

Abstract
There are significant associations between possession of certain HLA class I alleles and rate of progression to AIDS. Immunological data provide an explanatory mechanism for this relationship. Patients with HLA types associated with rapid disease progression recognize a significantly smaller fraction of their known repertoire of viral epitopes than do patients with HLA types associated with slow progression. Population frequency of HLA types (or supertypes) and their capacity to elicit cytotoxic T lymphocyte responses are also negatively correlated. These data provide an immunological mechanism to explain HLA-related risk of progression to AIDS and emphasize the central role of viral evolution in the pathogenesis of HIV.
Why is it that, after infection with HIV, some people become ill quite rapidly while others remain well for more than a decade? Despite 20 years of fruitful research into HIV and AIDS (1), this simple question remains unanswered. It is clear that the length of the asymptomatic period is determined, in some way, through the interaction of the infecting virus and the immune response of the host. This understanding is solidly underpinned by two long-standing observations. First, patients with higher viral loads progress to disease more quickly (2, 3). Second, the possession of certain human leukocyte antigen (HLA) class I molecules (e.g., HLA B35) predisposes patients to rapid disease progression, whereas others (e.g., HLA B27 and HLA B58) endow them with a longer asymptomatic period (4–9).
HLA class I molecules are present on the surface of all nucleated cells where they present short viral peptide fragments, called epitopes, that elicit immune responses from cytotoxic T lymphocytes (CTLs). Each HLA class I molecule is able to present only a limited range of peptides. The HLA class I genotype of a patient therefore dictates the repertoire of CTL responses he or she is able to mount, which translates into different abilities to cope with an HIV infection. Patients who are heterozygous for their HLA class I molecules are at a significant advantage if infected with HIV, implying that the quantity and breadth of the immune response determines the success or failure of viral control (4). However, counts of HIV-specific CTLs cannot be simply correlated with viral load or viral clearance rate (10), in contrast to earlier reports (11). This lack of correlation between virus load and HIV-specific CTLs implies that other characteristics of the cellular immune response, not only its quantity, underlie the relationship between HLA class I alleles and HIV control. The present study sheds light on this controversy by explaining the association between HLA alleles and HIV disease progression through quantifiable CTL function.
HLA class I alleles can be reclassified into nine major supertypes based on their peptide-binding properties (12). Under this classification, the HLA supertype of an individual is highly predictive of his or her viral load (13). Furthermore, this classification scheme reveals a rare allele advantage, because less common HLA supertypes (HLA B58s and HLA B62s) are associated with the lowest viral loads, whereas more common HLA supertypes (HLA A2s and HLA B7s) are associated with higher viral loads.
Why should rare alleles be advantageous? A hallmark of HIV infection is its ability to generate immune escape mutations in epitopes (14–17) and their flanking regions (18, 19). CTL escape mutants have been found in population studies, in which they correlate with higher viral loads (20), and they can be transmitted from mother to child (21, 22) as well as between sexual partners (ref. 18 and C. Edwards, H. T. Zhang, and A. Milicic, personal communication). Escape mutations tend to be maintained after transmission into a new host who shares the restricting HLA molecule. Reversion has been described after HIV is transmitted to a new host in whom there is HLA mismatch (18, 23). However, the extent to which escape mutations revert to wild-type sequence will depend on the fitness cost of the escape mutation (23, 24). The accumulation of transmitted immune escape mutants probably explains the relative paucity of epitope regions in more variable parts of the HIV genome (25). Therefore, having rare HLA alleles is advantageous, because such individuals are less likely to be infected with HIV that is preadapted to the CTL responses they can make.
Hence, two questions arise. One, do HLA alleles associated with slow disease progression elicit detectable CTL responses in a higher proportion of patients than HLA alleles associated with rapid disease progression? Two, do rare HLA alleles elicit CTL responses in a greater proportion of patients than more common HLA types? These have to be treated as two separate questions because, for classically defined HLA types, there is no significant relationship between relative hazard and population frequency. The results of Trachtenberg et al. (13) raise a third question: Do rare HLA supertypes elicit CTL responses in a greater proportion of patients than more common HLA supertypes? We present data and simple regression analyses to answer these three questions.
