

Abstract
Neural networks engaged in high-frequency activity rely on sustained synaptic vesicle recycling and coordinated recruitment from functionally distinct synaptic vesicle (SV) pools. However, the molecular pathways matching neural activity to SV dynamics and release requirements remain unclear. Here we identify unique roles of SNARE-binding Tomosyn1 (Tomo1) proteins as activity-dependent substrates that regulate dynamics of SV pool partitioning at rat hippocampal synapses. Our analysis is based on monitoring changes in distinct functionally defined SV pools via V-Glut1-pHluorin fluorescence in cultured hippocampal neurons in response to alterations in presynaptic protein expression. Specifically, we find knockdown of Tomo1 facilitates release efficacy from the Readily Releasable Pool (RRP), and regulates SV distribution to the Total Recycling Pool (TRP), which is matched by a decrease in the SV Resting Pool. Notably, these effects were reversed by Tomo1 rescue and overexpression. Further, we identify that these actions of Tomo1 are regulated via activity-dependent phosphorylation by cyclin-dependent kinase 5 (Cdk5). Assessment of molecular interactions that may contribute to these actions identified Tomo1 interaction with the GTP-bound state of Rab3A, an SV GTPase involved in SV targeting and presynaptic membrane tethering. In addition, Tomo1 via Rab3A-GTP was also observed to interact with Synapsin 1a/b cytoskeletal interacting proteins. Finally, our data indicate that Tomo1 regulation of SV pool sizes serves to adapt presynaptic neurotransmitter release to chronic silencing of network activity. Overall, the results establish Tomo1 proteins as central mediators in neural activity-dependent changes in SV distribution among SV pools.
SIGNIFICANCE STATEMENT Although information transfer at central synapses via sustained high-frequency neural activity requires coordinated synaptic vesicle (SV) recycling, the mechanism(s) by which synapses sense and dynamically modify SV pools to match network demands remains poorly defined. To advance understanding, we quantified SV pool sizes and their sensitivity to neural activity while altering Tomo1 expression, a putative regulator of the presynaptic Readily Releasable Pool. Remarkably, we find Tomo1 actions to extend beyond the Readily Releasable Pool to mediate the Total Recycling Pool and SV Resting Pool distribution, and this action is sensitive to neural activity through Cdk5 phosphorylation of Tomo1. Moreover, Tomo1 appears to exert these actions through interaction with Rab3A-GTP and synapsin proteins. Together, our results argue that Tomo1 is a central mediator of SV availability for neurotransmission.
Keywords: Cdk5, hippocampal neuron, neurotransmitter release, plasticity, synaptic vesicle, tomosyn
Introduction
Neurotransmitter release from presynaptic elements closely follows action potential (AP) invasion, activation of voltage-gated Ca2+ channels and SNARE protein-mediated synaptic vesicle (SV) fusion (Jahn and Fasshauer, 2012; Südhof, 2013; Zhou et al., 2015). Essential to functional operation of synapses is the dynamic recruitment and cycling of SVs (Holtzman, 1977; Poodry and Edgar, 1979; Hummon and Costello, 1987). Indeed, sustenance of release to continued high-frequency activity or bursts of APs demands active recruitment of SVs into a fusion competent Readily Releasable Pool (RRP), and specific forms of synaptic plasticity are strictly defined by SV cycling into the RRP (Zucker and Regehr, 2002). Defining the regulatory controls on the cycling of SVs and their transition between functionally distinct SV pools is therefore of key importance.
In hippocampal neurons SVs have been found to occupy three functionally distinct pools, including the following: an RRP (5–8 vesicles) comprised of active zone SVs fully primed for immediate release; a Recycling Pool (RP; ∼20%-60% of SVs) that undergoes continuous exocytotic/endocytotic cycling; and a SV Resting Pool (ResP) (∼40%-80% of SVs) that is recalcitrant to activity-driven SV cycling (Rizzoli and Betz, 2005; Alabi and Tsien, 2012). Importantly, proportional reallocation of vesicles among the functionally defined pools is central to activity-dependent changes in synaptic strength, a parameter defined as the product of individual vesicle fusion probability and number of release ready vesicles. Notably, the RRP scales with the size of the RP during high-frequency AP firing (Waters and Smith, 2002), and recruitment of ResP vesicles into the RP impacts expression of Hebbian as well as homeostatic forms of synaptic plasticity at cortical and hippocampal synapses (Virmani et al., 2006; Kim and Ryan, 2010; Ratnayaka et al., 2012; Rose et al., 2013). Despite the importance of regulatory controls on distribution of SVs among these pools, the molecular mechanisms dictating transition of SVs from inactive/noncycling ResP to the active total recycling pool (Total Recycling Pool [TRP], i.e., RRP + RP) are poorly defined. To date, cyclin-dependent kinase 5 (Cdk5) is the single catalytic mediator described to induce changes in TRP/ResP partitioning (Kim and Ryan, 2010), perhaps via synapsin phosphorylation altering actin association (Verstegen et al., 2014). However, alternative mechanisms are likely as SV pool partitioning has also been reported to be independent of actin dynamics (Marra et al., 2012; Orenbuch et al., 2012).
Here we examine the role of Tomosyn-1 (Tomo1) family proteins in distribution of SV among functional presynaptic pools. A role for Tomo1 proteins in neurotransmission is substantiated by behavioral effects in mice (Barak et al., 2013), Drosophila (Chen et al., 2011), and C. elegans (Gracheva et al., 2007a) when its expression level is altered. Moreover, Tomo1 proteins have been genetically linked to autism spectrum disorders, mental retardation, and seizures (Davis et al., 2009; Matsunami et al., 2013; Cukier et al., 2014). Mechanistically, Tomo1 proteins act as potent inhibitors of evoked transmitter release from the RRP in neuroendocrine cells (Yizhar et al., 2004), rat superior cervical ganglion (Baba et al., 2005) and C. elegans neurons (Gracheva et al., 2007b; Gracheva et al., 2010), via interaction of their C-terminal R-SNARE domain with Syntaxin and SNAP25 to produce nonfusogenic SNARE complexes (Fujita et al., 1998; Hatsuzawa et al., 2003; Gladycheva et al., 2007). Yet, recently, Tomo1 was suggested to exert effects beyond inhibition of SV priming into the RRP, as loss-of-function mutations or targeted knockdown (KD) of Tomosyn enhanced EGTA-sensitive, delayed release of SVs at Drosophila and C. elegans neuromuscular junctions (McEwen et al., 2006; Chen et al., 2011). In addition, Tomosyn orthologs in yeast, Sro7p/Sro77p, result in accumulation of nonfusogenic vesicle clusters when overexpressed (Lehman et al., 1999; Rossi and Brennwald, 2011; Rossi et al., 2015). In the present study we uncover a novel site, mechanism, and activity-dependent regulatory pathway through which Tomo1 clamps SVs in the ResP, thereby reducing release by preventing SV transition into the TRP. These data indicate that Tomo1 proteins serve as central presynaptic regulators of release probability.
Materials and Methods
Antibodies.
The antibodies used included the following: anti-Synapsin 1 (SYSY; rabbit, #106011, 1:1000), anti-Tomo1 (SYSY, rabbit, #183103, Western blot 1:1000, immunocytochemistry [ICC] 1:400; BD Biosciences, mouse, #611296, Western blot 1:400), anti-Rab3A (SYSY, mouse, #107011, Western blot 1:1000; SYSY, rabbit, #107003, ICC 1:1000), anti-Cdk5 (Santa Cruz Biotechnology; rabbit, #SC173, Western blot 1:200; mouse, #SC6247, Western blot 1:200); anti-phospho-specific Cdk5 (Santa Cruz Biotechnology, rabbit, #SC12919, ICC 1:100), anti-synaptophysin (Sigma-Aldrich, mouse, #S5768, ICC, Western blot 1:250), anti-RIM (SYSY, rabbit, #140003, Western blot, ICC 1:500), proximity ligation assay (PLA) (Sigma-Aldrich, DUO92102), anti-actin (Sigma-Aldrich, mouse, #A2228#, Western blot 1:5000), anti-mouse IRDye800CW and anti-rabbit IRDye680LT (LI-COR, 1:5000), and anti-rabbit/mouse Alexa-488 and Alexa-594 secondaries (Invitrogen). Immunocytochemical mounts were treated with Vectashield containing DAPI (Vector Laboratories, #H-1200).
Plasmid constructs and lentiviral vectors.
The pCAGGS super ecliptic vGLUT1-pHluorin (vGpH) construct was obtained from Robert Edwards (University of California at San Francisco) (Voglmaier et al., 2006). mCherry (mCh) was subcloned in frame to the C terminus of vGLUT1-pHluorin to create vGLUT1-pHluorin-mCh with mCh exposed to the cytoplasm upon expression. Additional recombinant expression constructs included the following: pLP-mCh vector (CMV promoter); pLP-mCh-mTomo1 (mouse); pDNR-mTomo1-ΔCT, that is Tomo1 with deletion of C-terminal SNARE domain residues 1067–1131 (Williams et al., 2011); pCDNA CAPTEV-CT containing mTomo1 (rat; Invitrogen vector); and pLenti (synapsin promoter) containing YFP-mTomo1 (Barak et al., 2013); pcDNA3.1-Cdk5 (D144N), a dominant negative Cdk5 mutant (Shuang et al., 1998). Lentiviral vectors encoding a short hairpin RNA for targeted knockdown of all Tomo1 isoforms or a nontargeted scrambled shRNA were obtained from Origene. The shRNA sequences are as follows: KD1, ACTGCTTCAGCCAGTGATTGTGTCTCCAA; KD2, CCGTATGCTGTGGTTGTTCTCCTGGAGAA, scrambled control (Scr), CAGGAACGCATAGACGCATGA and were independently expressed (U6 promoter) using pGFP-C-shLenti or tRFP-CB-shLenti. Lentiviral plasmids were submitted to the University of Michigan Vector Core for high-titer lentiviral production. Briefly, HEK293T cells were transfected by Lipofectamine (Invitrogen) with vectors encoding REV, MDL, pvSVG, and a lentiviral plasmid containing expression construct. Forty-two hours after transduction, the medium containing the viruses was collected, filtered through a 0.45 μm filter to remove cell debris, then ultracentrifuged at 42,152 × g at 4°C for 2 h. The supernatant was discarded and virus pellet gently resuspended in 10 ml of NbActiv4 (BrainBits, Nb4). Aliquots containing 500 μl of NB4Activ with viruses were quickly frozen and stored at −80°C.
Primary hippocampal neuronal culture and transfections/infections.
