


Abstract
Protein-primed replication of hepatitis B viruses (HBVs) is initiated by the chaperone dependent binding of the reverse transcriptase (P protein) to the bulged ε stem-loop on the pregenomic RNA, and the ε-templated synthesis of the 5′ terminal nucleotides of the first DNA strand. How P protein recognizes the initiation site is poorly understood. In mammalian HBVs and in duck HBV (DHBV) the entire stem-loop is extensively base paired; in other avian HBVs the upper stem regions have a low base pairing potential. Initiation can be reconstituted with in vitro translated DHBV, but not HBV, P protein and DHBV ε (Dε) RNA. Employing the SELEX method on a constrained library of Dε upper stem variants, we obtained a series of well-binding aptamers. Most contained C-rich consensus motifs with very low base pairing potential; some supported initiation, others did not. Consensus-based secondary mutants allowed to pin down this functional difference to the residues flanking the conserved loop, and an unpaired U. In vitro active consensus sequences also supported virus replication. Hence, most of the upper stem acts as a spacer, which, if not base paired, warrants accessibility of relevant anchor residues. This suggests that the base paired Dε represents an exceptional rather than a prototypic avian HBV ε signal, and it offers an explanation as to why attempts to in vitro reconstitute initiation with human HBV have thus far failed.
INTRODUCTION
Human hepatitis B virus (HBV), the causative agent of B-type hepatitis (1), is the type member of the hepadnaviruses which infect a few other mammalian and some bird species. Their 3 kb DNA genome is generated by reverse transcription of the pregenomic RNA (pgRNA), which also serves as mRNA for the capsid protein and the reverse transcriptase, called P protein [for reviews: (2–4)]. P protein contains, in addition to the evolutionarily conserved DNA polymerase and RNase H domains (5–7), a unique terminal protein (TP) domain at its N-terminus which acts as a protein primer for reverse transcription (8). Replication is initiated by the binding of P to an about 60 nt bulged stem-loop, ε, close to the 5′ end of the pgRNA (Figure 1); this interaction also mediates selective pgRNA packaging into capsids (9,10). The product of this priming reaction is a 3 or 4 nt DNA which is templated by ε and whose 5′end is covalently linked to a Tyr-residue in TP (11); this oligonucleotide is subsequently used to prime synthesis of a full-length (−)-DNA from a 3′-proximal RNA element. P binding to ε is dependent on cellular chaperones (12–15). Hsp40 and Hsc70 plus ATP are absolutely indispensable (16), and Hop plus Hsp90 enhance the reaction. The only cell-free system to reconstitute the priming reaction in the test tube was, until very recently (16,17), in vitro translation of duck HBV (DHBV) P protein in rabbit reticulocyte lysate (RL) which also provides the chaperones and, probably, further factors that enable the P protein to bind to its cognate Dε signal (8). Upon addition of dNTPs, the complex carries out synthesis of the oligonucleotide primer; using α-32P-labelled dNTPs the reaction can sensitively be monitored via the covalent linkage of the labelled primer to the P protein.
Figure 1.
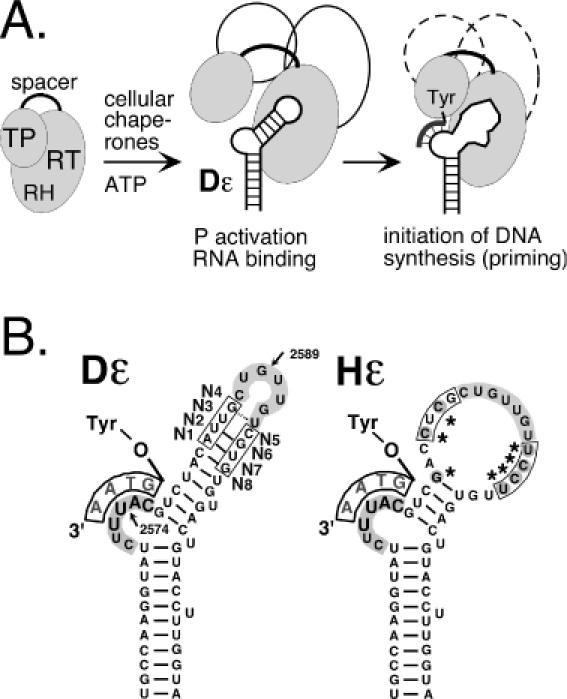
Hepadnavirus replication initiation. (A) Chaperone-dependent activation of P protein. P protein consists of TP, reverse transcriptase (RT) and RNaseH (RH) domains and a non-essential spacer. Cellular chaperones (symbolized by open ovals) are essential to enable P to bind its cognate ε RNA. A structural alteration in the upper stem allows a Tyr-residue in TP to act as acceptor for the 5′ terminal nucleotide of a short DNA oligonucleotide (thick line attached to Tyr) that is templated by the bulge region. This priming reaction can be reconstituted in RL. (B) Secondary structures of duck (DHBV) and heron HBV (HHBV) ε signals. Both Dε and Hε consist of a lower stem, a bulge and an upper stem, which is largely base paired in Dε but not in Hε. The conserved apical sequence, highlighted by grey background, is commonly referred to as loop although in Dε the 3′ terminal U is most probably base paired as shown; in Hε it is part of a large region lacking conventional base pairs due to various nucleotide exchanges (marked by asterisks). The nucleotide positions most sensitive to nucleases are indicated by arrows, and the randomized positions are marked N1 to N8. Equivalent Hε residues are boxed.
In the absence of a nucleic acid primer precise initiation of hepadnaviral DNA synthesis (for Dε at C2576; Figure 1) must be guided by information stored in the sequence and/or structure of ε. Stuctural features shared by all hepadnaviral ε elements (Figure 1B) are (i) a central bulge, part of which acts as template for the DNA primer; (ii) a lower stem; (iii) an upper stem with an apical loop whose sequence is conserved in all avian, and with a slightly different sequence, in all mammalian hepadnaviruses. This overall structure is important because deletions of the subelements abolish ε function. More subtle mutations, mostly studied with in vitro translated DHBV P protein, defined some positions in Dε where the specific bases are important, such as U2590 in the loop (18) and the 3 bp underlying the bulge (19). Often, however, the effects of nucleotide exchanges are indirect in that they cause the RNA to adopt stable non-wild-type-like structures that prevent P binding. Moreover, P binding is not sufficient for an RNA to be a suitable priming template, as there are mutants which bind to P but are priming-incompetent (18). Hence replication initiation is probably a two-step process in which the initial physical RNA binding requires a subsequent structural rearrangement for its use as template (20).
The role of the upper stem separating the bulge and loop elements is poorly understood. In HBV ε, this region is completely base paired, except for a single unpaired U, and according to NMR data is capped by a stable tri-loop (21). The primary sequence of the entire stem-loop and hence the potential to form such a rigid structure is highly conserved in all mammalian hepadnaviruses, suggesting that it is important for recognition by P protein. The Dε structure, although less stable, resembles that of HBV ε (18) which is, however, not accepted by DHBV P protein as template. Sequence data from other avian hepadnaviruses, initially restricted to heron HBV (HHBV; (22)) but recently expanded to viruses from other avian hosts, reveal a substantial variability in this region, mostly incompatible with extensive base pairing in the upper stem (Table 1). Remarkably, the Hε signal (Figure 1B) of HHBV can productively interact with DHBV P protein (23) although the top half of its upper stem lacks any conventional base pairs. Hence, in avian hepadnaviruses either a stably structured upper stem is not crucial for P recognition, or the different sequences generate common structural features that are not adequately described by canonical base pairing.
Table 1. Natural sequence variability in avian hepadnavirus ε signals.

The left column gives the virus designation, or the host organism; numbers indicate specific isolates. The N1 to N4 and N5 to N8 positions are shown in underlined capitals. Genbank accession codes for each sequence are given on the right.
Defining by site-directed mutagenesis the features which distinguish binding from non-binding and priming-competent from priming-deficient RNAs has been hampered by the size of Dε, its propensity to adopt substantially different secondary structures by even single nucleotide exchanges (18,20), the low amounts of P protein from in vitro translation (in the range of 1 ng per microlitre RL) and the only transient association of chaperones with substrate proteins (24).
