and the products analysed by gel electrophoresis (Materials and Methods). Substrate cleavage with Type II restriction enzymes was assayed in parallel to identify the fragments produced. The various products that can result from the plasmid or catenane substrates are shown in Figure 2a and b. Digestion of any of the plasmids in Figure 1 with SmaI produces one double-strand break in the DNA. The resulting full-length linear fragment (Lin) can be separated from the supercoiled (CCC) and nicked (OC1) forms of the plasmid. For a supercoiled catenane (CCcat), a number of different species can arise depending on the location and number of strand breaks. Digestion of any of the catenanes in Figure 1 with PstI produces a double-strand break in the small DNA ring. This produces a small linear fragment (Lins) and liberates the large ring in supercoiled (CCCL) and/or nicked (OCL) forms. Digestion with SmaI produces a double-strand break in the large ring. This produces a large linear fragment (LinL) and liberates the small ring in supercoiled (CCCs) and/or nicked (OCs) forms. Each of these species has a distinct electrophoretic mobility. In addition, a nicked catenane can exist in three forms, cut in; both rings (OC4); the large ring alone (OC3); or the small ring alone (OC2). Each of these species has an increasing electrophoretic mobility and can be distinguished from the other DNA species. Under some electrophoresis conditions, the following pairs of species comigrate: CCC and CCcat; OC and OC4; and OCs and Lins.
Figure 2.
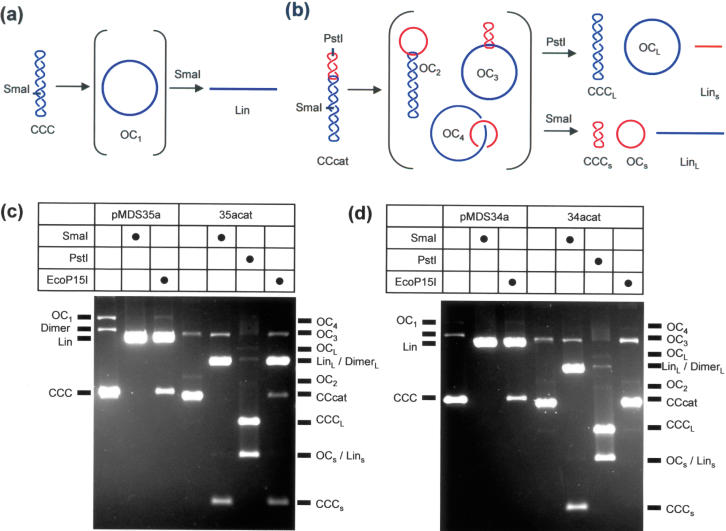
Cleavage of plasmid and catenane substrates by EcoP15I. The outcome of single- or double-strand cleavage of generic plasmid (a) or catenane (b) substrates is shown, alongside the corresponding acronyms for each fragment. Dimeric forms of the plasmid (Dimer) and the large and small rings of the catenane (DimerL and DimerS, respectively) are not illustrated. (c) Cleavage of pMDS35a and 35acat. (d) Cleavage of pMDS34a and 34acat. In each case, 10 nM DNA was incubated for 1 h with EcoP15I, SmaI or PstI as indicated (by a black circle) and the resulting fragments separated by agarose gel electrophoresis (see Materials and Methods and text for further details). Markers for the plasmid and catenane fragments are on the left and right, respectively, of each gel. Gel images were adjusted to maximize fragment brightness, for the purposes of visualization only, using a linear intensity scale.