Neuronal cultures were prepared from postnatal day 1–2 rat hippocampi of either sex as previously reported (Jakawich et al., 2010). Hippocampal neurons were plated on poly-d-lysine coated, 22-mm-diameter, #1.5 thickness coverglass (Neuvitro, GG-22) or on 14 mm microwell glass-bottom 35 mm culture dishes (MatTek) at a density of 400 cells/mm2. Neurons were cultured in NBActiv4. Cells were maintained in a 37°C, 95% O2/5% CO2 humidified incubator, and half the media was exchanged every 3–4 d with fresh NBActiv4 media. Neuronal cultures were transfected at 10–14 DIV. Transfection was achieved using Lipofectamine 2000 (Invitrogen) with 0.8–1 μg of selected plasmid DNA mixed with 1 μl of Lipofectamine in 200 μl of NBActiv4 and allowed to stand for 30 min before being dripped onto the cell cultures. Cultures were incubated for 1 h with the Lipofectamine/DNA mix, after which media was exchanged with fresh NBActiv4 media. For lentiviral infections, the stock virus (∼1 × 107 MOI/ml) stored at −80°C was thawed and diluted 1:10 in NBActiv4 culture media and added to neuronal cultures (5–9 DIV). After 12 h, additional NBActiv4 media was added diluting the virus 1:2. After 2 d, the culture media was exchanged with fresh NBActiv4 and neurons were used for experimentation between 7 and 14 d after infection.
vGpH image acquisition and analysis.
vGpH imaging was performed on individual neurons 3–10 d after transfection (14–24 DIV). Glass coverslips containing the neuronal cultures were mounted into a chamber outfitted for field stimulation (Warner Instruments, RC-49MFS) and perfused (0.5 ml/min) for rapid solution exchange (chamber volume, 100 μl) with temperature controlled (37°C; Warner Instruments, TC-324B) physiological saline containing the following (in mm): NaCl 136, KCl 2.5, HEPES 10, glucose 10, CaCl2 2, MgCl2 1.3,DL-AP5 0.05 (Tocris Bioscience, 3693), CNQX 0.01 (Tocris Bioscience, 1045), that was adjusted to pH 7.4. APs were evoked by field stimulation with 2 ms current pulses applied through platinum-iridium electrodes spatially placed within the chamber to achieve 10 V/cm fields. The frequency and period of stimulus application were controlled (Winston Electronics, A65) and synchronized to MetaMorph image acquisition software (Molecular Devices). Transfected neurons were spatially localized located on coverslips by mCh fluorescence of vGpH-mCh expression (Kim and Ryan, 2010). In cases where neurons were cotransfected with vGpH-mCH and an additional protein (i.e., tRFP-shTomo1, mCH-Tomo1), cotransfection was considered positive when the red channel fluorescence was diffuse and filled the neurons rather than the characteristic punctate localization of vGpH-mCH alone. The effectiveness of each applied stimulus within a train to evoke APs was confirmed on initial sets of Fluo4-treated neuronal cultures by measurement of time-synchronized alterations in intracellular free calcium. vGpH responses to field stimulation were always conducted in the presence of 1 μm bafilomycin-A1 (LC Laboratories, B-1080) to isolate exocytosis from endocytosis. Acquired vGpH fluorescent signals preceding and during stimulation were normalized to maximal vGpH fluorescence (FMAX). This was done by brief application of a bath solution containing the following (in mm): NH4Cl 50, NaCl 86, KCl 2.5, HEPES 10, glucose 10, CaCl 2 2, MgCl2 1.3,adjusted to set pH at 7.4 and osmolality between 290 and 300 mmol/kg, which leads to alkalization of the total vesicle pool (Sankaranarayanan et al., 2000). Fluorescence imaging was performed on an Olympus IX81 microscope with Plan-Apochromat 100× 1.4 NA or ApoN 60× 1.49 oil-immersion objectives (Olympus America). Illumination was provided by a Lambda LS 300 Watt Xenon arc lamp (Sutter Instruments) coupled to an electronically shuttered liquid light guide for controlled transmission of light to the microscope optics. Images were acquired using an ImagEM EM-CCD Hamamatsu (C9100-23B) camera. Optical filter-sets designed for mCh and eGFP were used, respectively: Ex, 472/30; 416/25 and Em, 520/35; 464/23. vGpH time-series images were acquired at 5 Hz, using a 40 ms integration time, not binned (512 × 512), with gain set to achieve the full dynamic 16-bit depth. A single image in the mCh channel was acquired at the beginning and end of the time series.
Spatial and temporal changes in fluorescence intensity within acquired vGpH time series image stacks were measured using FIJI (Schindelin et al., 2012), an open source distribution of ImageJ (http://imagej.nih.gov/ij/) focused on life sciences. Regions of interest (ROIs) were defined as puncta of fluorescent intensity localized along neuronal processes in images during NH4Cl treatment. Binary ROI masks were created by manual thresholding of a ΔFNH4Cl image. The ΔFNH4Cl image was calculated as FNH4Cl − Fpre, where FNH4Cl is defined as an average of 10 frames during NH4CL application and Fpre is defined as an average of 10 frames before application of any electrical stimulus in normal physiological saline. The resulting ΔFNH4Cl image was then subjected to a watershed protocol to create distinct ROIs for closely adjoining puncta. The resulting ROIs were then applied to each image within the time series, and the average fluorescent intensity within each ROI per image was determined using a custom-built plugin (Balaji and Ryan, 2007). The resulting time series of averaged intensity for each ROI was imported into Igor Pro (Wavemetrics) and corrected for photobleach decay by fitting a double exponential to fluorescence measurements taken from full-frame images across the time-series and subtracting it from the averaged intensity for each ROI (Royle et al., 2008). The magnitude of the correction never accounted for >15% of the measured fluorescence change in response to neural stimuli. The baseline intensity of each ROI was then subtracted (Fpre), and the data of each ROI were normalized to its maximal intensity (FNH4Cl). Time-series data for ROIs in each cell were averaged per time point to identify fractional TRP and ResP sizes. ROI time-series data were also grouped into ensembles for each treatment to generate cumulative frequency distributions of fractional TRP size. Data were plotted and statistical analysis performed using Prism 6 (Graphpad).
Immunoprecipitation.
Neuronal cultures (14–26 DIV) on glass-bottom dishes were rinsed twice with PBS (Invitrogen) and cells lysed in 70 μl immunoprecipitation (IP) lysis buffer per culture dish at 4°C using a cell scraper. IP lysis buffer contained the following (in mm): Tris-HCl 25, NaCl 50, MgCl2 2, CaCl2 1, 0.5% NP-40 adjusted to pH 7.4 and with protease and phosphatase inhibitors (Cell Signaling Technology, 5872) added immediately before use. Collected lysates were precleared via centrifugation at 2000 × g for 3 min. Magnetic Protein A beads (Invitrogen 10002D) prebound to 0.5–1 μg of antibody were then mixed with lysate and rotated at 4°C for 1 h, after which beads were collected using a magnetic system (Invitrogen) and rinsed 3× with PBS at 4°C. IP pellet, supernatant, and final wash samples were fractionated by SDS-PAGE and Western blotted onto a nitrocellulose membrane. Immunoreactivity was assessed using indicated primary antibodies (1 h, room temperature) and species-specific secondary antibodies (45 min, room temperature) and scanned using an Odyssey CLx (Li-Cor Biosciences). Fluorescence density was measured using the Gel Analyzer plugin in Fiji. For IP experiments that evaluated binding efficiency of Tomo1 to specific nucleotide-bound states of Rab3A, cells were lysed in IP lysis buffer containing 4.0 μm EDTA to release endogenous nucleotide binding. Precleared lysates were then incubated at 37°C for 10 min with a saturating concentration of nonhydrolysable GTP (GDP-γ-S, 200 μm, Sigma-Aldrich) or GDP (GDP-β-S, 200 μm, Sigma-Aldrich) added to the supernatant, and MgCl2 to a final concentration of 20 mm.
Phosphorylation of Tomo1.
For in vitro Cdk5 phosphorylation of Tomo1, NativePure streptavidin-agarose affinity purification was used to purify N-terminal biotin tagged mTomo1 protein from HEK293T cells transfected with pCDNA capTEV-mTomo1 according to the manufacturer's protocol (Invitrogen). As a control, HEK293T cells were transfected with the empty pCDNA capTEV vector and subjected to an identical purification procedure. In brief, five dishes (10 cm) of HEK293T cells at 70% confluency were transfected with 12 μg of DNA/dish using Lipofectamine 2000 (Invitrogen). At 48 h after transfection, cells were collected in NativePure lysis buffer, subjected to 3 liquid nitrogen freeze/thaw cycles, and then centrifuged at 10,000 × g for 10 min at 4C. Strepavidin beads were added to the postnuclear supernatant and rotated for 3 h at 4°C. Beads were then collected (1000 × g for 2 min), rinsed, and the purified Tomo1 protein stored (4°C) in lysis buffer with 0.1 mm sodium azide. SDS-PAGE followed by Commassie staining was used to assess and quantify protein yield and purification. For in vitro Cdk5/p25 phosphorylation reactions, 11.5 μg of mTomo1 on streptavidin beads was rinsed in phosphorylation buffer containing the following (in mm): 25 MOPS, pH 7.2, 12.5 β-glycerol phosphate, 25 MgCl2, 5 EGTA, 2 EDTA, 0.25 DTT, and 250 μg of active Cdk5/p25 kinase (Signal Chem, C33–10G). Reactions were initiated by adding 50 μm γ-[32P]ATP (PerkinElmer, NEG002A) with incubation at 30°C and intermittent mixing for 15 min. Reactions were terminated by centrifugation at 3000 × g for 2 min, followed by 3 washes of bead pellet in cold kinase buffer (4°C) lacking Cdk5/p25 and γ-[32P]ATP. Bead pellet samples were then fractionated by SDS-PAGE, blotted, and the 32P labeled Tomo1 detected using a Molecular Imager FX (Bio-Rad). Tomo1 on the nitrocellulose membrane was subsequently immunolocalized by Western blotting. Controls included reactions with purified mTomo1 protein containing 1 mm Roscovitine (Rosco), purified mTomo1 with no Cdk5/p25 kinase added, and streptavidin beads used in empty vector purification.
To measure tomosyn phosphorylation, in situ neuronal cultures plated on 14 mm microwell glass bottom dishes were rinsed 3× in warmed phosphate deficient DMEM with high glucose (#11971-025, Invitrogen). Cultures were then incubated in 50 μl of DMEM containing 500 μCi/ml of 32P orthophosphate for 4 h in a 37°C, 95% O2/5% CO2 humidified incubator to achieve 32P labeling of cytosolic ATP. Treatments included ± Rosco (100 μm, 4.5 h) with ± picrotoxin (50 μm, 4 h). Picrotoxin was applied 30 min following treatment ± the Cdk5 inhibitor Rosco. Cultures were then rinsed 3× with ice-cold TBS containing the following (in mm): Tris-HCl 50, NaCl 100, Na2HPO4 20, with pH adjusted to 8.0) and lysed in IP lysis buffer. Tomosyn was immunoprecipitated, subjected to SDS-PAGE fractionation, and blotted onto nitrocellulose. To detect and quantify 32P incorporation, the Tomo1 blots were imaged using a Molecular Imager FX (Bio-Rad) and subsequently immunoblotted for Tomo1.
Electrophysiological recordings in cultures.
Whole-cell patch-clamp recordings were performed with a HEKA 10 amplifier and Patchmaster Software (HEKA Elektronik, Dr. Schulze GmbH) from 17 to 24 DIV cultured hippocampal pyramidal-like neurons. The extracellular bath solution for recording miniature EPSCs (mEPSCs) contained the following (in mm): NaCl 119, KCl 5, CaCl2 2, MgCl2 2, glucose 30, and HEPES 10, that was adjusted to pH to 7.4 with NaOH. TTX (1 μm) and the GABAA receptor antagonist bicuculine (10 μm) were added just before use. The extracellular solution for recording evoked EPSCs was similar, with the exceptions that CaCl2 was reduced to 0.5 mm, MgCl2 increased to 3.5 mm, and TTX and bicuculine were omitted. The internal pipette solution contained the following (in mm): Cs-gluconate 105, MgCl2 5, EGTA 0.2, ATP 2, GTP 0.3, and HEPES 40, adjusted to pH to 7.2 with CsOH. Pipette resistance ranged from 2 to 5 mΩ. Neurons were voltage-clamped to −70 mV. Evoked EPSCs were produced by positioning 25-μm-diameter bipolar stimulating electrode close to the neuron (100–200 μm) and applying 0.4 ms current pulses of constant amplitude (WPI, A360 Stimulus Isolater). Measurement of amplitude and frequency of mEPSC and amplitudes of evoked EPSC were performed offline using Patcher's Power tools (Dr. Francisco Mendez and Frank Würriehausen, Max-Planck-Institute) and Taro Tools (Jikei University School of Medicine) plugins for Igor Pro (Wavemetrics).