A potential alternative is the SELEX (systematic evolution of ligands by exponential enrichment) procedure (25,26) in which a pool of sequences is subjected to successive cycles of binding to a ligand [for reviews see (27,28)]. As yet, it has not been applied to the hepadnaviral P-ε system. However, in vitro translated His-tagged P protein can be bound as a chaperone-activated, priming-competent Dε-containing complex to Ni2+-NTA agarose (29), prompting us to investigate this possibility. Because such a selection would solely be based on P protein binding, we anticipated to find both priming-competent and priming-incompetent binders; a functional comparison should then allow one to distinguish residues that are crucial for mere P binding from those with a specific role in priming. As a first step, we generated a pool of nominally 48 (65,536) Dε RNAs in which the four nucleotide positions preceding (N1–N4), and those four following the apical loop (N5–N8) were randomized (Figure 1B). Except for the N4 and N5 positions these two regions are hot-spots of natural variability (Table 1). Maintaining the conserved Dε subelements was expected to minimize selection of aptamers that bind to the P protein complex at functionally irrelevant sites. Below we report on the characterization of individual variants obtained by this approach and their use as a new sequence framework in which the role of defined nucleotide positions can be studied, decoupled from interference by concomitant changes in secondary structure.
MATERIALS AND METHODS
Bacterial strains and eukaryotic cells
All plasmids were propagated in Escherichia coli Top 10 cells (Invitrogen). For cell culture studies, the chicken hepatoma cell line LMH was used. Cells were maintained as previously described previously (30).
Plasmid constructs
For in vitro translation of His-tagged DHBV P protein plasmid pT7AMVpolDHBV16-N-His6Δ3′eps was used (29). In brief it contains, behind the T7 promoter, a leader sequence from alphalpha mosaic virus for enhanced translation, the complete P open reading frame from DHBV 16 (nucleotide positions 170–2530) plus an insert of 6 His codons between codon 2 and 3. Constructs for expression of complete DHBV genomes were based on plasmid pCD16 (31) which contains a slightly overlength DHBV16 genome (nucleotide positions 2520–3021/1 to2816) under control of the cytomegalovirus (CMV) IE enhancer/promoter. pCD16 derivatives containing selected aptamer sequences were obtained through PCR-mediated mutagenesis. Wild-type Dε RNA was in vitro transcribed from plasmid pBDεwt that contains DHBV16 sequence from position 2520–2652 (19).
In vitro translation
In vitro translations were performed in rabbit RL using the TNT T7 Quick Coupled Transcription/Translation system (Promega) programmed with plasmid pT7AMVpolDHBV16-N-His6Δ3′eps. For 35S-labelling, 35S methionine (specific activity 1000 Ci/mmol; Amersham/Pharmacia) was used at 25 μCi per 50 μl reaction.
In vitro transcription
T7 RNA polymerase mediated run-off transcription was performed as described previously (18). The starting RNA pool was generated by annealing the (minus)-polarity oligo DepsN4LN4(-) with a plus-polarity T7 promoter oligo, and the resulting partial duplex DNA was used as a template for transcription using the T7 MEGAshortscript kit (Ambion). The complementary sequence of oligo DepsN4LN4(-) was GAATTAATACGACTCACTATAGGGCTGCCAAGGTATCTTTACGTCTACNNNNCTGTTGTNNNNTGTGACTGTACCTTTGGTACCCTTT, with the Dε sequence given in bold face, and the T7 promoter in italics; randomized positions are indicated by underlined N. The products were analysed by electrophoresis in 12% denaturing polyacrylamide gels, followed by silver staining; RNA concentrations were determined by measuring the absorbance at 260 nm. Subsequent RNA pools were produced analogously, but using the duplex RT–PCR products as template.
In vitro priming reactions
Assays were performed as previously described in (18). In brief, P protein was in vitro translated for 1 h at 30°C; reaction scales were chosen so as to allow using aliquots from the same translation reaction for the desired number of parallel priming assays. Wild-type or variant Dε RNA were added (final concentration 1 μM unless indicated otherwise) and incubated for another hour at 30°C to allow for the chaperone-dependent formation of P–Dε complexes. Thereafter, 10 μl aliquots were mixed with an equal volume of 2 × priming buffer (final concentrations: 10 mM Tris–HCl pH 8.0, 6 mM MgCl2, 10 mM NH4Cl, 2 mM MnCl2, 0.2% NP-40, 0.5 mM spermidin, 0.06% β-mercaptoethanol) containing unlabelled dCTP, dGTP, and dTTP plus 0.5 μl α-32P dATP (3000 Ci/mmol; Amersham). After 1 h at 37°C, the reactions were stopped by adding SDS containing sample buffer. Aliquots were separated by SDS–PAGE (7.5% polyacrylamide, 0.1% SDS) using the Laemmli system. Gels were exposed on X-ray film or a Fuji BAS 1500 phosphoimager; semiquantitations were performed using MacBas Software.
Radioactive labelling of RNA
RNAs were internally labelled by in vitro transcription in the presence of [α-32P]UTP, 5′end-labelled RNAs were obtained by dephosphorylation and rephosphorylation with [γ-32P]ATP (5000 Ci/mmol) as described in (18); non-incorporated NTPs were removed using Quick Spin columns (Roche). For enzymatic structure probing RNAs were purified by denaturing gel electrophoresis and dissolved at a final concentration of 5 μM in TE buffer (specific radioactivity about 2 × 105 cpm/pmol).
In vitro selection of P binding RNAs
To a 50 μl in vitro translation reaction, pool RNA was added to a final concentration of 1 μM and incubated for 1 h at 30°C. For P protein negative controls the P encoding plasmid was omitted. Subsequently, 400 μl binding buffer (0.1 M sodium phosphate, pH 7.4, 150 mM NaCl, 20 mM imidazol, 0.1% (v/v) NP-40, 100 μg/ml yeast tRNA) containing 50 μl (gel bed) Ni2+ NTA agarose beads (Qiagen) were added (20), and incubated for one more hour. To remove unbound RNA and RL components, the beads were washed twice with 1 ml each of ice cold binding buffer, then twice with 1 ml each of TMK buffer (50 mM Tris/HCl, pH7.5, 10 mM MgCl2, 40 mM KCl, 100 μg/ml yeast tRNA). Finally the beads were suspended in 100 μl TMK buffer and P protein bound RNA was purifed by phenol extraction. RNAs were precipitated, and dissolved in 20 μl TE buffer.
RT–PCR amplification
An aliquot of 1 μl of the RNA aptamer solution was reverse transcribed using Superscript II (Gibco/BRL) reverse transcriptase as recommended by the supplier. The RNA template was degraded by alkaline hydrolysis and an aliquot of this solution was amplified using Taq DNA polymerase (Promega). RT–PCR products were directly sequenced using the Thermo Sequenase Cycle Sequencing Kit (USB, Cleveland, OH). Initially, the products from the ninth selection round were cloned via the terminal Bam HI and Eco RI sites into pUC 18, and plasmid DNAs from 15 individual colonies were sequenced (s1–s15); s9 was heterogeneous and not further analysed. For a more thorough characterization, RT–PCR products from rounds 2, 3, 4, 6 and 9 were amplified using Pfx polymerase (Invitrogen), cloned into pUC 19, and between 22 and 36 plasmid DNAs from each round were sequenced.
Competition assay for RNA binding to P protein
RNAs were bound to in vitro translated P protein as described above except that the P protein was labelled using 35S-Met. After 1 h at 30°C, a mixture of internally 32P-labelled wt Dε RNA (50 nM final concentration) plus 1 μM unlabelled aptamer RNA was added and incubated for 1 h; then the P protein and bound RNAs were enriched using Ni-NTA agarose beads. After the fourth wash, the beads were mixed with 200 μl SDS-PAGE sample buffer, 80 μl 1 M imidazol pH7.5 and 80 μl 0.5 M EDTA pH 8.0 to release the RNA. 20 μl aliquots were analysed by SDS-PAGE (14% polyacrylamide, 0.1% SDS). 32P-labelled RNA and 35S labelled P protein were quantitated by phosphorimaging.
Direct RNA-binding assays
The assays were performed essentially as described above except that internally 32P-labelled RNAs (50 nM final concentration) were added to the in vitro translation reactions. The radioactivity in the supernatants, and in the Ni2+-NTA agarose beads after each washing step was determined by Cerenkov counting. After the fourth washing step, bound RNAs were analysed by denaturing polyacrylamide gel electrophoresis. Bands were visualized and quantified by phosphorimaging.
