

Abstract
BNIP1, a member of the BH3-only protein family, was first discovered as one of the proteins that are capable of interacting with the antiapoptotic adenovirus E1B 19-kDa protein. Here we disclose a totally unexpected finding that BNIP1 is a component of the complex comprising syntaxin 18, an endoplasmic reticulum (ER)-located soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptor (SNARE). Functional analysis revealed that BNIP1 participates in the formation of the ER network structure, but not in membrane trafficking between the ER and Golgi. Notably, a highly conserved leucine residue in the BH3 domain of BNIP1 plays an important role not only in the induction of apoptosis but also in the binding of α-SNAP, an adaptor that serves as a link between the chaperone ATPase NSF and SNAREs. This predicts that α-SNAP may suppress apoptosis by competing with antiapoptotic proteins for the BH3 domain of BNIP1. Indeed, overexpression of α-SNAP markedly delayed staurosporine-induced apoptosis. Our results shed light on possible crosstalk between apparently independent cellular events, apoptosis and ER membrane fusion.
Keywords: α-SNAP, apoptosis, BH3 domain, endoplasmic reticulum, membrane fusion
Introduction
The endoplasmic reticulum (ER), the largest membrane system within eukaryotic cells, consists of the reticular network of membrane tubules, sheet-like structures and lamellae (Baumann and Walz, 2001; Voeltz et al, 2002). The ER membranes differentiate into several morphologically and functionally distinct domains, such as the nuclear envelope and the rough and smooth domains. Despite this structural complexity, the ER is a highly dynamic organelle. During mitosis, it is partitioned between daughter cells, and subdomains are reconstructed. Even in interphase, new membrane tubules are continuously produced and fused with other tubules to form three-way junctions in a microtubule-dependent fashion (Lee and Chen, 1988; Allan and Vale, 1991; Dreier and Rapoport, 2000; Lippincott-Schwartz et al, 2000).
The ER has many diverse functions, including the translocation of proteins across the membrane, the folding, modification and export of secretory and membrane proteins, the retrieval of ER-resident proteins that have escaped the ER, stress response, lipid synthesis and Ca2+ storage. Although each function has been studied in great detail, crosstalk between the different functions has been so far poorly investigated. How and to what degree the different functions of the ER are correlated is totally unknown. Nevertheless, there must be mechanisms that allow the ER to manage its versatile functions. Indeed, a pioneer work of Brown, Goldstein and co-workers has demonstrated the linkage between stress response and the export of the membrane-bound transcription factor ATF6 from the ER (Ye et al, 2000).
Recently, the ER has emerged as an organelle involved in apoptotic cell death, although it is well known that mitochondria are central to this process (Breckenridge et al, 2003a; Kuwana and Newmeyer, 2003; Thomenius and Distelhorst, 2003). Antiapoptotic proteins, Bcl-2 and Bcl-xL, and proapoptotic proteins, Bax and Bak, are located in the ER as well as in mitochondria, and inhibit and initiate apoptosis, respectively (Krajewski et al, 1993; Ng and Shore, 1998; Häcki et al, 2000; Zong et al, 2003). These proteins appear to regulate apoptosis by affecting ER Ca2+ storage: Bcl-2 can reduce the releasable pool of ER Ca2+, whereas Bax and Bak act in the opposite way (Foyouzi-Youssefi et al, 2000; Pinton et al, 2000; Nutt et al, 2002; Scorrano et al, 2003). In addition to Bax and Bak, members of the proapoptotic BH3-only protein family (Bouillet and Strasser, 2002), such as BNIP1 (Boyd et al, 1994) and spike (Mund et al, 2003), are located in the ER.
Here we disclose a totally unexpected finding that the BH3-only protein BNIP1 is a component of the syntaxin 18 complex, a membrane fusion machinery on the ER membrane. Functional analysis revealed that BNIP1 is required for maintaining the integrity of the ER network. Importantly, the BH3 domain of BNIP1, the domain responsible for the induction of apoptosis, provides a binding site for α-soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (α-SNAP), raising the possibility that BNIP1 plays a pivotal role in crosstalk between apoptosis and membrane fusion.
Results
BNIP1 exhibits sequence similarity with yeast Sec20p
Membrane fusion is mediated by specific pairing between vesicle-associated SNAP receptors (v-SNAREs; the VAMP family) and target membrane-associated SNAREs (t-SNAREs; the syntaxin and SNAP-25 families) (Weber et al, 1998; Lin and Scheller, 2000; Jahn et al, 2003). We have recently isolated a syntaxin 18 complex from rat liver membranes and determined its subunit composition (Hirose et al, 2004). Syntaxin 18 is an ER-located SNARE involved in membrane trafficking between the ER and Golgi (Hatsuzawa et al, 2000), and most likely the ortholog of yeast Ufe1p, which is involved in both ER homotypic fusion (Patel et al, 1998) and retrograde transport from the Golgi to the ER (Lewis and Pelham, 1996). We found that syntaxin 18 is complexed with several proteins including ZW10 (a kinetochore-associated protein) (Chan et al, 2000), RINT-1 (a Rad50-interacting protein) (Xiao et al, 2001) and p31 (a mammalian homolog of yeast Use1p/Slt1p) (Belgareh-Touze et al, 2003; Burri et al, 2003; Dilcher et al, 2003). Yeast Ufe1p is known to bind Sec22p, Sec20p, Tip20p and Use1p/Slt1p (Lewis et al, 1997; Burri et al, 2003; Dilcher et al, 2003). Although the putative mammalian orthologs of Sec22p, Tip20p and Use1p/Slt1p (namely Sec22b, RINT-1 and p31, respectively) were found to exist in the isolated syntaxin 18 complex, no counterpart for Sec20p was discovered (Hirose et al, 2004).
To identify a possible mammalian Sec20p homolog, we searched the NCBI database. The search result showed that BNIP1 exhibits very low but significant sequence similarity to Sec20p (Figure 1A). BNIP1 is an ER-located BH3-only protein that was first discovered as one of the proteins capable of interacting with the antiapoptotic adenovirus E1B 19-kDa protein (Boyd et al, 1994). Sequence motif analysis of BNIP1 revealed the presence of an N-terminal putative coiled-coil region, a BH3 domain and a Sec20p homology region comprising a t-SNARE motif, followed by a C-terminal transmembrane domain (TMD) (Figure 1B).
Figure 1.

Sequence similarity between BNIP1 and Sec20p. (A) Related proteins were found in Homo sapiens (Hs. BNIP1), Saccharomyces cerevisiae (Sc. Sec20p), Schizosaccharomyces pombe (Sp. CAA16989), Candida albicans (Ca. Sec20p), Drosophila melanogaster (Dm. AAF51945) and Arabidopsis thaliana (At. BAB02931). Stars represent identical amino acids between BNIP1 and yeast Sec20p. (B) Domain structure of BNIP1. Stars represent highly conserved amino-acid residues in the BH3 domain.
