

Abstract
Kelch-like 3 (KLHL3) is a component of an E3 ubiquitin ligase complex that regulates blood pressure by targeting With-No-Lysine (WNK) kinases for degradation. Mutations in KLHL3 cause constitutively increased renal salt reabsorption and impaired K+ secretion, resulting in hypertension and hyperkalemia. Although clinical studies have shown that dietary K+ intake affects blood pressure, the mechanisms have been obscure. In this study, we demonstrate that the KLHL3 ubiquitin ligase complex is involved in the low-K+-mediated activation of Na-Cl cotransporter (NCC) in the kidney. In the distal convoluted tubules of mice eating a low-K+ diet, we found increased KLHL3 phosphorylation at S433 (KLHL3S433-P), a modification that impairs WNK binding, and also reduced total KLHL3 levels. These changes are accompanied by the accumulation of the target substrate WNK4, and activation of the downstream kinases SPAK (STE20/SPS1-related proline-alanine-rich protein kinase) and OSR1 (oxidative stress-responsive 1), resulting in NCC phosphorylation and its accumulation at the plasma membrane. Increased phosphorylation of S433 was explained by increased levels of active, phosphorylated protein kinase C (but not protein kinase A), which directly phosphorylates S433. Moreover, in HEK cells expressing KLHL3 and WNK4, we showed that the activation of protein kinase C by phorbol 12-myristate 13-acetate induces KLHL3S433-P and increases WNK4 levels by abrogating its ubiquitination. These data demonstrate the role of KLHL3 in low-K+-mediated induction of NCC; this physiologic adaptation reduces distal electrogenic Na+ reabsorption, preventing further renal K+ loss but promoting increased blood pressure.
Keywords: hypokalemia, distal nephron, ubiquitin proteasome pathway, post-translational modification
Introduction
Hypertension affects one billion people worldwide, contributing to death from stroke, cardiovascular disease, and end-stage kidney disease. Genetic studies of Mendelian forms of hypertension have demonstrated the key role of Na-Cl handling in the distal nephron in the long-term control of blood pressure [1,2].
Among the monogenic forms of hypertension, pseudohypoaldosteronism type II (PHAII; also known as familial hypertensive hyperkalemia, Gordon syndrome, OMIM no. 145260) is of particular interest because of the unusual phenotype of hypertension and hyperkalemia. Mutations in four genes have been identified to cause PHAII [2–4]. Two genes encode serine threonine kinases With-No-Lysine 1 (WNK1) and WNK4, and the other two genes encode cullin-3 (CUL3) and Kelch-like 3 (KLHL3), elements of a RING (really interesting new gene) E3 ubiquitin ligase complex. Although their roles in blood pressure regulation were not known at the time of the discovery, subsequent biochemical analysis revealed their contribution to renal electrolyte homeostasis. WNK4’s kinase activity is activated by decreased intracellular Cl− levels [5,6], and directly phosphorylates the kinases SPAK (STE20/SPS1-related proline-alanine-rich protein kinase) and OSR1 (oxidative stress-responsive 1) [7]. SPAK and OSR1, in turn, phosphorylate and activate the thiazide-sensitive, Na-Cl cotransporter (NCC) in the distal convoluted tubules (DCT) [8]. Other targets of WNK4 have also been identified, including ROMK (renal outer medullary K+ channel), which is inhibited independently of the kinase activity [9]. The KLHL3/CUL3 ubiquitin ligase binds and targets WNK4 and WNK1 for degradation, regulating their levels [10,11]. In PHAII patients, missense mutations in the Kelch domain of KLHL3, and in an acidic domain of WNK4, both abrogate the association of KLHL3 and WNK4, preventing WNK4 degradation and resulting in constitutive Na-Cl reabsorption with impaired K+ secretion [2,4,10,11].