Materials and Methods
Patient Cohort. The Swiss-Spanish Intermittent Therapy Trial was a large study of structured treatment interruptions that assessed the clinical, virological, and immunological outcome of planned short breaks in the chemotherapeutic regimen of chronically HIV-infected patients. The study yielded cross-sectional and longitudinal data on CTL responses detected by IFN-γ enzyme-linked immunospot assays in 84 patients followed for an average of 14 months (range, 3–19 months) (Table 1). The patient group is described in detail elsewhere (26), as are the dynamics of the breadth and magnitude of their CTL responses and viral loads (10, 27).
Table 1. Details of the cohort recruited to the Swiss–Spanish Intermittent Therapy Trial (SSITT).
| No. of patients recruited to SSITT | 133 |
| No. of patients analyzed in this study* | 84 |
| Sex (male/female) | 52/32 |
| Details of therapy | |
| No. of patients on dual therapy | 9 |
| No. of patients on triple therapy | 75 |
| Patient age,† years | 40 (22-68) |
| Therapy duration,† days | 808 (254-1,337) |
| VL undetectable duration,† days | 707 (187-1,285) |
| CD4 pre-therapy,† cells per μl | 359 (1-1,035) |
| Viral load pre-therapy,† log10 RNA copies per ml | 4.41 (2.23-6.11) |
Immune responses were measured for 97 patients. Twelve patients were excluded because they had no immune responses detected at any time point measured, and one patient was excluded because he had excessive responses for most peptides measured, leaving 84 patients in the data set.
Data are median values, with the ranges shown in parentheses.
Epitopes Tested. Each patient was HLA-typed and tested repeatedly (mean number of times tested, 16; range, 3–26) to assess the frequency of responsive HIV-specific CTLs in their peripheral blood lymphocytes. Patients were tested for CTL responses with synthetic peptides corresponding to previously described HLA class I-restricted optimal HIV CTL epitopes. The epitopes tested are described in the Los Alamos database (www.hiv.lanl.gov/content/immunology/tables/ctl_summary.html) in the context of HIV infection and are listed in Table 2, which is published as supporting information on the PNAS web site.
Because the HLA class I type of a patient determines the repertoire of known epitopes they might be expected to recognize, their cells were tested against a panel of peptide epitopes designed to match their individual HLA type (median number tested, 16; range, 2–31).
Relative Hazards of Disease Progression. Because large patient cohorts are needed to establish genetic associations and the patients in this study were given potent antiretroviral treatment, we could not derive relative hazards of disease progression from this data set. Instead, we used the relative hazards as calculated by O'Brien et al. (8) for a large cohort of HIV-infected white patients. The full list of relative hazards for the O'Brien et al. cohorts can be accessed at http://home.ncifcrf.gov/ccr/lgd/datatables/gao1_01.htm. Population HLA frequencies for class I A and B alleles are very highly correlated between the Swiss and U.S. white patients (ρ = 0.99 and 0.97, respectively) (28, 29).
Statistical Analysis. Data were analyzed by using minitab statistical software [release 13, Minitab Statistical Software, State College, PA (2000)].
Weighted Regressions. In a weighted least-squares regression, the weighted error sum of squares,
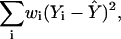 |
is minimized (wi are the weights). In this context, the result is to deemphasize the HLAs for which we fear we are missing most information: those with few known epitopes. An alternative strategy, just excluding the HLAs tested with only one epitope, gives similar results.
