

Abstract
Current pharmacotherapies for alcohol used disorder (AUD) are few and relatively ineffective illustrating the need for the development of new, effective medications. Using a translational approach, our laboratory reported that ivermectin, an FDA-approved, human and animal anti-parasitic agent, can significantly reduce ethanol intake in male and female mice across different drinking paradigms. Extending this line of investigation, the current paper investigated the utility of moxidectin (MOX), an analogue of ivermectin, to reduce ethanol intake. Notably, MOX is widely held to have lower neurotoxicity potential and improved margin of safety compared to ivermectin. Using a 24-h-two-bottle choice paradigm, MOX significantly reduced ethanol intake in a dose dependent manner in both male and female C57BL/6J mice, respectively (1.25 – 7.5 mg/kg) and (1.25 – 10 mg/kg). Further, multi-day administration of MOX (2.5 mg/kg; intraperitoneal injection) for 5 consecutive days significantly reduced ethanol intake in both the 24-h-two-bottle choice and Drinking-in-the-Dark paradigms in female mice. No overt signs of behavioral toxicity were observed. Notably in both male and female mice, MOX significantly reduced ethanol intake starting approximately 4 h post-injection. Using a Xenopus oocyte expression system, we found that MOX significantly potentiated P2X4 receptors (P2X4R) function and antagonized the inhibitory effects of ethanol on ATP-gated currents in P2X4Rs. This latter finding represents the first report of MOX having activity on P2X4Rs. In addition, MOX potentiated GABAA receptors, but to a lesser degree as compared to ivermectin supporting the hypothesis that MOX would be advantageous (compared to ivermectin) with respect to reducing contraindications. Overall, the results illustrate the potential for development of MOX as a novel pharmacotherapy for the treatment of AUD.
Keywords: Alcoholism therapy, medication development, drug repurposing
1. Introduction
Alcohol use disorder (AUD) has major health implications in the United States, affecting over 17 million people, causing more than 100,000 deaths and costing over $200 billion annually (Bouchery et al., 2011; Grant et al., 2004; Hardwood, 2000). Despite an ongoing effort focusing on the development of new medications for AUD, there are only three-FDA approved pharmacotherapies available (disulfiram, naltrexone, and acamprosate), all which have yielded limited success (Harris et al., 2010; Litten et al., 2012). This is evident by the continual prevalence of high rates of uncontrolled heavy drinking and high relapse rate in patients even after long-term inpatient treatment and support (Substance Abuse and Mental Health Services Administration, National Survey on Drug Use and Health, 2013). As such, the development of effective medications to treat AUD is an important public health goal (Bouchery et al., 2011; Heilig and Egli, 2006; Johnson et al., 2007; Johnson, 2010; Steensland et al., 2007).
Our laboratory has been investigating the utility of different compounds from the avermectin family of macrocyclic lactones (eg., ivermectin, abamectin, selamectin) to be developed into novel pharmacotherapies for AUD. This class of compounds is already recognized for their ability to act on several CNS receptor targets (eg., GABAA receptors [GABAARs], glycine receptors, and nicotinic acetylcholine receptors) in mammals, all of which have been linked to the behavioral effects of ethanol (Dawson et al., 2000; Shan et al., 2001; Spinosa et al., 2002). Using a combination of electrophysiology methods and rodent drinking models, we observed that the ability of these avermectins to reduce ethanol intake in mice appeared to be related to their ability to significantly reduce or eliminate the inhibitory effects of ethanol on ATP-gated P2X4 receptor (P2X4R) function in vitro (Asatryan et al., 2014). This initial in vivo-in vitro correlation implicates the use of P2X4R as a screening platform for the development of avermectins and other related analogues into novel AUD therapeutics.
Since ivermectin is already an FDA-approved drug that has been safely used in humans for several decades for the treatment of parasites (Guzzo et al. 2002; Omura, 2008), we have been investigating the feasibility of repurposing ivermectin into a novel pharmacotherapy for AUD. In support of this hypothesis, we demonstrated that ivermectin can significantly antagonize the inhibitory effects of ethanol on P2X4R function in vitro (Asatryan et al., 2008, 2010, 2014) and significantly reduce ethanol intake in multiple drinking paradigms in both male and female mice (Asatryan et al., 2014; Yardley et al., 2012, 2014, 2015; Wyatt et al., 2014). Notably, doses of IVM as high as 10 mg/kg did not appear to cause any overt signs of organ toxicity or exert rewarding properties indicating that the psychotropic effects of IVM are dissociated from any addiction liability that can affect therapeutic compliance (Bortolato et al., 2013; Yardley et al., 2015).
Although we have established that ivermectin is an effective agent for reducing ethanol intake in mice, recently we have begun shifting our attention to moxidectin (MOX; an ivermectin analogue) due to a number of recent reports in the literature that suggests MOX exhibits lower neurotoxicity potential compared to ivermectin (Janko et al., 2012; Menez et al., 2012). This more favorable central nervous system (CNS) safety profile is thought to be due to: 1) MOX having lower potency on GABAARs as compared to ivermectin which should be advantageous with respect to reducing contraindications, and 2) the differential transport across the blood brain barrier (BBB), with MOX being a weaker P-glycoprotein (P-gp) transporter substrate and less dependence on P-gp for removal from the brain (Janko et al., 2012; Menez et al., 2012). As such, the potential chronic use of MOX as a long-term AUD therapy is less likely to have complications from excessive stimulation of GABAARs that can lead to CNS depression and potentially coma, and also less likely to result in brain accumulation due to a deficiency in P-gp function or drug-drug interaction with other concurrent medications that may also act as P-gp substrates (Balayssac et al., 2005; Edwards et al., 2003; Prichard et al., 2012;). Notably, MOX is now being developed as an alternative therapy to ivermectin as an anti-parasitic agent for humans. To date, no significant clinical abnormalities have been reported (Cotreau at al., 2003; Korth-Bradley et al., 2012). With MOX becoming approved for human use, it could represent another avermectin candidate that could be repurposed as a pharmacotherapy for AUD.
To begin to determine if MOX has the potential to be developed into a safe and effective pharmacotherapy for AUD, the present paper investigates the ability of MOX to reduce ethanol intake in male and female mice using a 24-h-two-bottle choice paradigm. The lower limit of alcohol consumed in this model suggests that it mimics social drinking (Blednov et al., 2010). We also utilized a Drinking-in-the-Dark (DID) paradigm where the amount of alcohol consumed is much higher in a short period of time (compared to the 24 h model) and results in blood ethanol concentrations (BECs) similar to human binge-like drinking (i.e., > 80 mg%) (Lowery et al., 2010; Neasta et al., 2010; Rhodes et al., 2005). Both of these models are well-validated and routinely used to assess changes in ethanol intake in rodents.
To begin to investigate the mechanism of MOX’s anti-alcohol effects, we used a two-electrode voltage clamp, Xenopus oocyte expression system (Davies et al., 2002; Davies et al., 2005) to test the ability of MOX to potentiate P2X4R function and to antagonize the inhibitory effects of ethanol on P2X4Rs as previously demonstrated for ivermectin and abamectin (Asatryan et al., 2014). We also compared the potentiating effects of MOX versus ivermectin on GABAARs. Overall, the findings support the development of MOX as a pharmacotherapy for the treatment of AUD.
