

Abstract
Antibiotic-resistant bacterial infections have seen a marked increase in recent years, while antibiotic discovery has waned. Resistance-modifying agents (RMA) offer an intriguing alternative strategy to fight against resistant bacteria. Here we report the discovery, antibiotic profiling, and structure-activity relationships of a novel class of RMAs, tetracyclic indolines. These selectively potentiate β-lactam antibiotics in methicillin-resistant Staphylococcus aureus (MRSA) without antibacterial or β-lactamase inhibitory activity on their own. The most potent analogue, 6a, showed strong potentiation of amoxicillin/clavulanic acid in a variety of hospital-acquired and community-acquired MRSA strains with low mammalian toxicity. These compounds may be further developed to extend the clinic life span of β-lactam antibiotics.
Keywords: β-lactam, antibiotic resistance, methicillin-resistant Staphylococcus aureus, resistance-modifying agent, structure-activity relationship, indoline alkaloids
Graphical Abstract
Introduction
Antibiotics revolutionized medicine in the twentieth century not only by saving countless lives from succumbing to infectious diseases, but also by facilitating progressions in medicine, such as dialysis and organ transplantation [1]. The first of these “miracle drugs” was penicillin, which was discovered by Alexander Fleming in 1928. However, as predicted by Fleming himself [2], penicillin resistance arose; β-lactamase-mediated resistance was observed in Staphylococcus aureus in 1947, just four years after mass production of penicillin. In the light of this development, methicillin, a semi-synthetic penicillin analogue that was resistant to β-lactamase inactivation, came into favor. However, it was not long before S. aureus developed resistance to methicillin through expression of an alternative penicillin-binding protein, PBP2a, which can catalyze the transpeptidation step of cell wall synthesis in the presence of β-lactamases [3]. Multi-drug resistant bacteria such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE), and drug-resistant Escherichia coli, are continuing to emerge. Notably, MRSA is the most frequently identified resistant pathogen in US hospitals [4, 5]. In addition to potential loss of life, antibiotic resistant bacteria also present a heavy economic burden [6, 7]. These issues illustrate the necessity for continuous discovery of antibiotic classes with novel structures and mechanisms of action. However, very few new classes of antibiotics have been brought to market in the past 50 years, pointing to the need for novel approaches to antibiotic discovery [8, 9].
An alternative strategy to combat bacterial resistance is the use of resistance-modifying agents (RMAs) in combination with established clinical antibiotics [10]. Unlike traditional antibacterials, RMAs do not directly kill or inhibit the growth of bacteria, but target bacterial non-essential genes/gene products [11–19] (e.g., quorum sensing systems [20–22], two-component system [23–27]). These are often metabolic burdens for bacteria and do not directly affect bacterial growth; hence, they pose no or little selective pressure on bacteria. The chance of developing resistance to this strategy is smaller than for conventional antibacterials [28]. For example, clavulanic acid is a β-lactamase inhibitor that was isolated from Streptococcus clavuligerus in 1972 [29]. Although lacking antibacterial activity itself, this molecule contains a β-lactam core and binds irreversibly to the active site of serine β-lactamases, thus deactivating the enzyme. When used in conjunction with β-lactam antibiotics (e.g., amoxicillin), it inhibits the breakdown of the antibiotic [29]. Although the synergistic effect of clavulanic acid is effective against serine β-lactamases, clavulanic acid is ineffective against metallo-β-lactamase-producing bacteria and does nothing to combat β-lactam resistance mediated by PBP2a, a prominent resistance mechanism in MRSA [30]. While research is underway for novel RMAs targeting bacterial growth pathways, such as cell wall biosynthesis [31], and other bacterial resistance mechanisms, such as efflux pumps [32], β-lactamase inhibitors remain the only class of RMAs in clinical use.
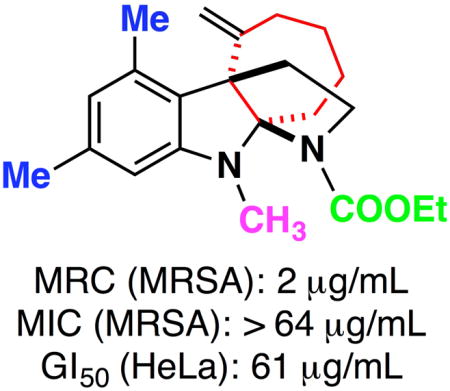
Recently, our group reported a series of novel anti-MRSA compounds, which were discovered by synthesizing and screening a highly diverse collection of polycyclic indole derivatives [33–38]. Further biological profiling studies showed that some hits from the screens, such as the ring-opened fused indoline Of1 (1, Figure 1) and tetracyclic indolenine 2, selectively re-sensitizes MRSA to β-lactam antibiotics [33]. Aza-tricyclic indoline 3 (Figure 1) was developed to optimize the physiochemical properties Of1 [39]. Herein, we report the antibiotic profiling and structure-activity relationship (SAR) studies of alkylated fused indoline Kf18 (4, Figure 1), another hit compound from our previous screens [33], with a distinct tetracyclic indoline scaffold. The SAR study of this compound shows promising developments in discovering novel RMAs for combatting epidemic of antibiotic resistance.
Figure 1.

Structures of a series of polycyclic indole derivatives that potentiate β-lactams in MRSA.
Result and Discussion
Antibiotic profiling of Kf18 (4)
To assess the scope of Kf18 to potentiate different classes of antibiotics, we used a nosocomial, multi-drug resistant MRSA strain, ATCC BAA-44, which was also used in our primary screen to identify methicillin potentiators [33]. The minimum inhibitory concentrations (MICs) of five β-lactam antibiotics (methicillin, amoxicillin/clavulanic acid, cefazolin, meropenem, and oxacillin) were determined in the absence of Kf18 and with the addition of 20 μM of Kf18 (entries 1–5, Table 1). From this experiment we confirmed that BAA-44 is highly resistant to all β-lactam antibiotics tested, and that Kf18 can significantly potentiate all five β-lactams. In addition to β-lactams, strain BAA-44 is also resistant to a number of other classes of antibiotics, such as tetracycline, ciprofloxacin, and clindamycin. The MICs of these antibiotics in the presence and absence of Kf18 were also tested using our established protocols [33, 35, 38]. As shown in Table 1 (entries 6–12), Kf18 does not potentiate these classes of antibiotics. Furthermore, the MIC of Kf18 in ATCC BAA-44 alone was >128 μg/mL, the highest concentration tested. These results suggested that Kf18 is a selective potentiator of β-lactam antibiotics in MRSA with a novel tetracyclic indoline scaffold.