RNA secondary structure probing
Enzymatic structure probing was performed essentially as described, using 5′end labelled transcripts (20). In brief, the RNAs were denatured and renatured, and aliquots containing about 5 × 104 cpm were dissolved in 50 μl TMK buffer containing 100 μg/ml of yeast tRNA and mixed at room temperature with one of the following nucleases: RNAse A (specific for unpaired pyrimidines; 1 ng per reaction, 1 min); RNase T1 (specific for unpaired G; 2 U per reaction, 10 min) or CL3 nuclease (specific for unpaired C residues; 4 U per reaction, 5 min). Reactions were stopped by phenol extraction, the products were precipitated and analysed by 8% denaturing PAGE and autoradiography. Alkaline ladders were obtained by treatment of 5′ labelled wt Dε RNA with 50 mM Na2CO3 (pH 9) at 90°C for 15 min. G-specific ladders were obtained by partial RNase T1 digestion under denaturing conditions.
Transfection of LMH cells
LMH cells were transfected with the appropriate pCD16 plasmids using FuGene6 (Roche) as previously described previously (7). Cells were harvested 4 days post transfection. For monitoring the formation of DNA-containing nucleocapsids, the cells were lysed, and aliquots corresponding to 20 μl of 1 ml cytoplasmic lysate from 6 × 106 cells were subjected to electrophoresis in 1% agarose gels (7). For protein detection, the gels were blotted onto a polyvinylidene fluoride (PVDF) membrane (Immobilon P; Millipore), and DHBV core protein was identified using a polyclonal rabbit antiserum against recombinant DHBV capsids, followed by a goat-anti-rabbit peroxidase conjugate and a chemiluminescent substrate (ECL+, Amersham). For detection of core-borne DNA gels run in parallel were blotted to a Nylon membrane (Roche). To release DNA from the capsids the membrane was soaked with 50 ml 0.2 N NaOH, 1.5 M NaCl for 15 s, neutralized by incubation in 50 ml 0.2 M Tris–HCl, pH 7.5, 1.5 M NaCl for 15 s, and washed twice with water for 1 min. After ultraviolet (UV) fixation, the membrane was probed with a 32P-labelled DHBV DNA fragment obtained by random priming. Bands were visualized by phosphorimaging.
Endogenous polymerase assay (EPA)
Endogenous polymerase assays were performed as previously described in (7). In brief, cytoplasmic capsids were immunoprecipitated, and incubated with dGTP, dCTP and dTTP, plus 0.5 μl [α-32P]dATP (3000 Ci/mmol). After 2 h at 37°C, and a 2 h chase with unlabelled dATP the proteins were digested with proteinase K and extracted with phenol. Non-incorporated dNTPs were removed from the aqueous phase by Quick Spin columns, and the reaction products were analysed by native agarose gel electrophoresis and autoradiography or phosphorimaging of the dried gels.
RESULTS
Experimental design and optimization of the selection scheme
The starting (round 0) RNA pool was generated by in vitro transcription from a partially duplex DNA oligonucleotide template. After incubation with in vitro translated His-tagged DHBV P protein in RL, P–Dε complexes were immobilized to Ni2+ NTA agarose beads. RNAs remaining bound after several washing steps were amplified by RT–PCR, using an upstream primer which reconstituted a T7 promoter such that the RT–PCR product could be directly used for a new round of in vitro transcription; the downstream primer provided additional restriction sites for later cloning (see Supplementary Figure 1 for a scheme).
Because of its complex dependence on chaperones, the affinity of wild-type Dε to P protein is not exactly known; estimates for an apparent Kd in the 25–100 nM range have been derived from the Dε RNA concentration required to generate a half-maximal priming signal (10,23). Hence, too extensive washing might result in unacceptable losses of bound RNAs, either directly due to dissociation from P or, indirectly, by loss of cellular factors; insufficient washing, in turn, might lead to the amplification of nonspecifically retained RNAs. As a preliminary test, a slightly longer in vitro transcript containing the HBV ε sequence between the identical PCR primer binding sites was mixed with Dε RNA and taken through the procedure; RT–PCR revealed that four washing steps were sufficient for its nearly complete removal (not shown). Second, the input amount of P protein-containing RL was varied. Expectedly, less RT–PCR product was obtained with less P protein. However, even without any P protein (P− reactions), RT–PCR products of the expected 103 bp size appeared with increasing numbers of PCR cycles (Supplementary Figure 2).
In all actual selection rounds, therefore, P− reactions were run in parallel, and only the material from the cycle giving the highest ratio of specific versus nonspecific products was used further. Direct sequencing of the RT–PCR product pools after the first few selection rounds revealed a strong preference for C at the N5 position, and a general preponderance of C at N1–N4, and C plus A at N6–N8 but no single winning sequence. Selection was therefore continued for a total of nine rounds.
Characterization of selected pool RNAs confirms successful selection
The RNA pools from rounds 0, 2, 4, 6, 8, and 9 were tested for their ability to compete with wild-type Dε RNA for P binding, and for their in vitro priming activity. For binding competition, radiolabelled wild-type Dε RNA was mixed with a 10-fold molar excess of pool RNA or of unlabelled Dε RNA, or no competitor, and was incubated with RL containing in vitro translated P protein; then the radioactivity remaining bound to the immobilized complexes after four washing steps was determined (Figure 2, hatched bars). Unlabelled wild-type Dε RNA reduced the signal to about 18% (5.4-fold reduction), the unselected pool RNA to only about 66% (1.5-fold); the competition potential increased quickly over round 2 (4.6-fold) to >8-fold in round 4, and then remained at a 5.7- 8.1-fold level.
Figure 2.
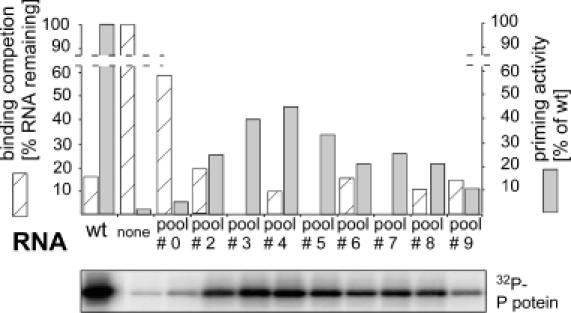
Functional characterization of SELEX-derived RNA pools. Binding to P protein was assessed by competition of the pool RNAs with radiolabelled wild-type Dε RNA (hatched bars); unlabelled wild-type Dε served as control. Complexes were immobilized to Ni2+-NTA agarose, washed, and the radioactivity remaining bound was measured by Cerenkov counting. The signal without competitor was set to 100%. Relative priming activities were determined by phosphoimaging of the priming signals (bottom panel) using the pool RNAs as template, and were normalized to the signal produced by the same concentration of wild-type Dε RNA set to 100% (grey shaded bars).
A similar course was seen when the pool RNAs were used as templates for in vitro priming (Figure 2, grey shaded bars). The priming signal with the starting pool RNA barely exceeded that of the background reaction in the absence of RNA, then increased strongly to reach a maximum with the round 4 RNA pool, at about half the intensity of the wild-type RNA control. The gradual decline with the later pool RNAs, despite their preserved binding competition potential, suggested that the fraction of binding-competent but priming-incompetent RNAs slowly increased. Together, these experiments confirmed that the selection scheme worked as expected.
Characterization of individual Dε aptamers from round 9
Reasoning that the latest pool would be the least heterogenous, we initially cloned out 14 individual sequences from round 9 (s1–s15; Table 2). Many contained C residues at the N1–N3 positions; and at N5–N8 the motifs CCAC (6 of 14) and CCAA (3 of 14) predominated. Nine clones had additional, probably PCR-acquired mutations at other sites. P-binding competence was first addressed by binding competition with wild-type 32P Dε RNA to 35S-P protein. RNAs retained on the immobilized complexes were separated by SDS-PAGE (14), and the 32P band intensities were normalized to the 35S signals in the corresponding lane (Figure 3A). While unlabelled wild-type RNA reduced binding to about 50% (not shown), aptamers s1, s2, s3, s5, s12 and s13 led to a stronger reduction (signals reduced to 15–35%); s4, s6 and s7 (not shown) competed with similar efficiency as unlabelled wild-type RNA (signals reduced to 50–60%); s11 and s14 reduced wild-type RNA binding to about 70% and s10 and s15 to about 90% of the control. Hence, except for s8 all sequences were able to compete with wild-type Dε for P protein-binding.