ICC and PLA imaging.
ICC was performed as previously reported (Glynn and McAllister, 2006). Primary antibodies were added at indicated dilutions in 3% BSA for 1 h followed by rinses (3% BSA in PBS, 5×, 5 min) and addition of secondary antibodies (diluted in PBS) for 45 min followed by five 5 min washes in PBS. PLA reactions were performed identical to ICC procedures through the addition of primary antibodies. In place of secondary antibodies, PLA+ and PLA− probes (Sigma-Aldrich) were added for 45 min at 37°C followed by three 5 min washes (0.2% BSA, 0.1% Triton X-100 in PBS). Next, PLA ligation and amplification solutions were sequentially added for 30 min and 100 min, respectively, with two washes after each.
Colocalization analysis of fluorescence images was performed using the JACoP plugin for FIJI (Bolte and Cordelières, 2006). The Pearson R correlation coefficient and Mander's overlap coefficient were generated for pairs of images using fluorescent intensities of spatially matched pixels. The pixels included in the analysis were determined by the Costes automatic thresholding method (Costes et al., 2004). For PLA density analysis, images were manually segmented using a bright-field channel to create an ROI that demarcates cell boundaries. Next, the particle analysis plugin in FIJI was used to automatically count the number of fluorescent puncta contained within the ROI that were at least 3 × 3 pixels in size. For comparisons of pCdk5 immunofluorescence intensity, ROIs were created around puncta positive for Synapsin 1-Alexa488 immunolabeling from images that were background subtracted with a 50-pixel rolling-ball radius, thresholded, and converted into binary masks. ROIs were then applied to the alternate channel of the same image field containing pCdk5-Alexa594 to extract individual fluorescence intensity per ROI. Straightened axon images were generated using the Straighten plugin for FIJI.
Statistical analysis.
All statistical analysis was done using GraphPad Prism. Where indicated, one-way ANOVA tests were used for comparisons of population means with significance set to a p < 0.05. Post hoc t tests with Tukey's correction were used for multiple comparisons between specific groups. Cumulative frequency distributions were compared using the Kolmogorov–Smirnov test; statistical significance was set to to a p < 0.05. Sample means throughout are presented as ± SEM.
Results
Tomosyn1 inhibits evoked but not basal synaptic transmission
All functional analyses of Tomo1 on presynaptic physiology and identification of its sites of action on SV pools were conducted on excitatory synapses of cultured rat hippocampal neurons (14–24 DIV). Tomo1 is predominantly expressed at excitatory synapses and is enriched presynaptically relative to Tomosyn2 family proteins, which have been reported by immunohistochemistry in brain slices to be primarily postsynaptic in the hippocampal mossy fibers (Barak et al., 2010). As shown in Figure 1A1, ICC of a straightened axon fragment demonstrated that Tomo1 immunofluorescence intensity spiked coincidently with that of Synaptophysin (Syp), an integral SV membrane protein. Quantitative analysis of pixel-by-pixel intensity profiles (n = 29) further confirmed that endogenous Tomo1 within cultured hippocampal neurons is colocalized with Syp (Fig. 1A2). To examine regulatory effects of Tomo1 on electrically evoked and basal neurotransmission, we used lentiviral infection to overexpress (OE) Tomo1 and shRNA to KD Tomo1 expression. Endogenous expression of Tomo1 was reduced on KD by 84.7% and 91.3% respective to the GFP only and shScramble expression conditions (Fig. 1B). To assess the reduction of Tomo1 on initial release probability (Pr), we measured short-term plasticity of EPSC measurements using a paired-pulse ratio (PPR) analysis, where the interval between external field stimuli was varied (Dobrunz and Stevens, 1997; Zucker and Regehr, 2002). As shown in Figure 1C–E, Tomo1 OE resulted in paired-pulse facilitation (EPSC2 > EPSC1) over interstimulus intervals (ISIs) ranging from 50 to 250 ms that was significantly greater than control (PPR at 50 ms 1.96 ± 0.18 Tomo1 OE vs 1.11 ± 0.05 control). By comparison, Tomo1 KD neurons demonstrated paired-pulse depression relative to control for the 50 ms ISI (0.79 ± 0.13 vs 1.11 ± 0.05, respectively). As paired-pulse facilitation is inversely proportional to the initial Pr (Zucker and Regehr, 2002), these data demonstrate that Tomo1 serves as a strong negative regulator of initial Pr at hippocampal synapses. We next asked whether Tomo1 exerts effects on spontaneous release at hippocampal synapses. To address this, we recorded mEPSC in the presence of the voltage-gated sodium channel blocker TTX to prevent AP firing and network activation, under Tomo1 OE and KD conditions compared with control. Consistent with the prior reports (McEwen et al., 2006; Chen et al., 2011; Williams et al., 2011), our data demonstrated no significant changes in frequency or amplitude of mEPSCs among the conditions tested (Fig. 1F). Together, the effects of Tomo1 on initial Pr associated with evoked EPSCs is consistent with its reported activity to depress SV transition into the RRP through formation of nonfusogenic SNARE complexes. Interestingly, this inhibitory action of Tomo1 did not translate to changes in spontaneous exocytotic fusion of SVs.
Figure 1.

Presynaptic localization of Tomo1 and inhibition of evoked neurotransmission in cultured hippocampal neurons. A1, Representative ICC images comparing endogenous Tomo1 with Syp, a presynaptic vesicular protein, within a straightened axon segment. Intensity line scans (A1, bottom) of axon segment show coincident immunofluorescent spikes of Tomo1 and Syp. A2, Fluorescence intensity relationship for spatially matched anti-Tomo1 and anti-Syp (Pearson r = 0.73, Mander's M1 = 0.8, M2 = 0.85 n = 29). B, Representative Western blot comparing Tomo1 immunoreactivity (anti-Tomo1) from lysate of lentiviral-infected GFP, YFP-Tomo1 (YFP-Tomo1), shRNA-Scramble (sh-Scr), or shRNA-Tomo1 (sh-Tomo1, top) hippocampal neurons. C, Representative paired-pulse stimulation EPSCs recorded from control (sh-Scramble), sh-Tomo1 KD, and YFP-Tomo1 OE lentiviral-infected conditions. PPRs to 50 ms ISI. D, Averaged PPR for all ISI values and (E) bar plots for the 50 ms and 850 ms ISI averages. F, Representative mEPSC records. G, Averaged mEPSC frequency and H, amplitude values (bottom) for lentiviral-infected neurons under control (sh-Scramble), sh-Tomo1 KD, and YFP-Tomo1 OE conditions. No significant differences were found in frequency or amplitude of spontaneous mEPSC between conditions. In this and all subsequent figures, graphs represent mean ± SEM. *p < 0.05.
Tomosyn1 inhibits multiple vesicle pools
Because Tomo1 may exert secretory effects beyond actions on the initial releasable pool of SVs (McEwen et al., 2006; Chen et al., 2011) we next used vGpH, as a direct optical reporter of SV cycling, to test for Tomo1 participation in partitioning of SVs at other presynaptic sites. For this assessment, we applied field stimulation protocols eliciting AP firing (Burrone et al., 2006; Alabi and Tsien, 2012) to drive release of SVs likely associated with the RRP (20 Hz, 40 pulses) (Rosenmund and Stevens, 1996; Schikorski and Stevens, 1997, 2001; Stevens and Williams, 2007) and TRP (20 Hz, 900 pulses) (Burrone et al., 2006; Voglmaier et al., 2006; Kim and Ryan, 2010), whereas the nonreleasable RestP vesicles were measured by a subsequent ammonium chloride alkalinization (Sankaranarayanan et al., 2000). All experiments were performed in the presence of the V-type ATPase inhibitor bafilomycin-A1 to restrict measurements to report net exocytosis by preventing reacidifcation of endocytosed SVs (Sankaranarayanan et al., 2000). Actions of Tomo1 were evaluated in vGpH-expressing neurons under Tomo1 OE and KD conditions compared with control (vGpH + empty vector and vGpH + sh-SCR). Relative to control, Tomo1 KD resulted in a significant enhancement in the release efficacy in response to the brief stimulus train targeting the RRP, whereas Tomo1 OE resulted in a significant decrease (Fig. 2A). These data are consistent with our PPR results, which indicated that Tomo1 KD and OE may alter the RRP size and/or the probability of release per vesicle (Pvr) in the RRP (Fig. 1C–E). Notably, evaluations of the TRP led to the discovery that Tomo1 KD resulted in significantly greater averaged TRP (0.678 ± 0.044, shTomo1-1) at the expense of the ResP compared with control (0.516 ± 0.036) and Tomo1 OE (0.367 ± 0.043) conditions (Fig. 2B,C). This change was not likely a result of nonspecific/off-target effects of shRNA KD construct, as it was observed using two distinct shRNA sequences (0.744 ± 0.036 shTomo1-2) and rescue with human Tomo1 OE returned TRP to control levels (0.543 ± 0.040). Furthermore, cumulative frequency distributions of TRP size indicated that regulation by Tomo1 of TRP vesicles was distributed across the entire population of synaptic boutons (Fig. 2D). Apparent from these distributions, Tomo-1 KD demonstrated a significant (p < 0.05) right shift toward greater TRP fractions (median = 0.67 and 0.72) and Tomo1 OE a left shift toward smaller TRP fractions (median = 0.41) relative to control (median = 0.54). No statistically significant differences were observed between conditions for averaged TRP depletion kinetics (τ, con 18.2 ± 3.15; KD 18.12 ± 4.96; OE 17.36 ± 6.05); or averaged total SV pool size (f NH4Cl − f 0, con 7007 ± 1325; KD 6622 ± 2278; OE 12191 ± 1813). The regulatory action of Tomo1 on TRP/ResP SV distribution is a newly identified site of Tomo1 action at central excitatory synapses. That is, Tomo1 action is not limited to inhibiting final stages of priming of SVs to release competency, but is also involved in a molecular mechanism that acts to clamp vesicles into the release-recalcitrant ResP.
Figure 2.