The outcome of the EcoP15I and Type II reactions with supercoiled pMDS34a, pMDS35a, 34acat and 35acat substrates are shown in Figure 2c and d. Incubation of either pMDS34a or pMDS35a with EcoP15I resulted in the formation of linear DNA cut adjacent to one or other of the Type III sites—no further cleavage can occur under the stringent buffer conditions used here [(4), data not shown]. Some CCC and OC1 DNA (∼5%) remained uncut at the end of the reactions, probably as a result of the competing DNA methylation (4). These results tally with the properties expected of a Type III enzyme. The outcome of EcoP15I reactions with the catenane substrates depended, however, on the relative locations of the Type III sites. For 35acat, where the sites are on the same DNA molecule (the large ring) in a head-to-head orientation (Figure 1a), cleavage of the large ring to produce LinL occurred as efficiently as cleavage of pMDS35a (only ∼5% CCcat and OC3 remained uncut) (Figure 2c). The small circle was liberated virtually intact (CCCs was observed predominantly with little or no OCs or Lins). Nicking of the catenane in the small ring was not observed (levels of OC2 and OC4 did not increase). These results indicate that while a catenane can still be cleaved by a Type III enzyme, cleavage is only observed in the DNA molecule carrying the sites. In contrast, when the EcoP15I reaction was repeated with 34acat, where the Type III sites are on separate rings (Figure 1a), single- or double-strand breaks were not observed in any of the rings (Figure 2d). The relative proportions of the CCcat, OC2, OC3 and OC4 species—were corroborated by scintillation counting—no changes were observed following treatment with EcoP15I (data not shown).
Identical results to those in Figure 2c and d were obtained with EcoPI and the corresponding substrates pMDS36a, pMDS37a, 36acat and 37acat (data not shown). To test if the topology of the supercoiled [−2] catenanes in some way reduced the probability of interactions between sites on separate rings, we relaxed pMDS36a, pMDS37a, 36acat and 37acat with topoisomerase I and re-tested the extent of cleavage with EcoPI. As above, 36acat was refractory to cleavage while all other substrates were cut in the DNA carrying the head-to-head sites (data not shown). Therefore, irrespective of topology, DNA cleavage between a pair of Type III sites can only occur when those sites are linked in 1D along a DNA contour. Given that the wild-type Type III enzymes are not stimulated by DNA supercoiling relative to relaxed or linear substrates, we did not test the reactions on catenanes with multiple interlinks (see Discussion).
Evidence that EcoPI does not form DNA loops, either by 3D capture or by 1D extrusion
The results presented above indicate that the interaction between a pair of Type III sites must be the result, principally, of 1D translocation between those sites. However, it has been suggested for Type I enzymes that in addition to translocation, DNA looping between pairs of sites can occur (18). While this looping is conditional and not absolutely required for cleavage (17), where it does occur it may have an effect on the rate and efficiency of translocation and on the distribution of cleavage sites. Accordingly, we sought to test the Type III enzymes for DNA-looping activity that may occur in parallel with 1D translocation.
The parental plasmids shown in Figure 1 can be used to directly analyse DNA loop formation (19–24). Generally, efficient recombination by Tn21 resolvase requires a negatively supercoiled DNA with two head-to-tail 21res sites in cis; it will fail to recombine 21res sites in any other arrangement. On pMDS34a and pMDS36a, where the 21res and Type III sites are interspersed, capture of the Type III sites would form a ‘figure-of-eight’ structure that, in turn, would segregate the 21res sites into isolated topological domains [see Figure 1 in (24)]. During the lifetime of the figure-of-eight structure, resolvase would be inhibited from carrying out its reaction. However, upon dissociation of the loops there is a high probability that resolvase will capture the 21res sites and proceed to recombine them. Measurable inhibition will only occur if the lifetime of the looped state is long (i.e. loop dissociation is slower than the slowest recombination step) or the formation of the looped state is significantly faster than 21res capture by Tn21 resolvase [in the order of tens to hundreds of milliseconds; (22)]. The inhibition of resolvase can be assigned specifically to a looping interaction, rather than to other spurious effects, by using pMDS35a or pMDS37a, where the Type III and 21res sites are in series. In these substrates, the 21res sites will remain in the same topological domain regardless of any Type III site interactions. Similar assays have been used successfully to measure DNA looping by the resolvases themselves (20–22), by transcriptional repressors (19), and by Type II restriction endonucleases (23,24). For a comprehensive description of the Tn21 resolvase inhibition assay and its analysis see (24).