Table 2. Sequences and binding and priming activities of selected round 9 aptamers.
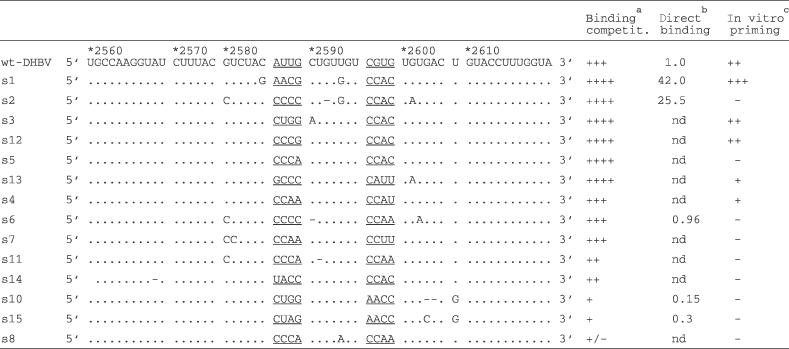
aCompetition efficiency as determined by the reduction of 32P-labelled wt-Dε RNA bound to P: +++, wt-like (40–50% reduction); ++++, > wt (65–85% reduction); ++, < wt (25–30% reduction); +, << wt (<15% reduction); +/− no detectable reduction. bCerenkov cpm of 32P-labelled RNA remaining bound to P after four washings; nd, not determined. cRelative intensity of priming signals. ++, wt-like; +++, > wt; +, < wt; −, no signal above background.
Figure 3.
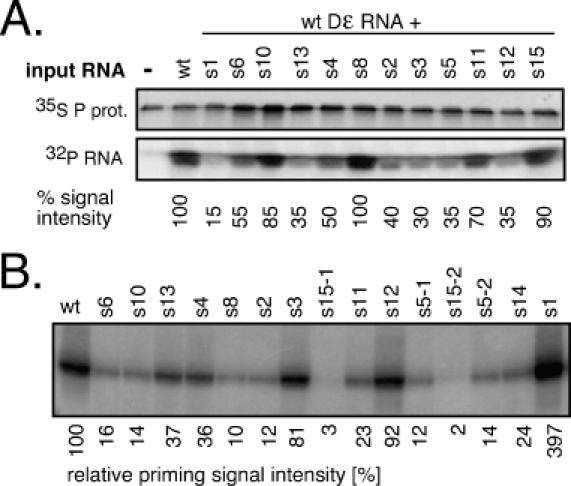
Characterization of individual round 9 aptamers. (A) Binding competition. 35S-Met labelled P protein was incubated with 50 nM 32P-labelled wild-type Dε RNA alone (lane wild-type), or plus the indicated aptamers at 2 μM concentration. Reaction products were separated by SDS-PAGE, and labelled protein and RNA were quantitated by phosphorimaging. The signal from the uncompeted reaction was set to 100%. (B) In vitro priming activities. In vitro transcribed aptamer RNAs were used as templates in priming assays. Reaction products were resolved by SDS-PAGE and 32P-labelled P protein was visualized by phosphorimaging. Aptamer identities are given on the top; for s5 and s15, two plasmid DNAs derived from separate bacterial colonies were used (s5-1 and s5-2; s15-1 and s15-2) to confirm reproducibility. Relative priming signal intensities, with that from wild-type Dε set at 100%, are shown below the gel.
In a direct-binding assay, 40- and 25-fold, respectively, more of the strongly competing aptamers s1 and s2 was retained on immobilized P complexes than of wild-type Dε RNA. The signal for s6 was similar (0.96-fold), and those for s15 and s10 (0.3- and 0.16-fold) were weaker than that of wild-type Dε (Supplementary Figure 3 and Table 2). Consistent data were obtained when the RNAs released from the beads were analysed by gel electrophoresis and autoradiography. The congruence between the direct and the competition data indicated that the aptamers bound to the same site on P as wild-type Dε.
The priming activities (Figure 3B) correlated for some RNAs with the binding data (strong signals in both assays for s1, s3, s12 versus weak signals for s10, s15) but not for others; in particular, the strongly binding aptamers s2 and s5 yielded only marginal, or very weak, priming signals. Hence, these data extend previous findings on the genetic separability of physical and productive P protein interaction.
Predictions and enzymatic probing (Supplementary Figure 4) suggested that s1 (strong-binding, strong-priming) has a similar secondary structure as wild-type Dε, with the region between the bulge and the apical loop being more accessible; s2 (strong-binding, no priming) gave a substantially different cleavage pattern. However, conclusions on whether the different structures were responsible for the different priming activities were hindered by their sequence divergence (Table 2). By contrast, aptamers s5 (strong-binding, weak-priming) and s12 (strong-binding, strong priming) differ only at the N4 position, which is G in s12 (as in wild-type Dε) but A in s5. Both produced very similar cleavage patterns that contained the typical bulge and loop signals U2574 and G2589 as wild-type RNA, however, many nucleotides between bulge and loop, as well as several nucleotides following G2589 in the loop were highly nuclease accessible (Figure 4). The unique G2586 in s12 gave a strong additional T1 product, indicating that it is not involved in base pairing. Hence, within a largely identical secondary structure context, the different priming phenotypes were related to the single G versus A exchange. However, whether this was a generalizable conclusion was not clear at this point (see below).
Figure 4.
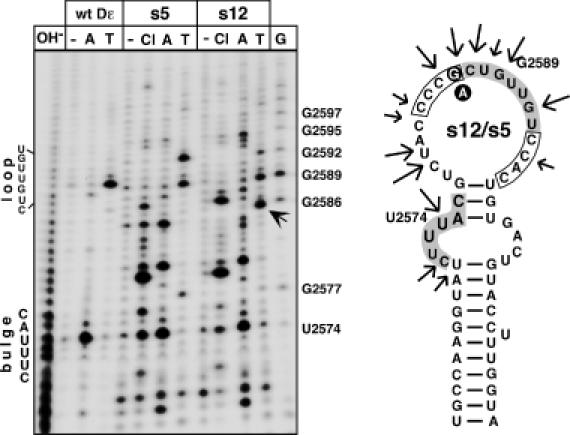
Secondary structure probing of round 9 aptamers s5 and s12. The 5′ abelled aptamers s12 (strongly priming) and s5 (weakly priming) were incubated with nuclease 3CL (Cl), RNase A (A), RNase T1 (T) or no enzyme (−). Reaction products were resolved by denaturing gel electrophoresis and visualized by autoradiography. The T1 product unique to s12 is highlighted by an arrow. Alkaline ladders (OH−) and G ladders (G) were obtained from wild-type Dε RNA. Positions of the bulge and loop in wild-type Dε are indicated on the left. On the secondary structure model the major nuclease sensitive sites are marked by arrows. The randomized regions are boxed, the bulge and loop residues as present in wild-type Dε are shown with grey background. The G versus an exchange is indicated by white letters on black background. The most stable M-Fold prediction has U2574 paired although it is highly accessible to RNase A; forcing it to be unpaired generates the structure shown. Note that both RNAs produce highly similar cleavage patterns. A corresponding analysis for aptamers s1 and s2 is shown as Supplementary Figure 4.