Table 1.
Tetracyclic indoline Kf18 (4) selectively potentiates β-lactam antibiotics in MRSA
| Entry | Antibiotic | MICa | MIC (+Kf18)a,b | Fold of potentiation |
|---|---|---|---|---|
| 1 | methicillin | 128 | 8 | 16 |
| 2 | amox/clav | 32 | 4 | 8 |
| 3 | cefazolin | 128 | 8 | 16 |
| 4 | meropenem | 32 | 2 | 16 |
| 5 | oxacillin | 64 | 8 | 8 |
| 6 | tetracycline | 32 | 16 | 2 |
| 7 | ciprofloxacin | 8 | 4 | 2 |
| 8 | clindamycin | 256 | 256 | 1 |
| 9 | erythromycin | 128 | 128 | 1 |
| 10 | gentamycin | 256 | 256 | 1 |
| 11 | streptomycin | 256 | 256 | 1 |
| 12 | daptomycin | 256 | 256 | 1 |
Minimum inhibitory concentration in μg/mL in MRSA strain ATCC BAA-44.
MIC value in the presence of 20 μM of Kf18.
In order to investigate the SAR of tetracyclic indoline Kf18, several series of structural analogues were synthesized and evaluated for their biological activities. Our evaluation included determining the minimum inhibitory concentrations (MICs) and the minimum re-sensitizing concentrations (MRCs) for two β-lactam antibiotics in two representative MRSA strains (i.e., ATCC BAA-44 and NRS-100 (a.k.a., COL)), as wells as assessing mammalian toxicity in human cervical adenocarcinoma (HeLa) cells. The two β-lactams used were cefazolin, a first-generation cephalosporin, and amox/clav (a.k.a., amoxicillin/clavulanic acid and Augmentin), an antibiotic/RMA combination. The MRC is defined as the minimum concentration of the test compound that, in combination with antibiotic, returned MRSA to below the Clinical Laboratory Standards Institutes (CLSI)-defined sensitive concentration [40]. The CLSI-defined sensitive concentrations used for our experiments were 8 μg/mL for cefazolin and 4 μg/mL for amox/clav (2:1 ratio).
SARs of the D-ring nitrogen functionality
The SAR studies began with the preparation of analogues with changes to the methyl carbamate affixed to the nitrogen of the D ring in Kf18 (Scheme 1). The methyl carbamate was removed in excellent yield by employing Me3SiI at an elevated temperature. The resulting amine 5 provided a facile entry point into a diverse array of analogues.
Scheme 1. Modifications of Kf18 (4).

Reagents and conditions: a) Me3SiI, toluene, 70 °C, 4 h; (b) for 6a, 6b, 6g, ClCOOEt, ClCOOBn, or ClSO2PhpCl, DIPEA, DMAP, DMF; for 6c, triphosgene, pyridine, CH2Cl2, tBuOH, Et3N; for 6d–f, Ac2O, (CF3CO)2O or succinic anhydride, DMAP, DIPEA, DMF, rt; for 6k, 1) N,N-di Boc-N-(trifluoromethane-sulfonyl)guani-dine, Et3N, ClCH2CH2Cl, 2) CH2Cl2, TFA; for 6h, benzyl isocyanate, Et3N, CH3CN; for 6i, ethyl isocyanate, DIPEA, CH2Cl2; for 6j, trichloroacetyl isocyanate, DIPEA, CH2Cl2.
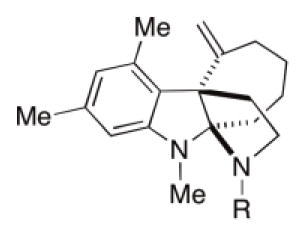
First, a few other carbamates were synthesized to explore the importance of methyl carbamate in Kf18. Ethyl and benzyl carbamates 6a and 6b were prepared from reacting 5 with their corresponding chloroformates. Tert-butyl carbamate, 6c, was obtained by sequential treatment of 5 with triphosgene and tert-butyl alcohol. Switching the methyl carbamate of Kf18 to the ethyl carbamate, 6a, improved the RMA activity by 8-fold for both cefazolin and amox/clav in both MRSA strains (entry 2, Table 2). 6a also showed significantly decreased mammalian toxicity. Substituting methyl with benzyl carbamate 6b, abolished activity (entry 3, Table 2). Further extension of the hydrophobic reach with tert-butyl carbamate 6c, only showed a modest increase in activity compared to Kf18 (entry 4, Table 2).
Table 2.
Biological Activities of Tetracyclic Indolines Kf18 (4), 5, and 6a-m
| ||||||
|---|---|---|---|---|---|---|
| Entry | Cmpd | R | MRC (cefazolin)a,b | MRC (amox/clav)a,c | MICa,b | GI50a,d |
| 1 | Kf18 | COOMe | 16 | 16 | >64 | 20 |
| 2 | 6a | COOEt | 2 | 2 | >64 | 61 |
| 3 | 6b | COOBn | >64 | 64 | >64 | >64 |
| 4 | 6c | COOtBu | 8 | 4 | >64 | 47 |
| 5 | 6d | COCH3 | 32 | 32 | >64 | 15 |
| 6 | 6e | COCF3 | >64 | >64 | >64 | >64 |
| 7 | 6f | COCH2CH2COOH | >64 | >64 | >64 | >64 |
| 8 | 6g | SO2PhpCl | >64 | >64 | >64 | >64 |
| 9 | 6h | CONHBn | >64 | >64 | >64 | >64 |
| 10 | 6i | CONHEt | >64 | >64 | >64 | 51 |
| 11 | 6j | CONH2 | 64 | 64 | >64 | 19 |
| 12 | 5 | H | 32 | 64 | >64 | 26 |
| 13 | 6k | C(NH)NH2 | 16 | 32 | >64 | 19 |
| 14 | 6l | Me | 8 | 16 | >64 | 20 |
| 15 | 6m | Et | 8 | 32 | >64 | 18 |
Numbers are in μg/mL.