Lack of Independence. The plots of relative hazard and HLA frequency against HLA capacity to elicit CTL responses are intuitively appealing but suffer from some lack of independence. Most patients contribute to more than one data point, because they bear several HLA alleles. The degree of correlation between HLA capacities to elicit CTL responses for a given patient varies widely across the six different pairwise comparisons, and none of the estimated correlation coefficients are significantly different from zero. To explore the impact of correlations between patients' individual HLA capacities to elicit CTL responses, we performed a randomization test. The data were subsampled such that each patient only contributed to the calculation of one HLA capacity to elicit CTL responses. This sampling of just one quarter of the information gathered was performed 100 times, new HLA capacities to elicit CTL responses were calculated, and the regressions shown in Fig. 2 were repeated. Both relationships were maintained under this very uncompromising test; the median P value for the relationship with relative hazard was P = 0.013, and for the relationship with HLA frequency the median P value was P = 0.031.
Fig. 2.

Both slowly progressing and rare class I alleles elicit CTL responses in a large proportion of patients bearing those alleles. (A) Class I-related relative hazard of disease progression is negatively correlated with HLA capacity to elicit CTL responses. In a weighted regression, the slope is highly significantly less than zero. (B) HLA allelic frequency in the cohort is negatively correlated with HLA capacity to elicit CTL responses. In a weighted regression, the slope is highly significantly less than zero. In both regression analyses the numbers of epitopes tested for each class I allele were used as weights in the regression, reflecting greater confidence in an average taken across many epitopes than across few (26).
Alternative Analyses. Our weighting scheme acts to acknowledge lack of confidence in the estimated capacity to elicit CTL responses for HLAs tested with few epitopes. An alternative strategy is to compare only the maximally targeted epitopes for each HLA (i.e., GEI for B8, EVI for A26, etc.) Under such a comparison, the relationship between relative hazard and maximum targeting frequency holds (R2 = 26%; P = 0.015). As one would expect from the relationship between number of epitopes tested and HLA allelic frequency (positive relationship, P = 0.04), the use of the maximum destroys the relationship between allelic frequency and targeting frequency (R2 = 2%; P = 0.5).
Results
We wished to know whether a patient with a particular HLA class I molecule could recognize known peptide antigens derived from the subtype B consensus sequence. For example, if a patient was found to bear HLA A2, peptide numbers 29–37 of Table 2 were tested in T cell assays (IFN-γ enzyme-linked immunospot assays) by using the patient's own peripheral blood lymphocytes (26). If that patient ever recognized one of those consensus peptide antigens, we scored the result as positive for that peptide. For each of 84 patients taking part in a study of structured therapy interruptions, this exercise was repeated at many time points (mean number of times tested, 16; range; 3–26) by using the relevant epitopes from the panel of peptides shown in Table 2. We used this optimal peptide approach so that when we detected a response, we could identify the restricting HLA. The use of overlapping peptide libraries would not have permitted such identification.
Many of the responses patients should be able to make given their HLA restriction are nevertheless absent: only one peptide (KAF restricted by B57) elicited responses in all patients with that allele (Fig. 1). HLA molecules vary greatly in their capacity to elicit responses to peptides. In what follows we explore patterns in that variability in capacity to elicit CTL responses.
Fig. 1.

HLA capacity to elicit CTL responses for all of the 22 different class I alleles tested in this cohort. HLA type and number of patients bearing that HLA type are indicated in the individual plots. Bars represent the fraction of all patients bearing that allele who ever, during the study, mounted a detectable CTL response to the optimal peptide indicated below the axis. The three-letter code indicates the first three amino acids of the peptide sequence (see Table 2). The horizontal lines spanning each plot are the average fraction responding across all epitopes tested for that allele. This value is the HLA capacity to elicit CTL responses, defined as the proportion of all possible patient/epitope combinations (for that HLA type) that ever elicit a response in patients bearing the appropriate HLA type. Alleles are ordered in terms of rising capacity to elicit CTL responses.