2. Materials
2.1. Drugs
MOX (10 mg/kg) solution (Boehringer Ingelheim, St. Joseph, MO) was diluted in a 0.9% sodium chloride solution (saline) to a concentration that would allow for an injection volume of 0.01 mL/g of body weight. We used saline as the diluent based on previous pilot work where we found that propylene glycol [(1,2 propanediol), (Alfa Aesar, Ward Hill, MA)], the solvent used by the manufacturer to dissolve MOX (when diluted in saline to equivalent concentrations as in the MOX [10mg/kg] solution) showed comparable drinking level as saline injection alone, and both the solvent and saline did not cause significant reduction in ethanol intake. 190 proof USP ethanol (Koptec, King of Prussia, PA) was diluted in water to achieve a 10% (10E) v/v solution or a 20% (20E) v/v solution (20E).
2.2. Animals
Studies were performed on drug-naïve male and female C57BL/6J mice purchased from Jackson Laboratory (Bar Harbor, ME). All studies were conducted with mice 10–12 weeks of age. Mice were first acclimated to the housing facility for one week and group-housed (4 mice per cage) in polycarbonate/polysulfone cages at a 12 h light/dark cycle (lights off at 12:00PM) with ad libitum access to food and water in rooms maintained at 22°C. All procedures in this study were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and all efforts were made to minimize animal suffering. The USC Institutional Animal Care and Use Committee approved the protocols.
3. Methods
3.1. 24-h-two-bottle choice paradigm
All experiments numbering noted in the methods section corresponds to the figures numbering in the results section for the ease of reading (i.e., Experiment 1a corresponds to Figure 1a).
Figure 1. Acute administration of MOX significantly reduced 10E intake in female and male C57BL/6J mice, in a dose-dependent manner, using a 24-h-two-bottle choice paradigm.

MOX was administered after habituation with saline injection and attaining stable drinking levels. Bars represent levels from the day prior to MOX injection (white; pre-MOX), the day of MOX injection (black; MOX), and the day after MOX injection (gray; post-MOX). a) MOX (5 mg/kg) significantly reduce 10E intake in female mice. Values represent mean ± SEM for 12 mice, * p < 0.05, # p < 0.0001 versus pre-MOX, Tukey multiple comparison post-hoc test. b) MOX (1.25, 2.5, 5, 7.5, and 10 mg/kg) significantly reduced 10E intake in female mice. Values represent mean ± SEM for 16 mice per dose group, * p < 0.05, # p < 0.0001 versus respective pre-MOX condition, Bonferroni’s post-hoc test. c) MOX (1.25, 2.5, 5, and 7.5 mg/kg) significantly reduced 10E intake in male mice. Values represent mean ± SEM for 12 mice per dose group, ** p < 0.01, # p < 0.0001 versus respective pre-MOX condition, Bonferroni’s post-hoc test.
For our initial investigations, we used a 24-h-two-bottle choice paradigm that is widely used to assess changes in drinking behaviors (Belknap et al., 1993; Yoneyama et al., 2008; Yardley et al., 2012) with modifications as presented previously (Yardley et al., 2012). Mice were individually housed 3 days before the start of the study and had 24 h access to two inverted water bottles (graduated cylinders) with metal sippers placed on the cage tops. Food was distributed near both bottles to avoid food associated tube preferences.
Experiment 1a
A within-subjects design was used with one group of female mice (n = 12) that were allowed free access to two bottles, one containing 10E and the other water. 10E position was alternated every other day during testing period to avoid for side preference. Body weights and food intake (weight of food on day 1 subtract weight of food on day 2 = food consumed for the 24 h period) were monitored daily during testing period. Fluid intake was recorded (fluid level on inverted cylinder on day 2 subtract fluid level on day 1 = fluid consumed for the 24 h period) daily during testing period. Daily observations on normal overt behaviors of mice were also noted (normal: constant active movement, responsive to experimenter intervention, and fur groomed; abnormal: lack of movement, hunched postured and hurdling in corner, unresponsive to experimenter intervention, and piloerection of fur). 10E position was also alternated every other day to avoid for side preference. Daily 10E intake was measured until it stabilized (± 10% variability from the mean of the last 3 days). Next mice were habituated to saline injections (intraperitoneal; i. p.) until 10E intake again stabilized. Mice then received one dose of MOX (5 mg/kg). Animals were injected with saline on the day following MOX injection. Change in drinking over the 24 h following each injection (saline or MOX) was measured. Pre-MOX is the day prior to MOX injection, and post-MOX is the day following MOX injection.
Experiment 1b followed similar procedures as described in experiment 1a. A new group of female mice (n = 16) were allowed free access to 10E and water. After saline habituation and obtaining stable baseline 10E intake, all mice received one injection (i. p.) of MOX (0.65, 1.25, 2.5, 5, 7.5, or 10 mg/kg). Animals were injected with saline on subsequent days after MOX injection until 10E drinking returned to baseline levels (which took about 1–2 days), and then mice were injected with another dose of MOX. This pattern of MOX administration continued until all doses of a particular study were completed. The doses were given in random fashion with no particular order.
Experiment 1c followed similar procedures as described in experiment 1b, except male mice (n = 12) were used in this study and the doses of MOX tested were 1.25, 2.5, 5, and 7.5 mg/kg
Experiment 2
We conducted an hour by hour evaluation using the two-bottle choice paradigm and a between-subjects design. A new group of female mice was habituated to saline injection. After establishing baseline drinking level, mice were split into two groups based on average 10E intake and subsequently received either one injection (i. p.) of MOX (2.5 mg/kg) or saline (controls) (n = 18 MOX versus 10 saline controls). Following drug injection, 10E intake was monitored for the two groups every hour up to the 9th h.
Experiment 3
We also conducted a multi-day MOX study using the two-bottle choice paradigm and a between-subjects design. All the female mice from experiment 2 received saline injection during the washout period until 10E intake returned to baseline, then received either daily injection (i. p.) of MOX (2.5 mg/kg) or saline (controls) for 5 consecutive days in the multi-day MOX study (n = 18 MOX versus 10 saline controls). 10E intake was monitored for 24 h period after each injection for 5 consecutive days.
3.2. Drinking-in-the-Dark (DID) paradigm
Experiment 4
The DID model is widely used to assess changes in binge-like drinking behaviors. A modified version of this procedure (Lowery et al., 2010; Neasta et al., 2010; Rhodes et al., 2005) was utilized during which mice had daily limited access (2 h) to one bottle containing 20E beginning at the third hour into the circadian dark phase, which results in mice reaching high BECs in a short period of time. A new group of female mice was used and following habituation to saline injection and establishing baseline drinking level, mice were split into two groups based on average 20E intake and subsequently received either one injection (i. p.) of MOX (2.5 mg/kg) or saline daily for 5 days (n = 18 MOX versus 10 saline controls). MOX or saline was administered 4 h prior to the start of the drinking session based on our time-course study for MOX which indicates that it took 4 h post-injection before significant reduction in ethanol intake was observed (Figure 2). 20E intake was monitored during the 2 h drinking session for 5 consecutive days. A single bottle of water was continuously available between ethanol access periods.
Figure 2. MOX (2.5 mg/kg) significantly reduced 10E intake approximately 4 h after administration in female mice.

The intake was measured on an hourly basis, up to the 9th h following MOX (square) or saline (circle) administration. The inset was reproduced from Figure 3 in Yardley et al., 2012, which shows ivermectin began to significantly reduced 10E intake approximately 9 h after administration. Values represent mean ± SEM cumulative intake for MOX (18 mice) and saline (10 mice), IVM (11 mice) and saline (10 mice), * p < 0.05, ** p < 0.01, # p < 0.0001 versus saline-treated group, Bonferroni’s post-hoc test.