All of the following experiments were carried out with EcoPI and pMDS36a. Initial reactions were performed in the presence of AdoMet but in the absence of ATP: these conditions should stabilize DNA-binding while precluding DNA-translocation. In brief, the reactions were carried out as follows (see Materials and Methods for full details). All components except the enzymes were pre-incubated at 20°C (this lower temperature was chosen to stabilize any EcoPI interactions while still allowing sensible rates of recombination). After ∼2 min, EcoPI was added at the concentrations indicated and allowed to pre-incubate with the DNA for a further 15 min. Tn21 resolvase was added and aliquots removed from the reactions at the indicated time points and quenched with EtBr (to stop the resolvase). The DNA was then cleaved with EcoRV, the reactions stopped by the addition of EDTA and the products analysed by gel electrophoresis. The relative levels of substrate (plasmid) and product (catenane) were assessed from the relative intensities of characteristic linear fragments generated by EcoRV. The amount of recombination is expressed as the percentage of product corrected for any unreacted nicked substrate (24).
The extent of inhibition of Tn21 resolvase recombination (and thus the extent of DNA looping by EcoPI) was assessed first at a stoichiometric concentration of EcoPI (one Res2Mod2 per site; Figure 3a). The recombination profiles in the presence of EcoPI did not differ significantly from those in the absence of endonuclease. To check if looping may be dependent on EcoPI concentration, we measured the extent of recombination at a set time point (30 s) with 0–50 nM EcoPI (0–5 Res2Mod2 per site; Figure 3a, inset). Inhibition of recombination was not observed at any concentration of EcoPI tested. If looping had occurred then maximum inhibition should have been achieved at ∼20 nM EcoPI (4). Either DNA loops are not formed under these conditions, or those loops which do form are unstable (24). In an attempt to enhance the DNA interactions, we introduced the non-hydrolysable ATP analogues AMP-PNP or ATPγS at 4 mM during pre-incubation. No DNA cleavage was observed under these conditions (data not shown). The recombination profiles in the presence of AMP-PNP are shown in Figure 3b. Again, inhibition was not observed. Identical profiles were obtained with ATPγS (data not shown). As above, we measured recombination at 30 s with increasing concentrations of EcoPI (Figure 3b, inset; in the presence of 4 mM ATPγS). The linear profile obtained indicates that DNA loops cannot be stabilized by increased concentrations of EcoPI in the presence of NTP analogues.
It is possible that DNA looping is only induced when NTP hydrolysis can also occur. We could not include ATP in the reaction with the wild-type enzyme, as the accompanying DNA cleavage would inhibit recombination permanently. Instead we utilized a previously characterized EcoPI variant, K918A (2,4). This enzyme is altered in its PD..D/EXK endonuclease motif (34). While Type III endonuclease mutants cannot by themselves cleave DNA, they have ATPase activities comparable to wild-type (2) and can participate in DNA nicking at distal Type III sites by activating a wild-type Res subunit bound at that site (2,4). This suggests that the ATPase activity of these mutants can still be coupled to translocation despite their lack of endonuclease activity. If these complexes extrude a DNA loop while moving on pMDS36a, then translocation would have the same effect as loop capture—it would sequester the 21res sites into distinct topological domains. Consequently, the inhibition of resolvase by EcoPI K918A with ATP could be indicative of energy-dependent 3D loop capture and/or 1D loop extrusion. In addition, we excluded AdoMet from these reactions—background methylation of the recognition site may contribute to a weaker binding affinity for the modified sites (4). Even under these conditions, where translocation can occur (2,4), no detectable inhibition of resolvase was observed upon pre-incubation with EcoPI K918A and ATP (Figure 3c).