The capacity of a given HLA allele to elicit a detectable anti-HIV response can be defined as the average frequency with which optimal epitopes, restricted to a particular HLA allele, elicit responses in patients carrying that allele. This frequency is a measure of the area under the curve shown in the bar charts in Fig. 1, adjusted for the number of epitopes tested, and is indicated with the horizontal line spanning each graph. For example, five optimal peptides restricted by B27 were tested in the nine patients bearing B27. Thus, there were 45 possible combinations for B27 to score as positive. Twenty-three positives were found, giving a capacity to elicit CTL responses of 51% for HLA B27.
To answer the first question (do “slow-progressor” HLA class I alleles elicit CTL responses to the peptides they restrict in a large proportion of the patients bearing that allele?), we performed a weighted linear regression with the number of peptides tested as the weights. The regression yields a highly significant negative relationship between the relative hazard of disease progression and the ability of the different HLAs to elicit a CTL response (Fig. 2A).
To answer the second question (do rare HLA class I alleles elicit CTL responses to the peptides they restrict in a large proportion of patients bearing those alleles?), we performed a weighted regression of HLA allelic frequency in this cohort against the ability of the different HLAs to elicit a CTL response with the number of peptides tested as the weights (Fig. 2B). Rare alleles elicit responses to a larger proportion of the known potential antigens as compared with common alleles. This relationship is also significant if Swiss-population HLA frequencies are used (R2 = 29%; P = 0.03).
To answer the third question (do rare HLA supertypes have a high capacity to elicit CTL responses?), we performed a linear regression (Fig. 3). The result was a significant negative relationship; patients bearing rare HLA supertypes have a higher probability of recognizing known optimal peptides restricted by the HLAs in that supertype.
Fig. 3.

The frequency of HLA supertypes within the cohort is negatively correlated with the supertype capacity to elicit CTL responses. High-resolution HLA typing was used to classify 284 of the 336 possible patient HLA alleles into nine supertypes according to the guidelines suggested by Sette and Sidney (12). Of the 88 optimal peptides (listed in Table 2), 58 had the structural motifs compatible with allocation to their respective supertype (12) and were subsequently used in the analysis. For each of the nine supertypes, the sum of the possible patient/epitope combinations for each constituent HLA type was calculated. The proportion of these combinations that ever elicited a response was calculated and defined as the supertype capacity to elicit CTL responses. For example, in this study, the constituent HLA types for supertype A1 are A1 and A32. Of patients whose supertype was A1, there were seven HLA A32 individuals tested against one epitope and 20 HLA A1 individuals tested against three epitopes. Therefore, there is a maximum of 67 [(20 × 3) + (7 × 1)] positive responses. Of these, only three responses were recorded by IFN-γ enzyme-linked immunospot, giving a supertype capacity to elicit CTL responses of 3 of 67 (4.48%). This regression is unweighted, because there is no simple equivalent of the number of epitopes tested.
Could these results be artifacts of the fact that we do not know all of the CTL epitopes of HIV? Overall, more optimal epitopes are known for common HLA types than for rare ones (P = 0.04). However, the number of optimal epitopes and the capacity of each HLA to elicit CTL responses do not correlate. Notice how the seven HLA types for which only one epitope was tested are scattered through Fig. 1. We weighted the regression analyses with the number of epitopes, thereby incorporating the fact that HLA alleles for which more epitopes are known have higher information content than those for which fewer epitopes are known (26).
We used a simple but robust measure of CTL function and compared it with the risk of disease progression across a large range of HLA types. Because the CTL assay was performed by using optimal peptides, absent responses may reflect the immense sequence variability characteristic of HIV. In agreement with Trachtenberg et al. (13), we would argue that patients with “rapid-progressor” class I alleles recognize a small proportion of their known repertoire because their infecting HIV carries a large number of CTL escape mutations relevant to the responses they might mount. This hypothesis is supported by our findings of an association between HLA frequency and immune response, because viral adaptation is likely to be influenced predominantly by the most common, HLA-dictated immune pressures. Widespread, transmitted immune selection would be expected to drive an increase in viral virulence as the virus adapts to the most common types of hosts. The fact that no such shift in the virulence of HIV has been observed is enigmatic.