3.3. Complementary RNA (cRNA) and complementary DNA (cDNA) injections in Xenopus laevis oocyte
Stage V or VI Xenopus oocytes (purchased from EcoCyte Bioscience, Austin, TX) were injected with 20 ng cRNA of rat p2rx4 gene or 1 ng cDNA for GABAAR (α1:β2:γ2:, 1:1:10) as described previously (Asatryan et al., 2010, 2014) using the Nanoject II Nanoliter injection system (Drummond Scientific, Broomall, PA). Injected oocytes were stored at 16°C in incubation medium co ntaining 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 5 mM HEPES, and 2.5 mM pyruvic acid with 1% heat inactivated HyClone® horse serum (VWR, San Dimas, CA) and 0.05 mg/ml gentamycin, adjusted to pH 7.5. Electrophysiological experiments were conducted 24 – 48 h after cRNA injections.
3.4. Whole Cell Two-Electrode Voltage Clamp Recordings
Two electrode voltage clamp recordings were performed using the OC-725C oocyte clamp (Warner Instruments, Hamden, CT) as previously described elsewhere (Davies et al., 2002, 2005). The oocytes were voltage clamped at −70 mV and the currents were recorded on a strip-chart recorder (Barnstead Instrument, Dubuqe, IA).
Experiment 5
P2X4R oocytes were continuously perfused at a rate of 3 – 4 mL/min at room temperature with modified Ringers buffer containing (in mM) 110 NaCl, 2.5 KCl, 10 HEPES and 1.8 BaCl2, pH 7.5, using a peristaltic pump (Rainin Instrument, Oakland, CA). To induce currents, submaximal concentrations (EC10) of adenosine 5′-triphosphate (ATP, Sigma, St. Louis, MO) were used, which produced 10% of maximal effect induced by 100 μM ATP. We have shown previously that the use of EC10 can maximize the effects of ethanol and minimize receptor desensitization (Asatryan et al., 2010; Davies et al., 2002, 2005). Ethanol and/or drugs were applied after stable response to ATP EC10 was obtained. A washout period of 5 min was allowed between each application to allow for re-sensitization of the receptor (Asatryan et al., 2010; Davies et al., 2002, 2005; Popova et al., 2013). Effects of ethanol (25 or 50 mM) or MOX ([0.5 and 1 μM], powder dissolved in in DMSO to 10mM stock solution, then dissolved to appropriate concentration in perfusion buffer) were tested alone and in combination during co-application with ATP. Pilot studies determined that the effects of ethanol and MOX on P2X4R were concentration-dependent and reversible (n = 4 – 6 oocytes).
Experiment 6
Effects of MOX (0.1, 0.5, and 1 μM) or ivermectin (0.1, 0.5, and 1 μM) on GABAARs were tested using GABA EC10 (Asatryan et al., 2014). Oocytes were perfused at a rate of 3 – 4 mL/min at room temperature with modified Bart’s saline containing (in mM) 83 NaCl, 1 KCl, 10 HEPES, 0.82 MgSO4, 2.4 NaHCO3, 0.91 CaCl2, and 0.33 Ca(NO3)2, pH 7.5. Drugs were applied after stable response to GABA EC10 was obtained. A washout period of 5 min was allowed between each GABA application. Due to irreversible and non-washable effects on GABA-induced currents, each oocyte was tested for one concentration of MOX or ivermectin (n = 4 – 12 oocytes per data point).
3.5. Statistical Analyses
Ethanol intake for all studies was calculated as g/kg [g of pure ethanol per kg of body weight; 10E intake = (volume of 10E consumed in mL x 0.07893 g/mL)/body weight in kg; 20E intake = (volume of 20E consumed in mL x 0.15786 g/mL)/body weight in kg]. The dependent variables include ethanol intake (g/kg), water (mL), food intake (g), and change in mouse weight (g). Experiment 1a: one-way ANOVA was used to assess the treatment effect of MOX with time [saline pre-treatment (pre-MOX), MOX dose, saline post-treatment (post-MOX)] as a repeated measures factor on the dependent variable. Experiment 1bc: two-way ANOVA was used to assess the treatment effect of MOX with each MOX dose (0.65 – 10 mg/kg) or time [saline pre-treatment (pre-MOX), MOX dose, saline post-treatment (post-MOX)] as a repeated measures factor on the dependent variable. Experiment 2: two-way ANOVA was used to assess the treatment effect of MOX between groups (MOX versus saline-injected) or with time (for each hour post-injection) as a repeated-measures factor on the dependent variables. Experiment 3–4: two-way ANOVA was used to assess the treatment effect of 5-day MOX administration between groups (MOX versus saline-injected) or with time (for each day of MOX administration) as a repeated-measures factor on the dependent variables. Experiment 5–6: two-tailed, unpaired, individual Student’s t-test was used to assess for difference between treatment groups.
Significant main effects and interactions of the ANOVAs were further investigated with post-hoc tests (i.e., Tukey’s for one way ANOVA and Bonferroni for two-way ANOVA). For all studies significance was set at p ≤ 0.05.
All graphs and statistical analyses were generated using Prism (GraphPad Software Inc., San Diego, CA).
4. Results
4.1. Acute administration of MOX (5 mg/kg) decreased 10E intake in female mice
Using female C57BL/6J mice, we tested the effects of acute administration of MOX (5 mg/kg) on ethanol intake using a 24-h-two-bottle choice paradigm (10E versus water). This dose was previously shown to be the lowest ivermectin dose that produced maximal reduction in ethanol consumption (Yardley et al., 2012). Baseline 10E intake was first obtained followed by habituation to saline injections.
As illustrated in Figure 1a, 10E intake stabilized at 14.14 g/kg following saline injections (pre-MOX). Acute administration of MOX (5 mg/kg) significantly reduced ethanol intake in excess of 44% when analyzed across time (pre-MOX, MOX, post-MOX) [F(2, 33) = 8.045, p = 0.0014], and 10E intake remained significantly lower than pre-MOX on the day following MOX treatment (by more than 20%, shown as post-MOX, p < 0.05) (Figure 1a).