Previously, we have reported phosphorylation at S433 in the Kelch domain (KLHL3S433-P) that prevents binding and degradation of WNK4 [12]. Importantly, angiotensin II signaling via protein kinase C (PKC) increases KLHL3S433-P, increasing WNK4 levels in vivo. In addition, recent data indicate that this site may also be regulated by vasopressin signaling [13]. These data, together with the fact that S433 is recurrently mutated in PHAII patients [2,4], show that the inactivation of KLHL3 either by phosphorylation or single amino acid substitution is sufficient to have a major impact on blood pressure levels and on Na+, K+, Cl− handling in the kidney.
Clinical studies have demonstrated the inverse relationship between K+ intake and blood pressure levels [14,15]. However, the biochemical pathways that low-K+ intake increases blood pressure are not entirely clear. Recent studies reported that NCC activation is likely to be a central mechanism, given that K+ depletion and hypokalemia increase NCC phosphorylation independently of plasma aldosterone [16,17]. The contribution of KLHL3/CUL3-based ubiquitin ligase in this process has been unknown. In this study, we show that low K+ suppresses KLHL3 function, which results in NCC activation by abrogating WNK4 degradation, and activating kinases SPAK and OSR1.
Materials and Methods
Creation of monoclonal antibodies against KLHL3S433-P
Human KLHL3 peptide C-NTRRSS*VGVG (*phospho-Ser), with cysteine at the N-terminus, was coupled to keyhole limpet hemocyanin, and was injected into mice (Biogate). Spleen cells from the injected mice were fused with mouse myeloma cells P3U1, and the hybridomas producing phospho-specific antibodies were selected by ELISA and further by dot blot assay [12,18]. The monoclonal phospho-antibody used in the study displays virtually no cross-reactivity with unphosphorylated KLHL3 peptide (Supplemental Figure 1) nor unphosphorylated KLHL3 protein (see Results section).
Animals
The present study was approved by the institutional review board (IRB) in the Teikyo University Review Board #14-018. Male C57BL/6 mice at 6 weeks of age were obtained from Tokyo Laboratory Animals Science (Japan). Dietary manipulations included a low (0.005%) or a normal (0.9%) K+ diet for the indicated period. Na-Cl content (0.3%) in low-K+ diet is identical to that in normal-K+ diet.
Western blot
Western blotting was performed as described previously [12,18]. Plasma membrane fraction was purified using plasma membrane isolation kit (Invent). Primary antibodies included anti-KLHL3 (Sigma) [12], anti-Flag (Sigma), anti-tubulin (Sigma), anti-WNK4 (produced in the lab; Supplemental Figure 2), anti-phospho SPAK/OSR1 (Millipore) [19], anti-SPAK (Cell Signaling Technology; CST) [19], anti-NCC [12], anti-NCC phosphorylated at Thr53 and Thr71 [20], anti-phospho PKC (bearing phosphorylation at Ser660 in PKCβII; the antibody also detects phosphorylated PKCα, βI, δ, ε, η, and θ; CST), anti-phospho PKCα/β (bearing phosphorylation at Thr638/641; Abcam), anti-total PKC, anti-phospho protein kinase A (PKA; bearing phosphorylation at Thr197 in the catalytic domain), and anti-PKA catalytic domain (CST).
Immunostaining
Immunofluorescence study was performed as described previously [12,18]. Primary antibodies included polyclonal anti-KLHL3S433-P [12], anti-aquaporin-2 (Santa Cruz Biotechnology), and anti-calbindin D28-K (Swant).
Cell culture, transient transfection and cell treatment
HEK cells were incubated as described previously [21]. Transient transfection was carried out using non-liposomal polymer (Mirus Bio) [12]. Where indicated, phorbol 12-myristate 13-acetate (PMA; 200 nM) and bisindolylmaleimide I (BIM; 2 μM) were added. Ubiquitination assay was performed as described previously [11].
Statistical analysis
The data are summarized as means ± SEM. Unpaired t test was used for comparisons between two groups. For multiple comparisons, statistical analysis was performed by ANOVA followed by Tukey post hoc tests. P value < 0.05 was considered statistically significant.