The associations observed between HLA class I alleles and time to progression to AIDS have long suggested a link between immune response and the ability to delay the onset of disease. Other arms of the immune response such as HLA class II-restricted T cells and humoral responses are also implicated in the control of infection. Recent evidence on viral mutations and population frequency of HLA alleles has supported this link without directly assessing the immune response, which provides the most plausible mechanistic explanation for these assertions (13, 20).
We have shown that CTL responses measured in patients with HLA alleles associated with rapid progression recognize only a small proportion of the known epitopes restricted by these HLA class I molecules. Conversely, CTL responses measured in patients with slow-progressor HLA alleles recognize a large proportion of the known mapped epitopes. These data provide a coherent immunological mechanism that explains HLA-related risk of disease progression in a model that links host genotype, host immune function, and viral evolution.
Supplementary Material
Acknowledgments
A.S. is grateful for the financial support of the Dutch Catharine van Tussenbroek Fonds and the Dutch VSB Fonds. D.A.P. is a Medical Research Council Clinician Scientist. The Swiss HIV Cohort Study is supported by Swiss National Science Foundation Grant 3345-062041. This work was supported in part by a Wellcome Trust program grant (awarded to R.E.P.).
Abbreviation: CTL, cytotoxic T lymphocyte.
Medizinische Universitäts Kinderklinik, Inselspital, CH-3010 Bern, Switzerland
Division of Infectious Diseases, Department of Internal Medicine, University Hospital Basel, Petersgraben 4, CH-4031 Basel, Switzerland
Ambulatorio Malattie Infettive, Ospedale Civico, Via Tesserete 46, CH-6903 Lugano, Switzerland
Kantonales Frauenspital Fontana, Luerlimachstrasse 118, CH-7000 Chur, Switzerland
Coordination and Data Center, Swiss HIV Cohort Study, Centre Hospitalier Universitaire Vaudois, Mont-Paisible 16, CH-1011 Lausanne, Switzerland
Nationales Zentrum für Retroviren, Gloriastrasse 30, CH-8028 Zurich, Switzerland
Department of Innere Medizin Infektiologische Sprechstunde, Kantonsspital St. Gallen, CH-9007 St. Gallen, Switzerland
Division of Infectious Diseases/Basel Institute for Clinical Epidemiology, Kantonsspital Basel, CH-4031 Basel, Switzerland
Service d'Immunologie et Allergie, Centre Hospitalier Universitaire Vaudois, BH 19-626, CH-1011 Lausanne, Switzerland
Swiss Forum for Migration and Population Studies, Université de Neuchâtel, Rue St. Honoré 2, CH-2000 Neuchâtel, Switzerland
Division de Maladies Infectieuses, Hôpitaux Universitaires de Genéve, CH-1211 Geneva 14, Switzerland
Hôpital de l'Enfance, Montétan 16, CH-1000 Lausanne 7, Switzerland
Service des Maladies Infectieuses, Médecine 2, Centre Hospitalier Universitaire Vaudois, BH 07, CH-1011 Lausanne, Switzerland
Frauenklinik, Kantonsspital St. Gallen, CH-9007 St. Gallen, Switzerland
Klinische Immunologie, Universitätsspital Zurich, Häldeliweg 4, CH-8044 Zurich, Switzerland
Institut für Sozialund Präventivmedizin, Finkenhubelweg 11, CH-3012 Bern, Switzerland
Institut für Medizinische Mikrobiologie, Petersplatz 10, CH-4003 Basel, Switzerland
Institut für Klinische Mikrobiologie und Immunologie, Frohbergstrasse 3, CH-9001 St. Gallen, Switzerland