4.2. MOX decreased 10E intake in a dose-dependent manner in female mice
We extended our initial single dose MOX study to one where several doses of MOX were tested in random fashion (saline, 5, 10, 2.5, 7.5, and 0.65 mg/kg) in order to begin to establish the dose characteristics of MOX in relation to 10E intake. Two-way ANOVA revealed a significant effect of MOX administration on ethanol intake when analyzed across time (pre-MOX, MOX, post-MOX) [F(2, 312) = 84.60, p < 0.001]. The analysis of the MOX doses (0.65 – 10mg) indicated that MOX significantly reduced ethanol intake in a dose-dependent manner in female mice (Figure 1b) [F(5, 312) = 4.299, p < 0.001]. The interaction between time and dose was significant [F(10, 312), 5.057, p < 0.001]. Bonferroni post-hoc comparisons between pre-MOX and MOX data indicated that 2.5 mg/kg MOX was the lowest dose that caused a maximum significant reduction in ethanol intake [~44% reduction, (t = 5.467, p < 0.001)]. For the MOX doses that significantly reduced 10E intake, we found that 10E intake returned to comparable pre-MOX ethanol intake levels within 1–2 days post-MOX injection (data not shown). The lowest dose of MOX tested (0.65 mg/kg) did not significantly reduce 10E intake compared to pre-MOX level. In addition, following saline habituation, further injections with saline alone did not significantly affect 10E intake (Figure 1b, dose 0.00 mg/kg). MOX administration did have significant impairment on food intake when analyzed across time (pre-MOX, MOX, post-MOX) [F(1, 242) = 84.85, p < 0.0001] and doses (0.65 – 10 mg/kg) [F(6, 242) = 9.710, p < 0.0001] (data not shown). The interaction between time and dose was significant [F(6, 242) = 25.33 p < 0.0001]. Bonferroni post-hoc comparisons between pre-MOX and MOX data revealed that only higher doses of MOX caused significant impairment on food intake [5 mg/kg (t = 5.918, p < 0.0001), 7.5 mg/kg (t = 8.776, p < 0.0001), 10 mg/kg (t=5.179, p < 0.0001)]. MOX (all doses tested, pre-MOX compared to MOX) did not cause significant changes in body weight (data not shown) or showed any signs of abnormal overt behaviors such as lack of movement with hunched postured and hurdling in corner, unresponsive to experimenter intervention, and piloerection of fur.
4.3. MOX decreased 10E intake in a dose-dependent manner in male mice
Extending our line of investigation, we also tested the utility of MOX using male C57BL/6 mice. Two-way ANOVA revealed a significant effect of MOX administration on ethanol intake when analyzed across time (pre-MOX, MOX, post-MOX) [F(1, 11) = 68.43, p < 0.0001]. The analysis of the MOX doses (1.25 – 7.5 mg/kg) indicated that MOX significantly reduced ethanol intake in a dose-dependent manner in male mice (Figure 1b) [F(4, 44) = 4.005, p < 0.01]. The interaction between time and dose was significant [F(4, 44), 6.434, p = 0.0004]. Bonferroni post-hoc comparisons between pre-MOX and MOX data indicated that 2.5 mg/kg MOX was the lowest dose that caused a maximum significant reduction in ethanol intake [~55% reduction, (t = 6.427, p < 0.0001)]. For the MOX doses that significantly reduced 10E intake, we found that 10E intake returned to comparable pre-MOX ethanol intake levels within 1–2 days post-MOX injection (data not shown). MOX administration did have significant impairment on food intake when analyzed across time (pre-MOX, MOX, post-MOX) [F(1, 11) = 24.75, p < 0.001] and doses (1.25 – 7.5 mg/kg) [F(4, 44) = 4.448, p < 0.01] (data not shown). The interaction between time and dose was significant [F(4, 44) = 4.700 p < 0.01]. Bonferroni post-hoc comparisons between pre-MOX and MOX data revealed that only higher doses of MOX caused significant impairment on food intake [5 mg/kg (t = 4.912, p < 0.0001), 7.5 mg/kg (t = 4.701, p < 0.001)]. MOX (all doses tested, pre-MOX compared to MOX) did not cause significant changes in body weight or showed any signs of abnormal overt behaviors.
4.4. Time course of the effect of MOX on ethanol intake in female mice
To gain insight to the pharmacokinetics of MOX distribution and its impact on ethanol intake, we evaluated the time course of the effect of MOX on hourly ethanol intake using a 24-h-two-bottle choice paradigm. Using this paradigm, we previously reported that the onset of significantly activity for ivermectin was approximately 9 h post-administration (Figure 2 inset) (Yardley et al., 2012). After stable 10E intake was obtained, either MOX (2.5 mg/kg, previously determined as the minimum effective dose) or saline was administered 1 h to female mice before the first reading. As expected, 10E intake increased significantly across time (hourly) [F(9,190) = 78.52, p < 0.001] and was significantly reduced following MOX treatment [F(1,190) = 36.33, p < 0.001] (Figure 2). The interaction between time and MOX dose was not significant. We conducted planned comparison which showed that ethanol intake was significantly decreased starting at 4 h after MOX administration (Figure 2).
4.5. Multiple day dosing of MOX administration reduced ethanol intake in female mice using a 24-h-two-bottle choice paradigm
Alcoholism is a chronic disorder. As such, an effective pharmacotherapy for AUD will mostly need to be taken chronically. To begin to investigate the utility of MOX for chronic use, we tested the effects of MOX (2.5 mg/kg) administered for 5 consecutive days in female mice using a 24-h-two-bottle choice paradigm. On the day prior to drug/vehicle treatment, there was no significant difference in average 10E intake for the MOX (12.26 g/kg) or control (12.68 g/kg) groups. We tested the effect of MOX versus saline, administered daily, on 10E intake over a 5-day period (Figure 3a). When analyzed across the 5-day treatment period, we found that MOX significantly reduced ethanol intake compared to the saline-injected control group [F(1,24) = 26.35, p < 0.001]. There was also a significant effect on 10E intake when analyzed across time (day) [F(4, 96) = 8.730, p < 0.001]. The interaction between treatment and time was not significant.
Figure 3. Daily administration of MOX (2.5 mg/kg x 5 days) significantly reduced 10E intake in female C57BL/6J mice using a 24-h-two-bottle choice paradigm.

Following habituation with saline injection and attaining stable drinking levels, MOX was administered for 5 consecutive days. Squares represent MOX and circles represent saline. a) MOX (2.5 mg/kg) significantly reduced 10E intake across the 5 treatment days. The effects of water intake, food intake, and weight are presented in panels, b, c, and d, respectively. Values represent mean ± SEM cumulative intake for MOX (18 mice) and saline (10 mice), ** p < 0.01, *** p < 0.001, # p < 0.0001 versus saline-treated group, Bonferroni’s post-hoc test.
MOX treatment did not significantly change either water or food intake between the two groups when analyzed across 5-day treatment period and time (day) (Figure 3b – 3c). There was a significant effect of MOX treatment [F(1, 24) = 24.4, p < 0.0001] and time (day) [F(4, 96) = 5.951, p < 0.001] on change in body weight with no significant interaction between treatment and time (Figure 3d).
4.6. Multiple day dosing of MOX administration reduced ethanol intake in female mice using a drinking-in-the-dark paradigm
We extended our multiple dosing of MOX administration using a modified version of the DID paradigm in female mice (Lowery et al., 2010; Neasta et al., 2010; Rhodes et al., 2005). One day prior to treatment, there was no significant difference in average 20E intake for the MOX (2.91 g/kg) and control (3.02 g/kg) groups. We tested the effect of MOX (2.5 mg/kg) versus saline, administered daily, 4 h prior to the start of the drinking sessions on 20E intake over a 5-day period (Figure 4a). MOX administration consistently reduced 20E intake across the testing period, and the reduction was significant when analyzed across time (day) [F(3,27) = 8.862, p < 0.001] but not across the 5-day treatment period. There was no interaction between treatment and time.
Figure 4. Daily administration of MOX (2.5 mg/kg x 5 days) reduced 20E intake in female C57BL/6J mice using a drinking-in-the-dark (DID) paradigm.

Following habituation with saline injection and attaining stable drinking levels, MOX was administered for 5 consecutive days. Squares represent MOX and circles represent saline. a) MOX (2.5 mg/kg) consistently reduced 20E intake across the 5 treatment days. The effects of food intake and weight are presented in panels, b and c, respectively. Values represent mean ± SEM cumulative intake for MOX (18 mice) and saline (10 mice).
