

Abstract
Background
Microarray studies in cancer compare expression levels between two or more sample groups on thousands of genes. Data analysis follows a population-level approach (e.g., comparison of sample means) to identify differentially expressed genes. This leads to the discovery of 'population-level' markers, i.e., genes with the expression patterns A > B and B > A. We introduce the PPST test that identifies genes where a significantly large subset of cases exhibit expression values beyond upper and lower thresholds observed in the control samples.
Results
Interestingly, the test identifies A > B and B < A pattern genes that are missed by population-level approaches, such as the t-test, and many genes that exhibit both significant overexpression and significant underexpression in statistically significantly large subsets of cancer patients (ABA pattern genes). These patterns tend to show distributions that are unique to individual genes, and are aptly visualized in a 'gene expression pattern grid'. The low degree of among-gene correlations in these genes suggests unique underlying genomic pathologies and high degree of unique tumor-specific differential expression. We compare the PPST and the ABA test to the parametric and non-parametric t-test by analyzing two independently published data sets from studies of progression in astrocytoma.
Conclusions
The PPST test resulted findings similar to the nonparametric t-test with higher self-consistency. These tests and the gene expression pattern grid may be useful for the identification of therapeutic targets and diagnostic or prognostic markers that are present only in subsets of cancer patients, and provide a more complete portrait of differential expression in cancer.
Background
Studies of differential expression of individual genes often find genes that are up-regulated in some tumors, and down-regulated in others. Microarray studies typically seek to identify differentially expressed genes using use fold-change [1], t-tests [2], and models [3-6]. Studies of global gene expression patterns in cancer have focused largely on the identification of novel cancer subtypes via classification [7-13] or the identification of differentially expressed genes [14-18]. Such studies typically use fold-change [1], t-tests [2], and models [3-6]. The methods of analysis for identifying differentially expressed genes in data from microarray experiments vary widely [20-45], but all are focused on the question of whether genes are over-expressed or under-expressed in samples in group A (e.g., tumor, or treatment, or metastastic, or responder) compared to samples in group B (e.g., normal, or control, or quiescient, or nonresponder). These patterns can efficiently be referred to as AB (overexpressed in A) and BA (underexpressed in A) patterns. Typically, researchers use study designs that favor biological replication to maximize the ability to detect reproducibly genes that are differentially expressed in a patient population, at a sacrifice of the ability to detect individual-specific patterns of differential expression with technical replication. Most cancers are diseases with heterogeneous etiologies; moreover, the development of every primary tumor in different individuals is a unique biological event. Thus, the expression levels of genes in the individual patient are also important; some important gene dysregulation may be highly specific to each individual. Statistical methods that average gene expression may hide important expressotypes (expression phenotypes). Current tests that compare mean group expression intensities are not likely to find genes that are in fact significantly dysregulated in only a proportion of the individuals in the case population, unless the magnitude of differential expression is very high in the subset of individuals. Unsupervised clustering can be used to attempt to identify unknown partitions, or subgroups within patients, but clustering is not a well-defined method for finding differentially expressed genes, and, upon discovery of novel groups, researchers are restricted to comparing group means, and cannot identify genes that may be dysregulated in subsets of patients where the combined patterns of dysregulation patterns do not suggest coherent subgroups.
Results
A remarkable pattern emerges when the PPST test is applied to published cancer data sets, including breast cancer [7], ovarian cancer [16], colon cancer (epithelial-rich normals only [17,47]), lymphoma [18], and lung cancer [19] at the 99th percentile. We find an abundance of AB and BA pattern genes, with roughly the same number of genes called significant under the parametric t-test. We also find large numbers of genes with significant ABA test scores, and some with 'BAB' pattern genes (Table 1). There is a marked tendency in most data sets for more ABA (cancer-normal-cancer) type genes than BAB pattern genes. These patterns are also reflected in 'expression pattern grids' of gene with significant s3(ABA) statistics (Fig. 1). These patterns are reproducible at more stringent levels of α (Table 1).
Table 1.