BNIP1 is associated with syntaxin 18
Compared to syntaxin 5 and other Golgi SNAREs, syntaxin 18 and its binding proteins exhibit relatively low sequence similarities with their yeast counterparts (Hirose et al, 2004). This fact in conjunction with the presence of a t-SNARE motif in BNIP1 encouraged us to explore the possibility that BNIP1 is a component of the syntaxin 18 complex. First, we expressed FLAG-tagged BNIP1 in 293T cells, solubilized the cells with Triton X-100 and performed immunoprecipitation experiments using an anti-FLAG antibody. As shown in Figure 2A, lane 4, syntaxin 18 was co-precipitated with FLAG-BNIP1. In addition, α-SNAP and two isoforms of syntaxin 5, 41-kDa ER and 34-kDa Golgi forms, were also co-precipitated. In contrast, other Golgi SNAREs such as GS27/membrin and GS15 or a plasma membrane-localized syntaxin, syntaxin 4, was not co-precipitated, demonstrating the specificity of immunoprecipitation.
Figure 2.
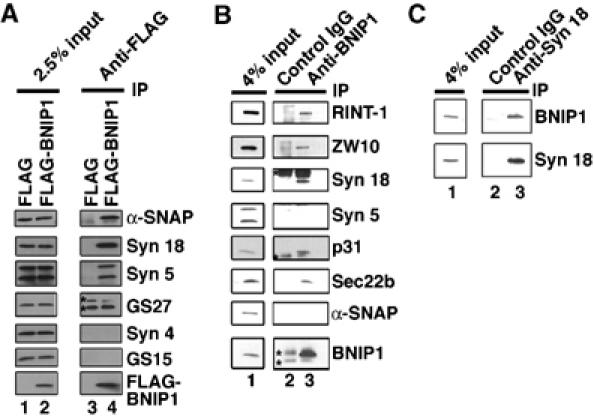
BNIP1 is a component of the syntaxin 18 complex. (A) 293T cells were transfected with 1 μg of pFLAG-BNIP1 (lanes 2 and 4) or pFLAG (lanes 1 and 3). After 24 h, the cells were lysed with Triton X-100 and immunoprecipitated with an anti-FLAG M2 monoclonal antibody with the aid of protein G-beads. The immunoprecipitated proteins were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and analyzed by immunoblotting with the indicated antibodies (lanes 3 and 4). Input (2.5% of total) was also analyzed (lanes 1 and 2). Stars denote immunoglobulin light chain. Note that GS27 was not co-precipitated with anti-FLAG. (B) 293T cell lysates were immunoprecipitated with a control IgG (lane 2) or an anti-BNIP1 antibody (lane 3). The co-precipitated proteins were analyzed by immunoblotting with the indicated antibodies. Input (4% of total) was also analyzed (lane 1). Stars denote immunoglobulin heavy or light chain. (C) 293T cell lysates were immunoprecipitated with a control antibody (lane 2) or a monoclonal anti-syntaxin 18 antibody (clone 1E1) (lane 3). The immunoprecipitated proteins were analyzed by immunoblotting with the indicated antibodies. Input (4% of total) was also analyzed (lane 1).
To confirm the association of BNIP1 with syntaxin 18, we raised an anti-BNIP1 antibody against a bacterially expressed protein lacking the TMD. The antibody specifically recognized BNIP1 in several cell lines (Supplementary Figure 1A) and, in agreement with a previous result (Boyd et al, 1994), stained the ER (Supplementary Figure 1B). When Triton X-100 extracts of 293T cells were incubated with the anti-BNIP1 antibody, not only syntaxin 18 but also RINT-1, ZW10, p31 and Sec22b, all of which are known to be components of the syntaxin 18 complex (Hirose et al, 2004), were co-immunoprecipitated (Figure 2B, lane 3). In contrast to the case of the immunoprecipitation of overexpressed BNIP1, neither syntaxin 5 nor α-SNAP was co-precipitated with endogenous BNIP1. This may imply a relatively weak interaction between BNIP1 and syntaxin 5 or α-SNAP. In a reciprocal immunoprecipitation experiment, BNIP1 was co-precipitated with a monoclonal anti-syntaxin 18 antibody (Figure 2C, lane 3), but not with a control antibody (lane 2). Similar immunoprecipitation results were obtained when 1% cholate or 1% octylglucoside was utilized to prepare cell lysates (data not shown). These results unequivocally demonstrated that BNIP1 is a component of the syntaxin 18 complex. Since the molecular mass of BNIP1 (∼26 kDa) is similar to that of immunoglobulin light chain, perhaps the identification of BNIP1 in the sequence analysis of immunoaffinity-purified syntaxin 18-binding proteins was hampered by contaminating immunoglobulin light chain (Hirose et al, 2004).
SNAREs are capable of forming a complex, and the complex is disassembled upon ATP hydrolysis catalyzed by NSF with the aid of α-SNAP (Söllner et al, 1993). This disassembly is believed to reflect the dissociation of the v-SNARE–t-SNARE complex after membrane fusion (Lin and Scheller, 2000; Jahn et al, 2003). In the case of the syntaxin 18 complex, however, a subcomplex comprising RINT-1, ZW10 and p31 is not disassembled even after they have dissociated from syntaxin 18 (Hirose et al, 2004). To examine whether or not BNIP1 is a component of the subcomplex, 293T cell lysates were incubated under conditions favoring SNARE complex disassembly. Like other conventional SNAREs, BNIP1 was released from syntaxin 18 in an α-SNAP-, NSF- and Mg2+-ATP-dependent manner (Figure 3A, lane 4), suggesting the functional linkage between BNIP1 and syntaxin 18. Moreover, BNIP1 did dissociate from RINT-1, ZW10 and p31, accompanied by the disassembly of the syntaxin 18 complex (lane 9). This indicates that BNIP1 is not a component of the RINT-1/ZW10/p31 subcomplex. This conclusion was corroborated by the results of similar immunoprecipitation experiments using antibodies against p31 and RINT-1 (Figure 3A, bottom panels).
Figure 3.
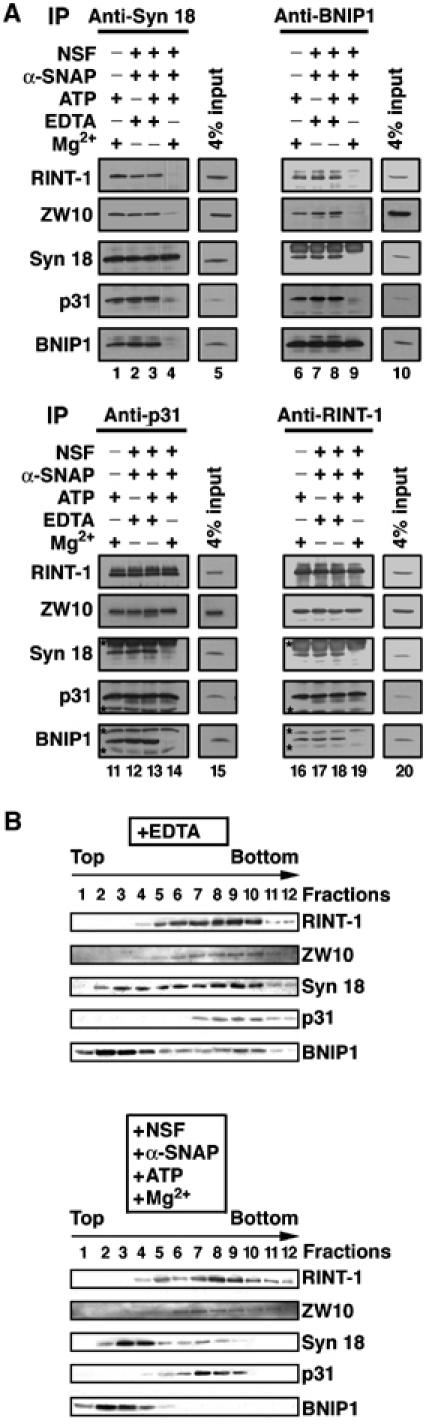
BNIP1 dissociates from syntaxin 18 and its binding proteins under conditions favoring SNARE complex disassembly. (A) 293T cell lysates were incubated under the indicated conditions (NSF: 10 μg/ml; α-SNAP: 5 μg/ml; ATP: 0.5 mM; MgCl2: 8 mM) at 16°C for 60 min. After incubation, the samples were immunoprecipitated with an antibody against syntaxin 18 (lanes 1–4), BNIP1 (lanes 6–9), p31 (lanes 11–14) or RINT-1 (lanes 16–19). The precipitated proteins were analyzed by immunoblotting with the indicated antibodies. Input (4% of total) was also analyzed (lanes 5, 10, 15 and 20). (B) 293T cell lysates that had been unincubated (top panel) or incubated (bottom panel) under conditions inducing SNARE complex disassembly were subjected to sedimentation analysis on 12–48% glycerol gradients (Hirose et al, 2004). Each fraction was analyzed by immunoblotting with the indicated antibodies.