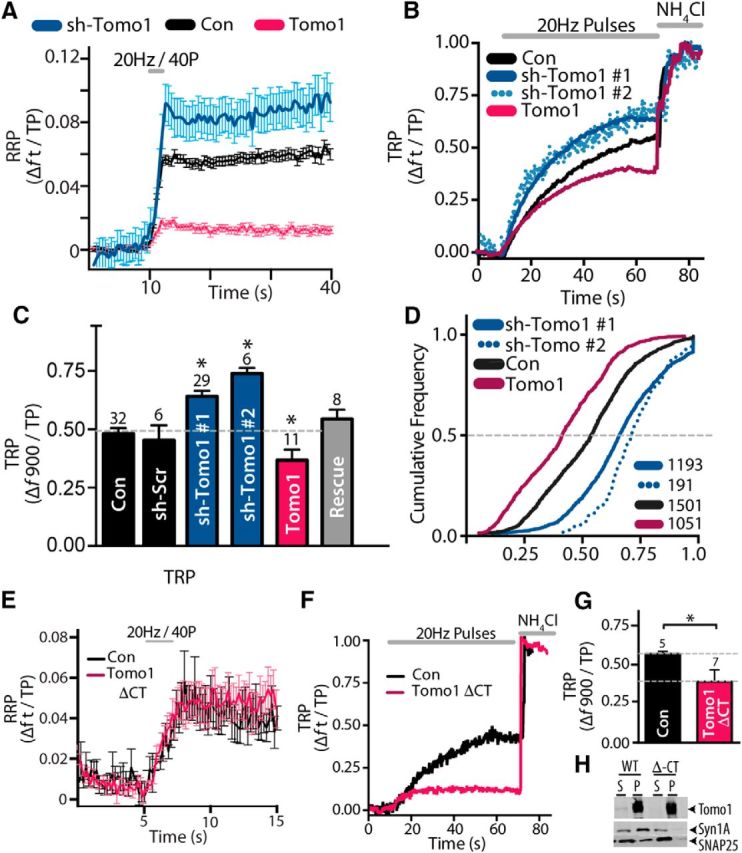
Effects of Tomo1 on SV partitioning among functionally defined SV pools. A, Comparison of averaged RRP for Tomo1 OE (mCh-Tomo1, magenta, n = 15), sh-Tomo1 KD (sh-Tomo1 + tRFP, blue, n = 9), and control (mCh empty vector, black, n = 10) conditions. RRP was defined as the change in vGpH fluorescence (Δft) to application of 40 field stimuli (10 V/cm) at 20 Hz, in saline containing 4 mm Ca2+ normalized to the total pool (TP) of SVs. B, Representative vGpH measurements of TRP (Δft/TP) from single neurons. The TRP is defined as Δf to a SV recycling pool-depleting stimulus (900 pulses at 20 Hz) normalized to the TP. C, Comparison of mean TRP for conditions indicated (n = cell number). D, Cumulative TRP frequency distributions for bouton ensembles of each condition (n = boutons). E, Comparison of averaged RRP on OE Tomo1 Δ-CT (magenta, n = 6) with control (black, n = 5). F, Representative vGpH measurement of TRP from neuron overexpressing Tomo1 lacking C-terminal SNARE domain (Tomo1 ΔCT) and from a control neuron (empty vector). G, Averaged TRP for control (black) and Tomo1 ΔCT conditions. Expression of Tomo1-wt (bottom dashed gray line, data from C) and Tomo1 ΔCT result in a comparable decrease in TRP. H, IP demonstrates that Tomo1 wt, but not Tomo1 Δ-CT, coprecipitates of Syntaxin1A and SNAP25 SNARE proteins from PC-12 cell lysate. The result confirms a central requirement of Tomo1's SNARE domain for Q-SNARE complex formation. For this figure and all subsequent Western blot images, lanes are as follows: S, Supernatant after immunoprecipitation; W, final wash; P, IP pellet. *p < 0.05.
Tomosyn was discovered as Syntaxin1A-binding protein (Fujita et al., 1998) that is capable of forming a ternary complex with Syntaxin and SNAP25 that mitigates VAMP on secretory vesicles from forming into SNARE complexes (Hatsuzawa et al., 2003; Pobbati et al., 2004; Bielopolski et al., 2014). Therefore, we next tested whether Tomo1's regulatory action on the RRP and TRP/ResP required the VAMP-like SNARE domain. As shown in Figure 2E, OE of a mTomo1 deficient in the C-terminal SNARE motif (Tomo1-ΔCT; mTomo1 (1–1069)) did not result in inhibition of the estimated RRP compared with control (Fig. 2E); however, it did significantly inhibit the TRP relative to control (Fig. 2F,G) and to a similar degree as OE of wt Tomo1 (compare withFig. 2C). IPs from PC-12 cell lysates expressing Tomo1-ΔCT versus wt Tomo1 confirmed that Tomo1-ΔCT fails to interact with Syntaxin1A and SNAP25 (Fig. 2H). These data suggest that there are two independent regulatory actions of Tomo1 on synaptic transmission: (1) regulation on efficacy of release associated with the RRP, which depends on the VAMP-like SNARE domain; and (2) Tomo1 regulation on distribution of SVs between the TRP and ResP, which is independent of the Tomo1 VAMP-like SNARE motif. Interestingly, the ability of Tomo1 to allocate vesicles to a nonreleasable ResP independent of its R-SNARE motif may reflect an evolutionary conserved function with its yeast ortholog Sro7, which lacks an R-SNARE motif and, when overexpressed, results in nonfusogenic vesicle clusters independent of the cognate SNARE sec9 (Hattendorf et al., 2007; Rossi and Brennwald, 2011).
Activity-dependent Cdk5 regulation and Tomosyn1 phosphorylation
The presynaptic partitioning of SVs among the TRP and ResP may be dynamically regulated in response to neuronal activity. Indeed, NMDA-receptor-dependent potentiation of neurotransmitter release in cultured hippocampal neurons drives an increase in the fraction of recycling vesicles (Ratnayaka et al., 2012). Moreover, engagement of forms of homeostatic plasticity driven by pharmacologically-induced changes in global network activity have been reported to be accompanied by compensatory shifts in SVs in the ResP within hippocampal slices and hippocampal and neocortical neuron cultures (Virmani et al., 2006; Kim and Ryan, 2010; Rose et al., 2013). While numerous presynaptic and postsynaptic mediators of homeostatic plasticity have been identified, particularly via forward genetic screens of the Drosophila neuromuscular junction (NMJ) (Frank et al., 2006, 2009; Dickman and Davis, 2009; Müller et al., 2011, 2012), the specific mammalian presynaptic signaling pathways that direct changes in the SV pool distribution during plasticity remain less defined. Among those established to exert effects on the TRP is Cdk5, which has been demonstrated via pharmacological blockade with Rosco to drive an increase in the presynaptic TRP, concomitant with a decrease in the ResP within hippocampal brain slices and cultured hippocampal neurons (Kim and Ryan, 2010; Marra et al., 2012). Cdk5 is a proline-directed Ser/Thr kinase whose activators (p35 and p39) are primarily brain-specific (Tsai et al., 1994; Tang et al., 1995; Xiong et al., 1997). Cdk5 has been ascribed multiple cellular functions, including primarily a neural-specific kinase critical to neuron differentiation and migration, axon guidance, synaptogenesis, membrane trafficking, and synaptic plasticity, with dysregulation of its activity also linked to neurodegenerative diseases, such as Alzheimer's, Parkinson's, and Huntington's diseases (Dhavan and Tsai, 2001; Lopes and Agostinho, 2011; Su and Tsai, 2011; Cheung and Ip, 2012; Yang et al., 2013).
Notably, learning and memory formation and hippocampal plasticity are also dependent on Cdk5 (Cheung and Ip, 2012; Lai et al., 2012). Of specific interest to the present study, Cdk5 has been reported to modulate TRP size (Kim and Ryan, 2010), and this modulation closely mirrored in extent the distributional changes we observed upon alterations of Tomo1 expression. As such, we evaluated whether Cdk5 actions on the TRP involved Tomo1; that is, are Tomo1 regulatory actions on SV TRP/ResP distribution governed by catalytic Cdk5 phosphorylation of Tomo1? For this assessment we initially tested whether alterations in neuronal activity within hippocampal cultures are reflected in alterations in the activation state of Cdk5. This was evaluated using a phosphorylation-specific Cdk5-Ser159 antibody, as Ser159 is phosphorylated upon Cdk5 activation (Sharma et al., 1999; Czapski et al., 2011). Figure 3A shows that treatment of cultures with Rosco (100 μm, 30 min) lowered averaged immunofluorescence intensity (0.75 ± 0.021), whereas cyclosporine A (CSA), an inhibitor of the presynaptic serine/threonine phosphatase calcineurin, which opposes the functional effects of Cdk5 on SV pools (Kim and Ryan, 2010), increased averaged intensity (1.234 ± 0.148) relative to immunofluorescence intensity of control cultures. These data indicate that anti-phospho-Cdk5-Ser159 immunofluorescence reports on relative activation of Cdk5. To assess the role of neural activity on Cdk5 activation, we compared treatment of cultures with TTX (1 μm, 24 h), to block global AP firing, to vehicle-treated controls. The results showed that blocking neuronal spiking with TTX led to strong reductions in averaged phosphorylated Cdk5 immunofluorescence intensity (0.7 ± 0.004) relative to control (Fig. 3B), suggesting that the activation state of Cdk5 is sensitive to neuronal activity.
Figure 3.

Tomo1 serves as catalytic substrate for neural activity-sensitive Cdk5. A1, Representative immunofluorescent images of Synapsin 1 (cyan, top) and phosphorylated Cdk5 (pCdk5; heat scale, bottom) in straightened axon segments from neurons treated (30 min) with CSA (50 μm), Rosco (100 μm), or control (DMSO, 0.5%). A2, Averaged pCdk5 immunofluorescence intensity relative to vehicle control. A3, Cumulative frequency of pCdk5 intensity across boutons shows that inhibition of Cdk5 by Rosco significantly reduces pCdk5 immunoreactivity, whereas CSA treatment enhances pCdk5 relative to control. B1–B3, pCdk5 immunofluorescence images comparing chronic silencing of neural activity by TTX (24 h, 1 μm) with control. Averaged pCdk5 intensity (B2) and cumulative frequency of pCdk5 (B3) intensity demonstrate that chronic dampening of neural activity significantly reduces Cdk5 activation. A, B, Analysis was performed on 45 images/preparation per condition and repeated on 3 neuronal preparations. C, Immunoblots showing coprecipitation of Tomo1 and Cdk5 from cultured hippocampal neuron lysates regardless of primary target of IP (Tomo1 or Cdk5). D, In vitro phosphorylation of affinity-purified Tomo1 by Cdk5/p25 as measured by 32P labeling. 32P-radioactive signal (top) and anti-Tomo1 immunoreactivity (bottom) on Western blot corresponding to the following conditions: absence of Cdk5/p25 kinase (H2O), Cdk5/p25 kinase (Cdk5), and Cdk5/p25 + 1 mm Rosco (Cdk5 + Rosco). *p < 0.05.
To determine whether Tomo1 is a Cdk5 phosphorylation substrate, intermolecular interaction was tested by coimmunoprecipitation from hippocampal neuronal culture lysate. The results established that Cdk5 coimmunoprecipitations with Tomo1, and that the coimmunoprecipitation is independent of whether Cdk5 or Tomo1 was the primary IP protein (Fig. 3C). We next tested whether Tomo1 is a phosphorylation target of Cdk5. For this, in vitro phosphorylation reactions were conducted using purified active p25 (an active proteolytic fragment of p35) plus Cdk5 in the presence of γ-[32P]-ATP, together with biotin-tagged mTomo1 protein that was affinity-purified from transfected HEK-293T cells to assure proper post-translational folding. Reactions showed a direct correlation between 32P-radiolabel and Tomo1 immunoreactivity by Western blot analysis (Fig. 3D). 32P-phosphorylation of Tomo1 was mediated by Cdk5, as radiolabeling was not observed in the empty-vector control samples or in Tomo1 p25/Cdk5 samples containing Rosco (1 mm) (Fig. 3D). Together, the data indicate that Tomo1 is a Cdk5 phosphorylation substrate.