Number of Genes with Significant Differences between Tumor and Normal Class in Various Cancer Types under the PPST and ABA tests and the parametric t-test (for comparison)
| α = 0.1 | α = 0.05 | ||||
| Data Set | PPST | ABA test (ABA/BAB) | PPST | ABA test (ABA/BAB) | parametric tα = 0.05 ** |
| breast7 | 572 | 40/5 | 326 | 28/49 | 313 |
| melanoma15 | 662 | 60/202 | 312 | 38/133 | 347 |
| colon45 | 1788 | 55/153 | 1558 | 46/55 | 1378 |
| ovarian16 | 3344 | 253/63 | 2060 | 189/22 | 1813 |
| lymphoma18 | 2077 | 286/42 | 1114 | 194/30 | 1370 |
| lung19 | 614 | 40/3 | 506 | 35/3 | 389 |
*A = tumor sample group, B = normal sample group **pooled variance t
Figure 1.
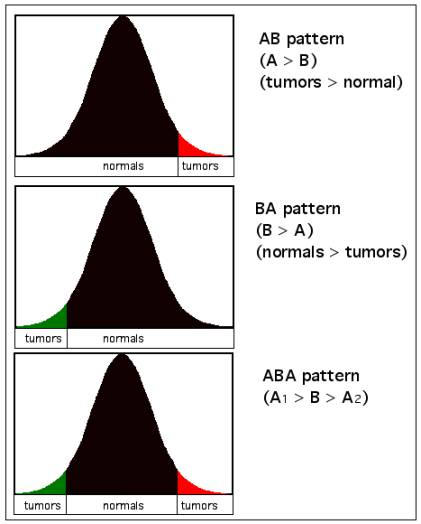
Conceptual representation of AB, BA, and ABA patterns of differential expression. The colored tails represent the placement of expression values of a given gene in tumors when compared to the distribution of expression values in normal samples. Standard AB and BA patterns are represented by red and black, respectively. Cases in which a surprising number of samples are distributed in both tails for a given gene are represented here as green (BA) and red (AB), respectively, and are painted similarly in Fig. 2 for specific samples.
The capability of the PPST test to identify genes that are in fact differentially expressed in only a subset of patients is made evident by a comparison of genes that are found to be significant under the PPST test, but missed by, for example, the t-test (even without Bonferroni-type adjustments). These are listed in Table 2, for the lymphoma data18, and notably include B-cell growth factor 1 (IL4; ABA pattern). Furthermore, 'classic' oncogenes such as cyclin D1 are found by the PPST test in the lung cancer data set [19] are not reported to be significant by the t-test. Cyclin D1 ranks 1009th among significant genes in the colon cancer data set under the t-test but ranks 90th under the PPST test (AB/BA pattern results only).
Table 2.
Exemplar Genes Found to the Significant (p < 0.05) under the PPST test in the lymphoma data set[18], but missed under the t-test.
| Gene | PPST Score* | Pattern | p-value under t-test |
| B-cell growth factor (IL-4) | 13 | ABA | 0.301 |
| CCND1 Cyclin D1 (PRAD1; parathyroid adenomatosis 1) | 13 | AB | 0.087 |
| homeobox protein Cdx2 mRNA | 12 | AB | 0.232 |
| LIF Leukemia inhibitory factor (cholinergic differentiation factor) | 10 | AB | 0.181 |
| tumor susceptiblity protein (TSG101) mRNA | -16 | BA | 0.526 |
| carcinoembryonic antigen | -18 | BA | 0.328 |
| CCND2 Cyclin D2 | -18 | BA | 0.076 |
| VIM Vimentin | -18 | BA | 0.976 |
*PPST Score = s1 or -s2 (for AB or BA pattern) and s3 (for ABA pattern)
Discussion
Our initial results are compelling in that they suggest that we can expect biomarkers of high clinical significance for subsets of patients to be important for distinct subsets of patients. This also suggests that clinical validation of the utility of biomarkers should examine panels of expression biomarkers, not individual biomarkers. Disruption of genomic function via these patterns cannot be studied in the population level biomarker framework for the simple reason that methods that compare, say, group means, will find no difference between the sample groups if the number of case samples found in the two tails are even approximately equal. This is a sensible approach even from within the framework of population-based hypothesis testing, because the PPST test can be expected to be more robust to one or two outliers that might mislead simple parametric tests. Note that a number of genes are 'nearly significant' under the t-test but are strongly significant under the PPST test for the AB/BA patterns (e.g., Table 2).