Results
Low-K+ diet increases KLHL3S433-P and decreases total KLHL3 levels
We produced a mouse monoclonal antibody that specifically recognizes KLHL3S433-P. Western blotting using this antibody with lysates from cells expressing KLHL3 recognized a signal of appropriate size that was absent in cells expressing no KLHL3 or KLHL3 carrying the S433A substitution (Figure 1A). This signal was also detected in lysates of mouse kidney, and was abolished by competition with the immunizing KLHL3 phosphopeptide, but not unphosphorylated peptide (Figure 1B). These data establish the specificity of the antibody in cells and in vivo, and provide further evidence that KLHL3S433 is phosphorylated in kidney [12].
Figure 1. K+ depletion increases KLHL3 phosphorylation at S433 and decreases total KLHL3 levels.
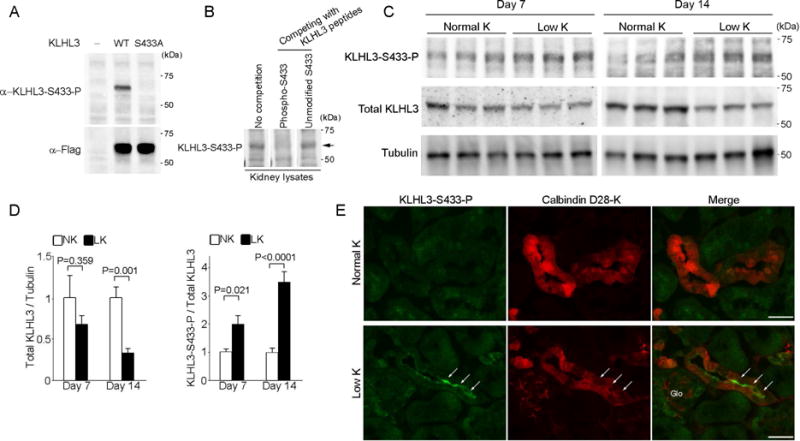
(A) Western blot analysis of HEK cells expressing no KLHL3, Flag-tagged KLHL3WT or KLHL3S433A incubated with monoclonal anti-KLHL3S433-P antibody (upper panel) and anti-Flag antibody (lower panel). The signal detected by the anti-KLHL3S433-P is abolished by the S433A substitution. (B) Total kidney lysates were prepared from mice eating a normal diet. Western blots of the lysates were incubated with anti-KLHL3S433-P antibody without or with competition with immunizing KLHL3 phosphopeptide and non-phosphopeptide. Competition with the KLHL3 phosphopeptide but not the non-phosphopeptide eliminates the antibody signal (arrow), confirming the specificity. (C) KLHL3S433-P and total KLHL3 levels in the kidneys of wild-type mice fed a normal K+ diet (NK) or a low-K+ diet (LK) determined by Western blot analysis in biological replicates. (D) Quantitation of total KLHL3 and KLHL3S433-P levels in the kidney described in (C) (n = 6 or 7 for day 7 and n = 5 for day 14). Data are expressed as means ± SEM. (E) Kidney sections stained for α-KLHL3S433-P (green, indicated by arrows) and α-calbindin D28-K (a marker for distal convoluted tubule cells, red) in mice eating a normal-K+ or a low-K+ diet. KLHL3S433-P is increased at the apical membrane of distal convoluted cells (which express NCC). Scale bars represent 50 μm. Glo, glomeruli.
To determine whether a change in K+ status alters KLHL3 function in vivo, we evaluated KLHL3S433-P and total KLHL3 levels by Western blotting in mice eating a low-K+ diet. As shown in Figure 1C and 1D, KLHL3S433-P levels were significantly elevated by the low-K+ diet at day 7 (P = 0.021 versus normal-K+ group), and also at day 14 (P < 0.001 versus normal-K+ group). To our interest, total KLHL3 levels were also significantly decreased by low K+ at day 14 (P = 0.001 versus normal-K+ group; Figure 1C and 1D). Given that KLHL3S433-P impairs the substrate binding ability [12], these data indicate that the activity of KLHL3/CUL3-based ubiquitin ligase is markedly suppressed by the net effects of KLHL3S433-P induction and the decrease in total levels.