Sequencing of RNAs from preceding pools reveals a selection of C-rich consensus motifs at the randomized positions
For a more systematic elucidation of common features in the selected aptamers, from each of the round 0, 2, 3, 4, 6 and 9 RNA pools between 22 and 36 unambiguous sequences were obtained. A comparison of the frequencies of each nucleotide at the randomized positions throughout selection (Supplementary Figure 5A) showed (i) a rapid and exclusive selection of C, as in wild-type Dε, at the N5 position—increasing from about 20% in the unselected pool to 100% in round 3, which remained constant except for the re-emergence of a small percentage of A in round 9; (ii) a clear but less absolute preference for C (in 60 to 80% of all sequences) at N1, N2, N3 and N6; (iii) a preference for A over C at N7; (iv) a slight opposite trend at N8; (v) no clear preference for a specific nucleotide at N4, suggesting a lack of selection pressure at this site. As these individual frequencies do not account for the sequence context, we also looked for the presence of specific tetranucleotide motifs. Indeed, two motifs were highly preferred, i.e. CCCX (X being any nucleotide) at N1–N4, and CCAC and CCAA (jointly termed CCAm below) at N5–N8, each accounting for 50% or more of all sequences (Supplementary Figure 5B); in fact, the round 9 aptamers s5 and s12 and, with the caveat of additional outside mutations, also s1 and s2 conformed to this consensus. The frequency of mutations outside the randomized positions increased, explaining the sequence divergence in the round 9 pool; one extra mutation (U2604>G) fortuitously present in a fraction of the starting pool sequences persisted throughout selection; because of its prominent location opposite the initiation site in the bulge (Figure 1B) it was included in the subsequent analyses (see below).
The accumulation of the C-rich motifs excluded a requirement for wild-type Dε sequence and secondary structure for efficient binding at most of the randomized positions (compare Figure 4 for s5 and s12 as consensus prototypes). Importantly, this lack of base pairing potential offered a common sequence framework to analyse the functional role of individual nucleotides without much interference from the formation of alternative secondary structures. We chose three targets for a more detailed analysis, namely N4, where a G was suspected to be important for priming (s12 versus s5); the C at position N5 which was strongly selected for; and the G replacing the U2604 residue opposite the bulge. All variants used contained no outside mutations; some were present among the cloned-out individuals, others were newly constructed; these aptamer sequences are summarized in Table 3.
Table 3. Characteristics of SELEX-sequence derived secondary generation Dε RNA variants.
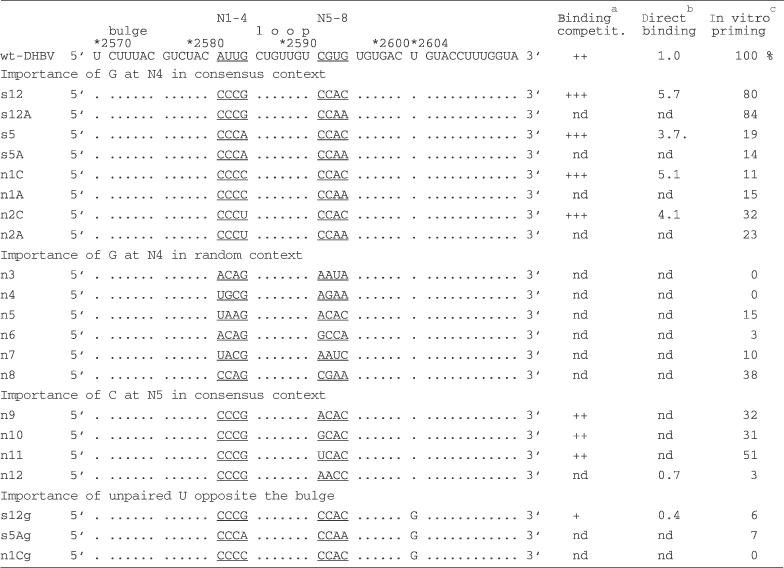
aCompetition efficiency as determined by the reduction of 32P-labelled wt-Dε RNA bound to P: ++, wt-like; +++, stronger competition than wt; +, weaker competition than wt. bRelative binding efficiency determined by Cerenkov cpm of 32P-labelled RNA remaining bound to P after four washings, with wt Dε RNA signal set to 1.0; nd, not determined. cRelative intensity of priming signals above background (no RNA), with wt-Dε signal set to 100%.
A G at N4 combined with a C at N5 is optimal for efficient priming
The importance of a G residue at N4 for priming was assessed in the context of the CCCX–CCAm consensus sequence; X was replaced from G to A, C and U; m was either C or A (Figure 5A). As before, s12 (X = G, m = C) produced a wild-type like priming signal whereas for all other X variants the signals were markedly reduced, with C and A at N4 giving the weakest (10–20% of wild type), and U giving slightly higher signals (25–30% of wild type). A versus C at N8 had no detectable effect. Hence, a G at N4 was crucial for efficient in vitro priming whereas at N8 both A and C were equally well tolerated. An M-Fold analysis substantiated the assumption that no major secondary structure changes were induced. All CCCX–CCAC and all CCCX–CCAA sequences produced identical most stable structures, with identical calculated free energies. In a direct-binding assay, both the well-priming X = G, m = C (i.e. s12) and the poorly priming X = C, m = C (i.e. n1C) variant showed nearly identical, increased affinities over wild-type Dε RNA (5.7- and 5.1-fold, respectively). Hence the different priming activities are clearly due to the nature of the base at N4.
Figure 5.
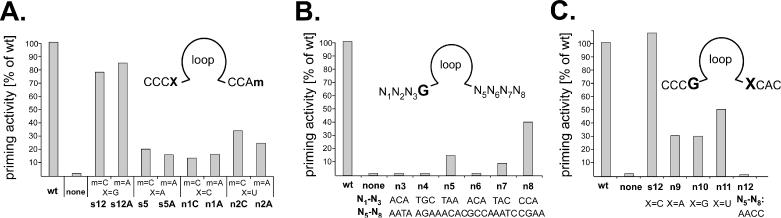
Priming activities of consensus-motif derived secondary generation aptamers. (A) Influence of the nucleotides at the N4 and N8 positions. Residue X (N4) in the consensus motif CCCX was changed from G to A, C and U, combined with either C or A at the m (N8) position in the downstream consensus motif CCAm. Nucleotides at the X and m positions, and the aptamer names are given below the graph; the X = G, m = C combination corresponds to s12. Bars show the relative priming signal intensities compared to wild-type Dε RNA set to 100%. (B) Context dependence of the priming-promoting activity of a G residue at N4. Aptamers with a G at N4 but various different sequences at N1 to N3 and N5 to N8 (shown below the graph) were subjected to in vitro priming assays. Note that only n8 with a sequence resembling the consensus motifs produced a significant signal. (C) Influence of the nucleotide at the N5 position. Within the priming-active CCCG–XCAC motif, the X residue (N5) was changed from C to A, G and U as indicated. Aptamer n12 is identical to s12A in (A), except that the sequence of the downstream CCAA motif is reversed. All priming assays shown in one graph were performed with one batch of P protein, and in parallel with positive (wild-type Dε RNA) and negative controls (no RNA).
All efficiently priming round 9 aptamers conformed to the N4 = G consensus (s1, s3 and s12) but not all variants with N4 = G were priming-active (s10 and s15). This suggested that the priming promoting effect of the G at N4 was context dependent. Therefore a collection of aptamers with a G at N4 but differing N1–N3 and N5–N8 sequences from the cloned out early round individuals was tested for priming (Figure 5B; n3–n8). Only n8 (CCAG–CGAA) which came closest to the CCCG - CCAm consensus produced a substantial signal; it was also the only aptamer tested with a C at N5. Next the importance of the N5 residue was analysed by replacing, in the context of the CCCG - CCAC consensus (i.e. s12), the C at N5 by A,G and U (Figure 5C; n9–n11). A and G reduced the activity to about 30%, U to about 50%, confirming that N4 = G plus N5 = C is the optimal combination. Mutant n12, finally, is identical to the well-priming aptamer s12A except that the order of the downstream CCAA motif is reversed to AACC. This change has no significant effect on secondary structure stability but completely ablated priming (Figure 5C) although binding to P was decreased only to about 70% of the wild-type Dε level (Table 3); hence the priming-defect of the round 9 aptamers s10 and s15 is probably also related to their AACC N5 to N8 motifs.
Replacement by G of the unpaired U opposite the bulge abolishes priming competence
To analyse a potential effect on priming of the fortuitous U2604G mutation, we chose U2604G variants of three of the above characterized aptamers, i.e. of s12 (called s12g), of s5A (s5Ag) and of n1 (n1g). The U2604G exchange did not improve the already poor priming performance of the latter two aptamers; strikingly, however, also s12g was virtually negative although the variant still bound to P with about half the affinity of wild-type Dε. Hence, even in the context of an otherwise optimal sequence context for priming this single substitution was detrimental for a productive interaction with P protein.