MRSA NRS-100.
MRSA ATCC BAA-44.
HeLa cells.
Next, non-carbamate functional groups were installed on the D-ring nitrogen. Acyl analogues 6d, 6e, and 6f were obtained by acylating amine 5 using their corresponding anhydrides in the presence of base (entries 5–7, Table 2). Acetamide 6d has slightly lower RMA activity compared to Kf18; other amides, 6e and 6f, were devoid of the RMA activity. It is likely that the acetyl group of 6d is similar but smaller to the methyl carbamate of Kf18. This fits with a “goldilock’s” trend in regard to the D-ring group size, where the relative sizes are: MeCO < MeOCO < EtOCO < tBuOCO < BnOCO, with the most active compound falling in the center.
The para-bromophenylsulfonamide motif has been an important component of other RMAs discovered in our lab (1–3, Figure 1). Hence, analogue 6g was prepared. Surprisingly, this derivative displayed >4-fold loss in RMA activity compared to Kf18 (entry 8, Table 2). These results prompted our return to structural analogues with greater similarity to Kf18. In this light, urea is a similar functional group to carbamate in size, yet with higher polarity and metabolic stability. Based on this notion, several urea analogues, 6h-j, were synthesized by reacting 5 with commercially available isocyanates (entries 9–11, Table 2). Unsurprisingly, benzyl urea 6h shows no detectable RMA activity, which is reminiscent of benzyl carbamate 6b. However, the ethyl urea 6i was also devoid of the RMA activity. The only urea tested that showed marginal activity was the unsubstituted analogue 6j (entries 10 and 11, Table 2). This is likely a result of additional hydrogen bond donors on 6j, which can substitute for favorable hydrophobic interactions of the ethyl carbamate in 6a. The result observed for 6j, prompted us to prepare an analogue with a guanidine group, 6k. This functional group is similar to unsubstituted urea, but it has additional hydrogen bonding capabilities. Furthermore, this group has been crucial to activity in aza-tricyclic indoline RMAs (3, Figure 1). This compound showed clear improvement in RMA activity compared to 6j, but it did not surpass that of Kf18.
To expand on this idea, two analogues were synthesized without the hydrogen bond accepting capabilities found in the methyl and ethyl carbamates. To accomplish this, lithium aluminum hydride was used to reduce compounds 6a and 6d, which gave analogues 6l and 6m, respectively (entries 14 and 15, Table 2). In fact, when compared to Kf18, these analogues demonstrate slightly improved RMA activity and similar mammalian toxicity. This initial entry into the SAR of Kf18 demonstrated that analogue 6a with an ethyl carbamate group at the D-ring nitrogen position shows the most potent RMA activity with an 8-fold improvement in MRC activity and a 3-fold improvement in mammalian toxicity over the original screening hit Kf18. Clearly, the ethyl carbamate is ideal for this series of compounds, and it was therefore included along with the methyl carbamate in subsequent SAR studies.
Additional Modifications of Kf18
In the next phase of the SAR study, the aim was to determine the optimal substitutions on the tetracyclic indoline core. Specifically, several series of analogues were prepared with variations in the aromatic region, the indoline nitrogen, as well as the size of the C ring. This was accomplished by using our previously developed synthetic strategy with minor modifications [33]. In brief, the aromatic substitution is altered by using commercially available phenyl hydrazines with various substitutions and different cyclic imines, 8a or 8b, to prepare their corresponding indoles, 9 or 10, using a modified version of our one-pot three-component Fischer indole synthesis (Scheme 2). These indoles were then cyclized with a cationic gold catalyst to give the characteristic tetracyclic indoline core structure. The aniline nitrogen of cyclized products 11 and 12 could then be alkylated with individual alkyl triflates to afford the desired tetracyclic indoline analogues 13 and 14, respectively.
Scheme 2. Synthesis of additional analogs of 4.

Reagents and conditions: (a) R2OCOCl, DMAP, DMF, 23 °C, 0.5 h; 8, 0.5 h; MsOH, then 7, 60–120 °C, 24 h; (b) 5 mol% Ph3PAuNTf2, 50 °C, toluene; (c) R3OTf, CH2Cl2, 23 °C, 7 h.
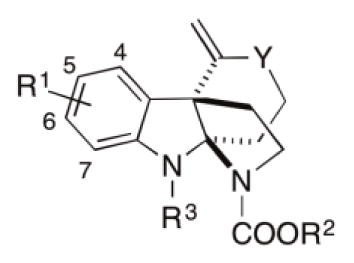
In our previous report of the SAR of the tricyclic indoline RMA 1 (Figure 1), we found that alkylating the indoline nitrogen abolishes RMA activity and thus wondered whether this trend would be observed for Kf18 [38]. To explore this, the methyl group of Kf18 was replaced with a larger ethyl group to give compound 13a. This change resulted in 4- to 8-fold improvement of the RMA activity compared to Kf18 (entry 2, Table 3). However, further increase of the size of the ethyl group to a propargyl group (i.e., 13b, entry 3, Table 3) was prohibited. Interestingly, removing this alkyl group altogether and replacing it with a mere hydrogen to give compound 11 slightly improved RMA activity, albeit less than 13a (entry 4, Table 3). This suggests that the R3 group is sensitive to the RMA activity, as both larger and smaller groups (ethyl and hydrogen) show improved RMA activity compared to the methyl group.
Table 3.