In the kidney, KLHL3 is present in distal convoluted tubule (DCT) cells, cortical thick ascending limb of Henle, and cortical collecting duct [22]. To determine the nephron segments and cell types in which low-K+ diet increases KLHL3S433-P, we performed immunofluorescence microscopy using a polyclonal antibody to KLHL3S433-P [12]. The results showed that KLHL3S433-P levels were increased in cells that were positive for calbindin D-28K (a marker for DCT cells; Figure 1E) [23], but were negative for aquaporin-2 (a marker for principal cells; data not shown), demonstrating that KLHL3S433-P is mainly increased in the DCT cells. In contrast, the increase in KLHL3S433-P was not evident in the principal cells (Supplemental Figure 3).
Increased KLHL3S433-P and decreased total KLHL3 result in increased WNK4 levels, stimulating SPAK/OSR1-NCC pathway in K+ depletion
Previous studies have shown that WNK4 levels are high in DCT cells [3]. If the activity of KLHL3-based ubiquitin ligase is suppressed by low-K+ diet, this should result in accumulation of the target substrate WNK4. We thus examined WNK4 levels in kidneys of mice eating a low-K+ diet. As shown in Figure 2A and 2B, low-K+ diet significantly increased WNK4 levels in the kidney (P = 0.018).
Figure 2. KLHL3S433 phosphorylation results in increased WNK4 levels, stimulating SPAK/NCC pathway in the kidney of mice on a low-K+ diet.
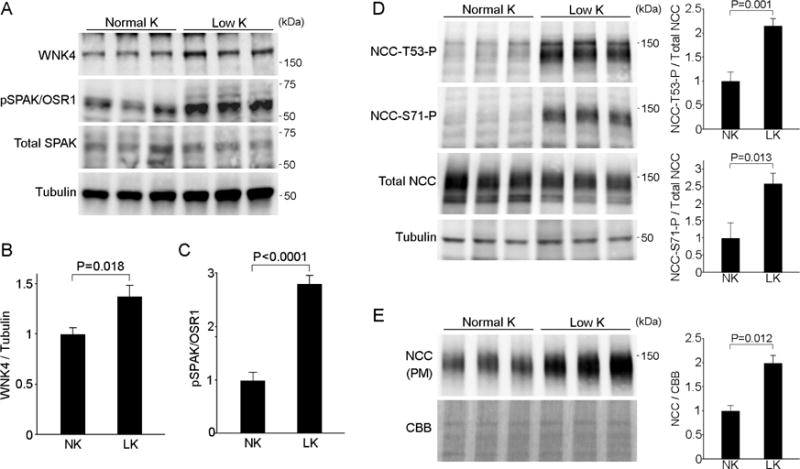
(A) Effects of low K+ diet on WNK4 levels and SPAK/OSR1 phosphorylation in the kidney. Blots show biological replicates. (B and C) Quantitation of WNK4 levels and SPAK/OSR1 phosphorylation levels described in (A). Data are expressed as means ± SEM; n = 7 for (B) and n = 5 for (C). (D) Effects of low-K+ diet on NCC phosphorylation at Thr53 and Ser71, and total NCC levels in whole cell lysates in the kidney. Blots show biological replicates. Bar graphs show the results of quantitation (n = 5 each group). (E) Expression of NCC in the plasma membrane fraction in the indicated animals (n = 3). Bar graphs show the results of quantitation. Data are expressed as means ± SEM.
When activated, WNK4 phosphorylates the kinases SPAK and OSR1, the positive regulators of NCC [7,8]. In PHAII patients, mutations in either KLHL3 or WNK4 result in the increased WNK4 levels, promoting hypertension and hyperkalemia that are correctable by thiazide. Consistently, we found that WNK4 accumulation in the kidney of mice on a low-K+ diet resulted in the increased phosphorylation of SPAK/OSR1 (Figure 2A and 2C), and NCC phosphorylation at both Thr53 and Ser71 (Figure 2D). Moreover, plasma membrane NCC levels were significantly increased in mice on a low-K+ diet (Figure 2E). These data are consistent with a recent report showing the importance of SPAK in stimulating NCC in hypokalemia [24], and demonstrate that KLHL3 inactivation is the upstream signal for SPAK/OSR1 activation in K+ depletion.