In vitro active aptamers are functional in the context of a complete DHBV genome
In the viral genome, the Dε sequence also acts, in concert with a downstream RNA element, as encapsidation signal for the pgRNA (32,33), and is part of the preC open reading frame from which a secretory, non-essential variant of the core protein (DHBeAg) is generated. Hence the authentic 5′ Dε element in the DHBV expression plasmid pCD16 was replaced by four aptamer sequences; s5 and s12 were chosen as prototypes of in vitro well priming and poorly priming members of the CCCX–CCAm consensus, s1 and s2 because of their high divergence from the wild-type Dε sequence.
The resulting plasmids were transfected into LMH cells, in parallel with the parental pCD16 plasmid. Formation of DNA containing intracellular nucleocapsids was assessed by native agarose gel electrophoresis where intact nucleocapsids run as distinct bands (34). The presence of nucleocapsids can be monitored by immunoblotting with anti-DHBV capsid protein antibodies, and encapsidated viral DNA can be detected by molecular hybridization. As shown in Figure 6 all constructs produced capsid-specific bands. Importantly, similar amounts of viral DNA as in wild-type capsids were present in the s1 and s12 derived samples; whereas the DNA signal was reduced in the s5 and absent from the s2 capsids. This was independently confirmed by an assay in which initiated DNA strands are extended in vitro by the encapsidated P protein (endogenous polymerase reaction) and, by inclusion of an [α-32P]-labelled dNTP, are radioactively labelled. Similar signals as with the wild-type genome, including full-length relaxed circular (RC) and linear (L) DNA, were obtained with s1 and s12 whereas the signals were decreased for s5 and absent from s2 (Figure 6, bottom). Hence, the in vitro priming phenotypes of all four aptamers were fully preserved in viral replication.
Figure 6.
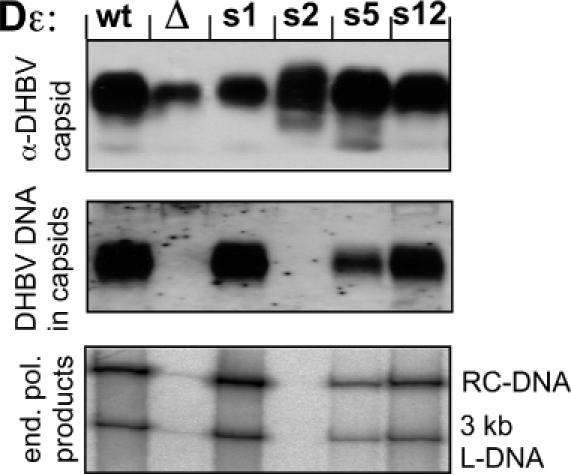
Functional analysis of selected aptamer sequences in a complete viral genome. LMH cells were transfected with DHBV expression plasmids carrying s1, s2, s5 and s12 derived Dε signals (s1, s2, s5 and s12), the wild-type genome (wt), or a modified wild-type genome lacking a functional ε signal (Δ). Cells were lysed and aliquots of the cytoplasmic lysate were run on a native agarose gel (upper two panels), blotted, and either probed with anti-DHBV capsid antibody (α-DHBV capsid) or hybridized with a DHBV specific DNA probe (DHBV DNA in capsids). Note that relative to the capsid signals, the DNA signals are increased in s1 and reduced in s5. Capsids immunoprecipitated from the lysates were subjected to an endogenous polymerase reaction; the products were separated by agarose gel electrophoresis and 32P-labelled viral genomes were visualized by phosphorimaging. The positions of the typical relaxed circular (RC) and linear (L) forms are indicated. The faint signal in lane Δ is due to contamination from the neighbouring lanes.
DISCUSSION
The multifunctional interaction between hepadnaviral P proteins and their cognate ε-elements is central to the virus life-cycle. Its inherent complexity has precluded direct structural analyses of the initiation complex, and both phylogeny and site-directed mutagenesis have provided only partial answers on the determinants of a productive interaction. The in vitro SELEX procedure, decoupling the basic requirements for P-binding from additional constraints on Dε for its functions as replication origin, encapsidation signal, and part of the precore open reading frame, appeared as an appropriate tool to address this question.
Methodical aspects
The SELEX concept (27,28) has recently been extended to complex targets such as in vitro translated proteins (35) and even whole cells (36–41); however, the peculiar dependence on cellular chaperones of the hepadnaviral P protein (12–17,20) has raised concern as to its applicability to this system. Several lines of evidence indicate that the approach worked as desired. The pool RNAs from successive rounds showed a clear increase in P-binding, and their priming capacity also increased strongly to round 4. At the N5 position, a C residue emerged quickly as the single predominant nucleotide. Consistent data were obtained with individual aptamers, and the congruency of the binding competition and the direct binding assays corroborated that the aptamers bound to the authentic Dε-binding site on the P protein.
Nonetheless, no single winning sequence emerged, and even the round 9 pool retained a certain heterogeneity. However, 11 out of 14 of the initially analysed individual sequences bound to P protein at least as strongly as wild-type Dε. The persistence of three relatively weakly binding aptamers might be due to the limited partitioning efficiency caused by the low abundance of RNA binding-competent P complexes.
Overall, however, these relative affinities are comparable to those in other SELEX studies. For SelB, a special elongation factor encoded by the fdhF gene that promotes selenocysteine incorporation (42), aptamers with 10-fold weaker to 50-fold stronger binding than the natural selenocysteine insertion sequence (SECIS) hairpin on fdhF mRNA have been reported (43,44); moreover, a variety of RNA sequences were able to bind to SelB but only some supported biological function (see below). For the phage PP7 operator the best aptamers bound only about 2- to 3-fold more strongly than the natural ligand (45). Notably, many exceptionally strong-binding aptamers such as a pseudoknotted RNA targeting the HIV-1 RT act as inhibitors (46–49); s1 and s12, in contrast, were functional templates for DNA primer synthesis by P protein. Obviously, therefore, a variety of sequence solutions exist for physical, and for productive, P-binding.
Though suggestive the initial round 9 aptamer analyses were limited by the accumulation of mutations outside the randomized regions. Hence, general conclusions could be derived only by including a much larger number of individual sequences. Cloned individuals from the preceding pools revealed a gradual loss of heterogeneity, contrasting with the rather quick increase in binding and priming capacity during the first four selection rounds. However, consensus motifs, i.e. CCCX at N1–N4 and CCAm at N5–N8, with two important characteristics emerged. First, their high C-content precludes base pairing and is also at variance with the naturally occurring sequences; therefore, neither a specific sequence nor a specific secondary structure is necessary for P-binding in these regions. Furthermore, a common non-conventional structure is highly unlikely; rather it is the absence of structure that seems important. Second, the consensus motifs provided a novel framework for a systematic mutational comparison of the role of the N4 and N5 positions in priming, decoupled from effects on overall structure.
Physical versus productive P protein-binding
Previous studies established the existence of Dε mutants, which bind to P protein but do not support priming (18,20). The selection of both priming-competent and incompetent aptamers allows one to generalize these individual observations. Of all CCCX - CCAm combinations, a G at the X position produced by far the strongest priming signals. As shown for aptamers s5 and s12 the different priming activities are not caused by different secondary structures. In the wild-type Dε context such a clear-cut conclusion would be obscured because the X = C and X = U mutations induce substantial differences in structure and stability. Together with the T1 nuclease accessibility this strongly suggests a direct role for the G at N4 in the formation of a priming-active RNP complex. Notably, also the s1 and s2 round 9 aptamers conformed to the consensus: both had the downstream CCAC motif, in the priming-competent s1 combined with a G at N4 (AACG), in the priming deficient s2 with a C at N4 (CCCC). Retrospectively, these data also provide an explanation for the binding-competent yet priming-inactive phenotypes of three site-directed mutants, K9, K10 and K12, in which the three positions in the upper stem part that are conserved between Dε and Hε (corresponding to N2, N4, and the nucleotide following N8) had simultaneously been replaced (18). At that time, it was unclear which of the mutations was responsible for the priming defect; now it is obvious that the lack of a G at N4 is the common denominator. Conversely, A or C at N8 made no significant difference, excluding both base-specific and secondary structure contributions for this nucleotide.