Biological Activities of Additional Analogues of Tetracyclic Indolines
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Cmpd | R1 | R2 | R3 | Y | MRC (cefazolin)a,b | MRC (amox/clav)a,c | MICa,b | GI50a,d |
| 1 | Kf18 | 4,6-diMe | Me | Me | CH2CH2 | 16 | 16 | >64 | 20 |
| 2 | 13a | 4,6-diMe | Me | Et | CH2CH2 | 2 | 4 | >64 | 30 |
| 3 | 13b | 4,6-diMe | Me | CH2CCH | CH2CH2 | 64 | 64 | >64 | 17 |
| 4 | 11 | 4,6-diMe | Me | H | CH2CH2 | 4 | 8 | >64 | 28 |
| 5 | 13c | H | Me | Me | CH2CH2 | >64 | >64 | >64 | 30 |
| 6 | 13d | 5-Me | Me | Me | CH2CH2 | 8 | 4 | >64 | 27 |
| 7 | 13e | 5-Cl | Me | Me | CH2CH2 | 8 | 4 | >64 | 53 |
| 8 | 6a | 4,6-diMe | Et | Me | CH2CH2 | 2 | 2 | >64 | 61 |
| 9 | 13f | 5-F | Et | Me | CH2CH2 | >64 | >64 | >64 | >64 |
| 10 | 13g | 5-F-6-MeO | Et | Me | CH2CH2 | >64 | >64 | >64 | >64 |
| 11 | 14a | 4,6-diMe | Me | Me | CH2 | 16 | 8 | >64 | 40 |
| 12 | 14b | 5-Me | Me | Me | CH2 | 16 | 16 | >64 | 15 |
| 13 | 14c | 5-Cl | Me | Me | CH2 | 16 | 8 | >64 | 41 |
| 14 | 14d | 5-Me | Et | Me | CH2 | 64 | 32 | >64 | 43 |
| 15 | 14e | 5-Cl | Et | Me | CH2 | 16 | 8 | >64 | 41 |
Numbers are in μg/mL.
MRSA NRS-100.
MRSA ATCC BAA-44.
HeLa cells.
Next, changes to the aromatic substitution of Kf18 were explored. Analogue 13c with no substitution on the phenyl ring has significantly reduced RMA activity. Replacing the 4,6-dimethyl group with 5-methyl or 5-chloro to deliver 13d or 13e respectively improves the RMA activity by at least 2-fold relative to Kf18 (entries 6 and 7, Table 3). Although the RMA activity was equivalent for these compounds, the 5-chloro derivative 13e is less toxic to mammalian cells. Further exploration of the substitutions on the 5-position of the indoline shows the 5-fluoro and 5-methoxy substituted analogues (13f, 13g, entries 9 and 10, Table 3) were devoid of RMA activity, despite having the more active ethyl carbamate on the D-ring nitrogen. These results suggest a prominent but unknown reliance on methyl or chloro groups in this class of RMA.
Kf18 has a fused seven membered ring that is not observed in other RMAs we have reported. In addition, this structural motif is a rarity in known indole alkaloid natural products. This information prompted us to reduce the C-ring size to a more common six-member ring 14a. This analogue shows slightly improved RMA activity compared to Kf18 (entry 11, Table 3). Next, the ring size was decreased with the concomitant change in the aromatic substitution pattern by swapping the 4,6-dimethyl groups of 14a with 5-methyl and 5-chloro to deliver 14b and 14c, respectively. Unexpectedly, these changes do not improve the RMA activity (entries 12 and 13, Table 3). Methyl and chloro groups have similar sizes, and it would be expected that their activity profiles should follow a similar trend; however, replacing the 5-methyl group with a chloro shows activity similar to the dimethyl analogue 14a (entry 11, Table 3). A notable difference is that the mammalian toxicity for the 5-methyl analogue 14b is comparable to its MRC in MRSA, while 14a and 14c have improved mammalian toxicity with the GI50 values well above their MRCs. We then asked if changes to the aromatic region would meld with our previously discovered optimal ethyl carbamate on the D ring nitrogen, and synthesized analogues 14d and 14e (entry 14 and 15, Table 3). However, these combinatorial changes did not further improve the RMA activity. The ethyl carbamate analogue 6a remains the best analogue in terms of its RMA activity and mammalian toxicity. It should also be noted that all tetracyclic indoline analogues showed no antibacterial activity at up to 64 μg/mL, the highest concentration tested.
MRSA Strain Scope Studies
In addition to the two hospital-acquired (HA) MRSA strains, ATCC BAA-44 and NRS100, the best analogue, 6a, was tested in several additional MRSA strains. MRSA NRS-45898 (a.k.a., NRS70 and N315) possesses the most prevalent U.S. hospital-acquired pulse-field type, USA100, and was endemic in many U.S. hospitals [41]. In recent years, community-acquired (CA) MRSA infections have consistently increased. Both NR-46070 (a.k.a., NRS384) [42] and NR-46231 (a.k.a., NRS702) are classified as USA300 isolates. USA300 isolates are among the most common types of CA-MRSA strains. The MICs of amox/clav in NRS-45898, NRS-46070, and NR-46231 are 16, 16, and 8 μg/mL, respectively, and the MIC of 6a was >64 μg/mL in all three strains. In combination with 4 μg/mL amox/clav, the MRCs of 6a were found to be 2 μg/mL in all three strains, consistent with the MRCs in two previously tested MRSA strains. These results demonstrate that compound 6a is a potent RMA for β-lactam antibiotics in both HA- and CA-MRSA strains.
In order to assess the synergistic action of 6a with amox/clav and cefazolin, a checkerboard fractional inhibitory concentration indexes (FICI) evaluation was performed using strain ATCC BAA-44 [43]. The FICIs of 6a with amox/clav and cefazolin were calculated as 0.127 and 0.078, respectively. Both of these are significantly less than 0.5, suggesting that 6a has a strong synergistic effect with both antibiotics. In addition, 6a was tested in a β-lactamase activity/nitrocefin assay using DMSO and clavulanic acid as negative and positive controls, respectively [44]. The results showed that 6a has no effect on β-lactamase activity at up to 32 μg/mL (i.e., 16xMRC), suggesting that 6a potentiates β-lactam antibiotics using a novel mechanism.