Tomosyn1 and Cdk5 share a common pathway in regulating SV pools
To determine whether Tomo1 actions on the TRP were under specific regulation by Cdk5, we evoked presynaptic release in control and Tomo1 KD neurons under conditions where Cdk5 was pharmacologically inhibited. As shown in Figure 4, inhibition of Cdk5 with Rosco (100 μm, 30 min) enhanced the fraction of recycling vesicles relative to that observed in carrier-treated (0.1% DMSO) controls (Fig. 4A, black trace left vs right). These findings are similar to those previously reported (Kim and Ryan, 2010). Importantly, we found that the increase in TRP size upon Cdk5 inhibition was dependent upon Tomo1, as Rosco-mediated enhancement of the TRP was absent in the Tomo1 KD condition (Fig. 4A, blue trace, right vs black trace, right). Tomo1 KD alone also led to an enhancement in the fraction of recycling vesicles (Fig. 4A, blue trace, left), similar to that observed with Cdk5 inhibition (Fig. 4A, black trace, right). As a complement to these findings, we next compared the effects in control versus Tomo1 KD neurons when Cdk5 activity was genetically depressed by expression of the catalytically inactive, dominant-negative Cdk5 mutant (Cdk5(D144N)). Notably, we observed an enhancement in the TRP fraction upon Cdk5-D144N expression that was comparable with that observed upon treatment with Rosco (Fig. 4A, magenta trace, left). Moreover, application of Rosco in the Cdk5-D144N expression condition did not significantly increase the TRP size (Fig. 4A, magenta trace, right), confirming that the effects of Rosco on the TRP are specific to Cdk5. The above experimental results demonstrate the following: (1) Tomo1 KD or ablation of Cdk5 catalytic activity led to a significant increase in the fraction of SVs in the TRP; and (2) downregulation of Tomo1 expression or Cdk5 activity occludes regulated increases in the TRP that are normally mediated by Cdk5 inhibition or Tomo1 KD, respectively (Fig. 4B). Moreover, consistent with prior reports (Kim and Ryan, 2010; Verstegen et al., 2014), we find that inhibition of Cdk5 by Rosco treatment significantly reduced the time constant for TRP depletion relative to control (Fig. 4D). The TRP depletion time constants in response to Rosco treatment in Tomo1 KD or Cdk5-D144N conditions were not significantly different from untreated control, although there appeared a trend toward lower tau values. Finally, analysis of cumulative frequency distributions of TRP size from bouton ensembles across cells show that genetic or pharmacological inhibition of Cdk5, KD of Tomo1, or downregulation of both proteins, results in a comparable greater TRP (right shift) relative to nontreated control cells (Fig. 4C). Together, these data strongly support Tomo1 and Cdk5 as acting within the same signal-transduction pathway that regulates the distribution of SVs to the TRP and ResP.
Figure 4.

Tomosyn and Cdk5 share a common signaling pathway regulating the TRP. A, Comparison of representative vGpH responses assessing the TRP in control (black), sh-Tomo1 (blue), or Cdk5 dominant-negative mutant (Cdk5 [DN], magenta) expressing neurons and measured in the absence (left) or presence (right) of Rosco (100 μm, 30 min pretreatment). B, Averaged TRP across expression conditions ± Rosco. Bottom dashed line indicates mean TRP to Tomo1 overexpression (Fig. 2C). C, Cumulative frequency distributions comparing TRP for bouton ensembles of each condition. Dashed distributions represent treatments (color-matched) with Rosco. sh-Tomo1 and Cdk5 DN expression significantly enhanced the TRP compared with control, with no further increase upon Rosco treatment. D, Averaged TRP depletion kinetics from same data shown in B. B, n = number of cells. C, n = number of boutons without, and with, Rosco treatment. *p < 0.05.
Identification of a novel Tomosyn1 effector interaction
How may Tomo1 act, independent of its SNARE motif, to modulate SV availability for release? Tomo1 as a nonintegral membrane protein is localized to the cytosol, plasma membrane and SVs in rat brain (Fujita et al., 1998; McEwen et al., 2006; Takamori et al., 2006). While Tomo1's association with the plasma membrane occurs via an interaction with Syntaxin1A (Gladycheva et al., 2007), the mechanism mediating Tomo1-SV association is unknown. In yeast, orthologs of Tomosyn (Sro7p/77p) interact with Sec4 (Rab GTPase), which is important in tethering/docking of vesicles and membrane association via C-terminal digeranylgeranylation (Farnsworth et al., 1991; Calero et al., 2003; Hutagalung and Novick, 2011). Based on a potential for evolutionary conservation of effector interactions, we therefore tested whether Tomo1 interacts with Rab3A GTPase. Rab3A is an isoform of the Rab3(A-D) GTPase family, which assumes primary targeting/docking roles at central synapses (Schlüter et al., 2004). Notably, Rab3A was observed to coprecipitate with endogenous Tomo1, as well as with expressed YFP-Tomo1, following neuronal lentiviral infection of hippocampal neuron cultures (Fig. 5A). Moreover, the interaction appeared specific, as coimmunoprecipitation of other synaptic (RIM1/2, Actin) or integral SV (Synaptophysin) proteins was not observed (Fig. 5A). Interestingly, the coimmunoprecipitation of Tomo1 and Rab3A without RIM proteins suggests that, when Rab3A interacts with Tomo1, it fails to also interact with RIM proteins. To test whether the nucleotide state of Rab3A affected Tomo1 binding, we next conducted IP assays from hippocampal lysates where Rab3A was locked into nucleotide-free, GTP-or GDP-bound states, in part using nonhydrolyzable nucleotide analogs. We observed that Tomo1 bound preferentially to Rab3A-GTP compared with its GDP or nucleotide free (Fig. 5B,C).
Figure 5.

Tomo1 interaction with Rab3A-GTP. A, IP of endogenous Tomo1 from cultured hippocampal neuronal lysates results in coprecipitation of Rab3A, but not Rim1/2, actin or synaptophysin (Syp). Bottom immunoblot demonstrates that IP of lentiviral OE YFP-Tomo1 also results in strong coprecipitation of Rab3A from neurons. B, Western blot showing IP of Tomo1 preferentially coprecipitates GTP-bound Rab3A. Neuronal lysates used for IP were pretreated to drive Rab3A into GTP (GTP γ-S), GDP (GDP β-S), or a nucleotide-free state (n = 3 cell preparations). C, Averaged Tomo1:Rab3A coimmunoprecipitation ratio for each nucleotide-loaded condition. D, Representative PLA images of Min6 cells assessing in-cell interaction of Tomo1 and Rab3A, compared with protein interaction controls (as indicated). Top row, DAPI staining overlaid on differential interference contrast bright-field to define plasma membrane periphery of cell cluster. Bottom row, PLA fluorescent puncta for each PLA interaction set. Insets, Enlarged spatially defined region of cytosol. E, Averaged PLA signal density for each protein interaction set (n = number of cell clusters). F, Min6 cell PLA Tomo1 + Rab3A interaction signal shown at high magnification indicates that fluorescent puncta are likely localized to dense-core secretory granules. G, Images of PLA fluorescent puncta on hippocampal neurons resulting from assay of indicated protein interactions. *p < 0.05.
To complement the IP results, the Tomo1/Rab3A interaction within a cell context was evaluated using a PLA. Insulin-secreting Min6 cells were used, as they possess large (∼270 nm diameter) secretory granules, use exocytotic machinery similar to that of neurons, and present a well-resolved cell periphery that promotes quantitative analysis of the PLA fluorescent puncta. PLA fluorescent puncta arise when different secondary antibodies containing unique short DNA strands react with primary antibodies against proteins that are in close proximity (<40 nm) to enable DNA ligation, amplification, and binding of fluorescently labeled oligonucleotides at sites of putative protein-protein interactions. Robust PLA signals between Rab3A and Tomo1 were observed (Fig. 5D,E; 0.378 ± 0.02 puncta/μm2) that appear localized with secretory granules (Fig. 5F). The density of PLA fluorescent puncta was not significantly different from that of independent tests for the previously established Rab3A/RIM2 (0.437 ± 0.015 puncta/μm2) interaction within presynaptic compartments (Wang et al., 1997; Dulubova et al., 2005). Negligible PLA signals were observed between Rab3A and the SV membrane protein Syp (0.077 ± 0.008 puncta/μm2) or when cells were probed for Rab3A alone (0.07 ± 0.007 puncta/μm2) (Fig. 5E). PLA experiments in cultured hippocampal cells also show robust signals between Tomo1 and Rab3A as well as Rim1/2 and Rab3A, but not Syp and Rab3A (Fig. 5G). The interaction between Tomo1 and Rab3A-GTP on SVs in hippocampal neurons may serve as a candidate molecular mechanism for clamping vesicles in the ResP, as studies in yeast show that Sro7 induced accumulation of nonfusongenic vesicles depends on Sec4-GTP (Rossi and Brennwald, 2011). In addition, mammalian studies on exocytosis have demonstrated that Rab3A-GTP acts as a clamp on SV and secretory granule release (Holz et al., 1994; Johannes et al., 1994; Star et al., 2005).
Synapsin 1 interaction with Tomosyn1/Rab3A complex and regulation by Cdk5
As Rab3A-GTP also binds Synapsin proteins (Giovedì et al., 2004), and as Synapsin phosphoproteins are major regulators of membrane tethering of SVs at, or adjacent to, the AZ (Greengard et al., 1994; Cesca et al., 2010), we next determined whether Synapsin 1 may interact with the Tomo1/Rab3A complex to potentially modulate Tomo1 regulatory actions on the TRP. Figure 6A demonstrates that Synapsin 1a and 1b immunoreactivity along with Rab3A was detected in Tomo1 immunoprecipitate samples prepared from cultured hippocampal neurons. By comparison, the use of rabbit IgG isotype-control yielded no detectable IP of Tomo1, Rab3A, or Synapsin 1 proteins (Fig. 6B). To test whether the Tomo1 and Synapsin 1 interaction occurred as a function of Rab3A binding, we next conducted Tomo1 IPs from neuronal lysates that were precleared of Rab3A protein using an initial anti-Rab3A antibody IP. Figure 6C shows that using Rab3A precleared supernatant (S), Synapsin 1 proteins were not coimmunoprecipitated with Tomo1, suggesting that Tomo1 forms a complex with Synapsin 1 proteins via its interaction with Rab3A.
Figure 6.

Interaction of Tomo1 with Rab3A and Synapsin 1 is regulated by Cdk5. A, Western blot showing that IP of Tomo1 from hippocampal neuronal lysates coprecipitates Synapsin 1 (a, b isoforms) and Rab3A. B, IP with isotype-control rabbit IgG, to verify the specificity protein anti-Tomo1 coprecipitation, does not precipitate Tomo1, Rab3A, or Synapsin 1. C, Rab3A was cleared from lysate by IP with a Rab3A antibody (top blot, S is IP cleared supernatant). Subsequent IP of Tomo1 from the Rab3A cleared supernatant (S) shows no detectable pulldown (P) of Rab3A or Synapsin1 (bottom blot). Note supernatant (S) after the IP retained Synapsin 1a,b and low-level Rab3A immunoreactivity. D, Effect of CSA (50 μm) and Rosco (100 μm) pretreatment (30 m) and vehicle control on Tomo1 coprecipitation of Rab3A and Synapsin 1 from neuronal lysates. E, Averaged Tomo1 coprecipitation of Rab3A and Synapsin 1, as normalized to control, for each condition. *p < 0.05.
To determine whether Cdk5 activity exerts functional regulation on Tomo1/Rab3A/Synapsin 1 interactions, the relative levels of Rab3A and Synapsin 1 were measured in Tomo1 immunoprecipitates prepared from lysate of hippocampal neurons pretreated with either Rosco, CSA, or vehicle (Fig. 6D,E). Notably, a significant increase in Rab3A and Synapsin 1 level was observed with CSA treatment, relative to control, a condition expected to result in enhanced levels of Cdk5 phosphorylation and subsequent decrease in the size of the TRP (Kim and Ryan, 2010, 2013). No significant change in Rab3A or Synapsin level was observed with Rosco treatment relative to control. The lack of a detectable decrease upon Rosco treatment is likely related to assay sensitivity, as even for the control condition immunoreactive signals are near the detection limit. The strong increase in Rab3A and Synapsin 1a, 1b coimmunoprecipitation with Tomo1 following CSA treatment suggests that the interaction may be sensitive to Cdk5 catalytic activity, although as calcineurin serves as a general phosphatase, the development of these interactions may also be sensitive to other targets of calcineurin (i.e., dynamin, amphiphysin, synaptojanin) or result from indirect effects of calcineurin. For example, calcineurin has been shown to modulate calcium influx (Kim and Ryan, 2013), the insertion of calcium-permeable AMPA receptors (Kim and Ziff, 2014) and regulate the synthesis of retinoic acid (Arendt et al., 2015), among others (Baumgärtel and Mansuy, 2012). Nonetheless, the discovery of a Tomo1/Rab3A/Synapsin 1 complex, together with well-established roles of Rab3 and Synapsin 1 in SV targeting and tethering, suggests a possibility for this molecular complex to modulate availability of SVs for evoked release.