Abteilung Infektionskrankheiten und Spitalhygiene, Universitätsspital Zurich, U RAE 54, Rämistrasse 100, CH-8091 Zurich, Switzerland
Department of Internal Medicine and Infectious Diseases, Zentrum für Infektionskrankheiten, Klinik im Park, Bellariastrasse 38, CH-8038 Zurich, Switzerland
Division Autonome de Médecine Préventive Hospitalière, Centre Hospitalier Universitaire Vaudois, BH 19-305, CH-1011 Lausanne, Switzerland
Klinik und Poliklinik für Infektiologie, Polikliniktrakt 2, B Inselspital, CH-3010 Bern, Switzerland
Institut für Infektionskrankheiten, Universität Bern, Postfach 61, CH-3010 Bern, Switzerland
Department of Gynecology, Ospedale Civico, CH-6903 Lugano, Switzerland
Division of Infectious Diseases, University Hospital Basel, Petersgraben 4, CH-4031 Basel, Switzerland
Universitäts-Frauenklinik, Schanzenstrasse 46, CH-4031 Basel, Switzerland
Clinique et Policlinique d'Obstétrique, Hôpitaux Universitaires de Genéve, CH-1211 Geneva 14, Switzerland
Laboratoire Central de Virologie, Hôpitaux Universitaires de Genéve, CH-1211 Geneva 14, Switzerland
Ostschweizer Kinderspital, Claudiusstrasse 6, CH-9006 St. Gallen, Switzerland
Department of Molecular Diagnostics, Institut für Medizinische Mikrobiologie, Petersplatz 10, CH-4003 Basel, Switzerland
Department of Gynäkologie/Geburtshilfe, Universitätsspital Zurich, Rämistrasse 100, CH-8091 Zurich, Switzerland
Universitäts Kinderklinik, Steinwiesstrasse 75, CH-8032 Zurich, Switzerland
Servizio Malattie Iinfettive, Ospedale Civico, Via Tesserete 46, CH-6903 Lugano, Switzerland
Institut Universitaire de Médecine Sociale et Préventive, Rue Bugnon 17, CH-1005 Lausanne, Switzerland
Service d'Immunologie et Allergie, Centre Hospitalier Universitaire Vaudois, BH 10-513, CH-1011 Lausanne, Switzerland
Istituto Cantonale di Microbiologia, Via Mirasole 22A, CH-6501 Bellinzona, Switzerland
National AIDS Therapy Evaluation Centre, Academic Medical Center, Building T, Room T0-123, Meibergdreef 9, 1105 AZ, Amsterdam, The Netherlands
Universitäts-Kinderspital Beider Basel, Römergasse 8, CH-4058 Basel, Switzerland
Division des Maladies Infectieuses, Unité VIH/SIDA Hôpitaux, Universitaires de Genéve, CH-1211 Geneva 14, Switzerland
Fachbereich Infektiologie, Kantonsspital St. Gallen, CH-9007 St. Gallen, Switzerland
Hôpital Intercantonale de la Broye, Avenue de la Promenade 4, CH-1530 Payerne, Switzerland
Service des Maladies Infectieuses, Centre Hospitalier Universitaire Vaudois, BH 07-865, CH-1011 Lausanne, Switzerland
Institut de Microbiologie, Centre Hospitalier Universitaire Vaudois, Rue du Bugnon 48, CH-1011 Lausanne, Switzerland
Laboratoire d'Épidémiologie et Santé Publique, Institut National de la Santé et de la Recherche Médicale, Unité 271, 8, Avenue Rockefeller, F-69373 Lyon Cedex 08, France
Department of Innere Medizin, Fachbereich Infektiologie/Spitalhygiene, Kantonsspital St. Gallen, CH-9007 St.Gallen, Switzerland
Department of Pediatrica, Ospedale Civico, Via Tesserete 46, CH-6903 Lugano, Switzerland
Universitäts-Frauenklinik des Inselspitals, Effingerstrasse 102, CH-3010 Bern, Switzerland
Hôpital des Enfants, 6, Rue Willy Donzé, CH-1211 Geneva 14, Switzerland
Infectious Disease Services, Hôpital Bichat, F-75877 Paris, France
HIV/AIDS-Forschungsförderung, Abteilung Biologie und Medizin, Schweizerischen Nationalfonds zur Förderung der wissenschaftlichen Forschung, Wildhainweg 12, CH-3001 Bern, Switzerland.