To gain another line of evidence for the dissociation of BNIP1 from the RINT-1/ZW10 /p31 subcomplex in a process coupled with NSF-mediated ATP hydrolysis, 293T cell lysates were unincubated or incubated with α-SNAP, NSF and Mg-ATP, and then subjected to glycerol gradient sedimentation analysis (Figure 3B). Without incubation, BNIP1 sedimented diffusely throughout many fractions (upper panel), suggesting the presence of its free form (fractions 1–4) and complexed forms (fractions 5–10). BNIP1 at fractions 7–10 likely represents form(s) complexed with RINT-1, ZW10, p31 and syntaxin 18. Incubation under conditions inducing SNARE complex disassembly caused all BNIP1, like syntaxin 18, to sediment at fractions corresponding to the free form, whereas RINT-1, ZW10 and p31 were only slightly shifted (bottom panel). The latter finding is consistent with the idea that the RINT-1/ZW10/p31 subcomplex does not dissociate concomitantly with SNARE complex disassembly. These results confirmed the immunoprecipitation results that BNIP1 dissociates from the subcomplex upon NSF-mediated ATP hydrolysis.
Our previous two-hybrid study revealed that RINT-1 directly interacts with ZW10, whereas neither of them binds syntaxin 18 or a syntaxin 18-interacting protein, p31 (Hirose et al, 2004), raising the question of how RINT-1/ZW10 forms a complex with syntaxin 18. We therefore tested by using a two-hybrid assay whether BNIP1 interacts with RINT-1 or ZW10. The results are summarized in Table I. BNIP1 was found to interact with RINT-1, syntaxin 18 and α-SNAP. The direct interaction between BNIP1 and RINT-1 is in line with the observation that their yeast counterparts, Sec20p and Tip20p, respectively, physically interact to form a stable complex (Sweet and Pelham, 1993).
Table 1.
Two-hybrid interaction between BNIP1 and proteins in the syntaxin 18 complex
| Gal4 DNA-binding domain | Gal4 activation domain | β-Galactosidase activitya |
|---|---|---|
| Syntaxin 18 | BNIP1 | + |
| α-SNAP | BNIP1 | + |
| α-SNAP | BNIP1 L114A | − |
| BNIP1 | RINT-1b | + |
| BNIP1 L114A | RINT-1 | + |
| ZW10 | BNIP1 | − |
| p31 | BNIP1 | − |
| Results were recorded as positive if blue color was developed within 4 h on filters. | ||
| To analyze the interaction between RINT-1 and BNIP1, the cDNA of RINT-1 was inserted downstream of the Gal4 activation domain because its insertion downstream of the Gal4 DNA-binding domain gave a positive signal without an interacting partner. | ||
Overexpression of BNIP1 causes aggregation of ER membranes
To assess the function of BNIP1, we first investigated organelle morphology in BNIP1-overexpressing cells. As shown in Supplementary Figure 2A, overexpression of BNIP1 caused aggregation of ER proteins, calnexin and prolyl 4-hydroxylase, in HeLa cells. A similar aggregated pattern was observed with green fluorescent protein-cytochrome b5 (GFP-b5), implying that the observed aggregates represent aggregated ER membranes and not protein aggregates. Electron microscopic analysis confirmed these immunofluorescence observations: there were highly aggregated membrane structures, occasionally whorl-shaped ones, with fairly intact Golgi structure in BNIP1-overexpressing cells (Supplementary Figure 2B). The whorl-shaped structure in BNIP1-overexpressing cells is reminiscent of the organized smooth ER (OSER), which is known to be formed upon overexpression of ER-associated membrane proteins that are capable of oligomerization (Snapp et al, 2003). Perhaps, BNIP1 induced ER aggregation by linking ER membranes via a homophilic interaction.
In contrast to this drastic change in the ER structure, no marked change was observed in the morphology of the ER exit sites (Sec23p), Golgi (GM130), early endosomes (EEA1) and mitochondria (MitoTracker) (Supplementary Figure 2C), suggesting the limited effect of BNIP1 overexpression on membrane structures.
Our previous study demonstrated that overexpression of syntaxin 18 causes not only ER aggregation but also a defect in membrane traffic from the ER (Hatsuzawa et al, 2000). The finding that overexpression of BNIP1 has limited effects on the morphology of the ER exit sites and the Golgi apparatus may suggest that BNIP1 does not participate in membrane transport. To test this idea, we measured the transport of VSVG-GFP, a chimera protein comprising GFP and vesicular stomatitis virus-encoded glycoprotein (VSVG) whose export from the ER takes place in a temperature-dependent manner (Presley et al, 1997). As shown in Supplementary Figure 3, no marked delay in VSVG-GFP transport from the ER to the plasma membrane through the Golgi was observed in BNIP1-overexpressing cells.
BNIP1 is required for the organization of the ER network
The finding that overexpression of BNIP1 induces ER aggregation but does not affect the morphology of other organelles including the Golgi apparatus may suggest that BNIP1 is involved in ER membrane fusion. To test this idea, we knocked down BNIP1 expression by RNA interference (RNAi) and examined the ER network structure in living HeLa cells. Analysis of living cells allows us to detect subtle changes in the ER network structure more clearly compared to fixed cells. A previous study demonstrated that inhibition of ER membrane fusion results in the loss of the three-way junctions of the ER network (Uchiyama et al, 2002). BNIP1 expression was successfully knocked down by small interfering RNA (siRNA (155–175)), as detected by immunoblotting (Figure 4A) and immunofluorescence analysis (Figure 4B). Concomitant with the reduction of BNIP1 expression, the reticular ER structure became disintegrated in some areas of HeLa cells, whereas mitochondria appeared to be fairly intact (Figure 4C and Supplementary Figure 4). This effect was most likely due to a decreased level of BNIP1 expression because no significant change in the ER structure was observed when cells were transfected without siRNA (mock-treated) or with an inefficient RNA duplex (siRNA (532–552)). Quantitative analysis showed that the number of three-way junctions was decreased by approximately 50% in BNIP1-depleted cells compared to control cells (Figure 4E).