Our re-analysis of two independently generated data sets on astrocytoma progression demonstrates the utility of extending analysis to include a search for genes that are differentially expressed in a subset of patients. Of the tests examined, the parametric t-test showed the least internal consistency, while the PPST exhibited the highest internal consistency in identifying progression markers. Comparison to the non-parametric t-tests demonstrates that PPST is most similar to the nonparameteric t-test, but is more self-consistent. While the ABA test showed the least internal consistency across populations, it also exhibited low overlap with any other test, so the genes reported are unique and tend not to be found by others tests, matching expectations.
Our results are consistent with Knudsen's 'two-hit' hypothesis on the genomic etiologies of cancer [49] with some insight into the diversity of genomic pathologies (functional 'hits') that may be relevant in patient populations. Studies of differential gene expression – and its role in the etiology of cancer and its responses to treatment – should seek these types of genes in addition to population-wide biomarkers, because they represent a subset of the genes that are expressed differentially in a significant subset of cancer patients. We recommend a major shift in perspective on the study on gene expression dysregulation away from the study of 'tumor populations' – which do not exist – toward the study of genomic pathologies in individual patients. For example, tumor subtypes are typically characterized by morphological characters, and these classifications may conflict with important expressotype subtypes that do not follow classical morphological tumor classes. Imposition of these subtypes on a study design may interfere with identifying expressotypes that provide high diagnostic, prognostic and theranostic value to the individual – and sets of individuals with similar expressotypes. This view is also consistent with the Hanahan-Weinberg model of oncogenesis [50], which envisions multiple possible mechanistic strategies to the acquisition of characteristics and capabilities of cancers including self-sufficiency in growth signals, insensitivity to anti-growth signals, tissue invasion & metastasis, limitless replicative potential, sustained angiogenesis and evasion of apoptosis. We also expect that individual cancers in different patients will be found to have evolved unique sets of solutions to each of these problems. Current prevailing methods for finding differentially expressed genes such as fold-change and t-tests do not allow for such complexities.
Our comparison of the methods (Table 3) highlights the uniqueness of the ABA test. It is an extension of the PPST test; it specifically focuses on genes that are differentially expressed in subsets of patients. This ability is extremely important in search of genes with expression patterns that correlate with drug response. The ABA and the two-tailed t-test are not comparable because the ABA test allows us to find genes that the t-test specifically cannot (genes that are simultaneously overexpressed in some patients while underexpressed in others). Such test will have high variance (leading to a low t-test score) and low mean difference, and will thus not be significant. The PPST and the ABA tests extend our abilities beyond the t-test. Other improvements or even superior alternatives to these tests may be possible. The performance of these tests and all tests described to date for the AB type patterns and now for ABA patterns should be compared using extensive numerical simulations and cross-validation. Developments are needed to determine how best to select a threshold to allow deliberate control of the false positive and false negative error rates.
Table 3.
Summary of the overlap study of the two astrocytoma progression marker data sets. A. Internal consistency of the methods under comparison. k = Khatua et al. data set51; vdb = van den Boom et al. data set52 B. Number of significant genes that overlap between the two data sets in the significant gene list for each method. C. Comparison (% overlap) of methods in the k data set. D. Comparison (% overlap) of methods in the vdb data set
| A | % overlap | k | |||
| test | t-test (p) | t-test (np) | PPST | ABA | |
| vdb | t-test | 5.307 | |||
| t-test (np) | 11.248 | ||||
| PPST | 15.24 | ||||
| ABA | 3.211 | ||||
| B | overlap | k | |||
| vdb | test | t-test (p) | t-test (np) | PPST | ABA |
| t-test | 35 | ||||
| t-test (np) | 118 | ||||
| PPST | 187 | ||||
| ABA | 11 | ||||
| C | Test 1 | ||||
| k | t-test (p) | t-test (np) | PPST | ABA | |
| Test 2 | t-test (p) | 1 | 78.527 | 74.394 | 2.579 |
| t-test (np) | 1 | 83 | 4.678 | ||
| PPST | 1 | 1.28 | |||
| ABA | 1 | ||||
| Test 1 | |||||
| D | v | t-test (p) | t-test (np) | PPST | ABA |
| Test 2 | t-test (p) | 1 | 46.382 | 26.649 | 0 |
| t-test (np) | 1 | 56.975 | 5.656 | ||
| PPST | 1 | 7.268 | |||
| ABA | 1 |
Conclusions
The two major conclusions these results suggest are (1) that the most commonly applied tests for identifying differentially expressed genes will miss important genes that are dysregulated in only a fraction of patients, and (2) that important aspects of differential expression may be, to a degree, highly individualistic in most cancers. Some potentially important genes with this form of unusual differential expression (ABA; Table 2) would be missed by methods that compare group means, because the means of the two sample groups would be approximately identical, and the variance in tumors would be high, leading to a large error term. The high internal consistency of PPST compared to the non-parametric t-test and our observation that the PPST test exhibited high consistency with the nonparametric t-test suggests that the PPST test may be of interest to researchers interested in identifying both population-level biomarkers and biomarkers important to a subset of patients.