DISCUSSION
Many biological processes rely on protein-mediated communication between two or more separate DNA sites. Two general mechanisms for this ‘action at a distance’ can be described (31): DNA looping or DNA tracking. The bacterial restriction enzymes demonstrate both sorts of behaviour (7). Many Type II enzymes have now been shown to capture DNA loops between pairs of recognition sites (23,24,35–40 and others, S.Halford, personal communication), while the Type I restriction endonucleases are clearly motor enzymes that couple ATP hydrolysis to DNA translocation between pairs of sites (1,3,10,17). The Type I enzymes also form DNA loops, both passively, prior to translocation (18), and actively, in the extrusion of a growing DNA loop during translocation (41–45). A similar loop extrusion model has been proposed to explain site communication by the ATP-dependent Type III enzymes (12). We sought to clarify the Type III mechanism first by comparing endonuclease activity on plasmids and singly interlinked catenanes that contained pairs of identical sites (Figure 1). Irrespective of the substrate topology, cleavage was only observed when two head-to-head oriented Type III sites were located in cis on the same DNA chain (Figure 2). A catenane with sites in trans on separate rings was not cleaved. While this inactivity rules out a simple looping mechanism, it should be noted that certain recombinases are also inactive when their recombination sites are on separate supercoiled rings of singly interlinked catenanes—they will only recombine two such rings when they are catenated by multiple interlinks (46–50). In these examples of complex DNA looping, supercoiling is a key driving force of the reaction and multiple catenane nodes can, to a large extent, substitute for supercoil nodes in the synaptic complexes. In contrast, supercoiling is not required for DNA cleavage by Type III enzymes. Circular and linear DNA molecules are cleaved with similar efficiencies as long as two head-to-head sites are present in cis (2,4,11,12). In light of the importance of the helicase motifs and of ATP hydrolysis, the more likely explanation for our results is that site communication occurs by 1D translocation along the DNA contour. We can also state that the Type III enzymes must remain in close contact with the DNA strands during the reaction. Any excursion from the DNA, however transient, would have resulted in interactions between linked rings of the catenanes. There was no evidence of such interactions.
Given the possibility that the Type III enzymes share mechanistic features with the Type I enzymes (1,3,12), we also tested the ability of EcoPI to form DNA loops, either by capture or extrusion routes. To achieve this, we measured the inhibition of Tn21 resolvase-catalysed recombination on supercoiled pMDS36a (Figure 1). No evidence for loop capture was obtained in the absence of DNA translocation (i.e. without ATP or with non-hydrolysable ATP analogues) (Figure 3a and b). Under the conditions used here, both Tn21 resolvase and EcoPI catalyse their reactions at approximately the same rate (i.e. in each case substrate depletion follows a similar time course). If a looped species played a key role in the Type III reaction it would most likely have a half-life greater than, or at least on par with, the cleavage rate. On this time-scale looping would inhibit Tn21 resolvase. Since this was not observed, any looped state must be short-lived relative to the recombination reaction and, accordingly, Type III translocation is more likely to occur during a longer-lived unlooped state.
What is perhaps more surprising given the expectations of the loop extrusion model (12) is that EcoPI K918A could not inhibit resolvase in the presence of ATP. It is possible that the K918A mutant does not translocate DNA, although this seems unlikely given previous studies (2,4). Moreover, equivalent PD..D/EXK mutations in the Type I enzymes do not affect rates of ATP hydrolysis or DNA translocation [(9,45,51); S. McClelland, D.Dryden and M.D.Szczelkun, unpublished data]. Alternatively, the extruded loop(s) may be relatively unstable once the endonucleases stop translocating (i.e. upon collision, which may have occurred by 15 min). One way to establish the presence of extruded loops is to follow transitory changes in DNA topology on circular substrates—where translocation follows the DNA helix, accompanying loop formation will partition the DNA into distinct positive and negative topological domains that can be distinguished by treatment with topoisomerases (45,52–54). Loop extrusion by the Type I enzyme EcoAI was confirmed using this approach (45). However, when the same assay was carried out with the Type III enzyme EcoPI, changes in DNA supercoiling were not observed (2). Alongside our results, this supports the intriguing possibility that DNA translocation by Type III enzymes is not accompanied by the formation of expanding DNA loops.