MOX treatment did not cause any significant changes in food intake between the two groups (Figure 4b) when analyzed across the 5-day treatment period and time (day). We did observe a significant effect of MOX treatment on body weight [F(1, 24) = 12.01, p < 0.001) but no significant effect of time (day) on change in body weight (Figure 4c), and there was no interaction between treatment and time.
4.7 MOX positively modulated ATP-gated P2X4Rs and antagonized the inhibitory effects of ethanol on P2X4R function
We investigated the effects of 0.5 μM and 1 μM MOX alone, and in combination with behaviorally relevant concentrations of ethanol (25 and 50 mM) on ATP-induced currents in P2X4Rs in vitro. In agreement with previous studies (Asatryan et al., 2010, 2014; Davies et al., 2005, 2010; Popova et al., 2010), ethanol (25 and 50 mM) significantly inhibited ATP-gated P2X4R currents (Figure 5a – 5b). In the presence of ATP, MOX (0.5 and 1 μM) produced a comparable degree of potentiation of P2X4R activity (Figure 5a – 5b). When tested in the presence of ethanol, 0.5 μM MOX significantly reduced the inhibitory effect caused by 25 mM (p = 0.003) but not 50 mM ethanol on ATP-gated P2X4R activated currents (Figure 5a). In contrast, 1 μM MOX eliminated the inhibitory effect of ethanol at both concentrations, 25 (p = 0.0028) and 50 mM (p = 0.029) (Figure 4b).
Figure 5. MOX (0.5 and 1 μM) antagonized the inhibitory effects of ethanol in P2X4Rs.

Exposure to a) 0.5 μM MOX and b) 1 μM MOX potentiates P2X4Rs and significantly eliminated the inhibitory effect of 25 and 50 mM ethanol on EC5 ATP-gated currents in P2X4Rs. Values represent mean ± SEM for 4 to 6 oocytes per data point, * p < 0.05, ** p < 0.01 versus respective control, two-tailed unpaired Student’s t-test.
4.8. MOX positively modulated GABAAR activity
We also tested the effects of MOX on GABAARs since we, and others have previously reported that ivermectin and other related avermectins have significant GABAergic activity (Asatryan et al., 2014; Janko et al., 2013; Menez et al., 2012). We used α1β2γ2 GABAARs due to their predominant expression in the CNS in mammals (Sigel and Steinmann, 2012). As illustrated in Figure 6, we found that MOX and ivermectin each significantly increased GABAAR function. There was no significant difference between the extent of potentiation of GABAAR function between MOX and ivermectin at 0.1 μM. However, the degree of MOX potentiation was significantly less (compared to ivermectin) when tested at higher concentrations [0.5 μM (p = 0.0072 and 1 μM (p = 0.00092)].
Figure 6. MOX (0.1, 0.5, and 1 μM) has a weaker modulatory activity in GABAARs than ivermectin.
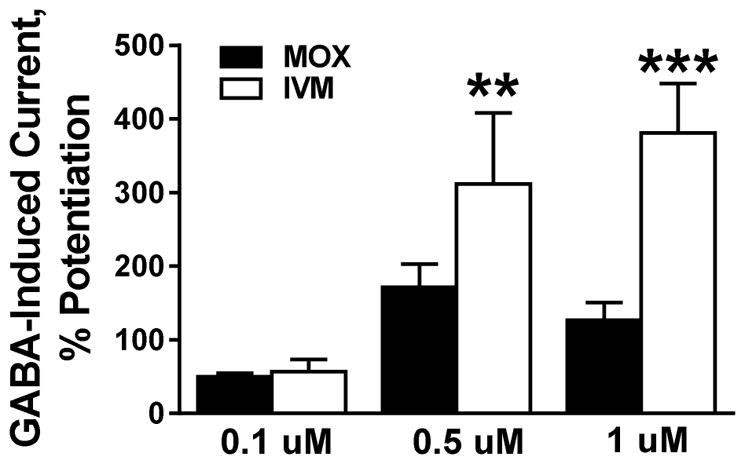
At low 0.1 μM concentration, MOX potentiated GABAARs to similar extent as ivermectin. At 0.5 μM and above, the effect of MOX was significantly smaller compared to that of ivermectin. Values represent mean ± SEM for 4 to 12 oocytes per data point, ** p < 0.01, *** p < 0.001 versus respective control, two-tailed unpaired Student’s t-test.
5. Discussion
The present study was the first investigation to test the utility of MOX as a pharmacotherapy for AUD. Several key findings came from this work. Testing the hypothesis that administration of MOX can reduce ethanol intake in male and female mice, we found that acute and repeated administration of MOX significantly reduced ethanol intake using two well-established drinking paradigms. Using a Xenopus oocyte expression system, we found that MOX significantly reduced the inhibitory effects of ethanol on P2X4Rs suggesting that at least a portion of MOX’s anti-ethanol effects may be linked to activity on P2X4Rs. Using a 5-day MOX administration treatment regimen with a 24-h-two-bottle choice paradigm, we found that the reduction in ethanol intake was significant and remained consistent over the 5-day period (with an average reduction of approximately 40% compared to saline-injected controls). Importantly, no significant decrease in food consumption or weight loss, or other abnormalities in overt behavior (e.g., lack of movement and constant hurdling in corner, unresponsive to experimenter intervention, and piloerection of fur) were noted. These findings are well aligned with our previous work demonstrating that chronic administration of ivermectin was efficacious in reducing ethanol intake and well tolerated (Yardley et al., 2012, 2014, 2015). Finally, MOX also consistently reduced ethanol intake when tested using a 5-day DID paradigm (average reduction approximately 30%).
Several reports have suggested that different rodent drinking models involve overlapping and distinct neurobiological mechanisms (Crabbe et al., 2011; McBride and Li, 1998). MOX has been purported to act several different neurotransmitter targets that can regulate ethanol behavior through multiple mechanisms (Menez et al., 2012, Wolstenholme and Rogers, 2005; Yamaguchi et al., 2012). Thus, it is possible that a higher dose of MOX may exert a different degree of activity on a single or combination of these targets leading to more pronounced reduction in ethanol intake using the DID paradigm. Collectively, these results support the development of MOX as a novel pharmacotherapy for the treatment of AUD and illustrate the potential for P2X4Rs as a novel target for AUD drug development.
The fact that the administration of MOX as illustrated in our multi-day studies did not result in any consistent, significant effect on water or food intake suggests the drug is well tolerated. As shown, we did find that MOX did cause some reduction in weight and the change remained stable over the 5-day period. We do not know the reason for this change, but it could be due to a loss of caloric intake linked to the decreased ethanol consumption that was caused by MOX. Notably, all animals appeared healthy and remained alive at the end of the study, suggesting that MOX does not have any unexpected interactions with ethanol that could lead to lethality or negative changes in behavior.
In the current investigation, we found that the reduction in ethanol intake reached significance approximately 4 h after MOX administration. This represents a significant improvement in onset of effect as compared to the 9 h window necessary for initial onset of ivermectin activity (Yardley et al., 2012). This finding suggests that MOX can reach effective therapeutic (anti-alcohol effect) brain concentrations more rapidly versus ivermectin and is consistent with previous findings suggesting that MOX has improved BBB penetration compared to ivermectin (Prichard et al., 2012, Kiki-Mvouaka et al., 2010).