K+ depletion increases PKC activity in vivo in kidney
We next evaluated the mechanism that increases KLHL3S433-P in the K+-depleted condition. We have previously shown that PKC directly phosphorylates KLHL3S433 in in vitro kinase assays. There is also a report showing the role of K+ in regulating PKC [25]. We thus determined PKC activity in the kidney of mice on a low-K+ diet. Of note, Western blotting analysis using antibody against the active, phosphorylated PKC (pan) revealed that PKC activity was significantly increased by K+ depletion in the kidney (1.8-fold increase; P < 0.001; Figure 3A). Given the evidence that KLHL3S433 is also phosphorylated by PKA [13], we additionally evaluated the levels of active, phosphorylated PKA (bearing phosphorylation at Thr197 in the catalytic domain). In mice eating a low-K+ diet, the levels of phosphorylated PKA were not significantly altered (Figure 3B).
Figure 3. Low-K+ diet increases protein kinase C activity in the kidney.
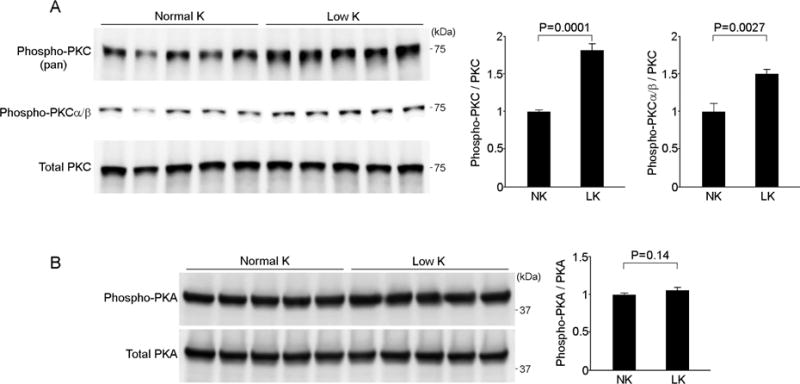
(A) The effects of K+ depletion on protein kinase C (PKC) phosphorylation in the kidney. The top panel shows active, phosphorylated PKC (pan) levels. The middle panel shows the levels of active, phosphorylated PKCα/β. The bottom panel shows the total PKC levels. Blots show biological replicates. Bar graphs show the results of densitometric quantitation. Data are expressed as means ± SEM; n = 5 each group. (B) The effects of K+ depletion on protein kinase A (PKA) phosphorylation in the kidney. Blots show biological replicates. Bar graphs show the results of densitometric quantitation. Data are expressed as means ± SEM; n = 5 each group.
Among the different PKC isozymes, KLHL3S433 is predominantly phosphorylated by conventional PKCα and PKCβ, and to a lesser extent by PKCε [12]. Recent studies demonstrated the role of PKCα in regulating electrolyte handling in the distal nephron [26]. We next performed Western blotting using phospho-specific antibody that recognizes active PKCα/β (bearing phosphorylation at Thr638/641). Consistently, active, phosphorylated PKCα/β is significantly increased in the kidney by low-K+ diet (P = 0.003; Figure 3A).
Activation of PKC increases KLHL3S433-P, which is sufficient to prevent KLHL3-mediated ubiquitination and degradation of WNK4
Finally, to show that the activation of endogenous PKC increases KLHL3S433-P, and that this event is sufficient to prevent the degradation of WNK4, we performed cell culture experiments. HEK cells expressing KLHL3 were stimulated with PMA, the PKC activator. Cells were then lysed, and the levels of KLHL3S433-P and total KLHL3 were quantitated. As expected, PMA increased the phosphorylated, active form PKC at 30 min (Figure 4A). Importantly, PMA significantly increased KLHL3S433-P levels (Figure 4B; 1.9-fold increase, P < 0.001).
Figure 4. PKC activator PMA increases KLHL3S433-P, preventing ubiquitination and degradation of WNK4 in HEK cells.