An online implementation of this test, it source code (Java), and that for many other methods of analysis, are accessible online in the Cancer Gene Expression Data Analysis tool http://bioinformatics.upmc.edu/. It is hoped that the development and application of more approaches like this will lead to a more complete representation of differential expression, leading to more meaningful and specific hypotheses of dysregulation, and thus a better comprehension of how diverse genomic pathologies contribute to the etiologies of cancers, and thereby facilitate the identification of targets that may lead to individual-specific therapies.
Methods
We have developed a test we call the Permutation Percentile Separability Test (PPST), which attempts to refute a null hypothesis that is slightly different from A = B, but which is capable of detecting AB, BA, ABA and BAB patterns. Under this test, we are interested in the question "are there are statistically significant number of samples in group A (e.g., tumor) that exhibit expression intensities beyond a particular percentile of the observed expression intensities in group B (e.g., normal)?" and vice versa. By 'statistically significant' we mean that the number of samples that exhibit apparent overexpression (or underexpression) exceeds that expected under the null distribution.
To test these hypotheses, we count the number of samples in both groups that are found beyond the nth percentile of the samples in the opposite group. This provides two scores, s1, and s2, for each gene (PPST scores). s1 is the number of samples in group A that are beyond the upper percentile (say, 95th) of group B plus the number of samples in group B that are below the lower 95th percentile of group A. This measure will tend to be large when all samples in both groups are significantly distinct from the alternate group in the same way (comparisons consistent with A > B). It can also be significant when a surprising number of samples in only one group varies from the expression levels in the alternate group. s2 is the number of samples with correspondingly opposite pattern (comparisons consistent with B > A). Sample class label permutations are used to generate an arbitrarily large number of permuted data sets. These scores s1 and s2 are calculated in each permuted data set to produce unique null distributions for each gene. For the sake of convenience of interpretation, we use -s2 when reporting s2 to denote underexpression. Genes with values of s1 beyond the specified acceptable Type 1 error risk (e.g., α = 5%) are determined to be significantly overexpressed in sample group A relative to B. Individuals in sample group A with expression intensity values over the 95th percentile of sample group B for a given gene may be considered overexpressed. Similarly, genes with values of s2 beyond the specified Type 1 risk for s2 are deemed underexpressed in sample group B relative to A. Varying the percentile threshold allows direct control over the false discovery rate.
Test for ABA patterns (ABA Test)
Genes that exhibit both significant s1 and s2 scores in this comparison may be considered 'ABA pattern genes' (Fig. 1); however, for stronger inference, permutation tests are also used to calculate s3, to determine, for a given gene, the number of samples from one group (A) that can expected to be distributed both in the upper and lower nth percentile tails of the intensity distribution of that gene in the other group (B); i.e., in the ABA (s3) or BAB (s4) pattern. These scores are not redundant to but rather allow for exploration of distribution-wise (upper and lower) false discovery rates. The application of the PPST test to find ABA patterns is called the 'ABA' test. Under the ABA test, differential expression of a gene may be deemed to be significant in both directions at once, i.e., simultaneously significantly over-expressed and under-expressed in a surprising number of patients in the case population. Both the PPST test and the ABA test will perform optimally when the variation in expression intensities in the normal sample population is well characterized.
A collection of published microarray data sets we have placed 'on-tap' in the caGEDA (Gene Expression Data Analysis) web application http://bioinformatics.upmc.edu/GE2/GEDA.html[51] were subjected to the PPST test and the ABA test. To avoid idiosyncracies that can result from the study of extreme values, we ran the tests at a fairly relaxed Type 1 error risk (α = 0.05 in both tails, or α = 0.10 overall). To compare the self-consistency of the parametric t-test, the nonparametric t-test, the PPST test and the ABA test, we re-analyzed two published data sets from independent astrocytoma progression studies [52,53]. Details of these studies are available in the original papers. In brief, Khatua et al. [52] studied global gene expression profiles from 6 early stage and 7 late-stage astrocytoma patients, while van den boom et al. [53] studied global gene expression profiles from 8 early stage and 8 late-stage astrocytoma patients. We calculated the overlap in the gene lists using our online Overlap tool http://bioinformatics.upmc.edu/GE2/Overlap.html.