The faster onset to the initial reduction of ethanol intake may be explained by the differences in structural features and physiochemical properties of ivermectin and MOX. Ivermectin and MOX share a common macrocyclic lactone ring and are distinguishable by specific substituents at the C13, C23, and C25 position (Figure 7). These substituents play a role in influencing the lipophilicity and affinity for P-gp transporters. The higher lipophilicity of MOX compared to ivermectin (log PMOX = 6, Pivermectin = 4.8) is consistent with its higher entrance into the brain, greater accumulation in adipose tissue, and longer retention time in the organism (Baoliang et al., 2006; Lanusse et al., 1997; Prichard et al., 2012). In addition, MOX has been shown to have a weaker affinity for P-gp compared to ivermectin and other avermectins based on structural and biochemical studies. The disaccharide sugar moiety found on ivermectin is absent on MOX. This moiety is thought to govern the affinity for P-gp (Lespine et al., 2007).
Figure 7. Structures of (a) ivermectin and (b) MOX.

Major structural differences are noted. C13: ivermectin contains a disaccharide while MOX is protonated; C23: MOX has a methoxime; C25: ivermectin is a mixture of C25-ethyl (~10%) or C25-methyl (~90%) groups while MOX has a dimethyl-butyl substituent.
Interestingly, despite reaching higher brain levels, MOX is predicted to have a better neurotoxicity profile when compared to ivermectin, which is thought to be due to 1) the differential transport across the blood brain barrier (BBB), with MOX being less dependent on P-gp for removal from the brain and less likely to accumulate due to a deficiency in P-gp function or drug-drug interaction arising from concurrent medications that may also act as P-gp substrates (Kiki-Mvouaka et al., 2010; Menez et al., 2012), and 2) the differential interaction with GABAARs, with MOX exhibiting lower activity at these receptors (Janko et al., 2012; Menez et al., 2012). In agreement with the latter, our results also indicated that the degree of MOX potentiation of GABAARs was significantly less as compared to ivermectin. At higher concentrations, we found that ivermectin continued to increase the degree of GABAARs activity whereas the effects of MOX quickly reached a plateau. This result, coupled with the aforementioned benefits of MOX (weak P-gp target; better BBB), suggests that chronic use of MOX as a long-term treatment for AUD should be more favorable as compared to ivermectin because there should be less complications arising from potential brain accumulation and/or over-stimulation of GABAARs that can lead to CNS depression and potentially coma. (Prichard et al., 2012; Balayssac et al., 2005).
Importantly, MOX is currently undergoing clinical development as an alternative to ivermectin for treating the parasite Onchocerca volvulus, and to date, no significant clinical abnormalities or serious adverse events have been reported over for these investigations (Cotreau et al., 2003; Korth-Bradley et al., 2012). The faster onset time, robust efficacy and favorable safety profile continue to support the development of MOX as a novel pharmacotherapy for the treatment of AUD.
In addition to our in vivo results, as reported above, we found that MOX positively modulated ATP-gated currents in P2X4Rs and antagonized the inhibitory effects of ethanol on P2X4R function. This is the first evidence demonstrating that a compound from the milbermycin subfamily of macrocyclic lactones can act on P2X4Rs in vitro. This finding is in good agreement with our earlier investigations where we demonstrated that both ivermectin and abamectin significantly antagonized the inhibitory effects of ethanol on P2X4R function (Asatryan et al., 2014).
In addition to activity on P2X4Rs, we (in this study) and others report that MOX can also act on other brain targets including GABAARs, glycine, and nAChRs (Menez et al., 2012, Wolstenholme et al., 2005; Yamaguchi et al., 2012). Notably, all of these receptors have been linked to the modulation of mesolimbic dopamine activity and regulation of ethanol behavior (Davies, 2003; Xiao et al., 2008). Thus, it is likely that the reduction of ethanol intake, by MOX, as presented in the current investigation reflects a cumulative effect of MOX activity on several different classes of receptors including P2X4Rs. Additional studies are necessary before definite conclusions can be drawn.
In that this was the first investigation focusing on MOX, several limitations should be noted. First, the majority of the results presented here were conducted using female mice and we did not monitor for the effects of estrous cycle, which could potentially confound the interpretation of the results. However, in our dose-response study we used both male and female mice (Figures 1b – 1c). As presented, there was a significant reduction of ethanol intake by MOX in both male and female mice and the degree of reduction was similar for both groups. In addition, we also utilized a saline-treated control group in both of the 5-day investigations using female mice (24-h-bottle-bottle-choice and DID studies), it is unlikely that the differences in 10E and 20E intake between the MOX and saline group could be attributed to the estrous cycle. This conclusion is similar to previous work where no systematic changes in ethanol intake across weeks of baseline consumption in female C57BL/6 mice were observed (Ford et al., 2008). Collectively, these results suggests that the estrous cycle is unlikely to have an impact on the anti-alcohol effects of MOX and further supports the utility of MOX as a pharmacotherapy for AUD in both male and female. In future investigations, we plan to continue evaluating the effects of MOX administration on ethanol intake in both male and female mice using long-term drinking paradigms.
Second, we did not test other tastants such as sucrose or quinine in this study. Previously, we reported that ivermectin produced a significant reduction in 24 h saccharin consumption, but did not significantly alter operant sucrose self-administration (Yardley et al., 2012). Based on this finding, it would not be unreasonable to predict that MOX may also act on other tastants, but this should not reduce the utility of MOX for its anti-alcohol effects. Third, in the DID investigation, we did not measure BECs. As such, we cannot definitively conclude that the BEC levels achieved in our DID study reached binge-like drinking levels. However, the levels of ethanol intake achieved in our study were similar to levels of ethanol intake reported by others where BECs were recorded (Wilcox et al., 2014; Rhodes et al., 2005).
6. Conclusion
The present findings support the development of MOX as a novel pharmacotherapy for the treatment of AUD. We presented solid evidence showing that both acute and repeated administration of MOX can reduce ethanol intake across different models of self-administered drinking paradigms. Importantly, we observed no signs of overt toxicity and all animals remained alive at the end of the study. Of note, the safety and initial efficacy of co-administration of a single 30-mg dose of ivermectin and intravenous alcohol infusion in alcoholic patients was recently tested. In this study, ivermectin (30 mg) was found to be safe and well tolerated where the number and severity of reported adverse effects were low and did not differ from the placebo session (Roche et al., 2016). Although ivermectin did not differ from placebo in regards to reducing alcohol cue-induced craving or basal alcohol craving, the study represented an important first step in developing this class of molecules as pharmacotherapies for AUD. In that MOX is currently being developed for use in humans as an alternative for ivermectin as an anti-parasitic coupled with the better pharmacokinetics and margin of safety (as compared to ivermectin) once fully approved for human use MOX should have the potential to be repurposed and rapidly advanced to the clinic for the treatment and/or prevention of AUD.
Highlights.
MOX reduces ethanol intake in both male and female mice without causing any overt toxicity.
MOX potentiates ATP-gated P2X4R function and antagonize the inhibitory effects of ethanol on the receptor.
At higher doses, MOX has minimal potentiating effect on GABAARs compared to ivermectin.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) AA022448 (D.L.D.), and the USC School of Pharmacy. This work was conducted as partial fulfillment of the requirements for the Ph.D. degree in Clinical and Experimental Therapeutics, School of Pharmacy, University of Southern California (N.H.). Results presented in this manuscript were partially presented at the 45th Annual Meeting of the Society of Neuroscience, Chicago, IL, October 2015, and at the 38th Annual Meeting of the Research Society on Alcoholism, San Antonio, TX, June 2015. The authors want to thank USC Master’s candidate Ruowei Liu for her assistance with the electrophysiology experiments, and USC undergraduates, Dustin Lieu and Jamie Thuy, for their assistance with the animal studies.