References
- 1.Anonymous (2003) Nat. Med. 9, 803.12835675 [Google Scholar]
- 2.Mellors, J. W., Rinaldo, C. R., Jr., Gupta, P., White, R. M., Todd, J. A. & Kingsley, L. A. (1996) Science 272, 1167–1170. [DOI] [PubMed] [Google Scholar]
- 3.Lyles, R. H., Munoz, A., Yamashita, T. E., Bazmi, H., Detels, R., Rinaldo, C. R., Margolick, J. B., Phair, J. P. & Mellors, J. W. (2000) J. Infect. Dis. 181, 872–880. [DOI] [PubMed] [Google Scholar]
- 4.Carrington, M., Nelson, G. W., Martin, M. P., Kissner, T., Vlahov, D., Goedert, J. J., Kaslow, R., Buchbinder, S., Hoots, K. & O'Brien, S. J. (1999) Science 283, 1748–1752. [DOI] [PubMed] [Google Scholar]
- 5.Carrington, M. & O'Brien, S. J. (2003) Annu. Rev. Med. 54, 535–551. [DOI] [PubMed] [Google Scholar]
- 6.Costello, C., Tang, J., Rivers, C., Karita, E., Meizen-Derr, J., Allen, S. & Kaslow, R. A. (1999) AIDS 13, 1990–1991. [DOI] [PubMed] [Google Scholar]
- 7.Migueles, S. A., Sabbaghian, M. S., Shupert, W. L., Bettinotti, M. P., Marincola, F. M., Martino, L., Hallahan, C. W., Selig, S. M., Schwartz, D., Sullivan, J. & Connors, M. (2000) Proc. Natl. Acad. Sci. USA 97, 2709–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Brien, S. J., Gao, X. & Carrington, M. (2001) Trends Mol. Med. 7, 379–381. [DOI] [PubMed] [Google Scholar]
- 9.Gao, X., Nelson, G. W., Karacki, P., Martin, M. P., Phair, J., Kaslow, R., Goedert, J. J., Buchbinder, S., Hoots, K., Vlahov, D., et al. (2001) N. Engl. J. Med. 344, 1668–1675. [DOI] [PubMed] [Google Scholar]
- 10.Oxenius, A., McLean, A. R., Fischer, M., Price, D. A., Dawson, S. J., Hafner, R., Schneider, C., Joller, H., Hirschel, B., Phillips, R. E., et al. (2002) J. Virol. 76, 10169–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogg, G. S., Jin, X., Bonhoeffer, S., Dunbar, P. R., Nowak, M. A., Monard, S., Segal, J. P., Cao, Y., Rowland-Jones, S. L., Cerundolo, V., et al. (1998) Science 279, 2103–2106. [DOI] [PubMed] [Google Scholar]
- 12.Sette, A. & Sidney, J. (1999) Immunogenetics 50, 201–212. [DOI] [PubMed] [Google Scholar]
- 13.Trachtenberg, E., Korber, B., Sollars, C., Kepler, T. B., Hraber, P. T., Hayes, E., Funkhouser, R., Fugate, M., Theiler, J., Hsu, Y. S., et al. (2003) Nat. Med. 9, 928–935. [DOI] [PubMed] [Google Scholar]
- 14.Price, D. A., Goulder, P. J., Klenerman, P., Sewell, A. K., Easterbrook, P. J., Troop, M., Bangham, C. R. & Phillips, R. E. (1997) Proc. Natl. Acad. Sci. USA 94, 1890–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips, R. E., Rowland-Jones, S., Nixon, D. F., Gotch, F. M., Edwards, J. P., Ogunlesi, A. O., Elvin, J. G., Rothbard, J. A., Bangham, C. R., Rizza, C. R., et al. (1991) Nature 354, 453–459. [DOI] [PubMed] [Google Scholar]