At the N5 position, analysed in the context of the upstream CCCG motif, the CCAC sequence outperformed the other combinations. Hence a C at N5, though not as crucial as a G at N4, significantly contributes to priming efficiency. That a U was second best to C may suggest an interaction with the G at N4. A canonical base pair is, however, unlikely, given that the G at N4 was easily accessible to RNase T1. One option is that N4 and N5 belong to one larger linear determinant, encompassing the loop, in which some anchor positions are important for physical binding as well as the rearrangement into a priming-active complex. This view would also explain why mutation of just one of the anchor residues does not entirely knock-out productive interaction with P protein. Alternatively, the classical loop sequence plus N4 and N5 might be involved in a specific three-dimensional structure that does not rely on Watson–Crick pairing but may be detectable by direct structural analyses.
In contrast, the U2604G mutation completely abrogated priming but not binding. A previously identified determinant for productive P protein-binding resides in the 3 bp underlying the bulge (19). U2604 is probably part of this recognition element but, because its replacement by G may lead to base pairing with the 5′ terminal C of the bulge, the structure as well as the sequence are affected; defining what is more important requires additional experimentation.
Together, the selection of both functionally active and inactive binders bears some resemblence to SELEX studies with the fdhF SECIS mRNA hairpin whose binding to SelB is required for efficient recognition of UGA as selenocysteine codon. In one class of selected aptamers the tip of the hairpin including the apical loop was highly conserved, and a mutant with various exchanges in the bottom part of the hairpin was active in UGA suppression. A second class of well-binding aptamers had the overall hairpin structure conserved but modified tip sequences; they were essentially inactive in suppression. This suggested the requirement, after binding, of a conformational change supported by the former but not the latter RNAs (43). In essence, we propose the same model for the P–ε system, with the apical loop and the region immediately underneath the bulge acting as two recognition elements that need to be properly spaced by the nucleotides forming the upper stem.
Role of the N1–N8 positions in hepadnavirus replication
Binding to in vitro translated P protein covers but one of the various functions of Dε, yet the results obtained with selected aptamer sequences within a complete DHBV genome completely mirrored the in vitro data: aptamers s1 and s12 supported virus replication at wild-type Dε levels, s5 showed a reduced activity and s2 was entirely inactive, confirming the absence of a specific sequence or secondary structure requirement for efficient priming except for the importance of a G at N4 combined with C at N5. Notably, all sequenced avian hepadnaviruses contain a G at N4 and many a C at N5; the only other nucleotide found at N5 is U which performed second best in in vitro priming.
The concept of the apical Dε loop being special because of its partial single-strandedness and conservation between DHBV and HHBV should therefore be extended to include the adjacent N4 and N5 positions. Similarly, the conjecture that base pairing in the upper stem part is important for P-binding and priming needs to be revised because not only Hε, which could have represented a rare exception, but a variety of in vitro selected as well as naturally occurring sequences are able of productive interaction with P. Their common denominator is, in fact, the absence of strong secondary structure, hence the base paired upper stem in Dε appears to be a tolerated exception rather than the rule. This also predicts that most of the other avian hepadnavirus ε elements are able to productively interact with DHBV P protein. This view is substantiated by reverse experiments in which the stability of the upper stem of wild-type Dε was deliberately increased. Exchanging U2584 (corresponding to N2) by C and U2585 by G generates N2–N6 and N3–N5 base pairs, strongly stabilizes the entire upper stem, and completely abolishes binding. A model summarizing these data is shown in Figure 7.
Figure 7.
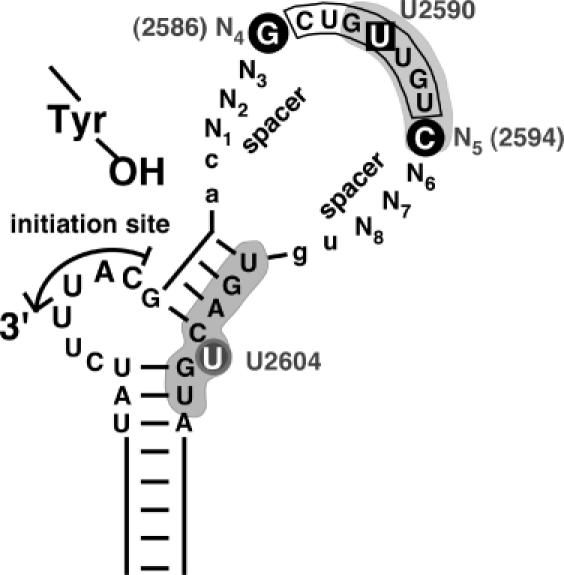
Revised view of relevant subelements in avian hepadnavirus ε signals. Based on this study and the expanded database of avian hepadnavirus sequences an open structure as in Hε, rather than Dε with its largely base paired upper stem, represents a prototypic ε signal. Functionally, the classical loop sequence (boxed) should be extended to encompass the adjacent N4 and N5 positions. N1 to N3 and N6 to N8, in contrast, serve mainly as spacers, with a lack of base pairing potential as a major requirement. Whether this also holds for the two preceding and the two following residues (lower case lettering) has not yet been experimentally addressed. Nucleotides with a specific role in priming are highlighted by black circles. Likely, they are part of a network of anchor residues, including the base pairs underlying the bulge and the unpaired U, that enable precise positioning of P over the initiation site. U2590 (boxed) in the loop sequence is one of the few residues whose mutation was previously shown to abrogate P-binding without affecting RNA secondary structure (18). Grey shading indicates a P protein footprint on the RNA (20). Notably, it includes the N5 position but spares N6 to N8 (and the two 3′ residues), in full accordance with the SELEX data and the natural sequence variability in this region.
That the exact SELEX-derived consensus motifs have not been found in nature may have several reasons. One is that transfection addresses only basic replication capacity whereas, in nature, small differences in replication efficicency would be amplified over many virus generations. In addition, the in vitro selected sequences affect the precore ORF which, in this part, encodes a signal sequence for the export of the precore protein precursor of DHBeAg. Though not essential, the conservedness of the ORF suggests some long-term advantage for the virus in vivo. The proline invariably encoded by the N2–N4 positions in the CCCX consensus (CCX) is unlikely to be a functionally neutral substitute for the leucine and serine residues present in all natural isolates. We are planning to address these issues by extending the SELEX approach established here to in vivo infections of ducks. Also, aptamers such as s2 that strongly bind to P protein but are priming-inactive lend themselves as decoys to redirect P protein from Dε on the pgRNA to replication-defective RNA molecules and thus to interfere with viral replication.
Finally, the open upper stem structure present in most avian hepadnavirus ε signals and its functionality with DHBV P protein shown in this study is in contrast to the extensive base pairing in the mammalian virus ε signals. At present, there is no in vitro system to directly test whether this is a coincidence or a fundamental difference. However, the high stability of the upper stem of the mammalian ε may hold a clue as to why HBV P protein, in vitro translated or recombinantly expressed, has never shown any priming activity when provided with HBV ε RNA. Rather than a factor acting on the reverse transcriptase, a factor acting on the RNA template, such as a helicase, may be missing from the reconstitution systems employed so far. Applying the SELEX approach as described here to HBV ε might therefore help to resolve this long-standing question.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG NA154/4-3) and by the Fonds der Chemischen Industrie. We thank Ida Wingert for excellent technical assistance.