(A) Plasmid encoding wild-type Flag-KLHL3 was transfected in HEK cells. An empty vector was used as control to equalize the total amount of DNA in transfection lacking KLHL3. Cells were incubated in the presence or absence of the PKC activator phorbol 12-myristate 13-acetate (PMA, 200 nM) for 30 min. Levels of phosphorylated PKC (pPKC) and total PKC were quantitated by Western blotting. Bar graphs show the results of quantitation. (B) Cell lysates described in (A) were subjected to Western blotting with monoclonal α-KLHL3S433-P, α-Flag (to detect total KLHL3), and α-tubulin. Bar graphs show the results of quantitation. Data are expressed as means ± SEM; n = 5 each group. (C) HEK cells expressing Flag-KLHL3 and WNK4-HA were incubated in the presence and absence of PMA (200 nM) for 24 hr. To confirm that the effect of PMA is mediated by PKC, one group was incubated with bisindolylmaleimide I (BIM), the PKC inhibitor. Bar graphs show the results of quantitation. Data are expressed as means ± SEM; n = 6 each group. (D) HEK cells expressing the indicated proteins were incubated in the presence or absence of PMA for 1 hr. Ubiquitinated WNK4 levels were analyzed by in vivo ubiquitination assay. The asterisk indicates the ubiquitinated WNK4. KLHL3-mediated WNK4 ubiquitination is abrogated by PMA treatment. Total WNK4 levels were consistently higher in cells incubated with PMA.
If this effect of PMA is of functional significance, KLHL3-mediated WNK4 degradation [11] should be prevented by the incubation of PMA. To test this, cells co-expressing KLHL3 and WNK4 were incubated with PMA for 24 hrs. Indeed, PMA almost completely prevented the KLHL3-mediated reduction in WNK4 levels in cells co-expressing KLHL3 and WNK4 (Figure 4C; KLHL3 versus KLHL3 +PMA, P < 0.0001; control versus KLHL3 + PMA, P = not significant). In addition, these effects of PMA were fully prevented by bisindolylmaleimide I, the selective inhibitor of PKC (Figure 4C).
Lastly, binding of WNK4 by KLHL3 leads to its polyubiquitination, leading to WNK4 degradation [11]. We tested whether PKC activation by PMA is capable of abrogating this effect. HEK cells expressing KLHL3 and WNK4 were incubated in the absence or presence of PMA. Cell lysates were then subjected to an in vivo ubiquitination assay [11]. The results demonstrated that WNK4 ubiquitination by KLHL3 was prevented by PMA (Figure 4D). These data demonstrate that the induction of KLHL3S433-P by PKC is sufficient to increase WNK4 levels by preventing KLHL3-mediated ubiquitination and degradation of WNK4.
Discussion
We here demonstrated that suppression of KLHL3/CUL3-based ubiquitin ligase mediates NCC activation by low-K+ diet. In the kidney, low-K+ diet results in increased PKCα/β activity and KLHL3S433-P levels in the DCT, leading to increased WNK4 levels. This, in turn, results in the activation of the kinases SPAK/OSR1, increasing NCC phosphorylation and activity. Indeed, we showed in cell culture that PKC activation is sufficient to increase WNK4 levels by phosphorylating KLHL3S433 and preventing WNK4 ubiquitination. Besides KLHL3S433-P induction, we also found that low-K+ diet results in reduced total KLHL3 levels. These data for the first time demonstrate the involvement of KLHL3/CUL3-based ubiquitin ligase in low-K+-mediated NCC induction.
It is well described and accepted that the reduced K+ intake is associated with hypertension [14,15]. Accumulating evidence indicates a key role of NCC in this process [16], and it is recently postulated that hypokalemia alters the resting membrane potential in DCT cells, which in turn changes intracellular Cl− levels, increasing the kinase activity of the Cl−-regulated kinase WNK4 [5,17]. Our findings provide an alternative or additional mechanism for this effect. The increase in WNK4 levels by KLHL3 inactivation and WNK4 kinase activation by the change in intracellular Cl− levels synergistically activate SPAK/OSR1-NCC pathway and Na-Cl transport in DCT cells. Given that WNK1 is a potent activator of NCC [27], and that KLHL3 binds and targets WNK1 for degradation [10,11,22], it is likely that WNK1 is also involved in this mechanism. Kinase-inactive WNK4 inhibits WNK1 [27]. We infer that WNK1 is similarly increased by the inactivation of KLHL3, which then becomes phosphorylated and activated by the decrease in intracellular Cl− levels, increasing NCC activity [27]. A very recent study also suggests a Cl− and SPAK/OSR1-independent component [28], adding an additional layer of NCC regulation by extracellular K+. These mechanisms act in concert to reduce distal electrogenic Na+ reabsorption by increasing NCC, preventing further renal K+ loss but promoting hypertension.