Figure 4.
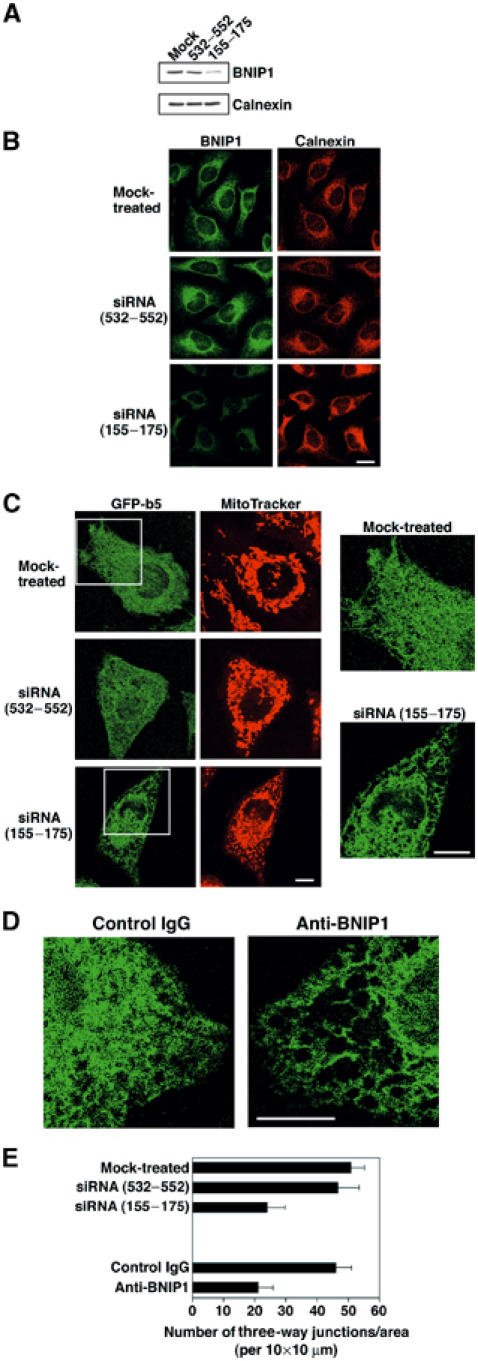
Loss of BNIP1 function leads to disruption of the ER network. GFP-b5-expressing HeLa cells (A) or nonexpressing cells (B) were transfected without (mock-treated) or with an inefficient RNA duplex (siRNA (532–552)) or an efficient one (siRNA (155–175)) and incubated for 24 h. Immunoblotting (A) and immunofluorescence microscopic analysis (B) showed a substantial reduction of BNIP1 expression in the cells transfected with siRNA (155–175). (C) Transfection was performed as described above. The ER morphology of living cells was investigated. Enlarged images of the boxed areas are shown on the right. Bar, 10 μm. (D) A control IgG or an antibody against BNIP1 was microinjected, and the ER morphology of living cells was investigated. Bar, 10 μm. (E) Quantitation of the number of the three-way junctions of the ER.
To gain another line of evidence that BNIP1 is required for the maintenance of the ER network structure, we microinjected an anti-BNIP1 antibody. As shown in Figure 4D and Supplementary Figure 5, injection of an anti-BNIP1 antibody resulted in disintegration of the ER network, especially at the cell periphery. This pattern was similar to that observed in BNIP1-depleted cells (Figure 4C). The number of three-way junctions was also decreased by approximately 50% in cells microinjected with the antibody (Figure 4E). Collectively, the results of functional analyses strongly suggest that BNIP1 is implicated in the organization of the ER network, likely by mediating ER membrane fusion.
BH3 domain of BNIP1 is required for the binding of α-SNAP
The fact that BNIP1 is a proapoptotic protein and also a component of the syntaxin 18 complex raises the intriguing possibility that this protein mediates crosstalk between apoptosis and membrane fusion. To address this possibility, we produced a series of BNIP1 constructs each with a point mutation at the BH3 domain and examined their interactions with syntaxin 18 and its binding proteins. As shown in Figure 5A, lane 19, the L114A mutant, in which Leu-114, one of the most conserved amino-acid residues in the BH3 domain (Figure 1B), was replaced with Ala, did not bind α-SNAP. A yeast two-hybrid assay (Table I) and a GST pull-down experiment (Figure 5B, lanes 9 and 10) demonstrated that BNIP1 interacts directly with α-SNAP, and that Leu-114 is essential for the binding. Other than α-SNAP, the L114A mutant bound RINT-1, ZW10, syntaxin 18 and p31 as efficiently as wild-type BNIP1 (Figure 5A). The L115A mutant also showed a decreased α-SNAP binding activity (lane 21). However, other constructs with a point mutation at the BH3 domain bound α-SNAP to similar extents as did wild-type BNIP1. To eliminate the trivial possibility that the L114A mutant had acquired an unintended, accidental second mutation during DNA manipulation, we produced a revertant by site-directed mutagenesis. The revertant construct bound α-SNAP to almost the same extent as wild-type BNIP1 (lane 20).
Figure 5.
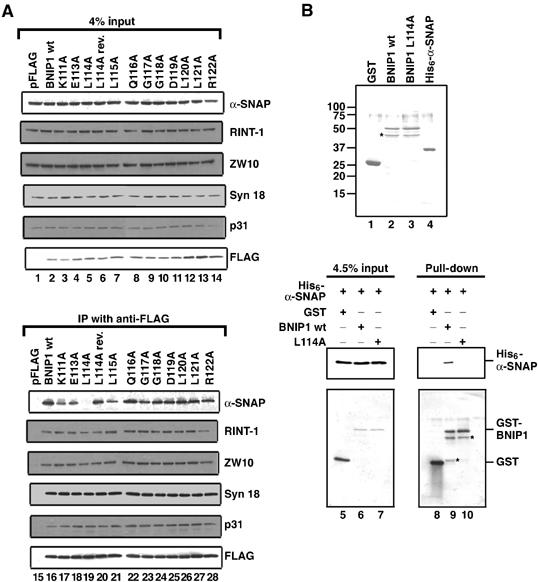
Leu-114 in the BH3 domain is required for α-SNAP binding. (A) 293T cells grown on six-well plates were transfected with pFLAG constructs encoding wild-type BNIP1 and the indicated BH3 domain mutants. At 24 h after transfection, cell lysates were prepared and FLAG-tagged proteins were immunoprecipitated with an anti-FLAG antibody. The immunoprecipitated proteins were separated by SDS–PAGE and analyzed by immunoblotting with the indicated antibodies (lower panel). Input (4% of total) was also analyzed (upper panel). (B) Coomassie staining of purified GST (lane 1), GST-BNIP1 wild type (lane 2), GST-BNIP1 L114A (lane 3) and His6-α-SNAP (lane 4). His6-α-SNAP (1 μg) was incubated with GST, GST-BNIP1 wild type or GST-BNIP1 L114A (1 μg each) at 4°C for 60 min. GST fusion proteins were pulled down with glutathione beads. The pulled-down proteins were separated by SDS–PAGE and analyzed by immunoblotting with antibodies against His tag and GST (lanes 8–10). Input (4.5% total) was also analyzed (lanes 5–7). Stars represent putative degradation products.