Summary
Antibiotic-resistant bacterial infections have seen a marked increase in recent years, while antibiotic discovery has waned. Resistance-modifying agents offer an intriguing alternative strategy to fight against resistant bacteria, as they may further extend the clinic life span of β-lactam antibiotics, one of the best classes of antibiotics developed. Here we report our recent discovery, antibiotic profiling, and structure-activity relationships of a novel class of resistance-modifying agents, tetracyclic indolines. These selectively potentiate β-lactam antibiotics in methicillin-resistant Staphylococcus aureus (MRSA) without antibacterial or β-lactamase inhibitory activity on their own. Several series of structural analogues of the screening hit Kf18 have been synthesized and their antibacterial activity, resistance-modifying activity, and mammalian toxicity have been evaluated. The most potent analogue, 6a, showed strong potentiation of amoxicillin/clavulanic acid in a variety of hospital-acquired and community-acquired MRSA strains with low mammalian toxicity. Although 6a showed strong synergistic effect with amox/clav and cefazolin, it did not inhibit β-lactamase activity directly, suggesting that the tetracyclic indolines potentiate β-lactams with a novel mechanism. Further development of this novel class of RMA for in vivo studies and mechanistic investigations are ongoing and will be reported.
Experimental Section
Bacterial Strains
Strains ATCC BAA-44 (MRSA) was a gift from the laboratory of Daniel Feldheim. Strains NRS100 (MRSA), NRS-45898 (MRSA), NRS-46070 (MRSA), and NRS-702 (MRSA) were purchased from ATCC (http://www.atcc.org).
Microdilution Tests for Minimal Inhibitory Concentration (MIC) Determination
MIC determination was performed as described previously.38 The minimal inhibitory concentrations (MICs) of active Kf18 analogues were determined by the broth microdilution method detailed in the CLSI handbook. All antimicrobial compounds were purchased from Sigma-Aldrich. The growth media used for all MIC experiments was Mueller–Hinton broth (MHB) purchased from HIMEDIA through VWR (cat. 95039-356). The inoculum was prepared by diluting a bacterial day culture (OD600 0.15–0.4) to OD600 0.002. This dilution was further diluted 2-fold when added to 96-well microplates (USA Scientific CytoOne 96-well TC plate, cat. CC7682-7596) for a final inoculum concentration of OD600 0.001. All plates were incubated at 37 °C with shaking for 18 h before results were interpreted.
Minimal Re-sensitizing Concentration (MRC) Determination
MRC screens were performed as described previously.38 Briefly, antibiotic MIC values where S. aureus is considered susceptible were determined from the CLSI handbook supplement. Kf18 analogues were diluted to 10 mg/mL in DMSO. Antibiotic was prepared at twice the intended final concentration in MHB. For amoxicillin/clavulanic acid, the initial concentration was 8/4 μg/mL and for cefazolin 16 μg/mL. A 50 μL portion of the antibiotic containing media was added to each well of 96-well plates, and 100 μL was added to the top row. A 6.4 μL portion of of 10 mg/mL alkaloid solution was added to the top row of each plate to afford a concentration of 64 μg/mL in the top row of each plate, and 2-fold serial dilutions were performed down the columns. Once the plates were prepared, a day culture of MRSA was diluted to OD600 0.002, and 50 μL was added to each well. The final concentration of MRSA added was OD600 0.001, the final concentration of amoxicillin/clavulanic acid was 4/2 μg/mL and the final concentration of cefazolin was 8 μg/mL, and the highest concentration of Kf18 tested was 64 μg/mL. Plates were incubated overnight at 37 °C with shaking. The MRC value was determined as the concentration of Kf18 analogue in the presence of antibiotic at which there was no observable overnight growth.
Cytotoxicity of Kf18 Analogues in HeLa Cells
To evaluate the cytotoxicity of Kf18 analogues in mammalian cells, a cell viability assay was carried out using a CellTiter-Glo luminescent cell viability assay kit (Promega). Human cervixcal adenocarcinoma HeLa cells were seeded on white, cell-culture-treated, 96-well plates (Corning 3917) with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin, at the densities of 20 000 cells/well. The medium volume for each well was 100 μL. Cells were incubated at 37 °C in 5% CO/95% air for 16 h. The medium was removed from each well and replaced with 99 μL of warmed, fresh medium. To each well, 1 μL of Kf18 analogue was added in DMSO to final concentrations of 0.5–32 μg/mL. Each series was performed in triplicate. After incubation at 37 °C for another 24 h, the plates were equilibrated to room temperature for 30 min, and 100 μL of CellTiter-Glo reagent (Promega) was added to each well and mixed for 2 min on an orbital shaker. The plate was incubated at room temperature for another 10 min to stabilize the luminescent signal. The luminescence of each sample was recorded with an Envision Multilabel Plate Reader (PerkinElmer).
β-Lactamase Activity Assays
β-lactamase Activity Assays were performed as described previously.43 An overnight culture of MRSA was grown in MHB and sub-cultured 1:100. Cells were grown to OD600 of ~0.5. Cells were devided into 5–6 10mL fractions in MHB and incubated 37 °C for 1 hour. Cells were placed on ice and media was separated from cells by centrifugation (3200xg, 4 °C, 30 min). In a UV-optical bottom 96-well plate 1 μl of DMSO; clavulanic acid at 2, 8, and 32 μg/mL; and 6a at at 2, 8, and 32 μg/mL (1×, 4×, and 16× MRC, respectively), was each added into separate wells, in triplicate. Then, 99 μl of media was added and equilibrated to room temperature before addition of the colorigenic agent, nitrocefin. To each well 1 μl of a 10 mM nitrocefin stock (in DMSO) was added (to afford a final, saturating, concentration of 100 μM). Nitrocefin hydrolysis was immediately monitored by tracking absorbance at 492 nm in real time using the Perkin Elmer EnVision plate reader. The initial rate of hydrolysis was obtained from the slope of the time course which was linear in the first several minutes of monitoring. This initial rate was used at a proxy from the relative concentration of B-lactamase in the media. DMSO was used a negative control and clavulanic acid was used as a positive control.