Tomosyn1 phosphorylation state is sensitive to neuronal activity and is important for synaptic scaling
Do changes in Cdk5 activation state mediated by neural activity lead to changes in presynaptic Tomo1 phosphorylation such that Tomo1 may act as a key homeostatic mediator of changes in neurotransmitter release? To address this question, we first determined whether Tomo1 is subject to in situ Cdk5 phosphorylation in neurons. This was performed by assessment of 32P radiolabeling of Tomo1 using hippocampal neuronal cultures in which cytosolic ATP was labeled by 32P orthophosphate. As shown in Figure 7A,B, IP of Tomo1 from these cultures demonstrated that Tomo1 was phosphorylated and that treatment with Rosco (100 μm) resulted in an ∼50% reduction in basal phosphorylation levels. Importantly, chronic elevation in neural activity induced by treatment with the GABAA receptor antagonist, picrotoxin (50 μm, 4 h), together with an elevation in extracellular calcium (4 mm) demonstrated an increased level of phosphorylated Tomo1 relative to control (Fig. 7B). Inhibiting Cdk5 by application of Rosco (100 μm) 30 min before and throughout the pharmacologically induced elevations in neural activity did not completely abrogate the increase in Tomo1 phosphorylation; but, notably, it did result in a decrease compared with chronic activity alone (Fig. 7B). Tomo1 is therefore an activity-dependent Cdk5 substrate, although Tomo1 may be subject additional kinases, such as PKA (Baba et al., 2005), during elevations in neuronal spiking. Finally, we evaluated whether Tomo1 is functionally important for presynaptic homeostatic elevations in SV release that occur following chronic silencing of neural activity (Jakawich et al., 2010; Henry et al., 2012). Figure 7C, D demonstrates that chronic silencing of network activity in the hippocampal cultures by treatment with competitive antagonists of AMPA/kainate glutamate receptors (CNQX, 40 μm) for 24 h resulted in greater vGpH responses to a 100P/10 Hz stimulus relative to vehicle-treated controls. Importantly, Tomo1 KD neurons lacked the ability to undergo these compensatory enhancements in neurotransmitter release relative to vehicle-treated shTomo1 neurons. Although KD of Tomo1 alone leads to a significant enhancement in SV release (Fig. 2), these results suggest that the ability of synapses to upregulate SV release within the context of prolonged activity blockade requires Tomo1. Together, our data indicate that the phosphorylation state of Tomo1 is sensitive to neural activity, phosphorylation of Tomo1 regulates Rab3A/Synapsin 1 interaction and that Tomo1 has a pivotal role in activity-dependent compensatory synaptic scaling.
Figure 7.

Activity-dependent phosphorylation of Tomo1 participates in synaptic scaling. A, IP of endogenous Tomo1 from cultured hippocampal neurons (bottom; Western) following in situ 32P labeling indicates that Tomo1 is phosphorylated (top; 32P radiolabel detection). The level of phosphorylation was affected by alterations in neural activity (± picrotoxin, 50 μm. 4.5 h) and Cdk5 activity (± Rosco, 100 μm, 4.5 h). B, Averaged extent of in situ Tomo1 phosphorylation relative to vehicle-treated control (0.5% DMSO, n = 3 cell preparations). Extent of phosphorylation was quantified from density of 32P-band corresponding to Tomo1 immunoreactivity. C, Averaged changes in presynaptic vGpH fluorescence to 100 pulse stimulus (10 Hz) following chronic silencing of neuronal activity (CNQX, 40 μm, 24 h) relative to control (vehicle only) in sh-Tomo1-transfected and control (empty vector) conditions. D, Averaged Δf − f0 for all boutons of specified conditions (n, con = 121, con + CNQX = 134, sh-Tomo1 = 750, sh-Tomo1 + CNQX = 409). *p < 0.05.
Discussion
Elucidating the signaling pathways that confer precise, dynamic recycling and partitioning of SVs in response to neural activity is of considerable importance, as sustained information transfer at central excitatory synapses requires the limited SVs to be rapidly recycled (Shepherd and Harris, 1998; Schikorski and Stevens, 2001; Micheva and Smith, 2005; Rizzoli and Betz, 2005; Denker and Rizzoli, 2010; Alabi and Tsien, 2012). In addition, Pr and short-term synaptic plasticity, as well as aspects of Hebbian and homeostatic forms of synaptic plasticity are strongly impacted by the distribution of the SVs into distinct presynaptic pools (RRP, RP, and ResP) (Kim and Ryan, 2010; Ratnayaka et al., 2012; Rose et al., 2013; Verstegen et al., 2014). Presynaptic boutons of a given neuron may also differ in distribution of SVs among releasable (TRP) vs nonreleasable (ResP) pools, thereby conferring the possibility of precise spatial regulation of Pr and presynaptic plasticity (Harata et al., 2001; Li et al., 2005; Micheva and Smith, 2005; Fernandez-Alfonso and Ryan, 2008; Fredj and Burrone, 2009; Welzel et al., 2011; Ratnayaka et al., 2012). In the present study, we establish that Tomo1, a soluble R-SNARE motif-containing protein, is a central mediator on the distribution of SVs between presynaptic pools. Interestingly, Tomo1's regulatory action on SV TRP and ResP distribution is independent of its C-terminally localized R-SNARE domain, proposing an action through downstream effector interaction with Tomo1's N-terminal β-propeller domains. Consistent with this, we identify a novel protein interaction between Tomo1 and Rab3A-GTP, which then promotes formation of a molecular complex with Synapsin 1 proteins. We also establish that: (1) Tomo1 is an activity-dependent phosphorylation substrate of Cdk5; (2) assembly of the Tomo1/Rab3A/Synapsin 1 complex is sensitive to the phosphorylation state of Tomo1; and (3) Tomo1 is required for dynamic changes in the TRP size. The development of Tomo1 complexes involving Rab3/Synapsin, whose assembly is regulated by Cdk5, may account for previously reported Cdk5 catalytic actions on transition of SVs between the TRP and ResP (Kim and Ryan, 2010, 2013).
Tomosyn proteins have been well characterized as potent inhibitors of evoked transmitter release in neuroendocrine cells (Hatsuzawa et al., 2003; Yizhar et al., 2004, 2007; Gladycheva et al., 2007), rat superior cervical ganglia (Baba et al., 2005), hippocampal dentate gyrus mossy fibers (Barak et al., 2013; Ben-Simon et al., 2015), as well as at C. elegans (McEwen et al., 2006; Gracheva et al., 2007a) and Drosophila (Chen et al., 2011) NMJs. The predominant mechanistic model of Tomo1 action is that it exerts inhibitory effects via its C-terminal R-SNARE domain, which competes with VAMP and Munc18 for interaction with reactive Syntaxin1A (Fujita et al., 1998; Gladycheva et al., 2007). Interaction of Tomo1 with Syntaxin1A and SNAP25 leads to nonfusogenic SNARE complexes, which are proposed to then limit the availability of reactive Q-SNAREs required for SV priming into the RRP (Hatsuzawa et al., 2003; Pobbati et al., 2004; Yu et al., 2014). Yet, Tomo1's N-terminal β-propeller domains have also been implicated in the inhibition of neuroendocrine and neuronal exocytotic activity (Yizhar et al., 2007; Sakisaka et al., 2008; Yamamoto et al., 2009; Burdina et al., 2011; Williams et al., 2011; Bielopolski et al., 2014). Inhibitory actions of Tomosyn outside of the RRP have also been reported for C. elegans and Drosophila NMJs (McEwen et al., 2006; Chen et al., 2011). Importantly, our data demonstrate that the large N-terminal β-propeller domains of Tomo1 comprise the region that is specific for regulating the distribution of SVs between the ResP and TRP. In addition, we find that Tomo1 is a binding partner of Rab3A-GTP, a discovery that is complementary to that of the yeast Tomo1 ortholog, Sro7p, which interacts with the secretory Rab Sec4-GTP to coordinate tethering and priming of vesicles at fusion sites (Lehman et al., 1999; Hattendorf et al., 2007; Rossi and Brennwald, 2011; Rossi et al., 2015). Indeed, Sro7p OE in yeast results in a pronounced growth defect and the formation of a large cluster of “ResP-like” nonreleasable post-Golgi vesicles within the cell, in a manner dependent upon Sec4-GTP (Rossi et al., 2015). In mammals, overexpression of a GTP-locked point mutant of Rab3A (Q81L) also impairs the size of the recycling pool (Star et al., 2005). As shown here, expression of Tomo1 leads to an increase in the ResP in cultures of mammalian hippocampal neurons. Notably, high-resolution structural/functional analysis has identified surface sites on Sro7 within its N-terminal β-propeller region that mediate interaction specificity among Rabs and are critically important to Sro7 function on polarized vesicle targeting and exocytosis (Watson et al., 2015). Based on close structural homology of Tomo1 and Sro7p (Williams et al., 2011), Rab3A-GTP interactions were predicted, but not reported, within the N-terminal region in Tomo1 (Watson et al., 2015). Moreover, structural analysis of the C. elegans NMJ shows that a loss-of-function mutation in Tomosyn leads to an increase in the number of SVs contacting the plasma membrane (Gracheva et al., 2006). Together, these results suggest that Rab3A-GTP interacting with Tomo1 may form a central component of a SV clamping mechanism.
Measurements of the TRP size from dissociated hippocampal neurons or in hippocampal brain slices account for an average of 50% of the total SV population (Li et al., 2005; Micheva and Smith, 2005; Fernandez-Alfonso and Ryan, 2008; Ratnayaka et al., 2012), although smaller percentages (Harata et al., 2001; Marra et al., 2012) and up to the entire population of SVs have been reported to cycle (Rose et al., 2013). One of the most prominently observed ResP functions has been as a locus for presynaptic scaling of neurotransmission. For example, changes in presynaptic strength that counter strong or chronic changes in synaptic activity have been associated with a dynamic reduction or expansion of ResP size (Kim and Ryan, 2010; Rose et al., 2013; Verstegen et al., 2014). Cdk5 was one of the first presynaptic protein effectors identified for activity-dependent synaptic scaling in hippocampal cultures that can regulate the fraction of ResP SVs. In cultured hippocampal neurons or excitatory hippocampal CA3 recurrent synapses in brain slice, a compensatory upregulation of neurotransmitter release occurs in response to chronic TTX-induced silencing that depends on Cdk5 (Kim and Ryan, 2010; Mitra et al., 2012). Interestingly, Kim and Ryan (2010) reported that this chronic silencing resulted in a 50% reduction of Cdk5 protein levels, whereas other presynaptic proteins were reported largely unaffected. However, notably, a 15% reduction of Tomosyn protein was observed. Here we establish that Tomo1 proteins are subject to activity-dependent Cdk5 phosphorylation and, importantly, that compensatory enhancements in release following chronic dampening of network activity (CNQX) require Tomo1 proteins. Interestingly, Synapsin proteins were also recently reported to be substrates for Cdk5 phosphorylation and that this promoted enhanced sequestration of vesicles to perisynaptic sites in a nonreleasable state (Verstegen et al., 2014). Our data establish that Tomo1 interacts with GTP-bound Rab3A and then into complex with Synapsin proteins. This SV-associated complex is likely to be important for restricting the release capability of SVs within the AZ cluster, as each of these proteins is capable of independently limiting SV release (Geppert et al., 1994; Greengard et al., 1994; Holz et al., 1994; Schlüter et al., 2004; Baba et al., 2005; Gracheva et al., 2007b; Yizhar et al., 2007; Cesca et al., 2010). Previously, both Cdk5 and Synapsin 1 have been shown to regulate the kinetics of TRP release (Kim and Ryan, 2010; Verstegen et al., 2014). We find that Tomo1 is capable of restricting TRP size, but it does not appear to affect the kinetics of TRP depletion, suggesting that Cdk5 exerts regulatory effects both dependent on and independent of Tomosyn function. Thus, it is likely that ResP SVs could be defined via multiple mechanisms and molecular pathways.