Footnotes
Conflict of interest
DLD and LA are inventors on a patent for the repurposing of ivermectin and related avermectins for the treatment of alcohol-use disorders. The authors have no other conflicts of interest and are entirely responsible for the scientific content of the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asatryan L, Popova M, Woodward JJ, King BF, Alkana RL, Davies DL. Roles of ectodomain and transmembrane regions in ethanol and agonist action in purinergic P2X2 and P2X3 receptors. Neuropharmacology. 2008;55:835–843. doi: 10.1016/j.neuropharm.2008.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Popova M, Perkins D, Trudell JR, Alkana RL, Davies DL. Ivermectin antagonizes ethanol inhibition in purinergic P2X4 receptors. J Pharmacol Exp Ther. 2010;334:720–728. doi: 10.1124/jpet.110.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asatryan L, Yardley MM, Khoja S, Trudell JR, Huynh N, Louie SG, Petasis NA, Alkana RL, Davies DL. Avermectins differentially affect ethanol intake and receptor function: implications for developing new therapeutics for alcohol use disorders. Int J Neuropsychopharmacol. 2014;17:907–916. doi: 10.1017/S1461145713001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balayssac D, Authier N, Cayre A, Coudore F. Does inhibition of P-glycoprotein lead to drug-drug interactions? Toxicol Lett. 2005;156:319–329. doi: 10.1016/j.toxlet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Baoliang P, Yuwan W, Zhende P, Lifschitz AL, Ming W. Pharmacokinetics of eprinomectin in plasma and milk following subcutaneous administration to lactating dairy cattle. Vet Res Commun. 2006;30:263–270. doi: 10.1007/s11259-006-3230-7. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Crabbe JC, Young ER. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology (Berl) 1993;112:503–510. doi: 10.1007/BF02244901. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Ozburn AR, Walker D, Ahmed S, Belknap JK, Harris RA. Hybrid mice as genetic models of high alcohol consumption. Behav Genet. 2010;40:93–110. doi: 10.1007/s10519-009-9298-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolato M, Yardley MM, Khoja S, Godar SC, Asatryan L, Finn DA, Alkana RL, Louie SG, Davies DL. Pharmacological insights into the role of P2X4 receptors in behavioural regulation: lessons from ivermectin. Int J Neuropsychopharmacol. 2013;16:1059–1070. doi: 10.1017/S1461145712000909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchery EE, Harwood HJ, Sacks JJ, Simon CJ, Brewer RD. Economic Costs of excessive alcohol consumption in the U.S. Am J Prevent Med. 2011;41:516–524. doi: 10.1016/j.amepre.2011.06.045. [DOI] [PubMed] [Google Scholar]
- Cotreau MM, Warren S, Ryan JL, Fleckenstein L, Vanapalli SR, Brown KR, Rock D, Chen CY, Schwertschlag US. The antiparasitic moxidectin: safety, tolerability, and pharmacokinetics in humans. J Clin Pharmacol. 2003;43:1108–1115. doi: 10.1177/0091270003257456. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Harris RA, Koob GF. Preclinical studies of alcohol binge-drinking. Ann N Y Acad Sci. 2011;1216:24–40. doi: 10.1111/j.1749-6632.2010.05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M. The role of GABAA receptors in mediating the effects of alcohol in the central nervous system. J Psychiatry Neurosci. 2003;28:263–274. [PMC free article] [PubMed] [Google Scholar]
- Davies DL, Machu TK, Guo Y, Alkana RL. Ethanol sensitivity in ATP-gated P2X receptors is subunit dependent. Alcohol Clin Exp Res. 2002;26:773–778. [PubMed] [Google Scholar]
- Davies DL, Kochegarov AA, Kuo ST, Kulkarni AA, Woodward JJ, King BF, Alkana RL. Ethanol differentially affects ATP-gated P2X(3) and P2X(4) receptor subtypes expressed in Xenopus oocytes. Neuropharmacology. 2005;49:243–253. doi: 10.1016/j.neuropharm.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Dawson GR, Wafford KA, Smith A, Marshall GR, Bayley PJ, Schaeffer JM, Meinke PT, McKernan RM. Anticonvulsant and adverse effects of avermectin analogs in mice are mediated through the gamma-aminobutyric acid A receptor. J Pharmacol Exp Ther. 2000;295:1051–1060. [PubMed] [Google Scholar]
- Edwards G. Ivermectin: does P-glycoprotein play a role in neurotoxicity. Filaria J. 2003;24:1–8. doi: 10.1186/1475-2883-2-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MM, Beckley EH, Nickel JD, Eddy S, Finn DA. Ethanol intake patterns in female mice: influence of allopregnanolone and the inhibition of its synthesis. Drug Alcohol Depend. 2008;97:73–85. doi: 10.1016/j.drugalcdep.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Dawson DA, Stinson FS, Chou P, Dufour MC, Pickering RP. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004;74:223–234. doi: 10.1016/j.drugalcdep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Guzzo CA, Furtek CL, Porras AG, Chen C, Tipping R, Clineschmidt CM, Sciberras DG, Hsieh JY, Lasseter KC. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J Clin Pharmacol. 2002;42:1122–1133. doi: 10.1177/009127002401382731. [DOI] [PubMed] [Google Scholar]
- Harris AH, Kivlahan DR, Bowe T, Humphreys KN. Pharmacotherapy of alcohol use disorders in the veterans health administration. Psychiatr Serv. 2010;61:392–398. doi: 10.1176/ps.2010.61.4.392. [DOI] [PubMed] [Google Scholar]
- Harwood HJ, Lewin G. Report prepared for the National Institute on Alcohol Abuse and Alcoholism. 2000. Updating Estimates of the Economic Costs of Alcohol Abuse in the United States: Estimates, Update Methods, and Data. [Google Scholar]
- Heilig M, Egli M. Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharmacol Ther. 2006;111:855–876. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Janko C, Geyer J. Moxidectin has a lower neurotoxic potential but comparable brain penetration in P-glycoprotein-deficient CF-1 mice compared to ivermectin. J Vet Pharmacol Ther. 2013;36:275–284. doi: 10.1111/j.1365-2885.2012.01424.x. [DOI] [PubMed] [Google Scholar]
- Johnson BA. Medication treatment of different types of alcoholism. Am J Psychiatr. 2010;167:630–639. doi: 10.1176/appi.ajp.2010.08101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Rosenthal N, Capece JA, Wiegand F, Mao L, Beyers K, McKay A, Ait-Daoud N, Anton RF, Ciraulo DA, Kranzler HR, Mann K, O’Malley SS, Swift R. Topiramate for treating alcohol dependence: a randomized controlled trial. J Am Med Assoc. 2007;298:1641–1651. doi: 10.1001/jama.298.14.1641. [DOI] [PubMed] [Google Scholar]
- Kiki-Mvouaka S, Menez C, Borin C, Lyazrhi F, Foucaud-Vignault M, Dupuy J, Collet X, Alvinerie M, Lespine A. Role of P-glycoprotein in the disposition of macrocyclic lactones: A comparison between ivermectin, eprinomectin, and moxidectin in mice. Drug Metab Dispos. 2010;38:573–80. doi: 10.1124/dmd.109.030700. [DOI] [PubMed] [Google Scholar]