Fractional Inhibitory Concentration Index (FICI) determination
FICI was calculated as described previously,42 with the formula FICI = FICA + FICB and where an FICI <0.5 indicates synergy. The FICI calculation for amox.clav with 6a was as follows: FICA equaled the MRC of amox.clav (4 μg/mL) in combination with 6a divided by the MIC of amox.clav alone (32 μg/mL); FICB equaled the MRC of 6a (2 μg/mL) in combination with amox.clav divided by MIC of 6a alone (>128 μg/mL). The FICI calculation for cefazolin with 6a was as follows: FICA equaled the MRC of cefazolin (8 μg/mL) in combination with 6a divided by the MIC of cefazolin alone (128 μg/mL); FICB equaled the MRC of 6a (2 μg/mL) in combination with cefazolin divided by MIC of 6a alone (>128 μg/mL).
Chemistry
Unless otherwise noted, reagents were obtained commercially and used without further purification. CH2Cl2 was distilled from CaH2 under a nitrogen atmosphere. THF was distilled from sodium–benzophenone under a nitrogen atmosphere. Toluene was distilled from sodium under a nitrogen atmosphere. All chemicals were purchased from Sigma-Aldrich, TCI America, or Alfa Aesar (United States) unless otherwise noted. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed on Dynamic adsorbents silica gel F-254 TLC plates. Flash chromatography was carried out on Zeoprep 60 ECO silica gel. 1H and 13C NMR spectra were recorded with Varian INOVA (400, 500 MHz) spectrometers. Infrared (IR) spectra were recorded on a Thermo Nicolet Avatar 370 FT-IR spectrometer. Compound purity (≥95%)was confirmed on the basis of the integration of the area under the UV absorption curve at λ = 254 or 210 nm signals using an Agilent 1260 series HPLC system coupled with a 6120 Quadrupole mass spectrometer (column: ZORBAX Narrow Bore SB-C18 RRHT, 2.1 × 50 mm, 1.8 μm, PN 827700-902). The system was eluted at 0.5 mL/min with a gradient of water/acetonitrile with 0.1% formic acid: 0–5 min, 5–95% acetonitrile; 5–7 min, 95% acetonitrile; 7–7.25 min, 95–5% acetonitrile; 7.25–8.5 min, 5% acetonitrile.
General Protocol for the One-Pot Three-Component Indole Synthesis (Method A)
The activating agent methyl chloroformate (1.2 equiv) was added to a solution of 4-dimethylaminopyridine (1.2 equiv) in anhydrous DMF at 0 °C. The reaction was stirred at 23 °C for 30 min. A solution of the alkynyl imine 8 (1.0 equiv) in anhydrous DMF was added and the reaction was stirred at the same temperature for 0.5 h. Methanesulfonic acid (3.0 equiv) was next added to the above mixture at 0 °C. The reaction was then stirred at 23 °C for 2 h. Aryl hydrazine 7 (1.5 equiv) was added and the mixture stirred for an addition 1 h at 23 °C. The reaction was then heated to 60–120 °C (60 °C for electron-sufficient hydrazines and 120 °C for electron-deficient aryl hydrazines) for 20 h. The reaction was cooled down to room temperature. The residue was then dissolved in ethyl acetate and washed with brine and a saturated aqueous solution of sodium bicarbonate. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a crude product, which was purified by column chromatography on silica gel to give the indole product 9 or 10.
Methyl 2-(2-(hex-5-ynyl)-4,6-dimethyl-1H-indol-3-yl) ethyl carbamate (9a)
The title compound was synthesized from (3,5-dimethylphenyl)hydrazine hydrochloride (7a), compound 8a and methyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; light yellow oil, 36%; 1H NMR (500 MHz, CDCl3) δ: 7.77 (s, 1H), 6.95 (s, 1H), 6.69 (s,1H), 4.78 (s, 1H), 3.68 (s, 3H), 3.39 (q, J = 6.8 Hz, 2H), 3.02 (t, J = 7.3 Hz, 2H), 2.74 (t, J = 7.7 Hz, 2H), 2.66 (s, 3H), 2.40 (s, 3H), 2.25 (td, J = 7.0, 2.7 Hz, 2H), 1.99 (t, J = 2.6 Hz, 1H), 1.82-1.76 (m, 2H), 1.61 (dt, J = 14.3, 7.1 Hz, 2H); ESI m/z calcd for C20H26N2O2 [M+H]+ 327.2, found 327.2; IR (thin film) 3387, 3350, 3309, 2947, 2865, 1700, 1525, 1454, 1376, 1253, 914, 836, 735, 635 cm−1.
Methyl (2-(2-(hex-5-yn-1-yl)-1H-indol-3-yl)ethyl) carbamate (9c)
The title compound was synthesized from phenyl hydrazine hydrochloride (7c), compound 8a and methyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; light yellow oil, 35%; 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.30-7.26 (m, 1H), 7.13 (td, J = 7.5, 1.4 Hz, 1H), 7.08 (td, J = 7.4, 1.3 Hz, 1H), 4.75 (s, 1H), 3.65 (s, 3H), 3.42 (q, J = 6.7 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H), 2.74 (t, J = 7.6 Hz, 2H), 2.21 (td, J = 7.0, 2.7 Hz, 2H), 1.96 (t, J = 2.7 Hz, 1H), 1.85-1.71 (m, 2H), 1.57 (p, J = 7.0 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 157.20, 136.14, 135.45, 128.43, 121.41, 119.47, 118.16, 110.54, 108.31, 84.20, 68.88, 52.14, 41.62, 28.97, 27.99, 25.61, 24.80, 18.33; ESI m/z calcd for C18H22N2O2 [M+H]+ 299.1, found 299.1; IR (thin film) 3402, 3298, 2940, 2862, 1700, 1525, 1465, 1246, 1011, 747 cm−1.