Abbreviations
PPST: permutation percentile separability test.
ABA test: a test that can detect genes with both A > B (gene is overexpressed in sample A compared to sample group B) and B < A (gene is overexpressed in sample B compared to sample group A) patterns.
s1, s2, s3, s4: measures of the number of samples that exhibit expression intensities beyond a specified percentile in an alternate group; used as scores in the determination of significance under the PPST and ABA tests.
Author contributions
JLW conceived of the PPST and ABA tests and executed the analyses, SP encoded the methods in caGEDA, TG provided direction, input and scientific motivation for pursuing a test with the capabilities of the PPST and ABA tests, MJB provided, direction, input and coordination of the research. All authors read and approved the final manuscript.
Figure 2.
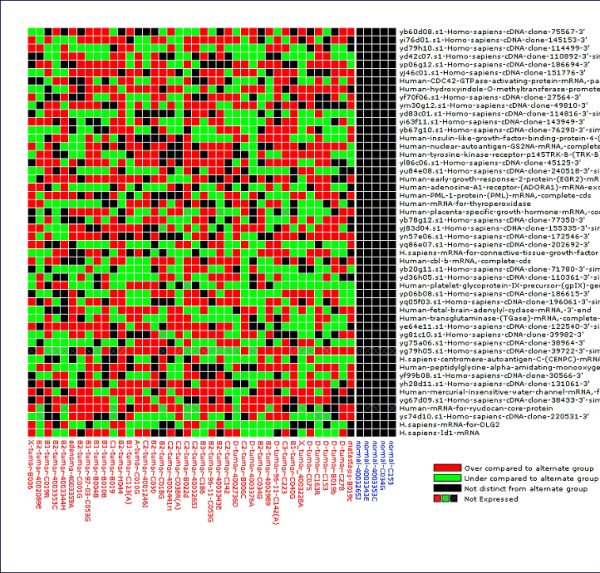
Gene Expression Pattern Grid of genes with significant ABA patterns from a comparison of epithelial-like normal colon tissue (blue samples) to colon cancers (red samples; Alon et al, 1999). We have previously determined that 5 samples in the Alon et al. data set were epithelial-like normal using unsupervised bootstrap cluster analysis and removed the remaining muscle-like normals from this analysis. These, and many other published cancer microarray data sets are 'on-tap' in our GEDA web application. The Gene Expression Pattern Grid, which is generated for any set of differentially expressed genes with the GEDA web application, summarizes the types of differential expression in a way that is related to the PPST test. Color signifies that an individual in one group exhibits an expression value that is significantly different from the expression pattern in the other group (red = overexpression; green = underexpression). Black signifies that an individual exhibits an expression value within the specified upper and lower percentiles in the other group. Tumor samples that fall within the upper and lower 95th %tiles of the distribution of expression values from the normal samples are labeled black, showing which genes for which a sample is are not different from normal. This representation includes information on both the population-level informativeness as well as which individuals appear to exhibit uniquely differentially expressed profiles. Samples within sample group are arranged according to their relative position in a hierarchical agglomerative clustering with pairwise distance = 1-Pearson's correlation coefficient. 'Not expressed' is a hypothesis generated in these graphs when the expression intensity value of that gene for that individual falls in the lower 95th %tile of the entire data set. Expression pattern grids were produced online with the Gene Expression Data Analysis web application http://bioinformatics.upmc.edu/.
Acknowledgments
Acknowledgments
This research was funded by JLW's faculty recruitment funds, provided by MB and Dr. Ronald Herberman using funds from a grant from the Claude Worthington Benedum Foundation (http://fdncentre.org/grantmaker/benedum). Eleanor Feingold is thanked for her critical read of the manuscript and of the implementation of the PPST and ABA tests.
Contributor Information
James Lyons-Weiler, Email: lyonsweilerj@upmc.edu.
Satish Patel, Email: Satish.Patel@gmail.com.
Michael J Becich, Email: becich@pitt.edu.