The role of Tomosyn as a presynaptic mediator of activity-dependent trans-synaptic modulation of neurotransmitter release is further supported by findings in C. elegans, where synaptic scaling mediated by a trans-synaptic signaling mechanism involving the cell-adhesion molecules Neurexin and Neuriligin led to a shunting neurotransmitter release (Simon et al., 2008; Hu et al., 2012). The inhibition of release depended on the actions of Tomosyn and correlated to increases in the protein levels of both Tomosyn and Rab3, with actions specifically inhibiting release of less calcium-sensitive (i.e., EGTA-sensitive) SVs (Simon et al., 2008; Hu et al., 2012). Moreover, increases in neurotransmitter release associated with synaptic scaling at the Drosophila NMJ require the function of Rab3-GAP, presumably because it releases Rab3 from a synaptic repressor by promoting GTP to GDP cycling (Müller et al., 2011). It remains to be tested whether Tomosyn functions as a repressor of nucleotide cycling when interacting with GTP bound Rab3A. Thus, several reports have coalesced to implicate Cdk5, Rab3 and Tomosyn as conserved regulators of presynaptic scaling in distinct model systems, further strengthening observations that TRP:ResP SV partitioning is a primary mechanism for altering presynaptic efficacy in response to neural circuit activity.
Footnotes
The work was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants NS053978 and NS097498 to E.L.S., Endowment for the Basic Science, University of Michigan to E.L.S., and National Institutes of Health, National Institute of Neurological Disorders and Stroke National Research Service Award NS077810 to V.A.C. We thank Christian Althaus and Cynthia Carruthers for technical advice on neuronal cultures.
The authors declare no competing financial interests.
References
- Alabi AA, Tsien RW. Synaptic vesicle pools and dynamics. Cold Spring Harb Perspect Biol. 2012;4:a013680. doi: 10.1101/cshperspect.a013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Zhang Z, Ganesan S, Hintze M, Shin MM, Tang Y, Cho A, Graef IA, Chen L. Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis. Proc Natl Acad Sci U S A. 2015;112:E5744–E5752. doi: 10.1073/pnas.1510239112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Sakisaka T, Mochida S, Takai Y. PKA-catalyzed phosphorylation of tomosyn and its implication in Ca2+-dependent exocytosis of neurotransmitter. J Cell Biol. 2005;170:1113–1125. doi: 10.1083/jcb.200504055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji J, Ryan TA. Single-vesicle imaging reveals that synaptic vesicle exocytosis and endocytosis are coupled by a single stochastic mode. Proc Natl Acad Sci U S A. 2007;104:20576–20581. doi: 10.1073/pnas.0707574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak B, Williams A, Bielopolski N, Gottfried I, Okun E, Brown MA, Matti U, Rettig J, Stuenkel EL, Ashery U. Tomosyn expression pattern in the mouse hippocampus suggests both presynaptic and postsynaptic functions. Front Neuroanat. 2010;4:149. doi: 10.3389/fnana.2010.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak B, Okun E, Ben-Simon Y, Lavi A, Shapira R, Madar R, Wang Y, Norman E, Sheinin A, Pita MA, Yizhar O, Mughal MR, Stuenkel E, van Praag H, Mattson MP, Ashery U. Neuron-specific expression of tomosyn1 in the mouse hippocampal dentate gyrus impairs spatial learning and memory. Neuromolecular Med. 2013;15:351–363. doi: 10.1007/s12017-013-8223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgärtel K, Mansuy IM. Neural functions of calcineurin in synaptic plasticity and memory. Learn Mem. 2012;19:375–384. doi: 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- Ben-Simon Y, Rodenas-Ruano A, Alviña K, Lam AD, Stuenkel EL, Castillo PE, Ashery U. A combined optogenetic-knockdown strategy reveals a major role of tomosyn in mossy fiber synaptic plasticity. Cell Rep. 2015;12:396–404. doi: 10.1016/j.celrep.2015.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielopolski N, Lam AD, Bar-On D, Sauer M, Stuenkel EL, Ashery U. Differential interaction of tomosyn with syntaxin and SNAP25 depends on domains in the WD40 beta-propeller core and determines its inhibitory activity. J Biol Chem. 2014;289:17087–17099. doi: 10.1074/jbc.M113.515296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Burdina AO, Klosterman SM, Shtessel L, Ahmed S, Richmond JE. In vivo analysis of conserved C. elegans tomosyn domains. PLoS One. 2011;6:e26185. doi: 10.1371/journal.pone.0026185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Li Z, Murthy VN. Studying vesicle cycling in presynaptic terminals using the genetically encoded probe synaptopHluorin. Nat Protoc. 2006;1:2970–2978. doi: 10.1038/nprot.2006.449. [DOI] [PubMed] [Google Scholar]
- Calero M, Chen CZ, Zhu W, Winand N, Havas KA, Gilbert PM, Burd CG, Collins RN. Dual prenylation is required for Rab protein localization and function. Mol Biol Cell. 2003;14:1852–1867. doi: 10.1091/mbc.E02-11-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chen K, Richlitzki A, Featherstone DE, Schwärzel M, Richmond JE. Tomosyn-dependent regulation of synaptic transmission is required for a late phase of associative odor memory. Proc Natl Acad Sci U S A. 2011;108:18482–18487. doi: 10.1073/pnas.1110184108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22:169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier HN, Dueker ND, Slifer SH, Lee JM, Whitehead PL, Lalanne E, Leyva N, Konidari I, Gentry RC, Hulme WF, Booven DV, Mayo V, Hofmann NK, Schmidt MA, Martin ER, Haines JL, Cuccaro ML, Gilbert JR, Pericak-Vance MA. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism. 2014;5:1. doi: 10.1186/2040-2392-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czapski GA, Gąssowska M, Songin M, Radecka UD, Strosznajder JB. Alterations of cyclin dependent kinase 5 expression and phosphorylation in amyloid precursor protein (APP)-transfected PC12 cells. FEBS Lett. 2011;585:1243–1248. doi: 10.1016/j.febslet.2011.03.058. [DOI] [PubMed] [Google Scholar]
- Davis LK, Meyer KJ, Rudd DS, Librant AL, Epping EA, Sheffield VC, Wassink TH. Novel copy number variants in children with autism and additional developmental anomalies. J Neurodev Disord. 2009;1:292–301. doi: 10.1007/s11689-009-9013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A, Rizzoli SO. Synaptic vesicle pools: an update. Front Synaptic Neurosci. 2010;2:135. doi: 10.3389/fnsyn.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;326:1127–1130. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/S0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Südhof TC, Rizo J. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J. 2005;24:2839–2850. doi: 10.1038/sj.emboj.7600753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth CC, Kawata M, Yoshida Y, Takai Y, Gelb MH, Glomset JA. C terminus of the small GTP-binding protein smg p25A contains two geranylgeranylated cysteine residues and a methyl ester. Proc Natl Acad Sci U S A. 1991;88:6196–6200. doi: 10.1073/pnas.88.14.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA. A heterogeneous “resting” pool of synaptic vesicles that is dynamically interchanged across boutons in mammalian CNS synapses. Brain Cell Biol. 2008;36:87–100. doi: 10.1007/s11068-008-9030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredj NB, Burrone J. A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci. 2009;12:751–758. doi: 10.1038/nn.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, Yokoyama S, Nishioka H, Matsuura Y, Mizoguchi A, Scheller RH, Takai Y. Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron. 1998;20:905–915. doi: 10.1016/S0896-6273(00)80472-9. [DOI] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Südhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- Giovedì S, Vaccaro P, Valtorta F, Darchen F, Greengard P, Cesareni G, Benfenati F. Synapsin is a novel Rab3 effector protein on small synaptic vesicles: I. Identification and characterization of the synapsin I-Rab3 interactions in vitro and in intact nerve terminals. J Biol Chem. 2004;279:43760–43768. doi: 10.1074/jbc.M403293200. [DOI] [PubMed] [Google Scholar]
- Gladycheva SE, Lam AD, Liu J, D'Andrea-Merrins M, Yizhar O, Lentz SI, Ashery U, Ernst SA, Stuenkel EL. Receptor-mediated regulation of tomosyn-syntaxin 1A interactions in bovine adrenal chromaffin cells. J Biol Chem. 2007;282:22887–22899. doi: 10.1074/jbc.M701787200. [DOI] [PubMed] [Google Scholar]
- Glynn MW, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat Protoc. 2006;1:1287–1296. doi: 10.1038/nprot.2006.220. [DOI] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Holgado AM, Berthelot-Grosjean M, Ackley BD, Hadwiger G, Nonet ML, Weimer RM, Richmond JE. Tomosyn inhibits synaptic vesicle priming in Caenorhabditis elegans. PLoS Biol. 2006;4:e261. doi: 10.1371/journal.pbio.0040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Touroutine D, Berthelot-Grosjean M, Parekh H, Richmond JE. Tomosyn negatively regulates both synaptic transmitter and neuropeptide release at the C. elegans neuromuscular junction. J Physiol. 2007a;585:705–709. doi: 10.1113/jphysiol.2007.138321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Touroutine D, Berthelot-Grosjean M, Parekh H, Richmond JE. Tomosyn negatively regulates CAPS-dependent peptide release at Caenorhabditis elegans synapses. J Neurosci. 2007b;27:10176–10184. doi: 10.1523/JNEUROSCI.2339-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Maryon EB, Berthelot-Grosjean M, Richmond JE. Differential regulation of synaptic vesicle tethering and docking by UNC-18 and TOM-1. Front Synaptic Neurosci. 2010;2:141. doi: 10.3389/fnsyn.2010.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Benfenati F, Valtorta F. Synapsin I, an actin-binding protein regulating synaptic vesicle traffic in the nerve terminal. Adv Second Messenger Phosphoprotein Res. 1994;29:31–45. doi: 10.1016/S1040-7952(06)80005-4. [DOI] [PubMed] [Google Scholar]
- Harata N, Ryan TA, Smith SJ, Buchanan J, Tsien RW. Visualizing recycling synaptic vesicles in hippocampal neurons by FM 1-43 photoconversion. Proc Natl Acad Sci U S A. 2001;98:12748–12753. doi: 10.1073/pnas.171442798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsuzawa K, Lang T, Fasshauer D, Bruns D, Jahn R. The R-SNARE motif of tomosyn forms SNARE core complexes with syntaxin 1 and SNAP-25 and down-regulates exocytosis. J Biol Chem. 2003;278:31159–31166. doi: 10.1074/jbc.M305500200. [DOI] [PubMed] [Google Scholar]
- Hattendorf DA, Andreeva A, Gangar A, Brennwald PJ, Weis WI. Structure of the yeast polarity protein Sro7 reveals a SNARE regulatory mechanism. Nature. 2007;446:567–571. doi: 10.1038/nature05635. [DOI] [PubMed] [Google Scholar]
- Henry FE, McCartney AJ, Neely R, Perez AS, Carruthers CJ, Stuenkel EL, Inoki K, Sutton MA. Retrograde changes in presynaptic function driven by dendritic mTORC1. J Neurosci. 2012;32:17128–17142. doi: 10.1523/JNEUROSCI.2149-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman E. The origin and fate of secretory packages, especially synaptic vesicles. Neuroscience. 1977;2:327–355. doi: 10.1016/0306-4522(77)90001-X. [DOI] [PubMed] [Google Scholar]
- Holz RW, Brondyk WH, Senter RA, Kuizon L, Macara IG. Evidence for the involvement of Rab3A in Ca(2+)-dependent exocytosis from adrenal chromaffin cells. J Biol Chem. 1994;269:10229–10234. [PubMed] [Google Scholar]