A previous study showed that deletion of the BH3 domain of BNIP1 results in a substantial loss of apoptotic-inducing activity (Yasuda and Chinnadurai, 2000). To determine if Leu-114 is important for apoptosis-inducing activity, we investigated the ability of the L114A mutant to suppress colony formation of MCF-7 cells. Consistent with the previous observation that BNIP1 displays a relatively weak proapoptotic activity (Yasuda and Chinnadurai, 2000), transfection with the plasmid for wild-type BNIP1 suppressed colony formation substantially but less efficiently than did transfection with the plasmid for Bad, another BH3-only protein (Bouillet and Strasser, 2002; Marsden and Strasser, 2003). The L114A mutant suppressed colony formation less significantly than wild-type BNIP1 (Figure 6A). Immunoblotting showed that the expression levels of wild-type BNIP1 and the L114A mutant were equivalent, ruling out the possibility that a poor apoptotic-inducing activity of the mutant is due to its low expression efficiency in cells. Importance of Leu-114 for apoptosis-inducing activity was confirmed by an experiment in which wild-type BNIP1 and the L114A mutant were transiently expressed in HeLa cells. The L114A mutant promoted apoptosis with very low efficiency (Figure 6B).
Figure 6.
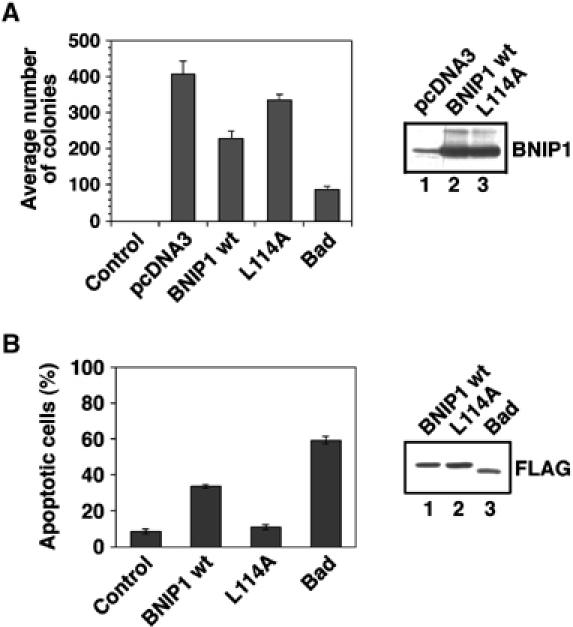
Leu-114 is important for apoptosis-inducing activity. (A) Suppression of colony formation by BNIP1 and the L114A mutant. MCF-7 cells were transfected with the indicated plasmids. The cells were cultured for 2 weeks in the presence of 0.8 mg/ml G418, fixed, stained with Giemsa solution and counted. Immunoblotting showed the equivalent expression of wild-type BNIP1 (lane 2) and the L114A mutant (lane 3). The band in lane 1 represents endogenous BNIP1. (B) HeLa cells were transfected with the plasmid for GFP (control) or FLAG-tagged proteins. At 24 h after transfection, nuclear morphology was visualized with Hoechst 33342 to count the number of live and apoptotic cells. The percentages of apoptotic cells among cells expressing GFP, wild-type BNIP1, the L114A mutant or Bad were determined. Immunoblotting using an anti-FLAG antibody showed the equivalent expression of wild-type BNIP1 (lane 1), the L114A mutant (lane 2) and Bad (lane 3).
Overexpression of α-SNAP delays the onset of staurosporine-induced apoptosis
The finding that the BH3 domain of BNIP1 undertakes both proapoptotic activity and α-SNAP binding prompted us to ask if overexpression of α-SNAP suppresses apoptosis by preventing BNIP1 from binding to antiapoptotic proteins. To this end, we established a Tet-on HeLa cell line that can express hemagglutinin (HA)-tagged α-SNAP in a doxycycline-dependent manner. At 48 h after induction of HA-α-SNAP expression, apoptosis was induced by incubation of the cells with 1 μM staurosporine for 3.5 h, and cell death was monitored by DNA staining and a caspase-3 activation assay. Although this apoptotic stimulus induced cell death of most control cells, many α-SNAP-expressing cells survived (Figure 7A). Quantitative analysis showed that only 10–17% of α-SNAP-expressing cells died under conditions where 65–90 % of nonexpressing cells died (Figure 7B). The fact that the percentages of apoptotic cells were lower when determined using the procaspase-3 activation method compared with the DNA staining method might be attributable to a lower sensitivity of the former method. Time course analysis revealed that α-SNAP overexpression does not completely inhibit staurosporine-induced apoptosis, but rather delays it. More than 65% of α-SNAP-expressing cells died upon a 7-h staurosporine treatment. The protective effect of α-SNAP was also observed when tunicamycin, an ER stress inducer, was used (data not shown).
Figure 7.
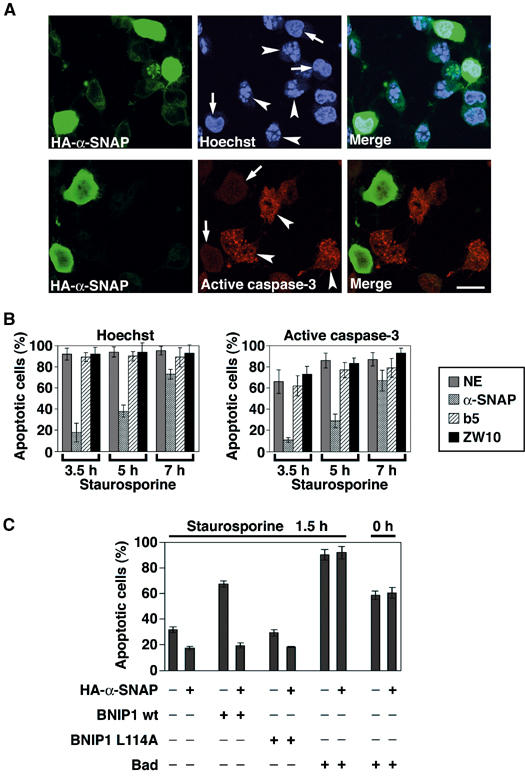
Overexpression of α-SNAP delays apoptosis. (A) HeLa Tet-on cells were grown on coverslips and expression of proteins was induced with 1 μg/ml doxycycline. At 48 h after induction, the cells were treated with 1 μM staurosporine for 3.5 h. In the case of Tet-On HeLa/HA-α-SNAP cells, cells were fixed and incubated with an anti-HA antibody followed by a fluorescein isothiocyanate-labeled secondary antibody. Nuclear morphology and the active form of caspase-3 were visualized with Hoechst 33342 and an antibody to active caspase-3, respectively. Arrows and arrowheads indicate live and apoptotic cells, respectively, in cells expressing ectopic proteins. Note that not all cells expressed α-SNAP. This allowed us to determine the percentage of apoptotic cells among cells nonexpressing or expressing α-SNAP under the same condition. Bar, 20 μm. (B) Time course of apoptosis in cells expressing ectopic proteins. Apoptosis was evaluated by Hoechst 33342 staining (left panel) or activation of caspase-3 (right panel). NE represents nonexpressing cells. (C) HeLa Tet-on cells in which α-SNAP expression had been induced for 40 h were transfected with the pTRE plasmid encoding FLAG-tagged BNIP1 wild type, the L114A mutant or Bad. At 16 h after transfection in the presence of 1 μg/ml doxycycline, the cells were treated with 1 μM staurosporine for 1.5 h. The cells were fixed, and nuclear morphology was visualized with Hoechst 33342.