Tony E Godfrey, Email: godfreyte@upmc.edu.
References
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;24:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Black MA, Doerge RW. Calculation of the minimum number of replicate spots required for detection of significant gene expression fold change in microarray experiments. Bioinformatics. 2002;18:1609–1616. doi: 10.1093/bioinformatics/18.12.1609. [DOI] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Siegel AF, Hood LE. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J Comput Biol. 2000;7:805–817. doi: 10.1089/10665270050514945. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, Poggio T, Gerald W, Loda M, Lander ES, Golub TR. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci USA. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Ross DT, Perou CM, van de Rijn M. Towards a novel classification of human malignancies based on gene expression patterns. J Pathol. 2001;195:41–52. doi: 10.1002/path.889. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Welford SM, Gregg J, Chen E, Garrison D, Sorensen PH, Denny CT, Nelson SF. Detection of differentially expressed genes in primary tumor tissues using representational differences analysis coupled to microarray hybridization. Nucleic Acids Res. 1998;26:3059–3065. doi: 10.1093/nar/26.12.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, Wilfond B, Borg A, Trent J. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Welsh JB, Zarrinkar PP, Sapinoso LM, Kern SG, Behling CA, Monk BJ, Lockhart DJ, Burger RA, Hampton GM. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Natl Acad Sci USA. 2001;98:1176–1181. doi: 10.1073/pnas.98.3.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci USA. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos J, Thykjaer T, Tarte K, Ensslen M, Raynaud P, Requirand G, Pellet F, Pantesco V, Reme T, Jourdan M, Rossi JF, Orntoft T, Klein B. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene. 2002;21:6848–6857. doi: 10.1038/sj.onc.1205868. [DOI] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ZJ, Hu XG, Cao GS, Tang Y. Analysis of gene expression profile of pancreatic carcinoma using cDNA microarray. World J Gastroenterol. 2003;9:818–823. doi: 10.3748/wjg.v9.i4.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushel PR, Hamadeh HK, Bennett L, Green J, Ableson A, Misener S, Afshari CA, Paules RS. Computational selection of distinct class- and subclass-specific gene expression signatures. J Biomed Inform. 2002;35:160–170. doi: 10.1016/S1532-0464(02)00525-7. [DOI] [PubMed] [Google Scholar]
- Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JG, Olson JM, Tapscott SJ, Zhao LP. An efficient and robust statistical modeling approach to discover differentially expressed genes using genomic expression profiles. Genome Res. 2001;11:1227–1236. doi: 10.1101/gr.165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Kulaeva O, Hoff B, Petrov A, Shams S, Tainsky MA. Noise sampling method: an ANOVA approach allowing robust selection of differentially regulated genes measured by DNA microarrays. Bioinformatics. 2003;19:1348–1359. doi: 10.1093/bioinformatics/btg165. [DOI] [PubMed] [Google Scholar]
- Welford SM, Gregg J, Chen E, Garrison D, Sorensen PH, Denny CT, Nelson SF. Detection of differentially expressed genes in primary tumor tissues using representational differences analysis coupled to microarray hybridization. Nucleic Acids Res. 1998;26:3059–3065. doi: 10.1093/nar/26.12.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IV, Chen E, Hasseman JP, Liang W, Frank BC, Wang S, Sharov V, Saeed AI, White J, Li J, Lee NH, Yeatman TJ, Quackenbush J. Within the fold: assessing differential expression measures and reproducibility in microarray assays. Genome Biol. 2002;24:3–62. doi: 10.1186/gb-2002-3-11-research0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Siegel AF, Hood LE. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. Journal of Computational Biology. 2000;7:805–817. doi: 10.1089/10665270050514945. [DOI] [PubMed] [Google Scholar]
- Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data, regularized t-test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- Broet P, Richardson S, Radvanyi F. Bayesian hierarchical model for identifying changes in gene expression from microarray experiments. J Comput Biol. 2002;9:671–683. doi: 10.1089/106652702760277381. [DOI] [PubMed] [Google Scholar]