Methyl (2-(2-(hex-5-yn-1-yl)-5-methyl-1 H-indol-3-yl)ethyl)carbamate (9d)
The title compound was synthesized from p-tolylhydrazine hydrochloride (7d), compound 8a and methyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; light yellow oil, 28%; 1H NMR (500 MHz, CDCl3) δ 7.76 (s, 1H), 7.29 (s, 1H), 7.18 (d, J = 8.2 Hz, 1H), 6.96 (dd, J = 8.2, 1.5 Hz, 1H), 4.72 (s, 1H), 3.66 (s, 3H), 3.43 (q, J = 6.7 Hz, 2H), 2.89 (t, J = 6.9 Hz, 2H), 2.74 (t, J = 7.6 Hz, 2H), 2.44 (s, 3H), 2.22 (td, J = 7.0, 2.7 Hz, 2H), 1.97 (t, J = 2.6 Hz, 1H), 1.86-1.70 (m, 2H), 1.61-1.55 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 157.21, 136.29, 133.75, 128.67, 122.86, 117.94, 110.21, 107.82, 84.22, 68.85, 52.11, 41.61, 28.97, 27.98, 25.64, 24.76, 21.64, 18.32; ESI m/z calcd for C19H24N2O2 [M+H]+ 313.18, found 313.2; IR (thin film) 3300, 2940, 2860, 1700, 1545, 1499, 1410, 1375, 1335, 1250, 1100, 1075, 1040, 945, 785, 630 cm−1.
Methyl (2-(5-chloro-2-(hex-5-yn-1-yl)-1H-indol-3-yl)ethyl)carbamate (9e)
The title compound was synthesized from 4-chlorophenylhydrazine hydrochloride (7e), compound 8a and methyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; light yellow oil, 24%; 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 8.5 Hz, 1H), 7.05 (dd, J = 8.5, 2.0 Hz, 1H), 4.83 (s, 1H), 3.67 (s, 3H), 3.39 (q, J = 6.8 Hz, 2H), 2.86 (t, J = 6.9 Hz, 2H), 2.71 (t, J = 7.6 Hz, 2H), 2.20 (td, J = 6.9, 2.7 Hz, 2H), 1.97 (s, 1H), 1.76 (p, J = 7.7 Hz, 2H), 1.55 (p, J = 7.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 157.21, 137.82, 133.77, 129.60, 124.97, 121.38, 117.53, 111.51, 108.07, 84.12, 68.92, 52.17, 41.58, 28.78, 27.92, 25.59, 24.60, 18.24; ESI m/z calcd for C18H21ClN2O2 [M+H]+ 333.1, found 333.1; IR (thin film) 3417, 3301, 2940, 2862, 1700, 1525, 1491, 1477, 1447, 1294, 1089, 829, 803, 780 cm−1.
Ethyl (2-(5-fluoro-2-(hex-5-yn-1-yl)-1H-indol-3-yl) ethyl)carbamate (9f)
The title compound was synthesized from 4-fluorophenyhydrazine hydrochloride hydrochloride (7f), compound 8a and ethyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; light yellow oil, 20%; 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 7.14 (ddd, J = 9.8, 5.9, 3.4 Hz, 2H), 6.83 (td, J = 9.1, 2.5 Hz, 1H), 4.84 (d, J = 6.1 Hz, 1H), 4.11 (tt, J = 7.1, 3.6 Hz, 2H), 3.37 (q, J = 6.7 Hz, 2H), 2.85 (t, J = 6.9 Hz, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.18 (td, J = 6.9, 2.7 Hz, 2H), 1.95 (t, J = 2.7 Hz, 1H), 1.75 (p, J = 7.6 Hz, 2H), 1.54 (p, J = 7.1 Hz, 2H), 1.29-1.22 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 171.41, 158.91, 156.83, 156.59, 138.21, 131.91, 128.90,128.80, 111.07, 110.97, 109.27, 109.01, 108.44, 103.15, 102.91, 84.12, 68.85, 60.80, 41.44, 28.79, 27.91, 25.63, 24.72, 21.11, 18.20, 14.71, 14.21; ESI m/z calcd for C19H23FN2O2 [M+H]+ 331.2, found 331.2; IR (thin film) 3406, 3290, 3271, 2951, 2858, 1704, 1525, 1495, 1461, 1447, 1264, 1234, 1104, 1033, 866, 780, 650, 553 cm−1.
Ethyl (2-(5-fluoro-2-(hex-5-yn-1-yl)-6-methoxy-1H-indol-3-yl)ethyl)carbamate (9g)
The title compound was synthesized from 4-fluoro-3-methoxyphe-nylhydrazine hydrochloride (7g), compound 8a and ethyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.0:1 v/v) Rf = 0.20; light yellow oil, 28%; 1H NMR (300 MHz, CDCl3) δ 8.27 (s, 1H), 7.16 (d, J = 11.7 Hz, 1H), 6.84 (d, J = 7.1 Hz, 1H), 4.87 (t, J = 6.0 Hz, 1H), 4.16-4.08 (m, 3H), 3.84 (s, 3H), 3.37 (q, J = 6.7 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.68 (t, J = 7.5 Hz, 2H), 2.18 (td, J = 6.9, 2.6 Hz, 2H), 1.95 (t, J = 2.6 Hz, 1H), 1.81-1.66 (m, 2H), 1.61-1.46 (m, 2H), 1.31-1.18 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 156.82, 150.35, 147.23, 144.18, 143.99, 135.93, 131.26, 121.28, 121.17, 108.17, 104.24, 103.97, 95.83, 95.81, 84.15, 68.80, 60.77, 56.81, 41.52, 28.94, 27.90, 25.58, 24.78, 18.20, 14.71; ESI m/z calcd for C20H25FN2O3 [M+H]+ 361.2, found 361.2; IR (thin film) 3301, 2940, 2862, 1696, 1521, 1484, 1357, 1327, 1249, 1160, 1037, 1070, 858, 780, 631 cm −1.
Methyl (2-(4,6-dimethyl-2-(pent-4-yn-1-yl)-1H-in-dol-3-yl)ethyl)carbamate (10a)
The title compound was synthesized from (3,5-dimethylphenyl)- hydrazine hydrochloride (7a), compound 8b and methyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; yellow oil, 35%; 1H NMR (400 MHz, CDCl3) δ 7.86 (s, 1H), 6.92 (s, 1H), 6.67 (s, 1H), 4.78 (s, 1H), 3.66 (s, 3H), 3.37 (q, J = 6.8 Hz, 2H), 3.01 (t, J = 7.2 Hz, 2H), 2.83 (t, J = 7.4 Hz, 2H), 2.64 (s, 3H), 2.38 (s, 3H), 2.22 (td, J = 6.9, 2.7 Hz, 2H), 2.05 (t, J = 2.6 Hz, 1H), 1.84 (p, J = 7.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 157.18, 136.24, 134.70, 131.25, 129.53, 124.42, 123.30, 109.01, 108.52, 83.94, 69.62, 52.17, 43.34, 28.57, 26.03, 24.51, 21.48, 20.18, 17.92; ESI m/z calcd for C19H24ClN2O2 [M+H]+ 313.1, found 313.2; IR (thin film) 3379, 3301, 2955, 1681, 1599, 1511, 1451, 1372, 1212, 1078, 1045, 914, 780 cm−1.