The physiological impact of the proposed pathway is underscored by the effects of point mutations in KLHL3 that alter S433 [2,4]. In PHAII patients, either KLHL3S433N or KLHL3S433G substitution is sufficient to cause hypertension and hyperkalemia that are correctable by thiazide [2,4]. The fact that angiotensin II signaling [12] and K+ depletion share a common mechanism via their effect on PKC also supports the critical importance of phosphorylation status at this site in regulating NCC activity. The increase in NCC activity in hypokalemia acts to minimize K+ excretion acutely by reducing Na+ delivery to the collecting duct, thereby reducing electrogenic Na+ reabsorption and blunting the electrical driving force for K+ secretion. Thus, this physiological mechanism to reduce renal K+ secretion in the setting of K+ depletion occurs at the expense of increased renal Na-Cl reabsorption.
Another important observation in this study is that total KLHL3 levels are decreased by low-K+ diet. Previous data by Ohta et al. have reported that CUL3 is capable of ubiquitinating KLHL3 [10]. There are also data showing that the PHAII-causing mutant CUL3 (CUL3 ∆403–459) displayed increased ubiquitination of KLHL3 [22,29]. Given these data, it is possible that low-K+ diet alters the function of CUL3, promoting KLHL3 ubiquitination. Alternatively, phosphorylation of KLHL3 not only impairs its substrate binding, but may also alter its stability. Additionally, the mechanisms whereby low-K+ diet increases PKC activity are currently unknown. Whether hypokalemia, as-yet-unidentified gut-kidney signaling [30], or both, play causative roles are areas for future investigation. Given the evidence that WNK4 amplifies the action of PKC [31], it may also be possible that WNK4 activation by intracellular Cl− depletion is involved in KLHL3S433-P induction.
In summary, our data demonstrate that the KLHL3/CUL3-based ubiquitin ligase is involved in the low-K+-mediated activation of NCC. Our data reveal a previously unrecognized pathway in which reduced K+ intake alters Na-Cl reabsorption in the kidney, and underscore the physiological importance of ubiquitin proteasome system in controlling fluid and electrolyte homeostasis. These data also highlight the importance of the appropriate management of K+ intake in hypertensive patients.
Supplementary Material
The study proposes a mechanism that low-K+ diet increases renal salt reabsorption.
K+ depletion inactivates KLHL3 by phosphorylation and by decreasing total levels.
Phosphorylation of KLHL3 by PKC is sufficient to prevent WNK4 degradation.
Suppression of KLHL3 by low K+ increases WNK4 and activates NCC via SPAK/OSR1.