Since the apoptotic pathways involve a number of protein–protein interactions, overexpression of proteins such as α-SNAP that are not related to the signaling pathways may block apoptosis by preventing protein–protein interactions in a nonspecific manner. To exclude this possibility, we examined the effect of overexpression of ZW10 and cytochrome b5 on apoptosis. The former, as well as BNIP1, is a component of the syntaxin 18 complex (Hirose et al, 2004) and the latter, like BNIP1, is an ER integral membrane protein. The two proteins were expressed as GFP fusion proteins in a Tet-on manner, and apoptosis was induced by staurosporine. As shown in Supplementary Figure 6, many GFP-b5- and GFP-ZW10-expressing HeLa cells died during a 3.5-h incubation with staurosporine. Quantitative data showed that the extents of cell death in GFP-b5- and GFP-ZW10-expressing HeLa cells are comparable to that of nonexpressing cells (Table II), implying the specificity of the effect of α-SNAP overexpression on apoptosis.
Table 2.
Inhibition of apoptosis by overexpression of α-SNAP
| Expression | None | HA-α-SNAP | GFP-b5 | GFP-ZW10 |
|---|---|---|---|---|
| Apoptotic cells (%)a | 80.1±9.2 | 17.4±4.2 | 79.3±4.8 | 77.4±10.2 |
| Tet-on HeLa/HA-α-SNAP, Tet-on HeLa/GFP-ZW10 and Tet-on HeLa/GFP-b5 cells grown on coverslips were incubated with 1 μg/ml doxycycline for 48 h. The cells were treated with 1 μM staurosporine for 3.5 h. Live and apoptotic cells were scored. | ||||
To provide a more direct linkage between α-SNAP, BNIP1 and apoptosis, we examined whether α-SNAP overexpression suppresses the proapoptotic effect of BNIP1, but not that of other BH3-only proteins. For this purpose, BNIP1 was expressed with or without α-SNAP in HeLa cells, and apoptosis was induced with staurosporine. As shown in Figure 7C, α-SNAP overexpression markedly suppressed apoptosis in BNIP1-expressing cells but not in cells expressing Bad, which was found not to interact with α-SNAP (data not shown). A similar result was obtained for Bad-expressing cells, when apoptosis was not induced by staurosporine. These results suggest the specific protective effect of α-SNAP on BNIP1-induced apoptosis.
Discussion
In the present study, we show that BNIP1, a proapoptotic BH3-only protein, is a component of the syntaxin 18 complex. Functional analysis revealed that BNIP1 is required for the maintenance of the ER network structure, but not for membrane traffic from the ER to the Golgi. BNIP1 appears to be the mammalian ortholog of yeast Sec20p (Sweet and Pelham, 1992), although it shows very low sequence similarity with Sec20p, and its molecular mass (26 kDa) is markedly different from that of Sec20p (44 kDa). Consistent with this idea, BNIP1 interacts directly with RINT-1 (the mammalian counterpart of Tip20p), as Sec20p does with Tip20p (Sweet and Pelham, 1993). The amino-acid conservation in proteins that constitute the ER fusion machinery is very low between yeast and mammals. Tip20p exhibits only 21% identity in amino acids 300–620 of RINT-1 (792 amino acids in total) (Sweet and Pelham, 1993; Hirose et al, 2004). The overall sequence identity between Ufe1p and syntaxin 18 is only 12% (Lewis and Pelham, 1996; Hatsuzawa et al, 2000). This situation is in marked contrast to that of Golgi syntaxins, yeast Sed5p and its mammalian counterpart, syntaxin 5. They show 30% amino-acid identity (Hardwick and Pelham, 1992; Bennett et al, 1993). The marked diversity in primary structure between the yeast and mammalian proteins may reflect distinct features of the ER in these organisms. In yeast, the nuclear envelope, which is contiguous to the ER membrane, is not disassembled in mitosis, and ER/nuclear membrane fusion during karyogamy occurs in a Ufe1p-dependent manner (Patel et al, 1998). In mammals, on the other hand, the nuclear envelope is disassembled at the onset of mitosis and reassembled at telophase.
The present and previous studies revealed that three membrane proteins (syntaxin 18, p31 and BNIP1) in the syntaxin 18 complex are capable of interacting with α-SNAP. Why do plural proteins in one complex bind α-SNAP? One possibility is that each α-SNAP-binding protein has a unique role. As shown in the present study, depletion of BNIP1 caused disintegration of the ER network. On the other hand, depletion of syntaxin 18 had no marked effect on the ER membrane structure (unpublished data). p31 may be involved in membrane transport between the ER and Golgi, rather than in ER membrane fusion, because ZW10, a component of a subcomplex comprising p31, is implicated in ER–Golgi membrane traffic (Hirose et al, 2004). Perhaps, comprehensive analysis of interactions between components of the syntaxin 18 complex may help in revealing the roles of the plural α-SNAP-binding proteins. In this regard, one progress in the present study is the finding that BNIP1 directly interacts with RINT-1. Our previous study failed to show the presence of a protein that can serve as a link between syntaxin 18 and the RINT-1/ZW10/p31 subcomplex (Hirose et al, 2004). The adaptor protein is most likely BNIP1. Dissociation of BNIP1 from the subcomplex under conditions favoring SNARE complex disassembly may imply that the direct association between BNIP1 and RINT-1 is eliminated concomitantly with the disassembly of the syntaxin 18 complex.
Our results demonstrated that the BH3 domain of BNIP1 is important not only for the induction of apoptosis but also for the binding of α-SNAP. Consistent with this observation, overexpression of α-SNAP markedly suppressed apoptosis induced by staurosporine. We could not exclude the possibility that α-SNAP suppresses apoptosis by binding to BH3-only proteins other than BNIP1. BNIP3, another protein capable of interacting with the adenovirus E1B 19-kDa protein, is its potential candidate. BNIP3, like BNIP1, possesses the BH3 domain, but is localized in mitochondria (Boyd et al, 1994). It is possible that α-SNAP blocks apoptosis by interacting with both ER and mitochondrial proteins.