- Domingos P, Pazzani M. On the optimality of the simple Bayesian classifier under zero-one loss. Machine Learning. 1997;29:103–130. doi: 10.1023/A:1007413511361. [DOI] [Google Scholar]
- Friedman N, Linial M, Nachman I, Pe'er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- Ibrahim JG, Chen MH, Gray RJ. Bayesian models for gene expression with DNA microarray data. Journal of the American Statistical Association. 2002;97:88–99. doi: 10.1198/016214502753479257. [DOI] [Google Scholar]
- Kendziorski CM, Newton MA, Lan H, Gould MN. On parametric empirical Bayes methods for comparing multiple groups using replicated gene expression profiles. Statistics in Medicine. 2003;22:3899–3914. doi: 10.1002/sim.1548. [DOI] [PubMed] [Google Scholar]
- Lee KE, Sha N, Dougherty ER, Vannucci M, Mallick BK. Gene selection: a Bayesian variable selection approach. Bioinformatics. 2003;19:90–97. doi: 10.1093/bioinformatics/19.1.90. [DOI] [PubMed] [Google Scholar]
- Townsend JP, Hartl DL. Bayesian analysis of gene expression levels: statistical quantification of relative mRNA level across multiple strains or treatments. Genome Biol. 2002;3:RESEARCH0071. doi: 10.1186/gb-2002-3-12-research0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theilhaber J, Bushnell S, Jackson A, Fuchs R. Bayesian estimation of fold-changes in the analysis of gene expression: the PFOLD algorithm. J Comput Biol. 2001;8:585–614. doi: 10.1089/106652701753307502. [DOI] [PubMed] [Google Scholar]
- Li Y, Campbell C, Tipping M. Bayesian automatic relevance determination algorithms for classifying gene expression data. Bioinformatics. 2002;18:1332–1339. doi: 10.1093/bioinformatics/18.10.1332. [DOI] [PubMed] [Google Scholar]
- Pan W. On the use of permutation in and the performance of a class of nonparametric methods to detect differential gene expression. Bioinformatics. 2003;19:1333–1340. doi: 10.1093/bioinformatics/btg167. [DOI] [PubMed] [Google Scholar]
- Huang X, Pan W. Comparing three methods for variance estimation with duplicated high density oligonucleotide arrays. Funct Integr Genomics. 2002;2:126–133. doi: 10.1007/s10142-002-0066-2. [DOI] [PubMed] [Google Scholar]
- Park PJ, Pagano M, Bonetti M. A nonparametric scoring algorithm for identifying informative genes from microarray data. Pac Symp Biocomput. 2001:52–63. doi: 10.1142/9789814447362_0006. [DOI] [PubMed] [Google Scholar]
- Troyanskaya OG, Garber ME, Brown PO, Botstein D, Altman RB. Nonparametric methods for identifying differentially expressed genes in microarray data. Bioinformatics. 2002;18:1454–1461. doi: 10.1093/bioinformatics/18.11.1454. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B, Tibshirani R. Empirical Bayes methods and false discovery rates for microarrays. Genet Epidemiol. 2002;23:70–86. doi: 10.1002/gepi.1124. [DOI] [PubMed] [Google Scholar]
- Storey J. A direct approach to false discovery rates. J Roy Stat Soc Ser B. 2002;64:479–498. doi: 10.1111/1467-9868.00346. [DOI] [Google Scholar]
- Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Long D, Lyons-Weiler J. Overcoming confounded controls in the analysis of gene expression data from microarray experiments. Applied Bioinformatics. 2004;2:197–208. We have previously determined that 5 samples in the Alon et al. colon cancer data set [17] were epithelial-like normal using unsupervised bootstrap cluster analysis and removed the remaining muscle-like normals from this analysis. [PubMed] [Google Scholar]
- For 72 additional studies of gene expression patterns in cancer, see the University of Pittsburgh Cancer Gene Expression Data Link Database http://bioinformatics.upmc.edu/Help/UPITTGED.html
- Knudsen AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Patel S, Lyons-Weiler J. caGEDA: A web application for the integrated analysis of global gene expression patterns in cancer. Applied Bioinformatics. 2004;3:49–62. doi: 10.2165/00822942-200403010-00007. [DOI] [PubMed] [Google Scholar]
- Khatua S, Peterson KM, Brown KM, Lawlor C, Santi MR, LaFleur B, Dressman D, Stephan DA, MacDonald TJ. Overexpression of the EGFR/FKBP12/HIF-2alpha pathway identified in childhood astrocytomas by angiogenesis gene profiling. Cancer Res. 2003;63:1865–1870. [PubMed] [Google Scholar]
- van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, Sommer C, Reifenberger G, Hanash SM. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163:1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]