What clues do the Type III enzymes give us about their mechanism of ATP-dependent DNA translocation? As with Type I enzymes, the Type III endonuclease reaction is not a genuine catalytic process; i.e. there is no turnover of the endonuclease once DNA cleavage has occurred (4,12). On a two-site, head-to-head substrate the dependence of cleavage upon endonuclease concentration follows a binomial relationship with respect to the number of recognition sites (4). Where a substrate is bound by only one endonuclease, or none, protein molecules that have taken part in reactions on other DNA molecules cannot be recruited to complete cleavage. Consequently, at sub-stoichiometric concentrations of endonuclease relative to recognition site, a DNA substrate is never 100% cleaved, even after extended digestion times (>1 h) [(4), unpublished data]. Our data point to an ‘irreversible’ interaction between the Mod subunit of the endonuclease and its recognition site following ATP binding and hydrolysis. Distinct from the Type I enzymes, DNA cleavage always occurs adjacent to the Type III recognition sequence [(1); A. Sears, L.J.Peakman and M.D.Szczelkun, unpublished data], which also suggests that contact with the site must be maintained. Furthermore, it is established that full endonuclease activity requires cooperation between Res subunits from complexes bound proximal to and distal from the cleavage site (2). Therefore, Type III enzymes must both remain in contact with their specific site and explore distant non-specific sites. While a Type I-like model certainly fills these criteria (12), there is, as explained above, no evidence of loop extrusion by Type III enzymes. The communication could also occur by a long-range DNA wrapping, similar to that suggested for some GTP-dependent restriction enzymes (55). However, since higher order wrapping should generate changes in the domain structure of a circular DNA, this model is also incompatible with the recombination data presented here and the topoisomerase data presented previously (2). Elucidation of the molecular mechanism of the Type III enzymes is currently at an impasse; while communication between cleavage sites is clearly via 1D tracking, the exact mechanism of translocation remains obscure (3). To address the contradictions posed by the different Type III models, there is an urgent need for assays to both observe directly and quantify the molecular events following DNA binding and during ATP hydrolysis.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Pavel Janscak for materials and comments, and Nigel Savery, Steve Halford, Sarah McClelland, Alice Sears, Louise Stanley et al. in the Bristol DNA–Protein Interactions Group for technical assistance, discussions and support. This work was funded by the Wellcome Trust. M.D.S. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Sciences.
REFERENCES
- 1.Bourniquel A.A. and Bickle,T.A. (2002) Complex restriction enzymes: NTP-driven molecular motors. Biochimie, 84, 1047–1059. [DOI] [PubMed] [Google Scholar]
- 2.Janscak P., Sandmeier,U., Szczelkun,M.D. and Bickle,T.A. (2001) Subunit assembly and mode of DNA cleavage of the type III restriction endonucleases EcoP1I and EcoP15I. J. Mol. Biol., 306, 417–431. [DOI] [PubMed] [Google Scholar]
- 3.McClelland S.E. and Szczelkun,M.D. (2004) The Type I and III restriction endonucleases: structural elements in molecular motors that process DNA. In Pingound,A. (ed.), Nucleic Acids and Molecular Biology—Restriction Enzymes. Springer-Verlag, Germany, Vol. 14, pp. 111–135. [Google Scholar]