Although the finding that BNIP1 has dual roles, that is, apoptosis and ER membrane fusion, is quite surprising, other BH3-only proteins may also have roles other than apoptosis. Bim, a BH3-only protein that is activated by signals including cytokine deprivation, is sequestered to the dynein complex by interacting with dynein light-chain DLC1/LC8 in healthy cells (Puthalakath et al, 1999). Similarly, Bmf, another BH3-only protein that confers cell death in response to anoikis, is sequestered to myosin V in healthy cells (Puthalakath et al, 2001). Although the interactions of these BH3-only proteins with cytoskeletal motors and their accessory proteins have been implicated in their sequesterization, they may have more positive roles in healthy cells, as demonstrated for BNIP1. In this context, it is noteworthy that BAP31, an ER membrane protein, is involved in both apoptosis and regulation of membrane traffic from the ER. In healthy cells, BAP31 appears to regulate export of proteins, such as cellubrevin (Annaert et al, 1997), MHC class I molecule (Spiliotis et al, 2000) and immunoglobulin D (Adachi et al, 1996). Stimulation of cell surface death receptors activates caspase-8, which results in the cleavage of BAP31. The p20 caspase cleavage fragment of BAP31 induces Ca2+ release from the ER, which enhances cytochrome c release from mitochondria (Breckenridge et al, 2003a, 2003b).
Materials and methods
Antibodies
To produce anti-BNIP1 antisera, bacterially expressed His6-BNIP1 lacking the TMD was purified using Ni-NTA agarose (Qiagen), mixed with Freund adjuvant (Difco Laboratories) and injected into Japan White rabbits. The antibody was affinity purified on beads bearing BNIP1ΔTMD. Polyclonal antibodies against α-SNAP, Sec22b, syntaxin 5, syntaxin 18, p31, RINT-1 and ZW10 and a monoclonal anti-syntaxin 18 antibody were prepared as described (Hatsuzawa et al, 2000; Hirose et al, 2004). Monoclonal antibodies against GS27, GS15, syntaxin 4, calnexin, GM130 and EEA1 were obtained from Transduction Laboratories. A polyclonal anti-Sec23p was purchased from Affinity BioReagents. A monoclonal anti-prolyl 4-hydroxylase β-subunit was obtained from Daiichi Fine Chemical. Monoclonal and polyclonal antibodies against FLAG were purchased from Sigma. Anti-HA and anti-GST were from Santa Cruz Biotechnology. Antibody against active caspase-3 was from Promega. Monoclonal antibodies against HA and penta His were from Roche Diagnostics Corp. and Qiagen, respectively.
Cell culture
HeLa cells were cultured in Dulbecco's modified minimal Eagle's medium supplemented with 50 IU/ml penicillin, 50 μg/ml streptomycin and 10% fetal calf serum. 293T cells and Vero cells were grown in Dulbecco's modified Eagle's medium supplemented with the same materials. MCF-7 cells were obtained from the Human Science Research Resource Bank and grown in Eagle's minimum essential medium supplemented with 10% fetal calf serum, 1 mM sodium pyruvate and 10 μg/ml bovine insulin (Sigma). Establishment of HeLa Tet-on cells stably expressing HA-α-SNAP and GFP-b5 was carried out similarly to that of HeLa Tet-on GFP-ZW10 stable cells (Hirose et al, 2004).
Plasmid construction and transfection
The cDNA of cytochrome b5 (kindly donated by Dr A Ito at Kyushu University) was inserted into pEGFP-C3 (Clontech). The full-length cDNA of BNIP1 was amplified by polymerase chain reaction using a human kidney cDNA library as template. The cDNA fragments encoding full-length BNIP1, the L114A mutant and full-length mouse Bad (kindly donated by Dr Y Gotoh at University of Tokyo) were inserted into pcDNA3 (Invitrogen), pFLAG-CMV-2 (Sigma) or pTRE (Clontech). The BH3 domain mutants were constructed by site-directed mutagenesis. Transfection was carried out using LipofectAMINE PLUS (Invitrogen) according to the manufacturer's protocol.
Immunoprecipitation
Approximately 90% confluent cells grown on 35 mm dishes were lysed with 0.5 ml of lysis buffer consisting of 25 mM HEPES–KOH (pH 7.2), 150 mM KCl, 2 mM EDTA, 1 mM dithiothreitol, 1% Triton X-100, 0.5 μg/ml leupeptin, 2 μM pepstatin, 2 μg/ml aprotinin and 1 mM phenylmethylsulfonyl fluoride, and centrifuged at 17 000 g for 10 min. Immunoprecipitation experiment was carried out as described (Hirose et al, 2004).
Cell death assays
Apoptosis-inducing activity was measured by the following two methods:
Suppression of colony formation: MCF-7 cells (approximately 1.0 × 105 cells/well) grown on six-well plates were transfected with the above constructs and plated into 100 mm dishes. At 48 h after transfection, 0.8 mg/ml G418 (Calbiochem) was added, and the cells were cultured for 2 weeks. G418-resistant colonies were stained with Giemsa solution and counted.
Transient expression assay: HeLa cells were transfected with 2 μg of pFLAG-CMV-2 (control), pFLAG-BNIP1 wild type, pFLAG-BNIP1 L114A or pFLAG-Bad. At 24 h after transfection, both attached and floating cells were recovered and fixed with 2% paraformaldehyde. The cells were stained with an anti-FLAG antibody and Hoechst 33342 to visualize expressed proteins and nuclear DNA, respectively. The numbers of live cells (intact nucleus) and apoptotic cells (condensed or fragmented chromatin) were counted. Alternatively, apoptosis was scored by activation of procaspase-3.
RNAi
BNIP1 was targeted with siRNA (155–175). The sequence is 5′-AACAGTTGCGTCACAGAATAC-3′, which corresponds to positions 155–175 relative to the start codon. As a control, an inefficient oligonucleotide duplex (siRNA (532–552)), the sequence of which corresponds to positions 532–552, was used. The siRNAs were purchased from Japan Bioservice. Transfection was performed using Oligofectamine (Invitrogen) according to the manufacturer's protocol.
Microinjection of antibody
Purified antibodies (control rabbit IgG ∼13 mg/ml or anti-BNIP1 ∼10 mg/ml) were injected into cells, and the cells were incubated for 20 h.
Quantitation of three-way junctions in the ER
The number of three-way junctions in the ER network was counted by confocal microscopy using living cells stably expressing GFP-b5. Two or three areas (each 10 × 10 μm), from the cell center (except the nucleus) to the periphery in each cell, were randomly selected, and three-way junctions in the ER were scored.
Immunofluorescence and electron microscopy
Immunofluorescence microscopy was performed as described (Tagaya et al, 1996). Cells were fixed with methanol at −20°C for 5 min for endogenous BNIP1 or with 4% paraformaldehyde for 20 min at room temperature for expressed proteins. Confocal microscopy was performed with a Fluoview 300 laser scanning microscope (Olympus) or a TCS SP2 AOBS (Leica Microsystems). Electron microscopy was performed as described (Yamaguchi et al, 1997)
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Acknowledgments
We are grateful to Dr A Ito and Dr Y Gotoh for cDNA clones. This work is supported in part by Grants-in-Aid Scientific Research 13680792, 10215205 and 11480183 from the Ministry of Education, Science, Sports and Culture of Japan. HH is a recipient of the Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists.