- Hu Z, Hom S, Kudze T, Tong XJ, Choi S, Aramuni G, Zhang W, Kaplan JM. Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science. 2012;337:980–984. doi: 10.1126/science.1224896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummon MR, Costello WJ. Induced disruption in the connectivity of an identified neuron in the Drosophila ts mutant shibire. J Neurosci. 1987;7:3633–3638. doi: 10.1523/JNEUROSCI.07-11-03633.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 2010;68:1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes L, Lledo PM, Roa M, Vincent JD, Henry JP, Darchen F. The GTPase Rab3a negatively controls calcium-dependent exocytosis in neuroendocrine cells. EMBO J. 1994;13:2029–2037. doi: 10.1002/j.1460-2075.1994.tb06476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ziff EB. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 2014;12:e1001900. doi: 10.1371/journal.pbio.1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. Balance of calcineurin Aalpha and CDK5 activities sets release probability at nerve terminals. J Neurosci. 2013;33:8937–8950. doi: 10.1523/JNEUROSCI.4288-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KO, Wong AS, Cheung MC, Xu P, Liang Z, Lok KC, Xie H, Palko ME, Yung WH, Tessarollo L, Cheung ZH, Ip NY. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci. 2012;15:1506–1515. doi: 10.1038/nn.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman K, Rossi G, Adamo JE, Brennwald P. Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. J Cell Biol. 1999;146:125–140. doi: 10.1083/jcb.146.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Burrone J, Tyler WJ, Hartman KN, Albeanu DF, Murthy VN. Synaptic vesicle recycling studied in transgenic mice expressing synaptopHluorin. Proc Natl Acad Sci U S A. 2005;102:6131–6136. doi: 10.1073/pnas.0501145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes JP, Agostinho P. Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol. 2011;94:49–63. doi: 10.1016/j.pneurobio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Marra V, Burden JJ, Thorpe JR, Smith IT, Smith SL, Häusser M, Branco T, Staras K. A preferentially segregated recycling vesicle pool of limited size supports neurotransmission in native central synapses. Neuron. 2012;76:579–589. doi: 10.1016/j.neuron.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunami N, Hadley D, Hensel CH, Christensen GB, Kim C, Frackelton E, Thomas K, da Silva RP, Stevens J, Baird L, Otterud B, Ho K, Varvil T, Leppert T, Lambert CG, Leppert M, Hakonarson H. Identification of rare recurrent copy number variants in high-risk autism families and their prevalence in a large ASD population. PLoS One. 2013;8:e52239. doi: 10.1371/journal.pone.0052239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Madison JM, Dybbs M, Kaplan JM. Antagonistic regulation of synaptic vesicle priming by Tomosyn and UNC-13. Neuron. 2006;51:303–315. doi: 10.1016/j.neuron.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Strong effects of subphysiological temperature on the function and plasticity of mammalian presynaptic terminals. J Neurosci. 2005;25:7481–7488. doi: 10.1523/JNEUROSCI.1801-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Mitra SS, Tsien RW. Heterogeneous reallocation of presynaptic efficacy in recurrent excitatory circuits adapting to inactivity. Nat Neurosci. 2012;15:250–257. doi: 10.1038/nn.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Pym EC, Tong A, Davis GW. Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron. 2011;69:749–762. doi: 10.1016/j.neuron.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Liu KS, Sigrist SJ, Davis GW. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J Neurosci. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenbuch A, Shalev L, Marra V, Sinai I, Lavy Y, Kahn J, Burden JJ, Staras K, Gitler D. Synapsin selectively controls the mobility of resting pool vesicles at hippocampal terminals. J Neurosci. 2012;32:3969–3980. doi: 10.1523/JNEUROSCI.5058-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobbati AV, Razeto A, Böddener M, Becker S, Fasshauer D. Structural basis for the inhibitory role of tomosyn in exocytosis. J Biol Chem. 2004;279:47192–47200. doi: 10.1074/jbc.M408767200. [DOI] [PubMed] [Google Scholar]
- Poodry CA, Edgar L. Reversible alteration in the neuromuscular junctions of Drosophila melanogaster bearing a temperature-sensitive mutation, shibire. J Cell Biol. 1979;81:520–527. doi: 10.1083/jcb.81.3.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnayaka A, Marra V, Bush D, Burden JJ, Branco T, Staras K. Recruitment of resting vesicles into recycling pools supports NMDA receptor-dependent synaptic potentiation in cultured hippocampal neurons. J Physiol. 2012;590:1585–1597. doi: 10.1113/jphysiol.2011.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rose T, Schoenenberger P, Jezek K, Oertner TG. Developmental refinement of vesicle cycling at Schaffer collateral synapses. Neuron. 2013;77:1109–1121. doi: 10.1016/j.neuron.2013.01.021. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rossi G, Brennwald P. Yeast homologues of lethal giant larvae and type V myosin cooperate in the regulation of Rab-dependent vesicle clustering and polarized exocytosis. Mol Biol Cell. 2011;22:842–857. doi: 10.1091/mbc.E10-07-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi G, Watson K, Demonch M, Temple B, Brennwald P. In vitro reconstitution of Rab GTPase-dependent vesicle clustering by the yeast lethal giant larvae/tomosyn homolog, Sro7. J Biol Chem. 2015;290:612–624. doi: 10.1074/jbc.M114.595892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royle SJ, Granseth B, Odermatt B, Derevier A, Lagnado L. Imaging phluorin-based probes at hippocampal synapses. Methods Mol Biol. 2008;457:293–303. doi: 10.1007/978-1-59745-261-8_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakisaka T, Yamamoto Y, Mochida S, Nakamura M, Nishikawa K, Ishizaki H, Okamoto-Tanaka M, Miyoshi J, Fujiyoshi Y, Manabe T, Takai Y. Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. J Cell Biol. 2008;183:323–337. doi: 10.1083/jcb.200805150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, Ryan TA. The use of pHluorins for optical measurements of presynaptic activity. Biophys J. 2000;79:2199–2208. doi: 10.1016/S0006-3495(00)76468-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter OM, Schmitz F, Jahn R, Rosenmund C, Südhof TC. A complete genetic analysis of neuronal Rab3 function. J Neurosci. 2004;24:6629–6637. doi: 10.1523/JNEUROSCI.1610-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Sharma M, Amin ND, Albers RW, Pant HC. Regulation of cyclin-dependent kinase 5 catalytic activity by phosphorylation. Proc Natl Acad Sci U S A. 1999;96:11156–11160. doi: 10.1073/pnas.96.20.11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3->CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuang R, Zhang L, Fletcher A, Groblewski GE, Pevsner J, Stuenkel EL. Regulation of Munc-18/syntaxin 1A interaction by cyclin-dependent kinase 5 in nerve endings. J Biol Chem. 1998;273:4957–4966. doi: 10.1074/jbc.273.9.4957. [DOI] [PubMed] [Google Scholar]
- Simon DJ, Madison JM, Conery AL, Thompson-Peer KL, Soskis M, Ruvkun GB, Kaplan JM, Kim JK. The microRNA miR-1 regulates a MEF-2-dependent retrograde signal at neuromuscular junctions. Cell. 2008;133:903–915. doi: 10.1016/j.cell.2008.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Newton AJ, Murthy VN. Real-time imaging of Rab3a and Rab5a reveals differential roles in presynaptic function. J Physiol. 2005;569:103–117. doi: 10.1113/jphysiol.2005.092528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Williams JH. Discharge of the readily releasable pool with action potentials at hippocampal synapses. J Neurophysiol. 2007;98:3221–3229. doi: 10.1152/jn.00857.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- Südhof TC. A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med. 2013;19:1227–1231. doi: 10.1038/nm.3338. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Verstegen AM, Tagliatti E, Lignani G, Marte A, Stolero T, Atias M, Corradi A, Valtorta F, Gitler D, Onofri F, Fassio A, Benfenati F. Phosphorylation of synapsin I by cyclin-dependent kinase-5 sets the ratio between the resting and recycling pools of synaptic vesicles at hippocampal synapses. J Neurosci. 2014;34:7266–7280. doi: 10.1523/JNEUROSCI.3973-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani T, Atasoy D, Kavalali ET. Synaptic vesicle recycling adapts to chronic changes in activity. J Neurosci. 2006;26:2197–2206. doi: 10.1523/JNEUROSCI.4500-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- Waters J, Smith SJ. Vesicle pool partitioning influences presynaptic diversity and weighting in rat hippocampal synapses. J Physiol. 2002;541:811–823. doi: 10.1113/jphysiol.2001.013485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson K, Rossi G, Temple B, Brennwald P. Structural basis for recognition of the Sec4 Rab GTPase by its effector, the Lgl/tomosyn homologue, Sro7. Mol Biol Cell. 2015;26:3289–3300. doi: 10.1091/mbc.E15-04-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welzel O, Henkel AW, Stroebel AM, Jung J, Tischbirek CH, Ebert K, Kornhuber J, Rizzoli SO, Groemer TW. Systematic heterogeneity of fractional vesicle pool sizes and release rates of hippocampal synapses. Biophys J. 2011;100:593–601. doi: 10.1016/j.bpj.2010.12.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AL, Bielopolski N, Meroz D, Lam AD, Passmore DR, Ben-Tal N, Ernst SA, Ashery U, Stuenkel EL. Structural and functional analysis of tomosyn identifies domains important in exocytotic regulation. J Biol Chem. 2011;286:14542–14553. doi: 10.1074/jbc.M110.215624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Pestell R, Rosner MR. Role of cyclins in neuronal differentiation of immortalized hippocampal cells. Mol Cell Biol. 1997;17:6585–6597. doi: 10.1128/MCB.17.11.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Mochida S, Kurooka T, Sakisaka T. Reciprocal intramolecular interactions of tomosyn control its inhibitory activity on SNARE complex formation. J Biol Chem. 2009;284:12480–12490. doi: 10.1074/jbc.M807182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang H, Zhang J, Luo F, Herrup K, Bibb JA, Lu R, Miller RH. Cyclin dependent kinase 5 is required for the normal development of oligodendrocytes and myelin formation. Dev Biol. 2013;378:94–106. doi: 10.1016/j.ydbio.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Matti U, Melamed R, Hagalili Y, Bruns D, Rettig J, Ashery U. Tomosyn inhibits priming of large dense-core vesicles in a calcium-dependent manner. Proc Natl Acad Sci U S A. 2004;101:2578–2583. doi: 10.1073/pnas.0308700100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Lipstein N, Gladycheva SE, Matti U, Ernst SA, Rettig J, Stuenkel EL, Ashery U. Multiple functional domains are involved in tomosyn regulation of exocytosis. J Neurochem. 2007;103:604–616. doi: 10.1111/j.1471-4159.2007.04791.x. [DOI] [PubMed] [Google Scholar]
- Yu H, Rathore SS, Gulbranson DR, Shen J. The N- and C-terminal domains of tomosyn play distinct roles in soluble N-ethylmaleimide-sensitive factor attachment protein receptor binding and fusion regulation. J Biol Chem. 2014;289:25571–25580. doi: 10.1074/jbc.M114.591487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, Alonso-Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Südhof TC, Brunger AT. Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature. 2015;525:62–67. doi: 10.1038/nature14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]