- 4.Peakman L.J., Antognozzi,M., Bickle,T.A., Janscak,P. and Szczelkun,M.D. (2003) S-Adenosyl methionine prevents promiscuous DNA cleavage by the EcoP1I type III restriction enzyme. J. Mol. Biol., 333, 321–335. [DOI] [PubMed] [Google Scholar]
- 5.Gorbalenya A.E. and Koonin,E.V. (1991) Endonuclease (R) subunits of type-I and type-III restriction-modification enzymes contain a helicase-like domain. FEBS Lett., 291, 277–281. [DOI] [PubMed] [Google Scholar]
- 6.Singleton M.R. and Wigley,D.B. (2002) Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol., 184, 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halford S.E., Welsh,A.J. and Szczelkun,M.D. (2004) Enzyme-mediated DNA looping. Annu. Rev. Biophys. Biomol. Struct., 33, 1–24. [DOI] [PubMed] [Google Scholar]
- 8.Garcia L.R. and Molineux,I.J. (1999) Translocation and specific cleavage of bacteriophage T7 DNA in vivo by EcoKI. Proc. Natl Acad. Sci. USA, 96, 12430–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies G.P., Kemp,P., Molineux,I.J. and Murray,N.E. (1999) The DNA translocation and ATPase activities of restriction-deficient mutants of EcoKI. J. Mol. Biol., 292, 787–796. [DOI] [PubMed] [Google Scholar]
- 10.Firman K. and Szczelkun,M.D. (2000) Measuring motion on DNA by the type I restriction endonuclease EcoR124I using triplex displacement. EMBO J., 19, 2094–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meisel A., Bickle,T.A., Krüger,D.H. and Schroeder,C. (1992) Type III restriction enzymes need two inversely oriented recognition sites for DNA cleavage. Nature, 355, 467–469. [DOI] [PubMed] [Google Scholar]
- 12.Meisel A., Mackeldanz,P., Bickle,T.A., Kruger,D.H. and Schroeder,C. (1995) Type III restriction endonucleases translocate DNA in a reaction driven by recognition site-specific ATP hydrolysis. EMBO J., 14, 2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saha S. and Rao,D.N. (1995) ATP hydrolysis is required for DNA cleavage by EcoPI restriction enzyme. J. Mol. Biol., 247, 559–567. [DOI] [PubMed] [Google Scholar]
- 14.Saha S. and Rao,D.N. (1997) Mutations in the Res subunit of the EcoPI restriction enzyme that affect ATP-dependent reactions. J. Mol. Biol., 269, 342–354. [DOI] [PubMed] [Google Scholar]
- 15.Stark W.M. and Boocock,M.A. (1995) Topological selectivity in site-specific recombination. In Sherratt,D.J. (ed.), Mobile Genetic Elements. Oxford University Press, Oxford, UK, pp. 101–129. [Google Scholar]
- 16.Kingston I.J., Gormley,N.A. and Halford,S.E. (2003) DNA supercoiling enables the type IIS restriction enzyme BspMI to recognize the relative orientation of two DNA sequences. Nucleic Acids Res., 31, 5221–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szczelkun M.D., Dillingham,M.S., Janscak,P., Firman,K. and Halford,S.E. (1996) Repercussions of DNA tracking by the type IC restriction endonuclease EcoR124I on linear, circular and catenated substrates. EMBO J., 15, 6335–6347. [PMC free article] [PubMed] [Google Scholar]
- 18.Berge T., Ellis,D.J., Dryden,D.T., Edwardson,J.M. and Henderson,R.M. (2000) Translocation-independent dimerization of the EcoKI endonuclease visualized by atomic force microscopy. Biophys. J., 79, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saldanha R., Flanagan,P. and Fennewald,M. (1987) Recombination by resolvase is inhibited by lac repressor simultaneously binding operators between res sites. J. Mol. Biol., 196, 505–516. [DOI] [PubMed] [Google Scholar]
- 20.Parker C.N. and Halford,S.E. (1991) Dynamics of long range interactions on DNA: the speed of synapsis during site-specific recombination by resolvase. Cell, 66, 781–791. [DOI] [PubMed] [Google Scholar]