References
- Adachi T, Schamel WW, Kim KM, Watanabe T, Becker B, Nielsen PJ, Reth M (1996) The specificity of association of the IgD molecule with the accessory proteins BAP31/BAP29 lies in the IgD transmembrane sequence. EMBO J 15: 1534–1541 [PMC free article] [PubMed] [Google Scholar]
- Allan VJ, Vale RD (1991) Cell cycle control of microtubule-based membrane transport and tubule formation in vitro. J Cell Biol 113: 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert WG, Becker B, Kistner U, Reth M, Jahn R (1997) Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J Cell Biol 139: 1397–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann O, Walz B (2001) Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol 205: 149–214 [DOI] [PubMed] [Google Scholar]
- Belgareh-Touze N, Corral-Debrinski M, Launhardt H, Galan JM, Munder T, Le Panse S, Haguenauer-Tsapis R (2003) Yeast functional analysis: identification of two essential genes involved in ER to Golgi trafficking. Traffic 4: 607–617 [DOI] [PubMed] [Google Scholar]
- Bennett MK, Garcia-Arraras JE, Elferink LA, Peterson K, Fleming AM, Hazuka CD, Scheller RH (1993) The syntaxin family of vesicular transport receptors. Cell 74: 863–873 [DOI] [PubMed] [Google Scholar]
- Bouillet P, Strasser A (2002) BH3-only proteins—evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci 115: 1567–1574 [DOI] [PubMed] [Google Scholar]
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D'Sa-Eipper C, Chinnadurai G (1994) Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 79: 341–351 [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC (2003a) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608–8618 [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003b) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burri L, Varlamov O, Doege CA, Hofmann K, Beilharz T, Rothman JE, Söllner TH, Lithgow T (2003) A SNARE required for retrograde transport to the endoplasmic reticulum. Proc Natl Acad Sci USA 100: 9873–9877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GK, Jablonski SA, Starr DA, Goldberg ML, Yen TJ (2000) Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat Cell Biol 2: 944–947 [DOI] [PubMed] [Google Scholar]
- Dilcher M, Veith B, Chidambaram S, Hartmann E, Schmitt HD, Fischer von Mollard G (2003) Use1p is a yeast SNARE protein required for retrograde traffic to the ER. EMBO J 22: 3664–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier L, Rapoport TA (2000) In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J Cell Biol 148: 883–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA 97: 5723–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcki J, Egger L, Monney L, Conus S, Rosse T, Fellay I, Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19: 2286–2295 [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Pelham HR (1992) SED5 encodes a 39-kD integral membrane protein required for vesicular transport between the ER and the Golgi complex. J Cell Biol 119: 513–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsuzawa K, Hirose H, Tani K, Yamamoto A, Scheller RH, Tagaya M (2000) Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis-Golgi vesicle trafficking. J Biol Chem 275: 13713–13720 [DOI] [PubMed] [Google Scholar]
- Hirose H, Arasaki K, Dohmae N, Takio K, Hatsuzawa K, Nagahama M, Tani K, Yamamoto A, Tohyama M, Tagaya M (2004) Implication of ZW10 in membrane trafficking between the endoplasmic reticulum and Golgi. EMBO J 23: 1267–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Lang T, Südhof TC (2003) Membrane fusion. Cell 112: 519–533 [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res 53: 4701–4714 [PubMed] [Google Scholar]
- Kuwana T, Newmeyer DD (2003) Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol 15: 691–699 [DOI] [PubMed] [Google Scholar]
- Lee C, Chen LB (1988) Dynamic behavior of endoplasmic reticulum in living cells. Cell 54: 37–46 [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Pelham HR (1996) SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell 85: 205–215 [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Rayner JC, Pelham HR (1997) A novel SNARE complex implicated in vesicle fusion with the endoplasmic reticulum. EMBO J 16: 3017–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RC, Scheller RH (2000) Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 16: 19–49 [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Roberts TH, Hirschberg K (2000) Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol 16: 557–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden VS, Strasser A (2003) Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol 21: 71–105 [DOI] [PubMed] [Google Scholar]
- Mund T, Gewies A, Schoenfeld N, Bauer MK, Grimm S (2003) Spike, a novel BH3-only protein, regulates apoptosis at the endoplasmic reticulum. FASEB J 17: 696–698 [DOI] [PubMed] [Google Scholar]
- Ng FW, Shore GC (1998) Bcl-XL cooperatively associates with the Bap31 complex in the endoplasmic reticulum, dependent on procaspase-8 and Ced-4 adaptor. J Biol Chem 273: 3140–3143 [DOI] [PubMed] [Google Scholar]
- Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ, Swisher SG (2002) Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J Biol Chem 277: 9219–9225 [DOI] [PubMed] [Google Scholar]
- Patel SK, Indig FE, Olivieri N, Levine ND, Latterich M (1998) Organelle membrane fusion: a novel function for the syntaxin homolog Ufe1p in ER membrane fusion. Cell 92: 611–620 [DOI] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol 148: 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J (1997) ER-to-Golgi transport visualized in living cells. Nature 389: 81–85 [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A (1999) The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3: 287–296 [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A (2001) Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 293: 1829–1832 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Snapp EL, Hegde RS, Francolini M, Lombardo F, Colombo S, Pedrazzini E, Borgese N, Lippincott-Schwartz J (2003) Formation of stacked ER cisternae by low affinity protein interactions. J Cell Biol 163: 257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362: 318–324 [DOI] [PubMed] [Google Scholar]
- Spiliotis ET, Manley H, Osorio M, Zuniga MC, Edidin M (2000) Selective export of MHC class I molecules from the ER after their dissociation from TAP. Immunity 13: 841–851 [DOI] [PubMed] [Google Scholar]
- Sweet DJ, Pelham HR (1992) The Saccharomyces cerevisiae SEC20 gene encodes a membrane glycoprotein which is sorted by the HDEL retrieval system. EMBO J 11: 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet DJ, Pelham HR (1993) The TIP1 gene of Saccharomyces cerevisiae encodes an 80 kDa cytoplasmic protein that interacts with the cytoplasmic domain of Sec20p. EMBO J 12: 2831–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagaya M, Furuno A, Mizushima S (1996) SNAP prevents Mg2+-ATP-induced release of N-ethylmaleimide-sensitive factor from the Golgi apparatus in digitonin-permeabilized PC12 cells. J Biol Chem 271: 466–470 [DOI] [PubMed] [Google Scholar]
- Thomenius MJ, Distelhorst CW (2003) Bcl-2 on the endoplasmic reticulum: protecting the mitochondria from a distance. J Cell Sci 116: 4493–4499 [DOI] [PubMed] [Google Scholar]
- Uchiyama K, Jokitalo E, Kano F, Murata M, Zhang X, Canas B, Newman R, Rabouille C, Pappin D, Freemont P, Kondo H (2002) VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J Cell Biol 159: 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, Rapoport TA (2002) Structural organization of the endoplasmic reticulum. EMBO Rep 3: 944–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
- Xiao J, Liu CC, Chen PL, Lee WH (2001) RINT-1, a novel Rad50-interacting protein, participates in radiation-induced G2/M checkpoint control. J Biol Chem 276: 6105–6111 [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Yamamoto A, Furuno A, Hatsuzawa K, Tani K, Himeno M, Tagaya M (1997) Possible involvement of heterotrimeric G proteins in the organization of the Golgi apparatus. J Biol Chem 272: 25260–25266 [DOI] [PubMed] [Google Scholar]
- Yasuda M, Chinnadurai G (2000) Functional identification of the apoptosis effector BH3 domain in cellular protein BNIP1. Oncogene 19: 2363–2367 [DOI] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6: 1355–1364 [DOI] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6