- 16.Borrow, P., Lewicki, H., Wei, X., Horwitz, M. S., Peffer, N., Meyers, H., Nelson, J. A., Gairin, J. E., Hahn, B. H., Oldstone, M. B., et al. (1997) Nat. Med. 3, 205–211. [DOI] [PubMed] [Google Scholar]
- 17.Harcourt, G. C., Garrard, S., Davenport, M. P., Edwards, A. & Phillips, R. E. (1998) J. Exp. Med. 188, 1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leslie, A. J., Pfafferott, K. J., Chetty, P., Draenert, R., Addo, M. M., Feeney, M., Tang, Y., Holmes, E. C., Allen, T., Prado, J. G., et al. (2004) Nat. Med. 10, 282–289. [DOI] [PubMed] [Google Scholar]
- 19.Yokomaku, Y., Miura, H., Tomiyama, H., Kawana-Tachikawa, A., Takiguchi, M., Kojima, A., Nagai, Y., Iwamoto, A., Matsuda, Z. & Ariyoshi, K. (2004) J. Virol. 78, 1324–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore, C. B., John, M., James, I. R., Christiansen, F. T., Witt, C. S. & Mallal, S. A. (2002) Science 296, 1439–1443. [DOI] [PubMed] [Google Scholar]
- 21.Goulder, P. J., Brander, C., Tang, Y., Tremblay, C., Colbert, R. A., Addo, M. M., Rosenberg, E. S., Nguyen, T., Allen, R., Trocha, A., et al. (2001) Nature 412, 334–338. [DOI] [PubMed] [Google Scholar]
- 22.Goulder, P. J., Pasquier, C., Holmes, E. C., Liang, B., Tang, Y., Izopet, J., Saune, K., Rosenberg, E. S., Burchett, S. K., McIntosh, K., et al. (2001) Immunol. Lett. 79, 109–116. [DOI] [PubMed] [Google Scholar]
- 23.Friedrich, T. C., Dodds, E. J., Yant, L. J., Vojnov, L., Rudersdorf, R., Cullen, C., Evans, D. T., Desrosiers, R. C., Mothe, B. R., Sidney, J., et al. (2004) Nat. Med. 10, 275–281. [DOI] [PubMed] [Google Scholar]
- 24.Altman, J. D. & Feinberg, M. B. (2004) Nat. Med. 10, 229–230. [DOI] [PubMed] [Google Scholar]
- 25.Yusim, K., Kesmir, C., Gaschen, B., Addo, M. M., Altfeld, M., Brunak, S., Chigaev, A., Detours, V. & Korber, B. T. (2002) J. Virol. 76, 8757–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fagard, C., Oxenius, A., Gunthard, H., Garcia, F., Le Braz, M., Mestre, G., Battegay, M., Furrer, H., Vernazza, P., Bernasconi, E., et al. (2003) Arch. Intern. Med. (Moscow) 163, 1220–1226. [DOI] [PubMed] [Google Scholar]
- 27.Oxenius, A., Price, D. A., Gunthard, H. F., Dawson, S. J., Fagard, C., Perrin, L., Fischer, M., Weber, R., Plana, M., Garcia, F., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 13747–13752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mori, M., Beatty, P. G., Graves, M., Boucher, K. M. & Milford, E. L. (1997) Transplantation 64, 1017–1027. [DOI] [PubMed] [Google Scholar]
- 29.Cavalli-Svorza, L. L. (1994) The History and Geography of Human Genes (Princeton Univ. Press, Princeton).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}