- 21.Grindley N.D.F. (1993) Analysis of a nucleoprotein complex: the synaptosome of γδ resolvase. Science, 260, 738–740. [DOI] [PubMed] [Google Scholar]
- 22.Oram M., Marko,J.F. and Halford,S.E. (1997) Communications between distant sites on supercoiled DNA from non-exponential kinetics for DNA synapsis by resolvase. J. Mol. Biol., 270, 396–412. [DOI] [PubMed] [Google Scholar]
- 23.Szczelkun M.D. and Halford,S.E. (1996) Recombination by resolvase to analyse DNA communications by the SfiI restriction endonuclease. EMBO J., 15, 1460–1469. [PMC free article] [PubMed] [Google Scholar]
- 24.Milsom S.E., Halford,S.E., Embleton,M.L. and Szczelkun,M.D. (2001) Analysis of DNA looping interactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol., 311, 515–527. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J.C. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.Yanisch-Perron C., Vieira,J. and Messing,J. (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene, 33, 103–119. [DOI] [PubMed] [Google Scholar]
- 27.Tomizawa J. (1985) Control of ColE1 plasmid replication: initial interaction of RNA I and the primer transcript is reversible. Cell, 40, 527–535. [DOI] [PubMed] [Google Scholar]
- 28.Tomizawa J. (1986) Control of ColE1 plasmid replication: binding of RNA I to RNA II and inhibition of primer formation. Cell, 47, 89–97. [DOI] [PubMed] [Google Scholar]
- 29.Vipond I.B., Baldwin,G.S., Oram,M., Erskine,S.G., Wentzell,L.M., Szczelkun,M.D., Nobbs,T.J. and Halford,S.E. (1995) A general assay for restriction endonucleases and other DNA-modifying enzymes with plasmid substrates. Mol. Biotechnol., 4, 259–268. [DOI] [PubMed] [Google Scholar]
- 30.Gowers D.M. and Halford,S.E. (2003) Protein motion from non-specific to specific DNA by three-dimensional routes aided by supercoiling. EMBO J., 22, 1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adzuma K. and Mizuuchi,K. (1989) Interaction of proteins located at a distance along DNA: mechanism of target immunity in the Mu DNA strand-transfer reaction. Cell, 57, 41–47. [DOI] [PubMed] [Google Scholar]
- 32.Embleton M.L., Siksnys,V. and Halford,S.E. (2001) DNA cleavage reactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol., 311, 503–514. [DOI] [PubMed] [Google Scholar]
- 33.Levene S.D., Donahue,C., Boles,T.C. and Cozzarelli,N.R. (1995) Analysis of the structure of dimeric DNA catenanes by electron microscopy. Biophys. J., 69, 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pingoud A. and Jeltsch,A. (2001) Structure and function of type II restriction endonucleases. Nucleic Acids Res., 29, 3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Topal M.D., Thresher,R.J., Conrad,M. and Griffith,J. (1991) NaeI endonuclease binding to pBR322 DNA induces looping. Biochemistry, 30, 2006–2010. [DOI] [PubMed] [Google Scholar]
- 36.Bath A.J., Milsom,S.E., Gormley,N.A. and Halford,S.E. (2002) Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem., 277, 4024–4033. [DOI] [PubMed] [Google Scholar]
- 37.Gormley N.A., Hillberg,A.L. and Halford,S.E. (2002) The type IIs restriction endonuclease BspMI is a tetramer that acts concertedly at two copies of an asymmetric DNA sequence. J. Biol. Chem., 277, 4034–4041. [DOI] [PubMed] [Google Scholar]
- 38.Reuter M., Kupper,D., Meisel,A., Schroeder,C. and Kruger,D.H. (1998) Cooperative binding properties of restriction endonuclease EcoRII with DNA recognition sites. J. Biol. Chem., 273, 8294–8300. [DOI] [PubMed] [Google Scholar]
- 39.Friedhoff P., Lurz,R., Luder,G. and Pingoud,A. (2001) Sau3AI, a monomeric type II restriction endonuclease that dimerizes on the DNA and thereby induces DNA loops. J. Biol. Chem., 276, 23581–23588. [DOI] [PubMed] [Google Scholar]