REFERENCES
- 1.Blumberg B.S. (1997) Hepatitis B virus, the vaccine, and the control of primary cancer of the liver. Proc. Natl Acad. Sci. USA, 94, 7121–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nassal M. (1999) Hepatitis B virus replication: novel roles for virus-host interactions. Intervirology, 42, 100–116. [DOI] [PubMed] [Google Scholar]
- 3.Nassal M. (2000) Macromolecular interactions in hepatitis B virus replication and particle assembly. In Cann,A.J. (ed.), DNA Virus Replication. Oxford University Press, Oxford, UK, Vol. 26, pp. 1–40. [Google Scholar]
- 4.Ganem D. and Schneider,R. (2001) Hepadnaviridae: the viruses and their replication. In Fields,B.N. et al. (eds) Fields Virology, 4th edn Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5.Eickbush T.H. (1994) Origin and evolutionary relationships of retroelements. In Morse,S.S. (ed.), The Evolutionary Biology of Viruses. Raven Press, New York, NY, pp. 121–160. [Google Scholar]
- 6.Das K., Xiong,X., Yang,H., Westland,C.E., Gibbs,C.S., Sarafianos,S.G. and Arnold,E. (2001) Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol., 75, 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck J., Vogel,M. and Nassal,M. (2002) dNTP versus NTP discrimination by phenylalanine 451 in duck hepatitis B virus P protein indicates a common structure of the dNTP-binding pocket with other reverse transcriptases. Nucleic Acids Res., 30, 1679–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G.H. and Seeger,C. (1992) The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell, 71, 663–670. [DOI] [PubMed] [Google Scholar]
- 9.Knaus T. and Nassal,M. (1993) The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res., 21, 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollack J.R. and Ganem,D. (1994) Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol., 68, 5579–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber M., Bronsema,V., Bartos,H., Bosserhoff,A., Bartenschlager,R. and Schaller,H. (1994) Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol., 68, 2994–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu J. and Seeger,C. (1996) Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl Acad. Sci. USA, 93, 1060–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J., Toft,D.O. and Seeger,C. (1997) Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J., 16, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J. and Anselmo,D. (2000) In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol., 74, 11447–11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck J. and Nassal,M. (2001) Reconstitution of a functional duck hepatitis B virus replication initiation complex from separate reverse transcriptase domains expressed in Escherichia coli. J. Virol., 75, 7410–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beck J. and Nassal,M. (2003) Efficient Hsp90-independent in vitro activation by Hsc70 and Hsp40 of duck hepatits B virus reverse transcriptase, an assumed Hsp90 client protein. J. Biol. Chem., 278, 36128–36138. [DOI] [PubMed] [Google Scholar]
- 17.Hu J., Toft,D., Anselmo,D. and Wang,X. (2002) In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol., 76, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beck J. and Nassal,M. (1997) Sequence- and structure-specific determinants in the interaction between the RNA encapsidation signal and reverse transcriptase of avian hepatitis B viruses. J. Virol., 71, 4971–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaaf S.G., Beck,J. and Nassal,M. (1999) A small 2′-OH- and base-dependent recognition element downstream of the initiation site in the RNA encapsidation signal is essential for hepatitis B virus replication initiation. J. Biol. Chem., 274, 37787–37794. [DOI] [PubMed] [Google Scholar]
- 20.Beck J. and Nassal,M. (1998) Formation of a functional hepatitis B virus replication initiation complex involves a major structural alteration in the RNA template. Mol. Cell. Biol., 18, 6265–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flodell S., Schleucher,J., Cromsigt,J., Ippel,H., Kidd-Ljunggren,K. and Wijmenga,S. (2002) The apical stem-loop of the hepatitis B virus encapsidation signal folds into a stable tri-loop with two underlying pyrimidine bulges. Nucleic Acids Res., 30, 4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sprengel R., Kaleta,E.F. and Will,H. (1988) Isolation and characterization of a hepatitis B virus endemic in herons. J. Virol., 62, 3832–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck J., Bartos,H. and Nassal,M. (1997) Experimental confirmation of a hepatitis B virus (HBV) epsilon-like bulge-and-loop structure in avian HBV RNA encapsidation signals. Virology, 227, 500–504. [DOI] [PubMed] [Google Scholar]
- 24.Hartl F.U. and Hayer-Hartl,M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science, 295, 1852–1858. [DOI] [PubMed] [Google Scholar]
- 25.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 26.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 27.Wilson D.S. and Szostak,J.W. (1999) In vitro selection of functional nucleic acids. Annu. Rev. Biochem., 68, 611–647. [DOI] [PubMed] [Google Scholar]
- 28.Brody E.N. and Gold,L. (2000) Aptamers as therapeutic and diagnostic agents. J. Biotechnol., 74, 5–13. [DOI] [PubMed] [Google Scholar]
- 29.Beck J. and Nassal,M. (1996) A sensitive procedure for mapping the boundaries of RNA elements binding in vitro translated proteins defines a minimal hepatitis B virus encapsidation signal. Nucleic Acids Res., 24, 4364–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Protzer U., Nassal,M., Chiang,P.W., Kirschfink,M. and Schaller,H. (1999) Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc. Natl Acad. Sci. USA, 96, 10818–10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obert S., Zachmann-Brand,B., Deindl,E., Tucker,W., Bartenschlager,R. and Schaller,H. (1996) A splice hepadnavirus RNA that is essential for virus replication. EMBO J., 15, 2565–2574. [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrow K.M. and Loeb,D.D. (2002) Characterization of the cis-acting contributions to avian hepadnavirus RNA encapsidation. J. Virol., 76, 9087–9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calvert J. and Summers,J. (1994) Two regions of an avian hepadnavirus RNA pregenome are required in cis for encapsidation. J. Virol., 68, 2084–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birnbaum F. and Nassal,M. (1990) Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J. Virol., 64, 3319–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox J.C., Hayhurst,A., Hesselberth,J., Bayer,T.S., Georgiou,G. and Ellington,A.D. (2002) Automated selection of aptamers against protein targets translated in vitro: from gene to aptamer. Nucleic Acids Res., 30, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris K.N., Jensen,K.B., Julin,C.M., Weil,M. and Gold,L. (1998) High affinity ligands from in vitro selection: complex targets. Proc. Natl Acad. Sci. USA, 95, 2902–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blank M., Weinschenk,T., Priemer,M. and Schluesener,H. (2001) Systematic evolution of a DNA aptamer binding to rat brain tumor microvessels. Selective targeting of endothelial regulatory protein pigpen. J. Biol. Chem., 276, 16464–16468. [DOI] [PubMed] [Google Scholar]
- 38.Cerchia L., Hamm,J., Libri,D., Tavitian,B. and de Franciscis,V. (2002) Nucleic acid aptamers in cancer medicine. FEBS Lett., 528, 12–16. [DOI] [PubMed] [Google Scholar]
- 39.Famulok M., Mayer,G. and Blind,M. (2000) Nucleic acid aptamers-from selection in vitro to applications in vivo. Acc. Chem. Res., 33, 591–599. [DOI] [PubMed] [Google Scholar]
- 40.Gold L. (2002) RNA as the catalyst for drug screening. Nat. Biotechnol., 20, 671–672. [DOI] [PubMed] [Google Scholar]
- 41.Daniels D.A., Chen,H., Hicke,B.J., Swiderek,K.M. and Gold,L. (2003) A tenascin-C aptamer identified by tumor cell SELEX: systematic evolution of ligands by exponential enrichment. Proc. Natl Acad. Sci. USA, 100, 15416–15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thanbichler M. and Böck,A. (2001) Functional analysis of prokaryotic SELB proteins. Biofactors, 14, 53–59. [DOI] [PubMed] [Google Scholar]
- 43.Klug S.J., Hüttenhofer,A., Kromayer,M. and Famulok,M. (1997) In vitro and in vivo characterization of novel mRNA motifs that bind special elongation factor SelB. Proc. Natl Acad. Sci. USA, 94, 6676–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klug S.J., Hüttenhofer,A. and Famulok,M. (1999) In vitro selection of RNA aptamers that bind special elongation factor SelB, a protein with multiple RNA-binding sites, reveals one major interaction domain at the carboxyl terminus. RNA, 5, 1180–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim F. and Peabody,D.S. (2002) RNA recognition site of PP7 coat protein. Nucleic Acids Res., 30, 4138–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burke D.H., Scates,L., Andrews,K. and Gold,L. (1996) Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J. Mol. Biol., 264, 650–666. [DOI] [PubMed] [Google Scholar]
- 47.Jaeger J., Restle,T. and Steitz,T.A. (1998) The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J., 17, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kensch O., Connolly,B.A., Steinhoff,H.J., McGregor,A., Goody,R.S. and Restle,T. (2000) HIV-1 reverse transcriptase-pseudoknot RNA aptamer interaction has a binding affinity in the low picomolar range coupled with high specificity. J. Biol. Chem., 275, 18271–18278. [DOI] [PubMed] [Google Scholar]
- 49.Chaloin L., Lehmann,M.J., Sczakiel,G. and Restle,T. (2002) Endogenous expression of a high-affinity pseudoknot RNA aptamer suppresses replication of HIV-1. Nucleic Acids Res., 30, 4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.