- Korth-Bradley JM, Parks V, Patat A, Matschke K, Mayer P, Fleckenstein L. Relative bioavailability of liquid and tablet formulations of the antiparasitic moxidectin. Clin Pharmacol in Drug Deve. 2012;1:2–37. doi: 10.1177/2160763X11432508. [DOI] [PubMed] [Google Scholar]
- Lanusse C, Lifschitz A, Virkel G, Alvarez L, Sánchez S, Sutra JF, Galtier P, Alvinerie M. Comparative plasma disposition kinetics of ivermectin, moxidectin and doramectin in cattle. J Vet Pharmacol Ther. 1997;20:91–99. doi: 10.1046/j.1365-2885.1997.00825.x. [DOI] [PubMed] [Google Scholar]
- Lespine A, Martin S, Dupuy J, Roulet A, Pineau T, Orlowski S, Alvinerie M. Interaction of macrocyclic lactones with P-glycoprotein: structure–affinity relationship. Eur J Pharm Sci. 2007;30:84–94. doi: 10.1016/j.ejps.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Litten RZ, Egli M, Heilig M, Cui C, Fertig JB, Ryan ML, Falk DE, Moss H, Huebner R, Noronha A. Medications development to treat alcohol dependence: a vision for the next decade. Addict Biol. 2012;17:513–527. doi: 10.1111/j.1369-1600.2012.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery EG, Spanos M, Navarro M, Lyons AM, Hodge CW, Thiele TE. CRF-1 antagonist and CRF-2 agonist decrease binge-like ethanol drinking in C57BL/6J mice independent of the HPA axis. Neuropsychopharmacology. 2010;35:1241–1252. doi: 10.1038/npp.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride WJ, Li TK. Animal models of alcoholism: neurobiology of high alcohol-drinking behavior in rodents. Crit Rev Neurbiol. 1998;12:339–369. doi: 10.1615/critrevneurobiol.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- Menez C, Sutra JF, Prichard R, Lespine A. Relative neurotoxicity of ivermectin and moxidectin in Mdr1ab (−/−) mice and effects on mammalian GABA(A) channel activity. PLoS Negl Trop Dis. 2012;6:e1883. doi: 10.1371/journal.pntd.0001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neasta J, Ben Hamida S, Yowell Q, Carnicella S, Ron D. Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc Natl Acad Sci USA. 2010;16:20093–20098. doi: 10.1073/pnas.1005554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura S. Ivermectin: 25 years and still going strong. Int J Antimicrob Agents. 2008;31:91–98. doi: 10.1016/j.ijantimicag.2007.08.023. [DOI] [PubMed] [Google Scholar]
- Popova M, Asatryan L, Ostrovskaya O, Wyatt LR, Li K, Alkana RL, Davies DL. A point mutation in the ectodomain-transmembrane 2 interface eliminates the inhibitory effects of ethanol in P2X4 receptors. J Neurochem. 2010;112:307–317. doi: 10.1111/j.1471-4159.2009.06460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard R, Menez C, Lespine A. Moxidectin and the avermectins: Consanguinity but not identity. Int J Parasitol Drugs Drug Resist. 2012;2:143–153. doi: 10.1016/j.ijpddr.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;31:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Roche DJ, Yardley MM, Lunny KF, Louie SG, Davies DL, Miotto K, Ray LA. A pilot study of the safety and initial efficacy of ivermectin for the treatment of alcohol use disorder. Alcohol Clin Exp Res. 2016;40:1312–1320. doi: 10.1111/acer.13064. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL, Lynch JW. Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J Biol Chem. 2001;276:12556–12564. doi: 10.1074/jbc.M011264200. [DOI] [PubMed] [Google Scholar]
- Sigel E, Steinmann ME. Structure, function, and modulation of GABAA receptors. J Bio Chem. 2012;287:40224–31. doi: 10.1074/jbc.R112.386664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinosa HS, Stilck SR, Bernardi MM. Possible anxiolytic effects of ivermectin in rats. Vet Res Commun. 2002;26:309–321. doi: 10.1023/a:1016094726033. [DOI] [PubMed] [Google Scholar]
- Steensland P, Simms JA, Holgate J, Richards JK, Bartlett SE. Varenicline, an {alpha}4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci USA. 2007;104:12518–12523. doi: 10.1073/pnas.0705368104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. National Survey on Drug Use and Health (NSDUH). Table 2.46B—Alcohol Use, Binge Alcohol Use, and Heavy Alcohol Use in the Past Month among Persons Aged 18 or Older, by Demographic Characteristics: Percentages, 2012 and 2013. 2013 Available: http://www.samhsa.gov/data/sites/default/files/NSDUH-DetTabsPDFWHTML2013/Web/HTML/NSDUH-DetTabsSect2peTabs43to84-2013.htm#tab2.46b.
- Wilcox MV, Cuzon Carlson VC, Sherazee N, Sprow GM, Bock R, Thiele TE, Lovinger DM, Alvarez VA. Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology. 2014;39:579–594. doi: 10.1038/npp.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme AJ, Rogers AT. Glutamate-gated chloride channels and the mode of action of the avermectin/milbemycin anthelmintics. Parasitology. 2005;131:85–95. doi: 10.1017/S0031182005008218. [DOI] [PubMed] [Google Scholar]
- Wyatt LR, Finn DA, Khoja S, Yardley MM, Asatryan L, Alkana RL, Davies DL. Contribution of P2X4 receptors to ethanol intake in male C57BL/6 mice. Neurochem Res. 2014;39:1127–1139. doi: 10.1007/s11064-014-1271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Shao XM, Olive MF, Griffin WC, Li KY, Krnjević K, Zhou C, Ye JH. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology. 2009;34:307–318. doi: 10.1038/npp.2008.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Sawa Y, Matsuda K, Ozoe F, Ozoe Y. Amino acid residues of both the extracellular and transmembrane domains influence binding of the antiparasitic agent milbemycin to Haemonchus contortus AVR-14B glutamate-gated chloride channels. Biochem Biophys Res Commun. 2012;419:562–566. doi: 10.1016/j.bbrc.2012.02.062. [DOI] [PubMed] [Google Scholar]
- Yardley MM, Wyatt L, Khoja S, Asatryan L, Ramaker MJ, Finn DA, Alkana RL, Huynh N, Louie SG, Petasis NA, Bortolato M, Davies DL. Ivermectin reduces alcohol intake and preference in mice. Neuropharmacology. 2012;63:190–201. doi: 10.1016/j.neuropharm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley MM, Neely M, Huynh N, Asatryan L, Louie SG, Alkana RL, Davies DL. Multiday administration of ivermectin is effective in reducing alcohol intake in mice at doses shown to be safe in humans. Neuroreport. 2014;13:1018–1023. doi: 10.1097/WNR.0000000000000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley MM, Huynh N, Rodgers KE, Alkana RL, Davies DL. Oral delivery of ivermectin using a fast dissolving oral film: Implications for repurposing ivermectin as a pharmacotherapy for alcohol use disorder. Alcohol. 2015;49:553–559. doi: 10.1016/j.alcohol.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama N, Crabbe JC, Ford MM, Murillo A, Finn DA. Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol. 2008;42:149–160. doi: 10.1016/j.alcohol.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]