- 40.Siksnys V., Skirgaila,R., Sasnauskas,G., Urbanke,C., Cherny,D., Grazulis,S. and Huber,R. (1999) The Cfr10I restriction enzyme is functional as a tetramer. J. Mol. Biol., 291, 1105–1118. [DOI] [PubMed] [Google Scholar]
- 41.Rosamond J., Endlich,B. and Linn,S. (1979) Electron microscopic studies of the mechanism of action of the restriction endonuclease of Escherichia coli B. J. Mol. Biol., 129, 619–635. [DOI] [PubMed] [Google Scholar]
- 42.Yuan R., Hamilton,D.L. and Burckhardt,J. (1980) DNA translocation by the restriction enzyme from E.coli K. Cell, 20, 237–244. [DOI] [PubMed] [Google Scholar]
- 43.Endlich B. and Linn,S. (1985) The DNA restriction endonuclease of Escherichia coli B. I. Studies of the DNA translocation and the ATPase activities. J. Biol. Chem., 260, 5720–5738. [PubMed] [Google Scholar]
- 44.Ellis D.J., Dryden,D.T., Berge,T., Edwardson,J.M. and Henderson,R.M. (1999) Direct observation of DNA translocation and cleavage by the EcoKI endonuclease using atomic force microscopy. Nature Struct. Biol., 6, 15–17. [DOI] [PubMed] [Google Scholar]
- 45.Janscak P. and Bickle,T.A. (2000) DNA supercoiling during ATP-dependent DNA translocation by the type I restriction enzyme EcoAI. J. Mol. Biol., 295, 1089–1099. [DOI] [PubMed] [Google Scholar]
- 46.Krasnow M.A. and Cozzarelli,N.R. (1983) Site-specific relaxation and recombination by the Tn3 resolvase: recognition of the DNA path between oriented res sites. Cell, 32, 1313–1324. [DOI] [PubMed] [Google Scholar]
- 47.Kanaar R., van de Putte,P. and Cozzarelli,N.R. (1989) Gin-mediated recombination of catenated and knotted DNA substrates: implications for the mechanism of interaction between cis-acting sites. Cell, 58, 147–159. [DOI] [PubMed] [Google Scholar]
- 48.Stark W.M., Sherratt,D.J. and Boocock,M.R. (1989) Site-specific recombination by Tn3 resolvase: topological changes in the forward and reverse reactions. Cell, 58, 779–790. [DOI] [PubMed] [Google Scholar]
- 49.Benjamin H.W. and Cozzarelli,N.R. (1990) Geometric arrangements of Tn3 resolvase sites. J. Biol. Chem., 265, 6441–6447. [PubMed] [Google Scholar]
- 50.Benjamin K.R., Abola,A.P., Kanaar,R. and Cozzarelli,N.R. (1996) Contributions of supercoiling to Tn3 resolvase and phage Mu Gin site-specific recombination. J. Mol. Biol., 256, 50–65. [DOI] [PubMed] [Google Scholar]
- 51.Janscak P., Sandmeier,U. and Bickle,T.A. (1999) Single amino acid substitutions in the HsdR subunit of the type IB restriction enzyme EcoAI uncouple the DNA translocation and DNA cleavage activities of the enzyme. Nucleic Acids Res., 27, 2638–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu L.F. and Wang,J.C. (1987) Supercoiling of the DNA template during transcription. Proc. Natl Acad. Sci. USA, 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ostrander E.A., Benedetti,P. and Wang,J.C. (1990) Template supercoiling by a chimera of yeast GAL4 protein and phage T7 RNA polymerase. Science, 249, 1261–1265. [DOI] [PubMed] [Google Scholar]
- 54.Koo H.S., Claassen,L., Grossman,L. and Liu,L.F. (1991) ATP-dependent partitioning of the DNA template into supercoiled domains by Escherichia coli UvrAB. Proc. Natl Acad. Sci. USA, 88, 1212–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pieper U., Groll,D.H., Wunsch,S., Gast,F.U., Speck,C., Mucke,N. and Pingoud,A. (2002) The GTP-dependent restriction enzyme McrBC from Escherichia coli forms high-molecular mass complexes with DNA and produces a cleavage pattern with a characteristic 10-base pair repeat. Biochemistry, 41, 5245–5254. [DOI] [PubMed] [Google Scholar]