Acknowledgments
We thank Ms. Hiromi Yamaguchi and Emiko Kuribayashi-Okuma for their technical supports. This work was supported in part by JSPS KAKENHI grants 15H04837 (S.S.) and 15H02538 (T.F.); Japan Agency for Medical Research and development, AMED; Banyu Life Science Foundation International (S.S.); Basic Science Research Projects from The Sumitomo Foundation (S.S.); Kanae Foundation for the Promotion of Medical Science (S.S.); Swiss National Centre of Competence in Research Kidney.CH (J.L.); NIH grant P01DK17433 (R.P.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 2.Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson FH, Disse-Nicodeme S, Choate KA, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 4.Louis-Dit-Picard H, Barc J, Trujillano D, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44:456–460. S451–453. doi: 10.1038/ng.2218. [DOI] [PubMed] [Google Scholar]
- 5.Piala AT, Moon TM, Akella R, et al. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7:ra41. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, et al. The Effect of WNK4 on the Na+-Cl− Cotransporter Is Modulated by Intracellular Chloride. J Am Soc Nephrol. 2015;26:1781–1786. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitari AC, Deak M, Morrice NA, et al. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson C, Rafiqi FH, Karlsson HK, et al. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci. 2008;121:675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 9.Kahle KT, Wilson FH, Leng Q, et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 10.Ohta A, Schumacher FR, Mehellou Y, et al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J. 2013;451:111–122. doi: 10.1042/BJ20121903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibata S, Zhang J, Puthumana J, et al. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A. 2013;110:7838–7843. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibata S, Arroyo JP, Castaneda-Bueno M, et al. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc Natl Acad Sci U S A. 2014;111:15556–15561. doi: 10.1073/pnas.1418342111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshizaki Y, Mori Y, Tsuzaki Y, et al. Impaired degradation of WNK by Akt and PKA phosphorylation of KLHL3. Biochem Biophys Res Commun. 2015;467:229–234. doi: 10.1016/j.bbrc.2015.09.184. [DOI] [PubMed] [Google Scholar]
- 14.Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med. 2001;344:3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 15.Mente A, O’Donnell MJ, Rangarajan S, et al. Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med. 2014;371:601–611. doi: 10.1056/NEJMoa1311989. [DOI] [PubMed] [Google Scholar]
- 16.Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, et al. Modulation of NCC activity by low and high K(+) intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol. 2014;306:F1507–1519. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terker AS, Zhang C, McCormick JA, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015;21:39–50. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata S, Rinehart J, Zhang J, et al. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013;18:660–671. doi: 10.1016/j.cmet.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Al-Qusairi L, Donnelly BF, et al. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest. 2015;125:3433–3448. doi: 10.1172/JCI75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Picard N, Trompf K, Yang CL, et al. Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J Am Soc Nephrol. 2014;25:511–522. doi: 10.1681/ASN.2012121202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shibata S, Nagase M, Yoshida S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 22.McCormick JA, Yang CL, Zhang C, et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J Clin Invest. 2014;124:4723–4736. doi: 10.1172/JCI76126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CT, Ng HY, Lee YT, et al. The role of calbindin-D28k on renal calcium and magnesium handling during treatment with loop and thiazide diuretics. Am J Physiol Renal Physiol. 2016;310:F230–236. doi: 10.1152/ajprenal.00057.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wade JB, Liu J, Coleman R, et al. SPAK-mediated NCC regulation in response to low-K+ diet. Am J Physiol Renal Physiol. 2015;308:F923–931. doi: 10.1152/ajprenal.00388.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sterling H, Lin DH, Chen YJ, et al. PKC expression is regulated by dietary K intake and mediates internalization of SK channels in the CCD. Am J Physiol Renal Physiol. 2004;286:F1072–1078. doi: 10.1152/ajprenal.00425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grahammer F, Nesterov V, Ahmed A, et al. mTORC2 critically regulates renal potassium handling. J Clin Invest. 2016;126:1773–1782. doi: 10.1172/JCI80304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadchouel J, Ellison DH, Gamba G. Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu Rev Physiol. 2016;78:367–389. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 28.Penton D, Czogalla J, Wengi A, et al. Extracellular K+ rapidly controls NCC phosphorylation in native DCT by Cl− -dependent and -independent mechanisms. J Physiol. 2016 doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schumacher FR, Siew K, Zhang J, et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7:1285–1306. doi: 10.15252/emmm.201505444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preston RA, Afshartous D, Rodco R, et al. Evidence for a gastrointestinal-renal kaliuretic signaling axis in humans. Kidney Int. 2015;88:1383–1391. doi: 10.1038/ki.2015.243. [DOI] [PubMed] [Google Scholar]
- 31.Cha SK, Huang CL. WNK4 kinase stimulates caveola-mediated endocytosis of TRPV5 amplifying the dynamic range of regulation of the channel by protein kinase C. J Biol Chem. 2010;285:6604–6611. doi: 10.1074/jbc.M109.056044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.