Methyl (2-(5-methyl-2-(pent-4-yn-1-yl)-1H-indol-3-yl)ethyl)carbamate (10b)
The title compound was synthesized from p-tolylhydrazine hydrochloride (7d), compound 8b and methyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.5:1 v/v) Rf = 0.20; yellow oil, 29%; 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 8.5 Hz, 1H), 7.04 (dd, J = 8.6, 2.0 Hz, 1H), 4.81 (bs, 1H), 3.65 (s, 3H), 3.38 (q, J = 6.7 Hz, 2H), 2.84 (dt, J = 16.5, 7.2 Hz, 4H), 2.18 (td, J = 6.9, 2.7 Hz, 2H), 2.09-2.00 (m, 1H), 1.83 (p, J = 7.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 157.21, 137.08, 133.82, 129.58, 125.04, 121.53, 117.62, 111.57, 108.53, 83.68, 69.72, 52.18, 41.60, 28.36, 24.66, 24.60, 17.87; ESI m/z calcd for C18H22N2O2 [M+H]+ 299.2, found 299.2.
Methyl (2-(5-methyl-2-(pent-4-yn-1-yl)-1 H-indol-3-yl)ethyl)carbamate (10c)
The title compound was synthesized from 4-chlorophenylhydrazine hydrochloride (7e), compound 8b and methyl chloroformate through Method A. TLC hexanes:ethyl acetate, 2.0:1 v/v) Rf = 0.20; yellow oil, 20%; 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 7.06 (dd, J = 8.6, 2.0 Hz, 1H), 4.83 (s, 1H), 3.67 (s, 3H), 3.40 (q, J = 6.7 Hz, 2H), 2.86 (dt, J = 16.5, 7.2 Hz, 4H), 2.20 (td, J = 6.9, 2.7 Hz, 2H), 2.11-1.99 (m, 1H), 1.85 (p, J = 7.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 157.21, 137.08, 133.82, 129.58, 125.04, 121.53, 117.62,111.57, 108.53, 83.68, 69.72, 52.18, 41.60, 28.36, 24.66, 24.60, 17.87; ESI m/z calcd for C17H19ClN2O2 [M+H]+ 319.1, found 319.2.
Ethyl (2-(5-chloro-2-(pent-4-yn-1-yl)-1H-indol-3-yl)ethyl)carbamate (10e)
The title compound was synthesized from 4-chlorophenylhydrazine hydrochloride (7e), compound 8b and ethyl chloroformate through Method A. TLC (hexanes:ethyl acetate, 2.0:1 v/v) Rf = 0.20; yellow oil, 23%; 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.14 (d, J = 8.5 Hz, 1H), 7.03 (dd, J = 8.5, 2.0 Hz, 1H), 4.90 (t, J = 6.1 Hz, 1H), 4.11 (qd, J = 7.1, 2.8 Hz, 2H), 3.38 (q, J = 6.7 Hz, 2H), 2.86 (t, J = 6.9 Hz, 2H), 2.80 (t, J = 7.5 Hz, 2H), 2.16 (td, J = 6.9, 2.7 Hz, 2H), 2.02 (t, J = 2.6 Hz, 1H), 1.82 (p, J = 7.1 Hz, 2H), 1.26-1.15 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 156.82, 137.10, 133.83, 129.52, 124.77, 121.28, 117.48, 111.54, 108.37, 83.65, 69.55, 60.82, 41.49, 28.30, 24.65, 24.55, 17.81, 14.66; ESI m/z calcd for C18H21ClN2O2 [M+H]+ 333.1, found 333.1.
General Protocol for Gold(I)-Catalyzed Tandem Cyclization (Method B)
To a suspension of Ph3PAuNTf2 (as the 2:1 toluene adduct) (0.05 equiv) in anhydrous toluene was added a solution of indole 9 or 10 (1.0 equiv) in anhydrous toluene. The suspension was heated to 50 °C until TLC showed that there was no starting material left (1–12 h) under argon atmosphere. The reaction mixture was then filtered through a short pad of silica gel. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on silica gel to afford tetracyclic indoline product 11 or 12.
Methyl 1,3-dimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)-cyclohepta[b] indole-13-carboxylate (11a)
The title compound was synthesized through Method B from compound 9a. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 67%; 1H NMR (500 MHz, CDCl3) δ 6.35 (s, 1H), 6.32 (s, 1H), 5.61 (s, 1H), 5.02 (s, 1H), 5.00 (s, 1H), 3.63 (s, 3H), 3.57 (dd, J = 10.2, 8.1 Hz, 1H), 3.06-3.01 (m, 1H), 2.60-2.54 (m, 2H), 2.31-2.26 (m, 1H), 2.24 (s, 3H), 2.19 (s, 3H), 2.09-2.06 (m, 1H), 1.98-1.89 (m, 1H), 1,84-1.81 (m, 1H), 1.73-1.68 (m, 1H), 1.61-1.53 (m, 2H), 1.29-1.20 (m, 1H); ESI m/z calcd for C20H26N2O2 [M+H]+ 327.2, found 327.2; IR (thin film) 3383, 2944, 2918, 2877, 2858, 1681, 1451, 1376, 1287, 1212, 1197, 1045, 981, 836, 780, 747 cm−1.
Methyl (5aR,10aS)-10-methylene-7,8,9,10-tetra hydro-5H,6H-5a,10a-(epiminoethano)-cyclohepta[b] indole-13-carboxylate (11c)
The title compound was synthesized through Method B from compound 9c. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; colorless oil, 74%; 1H NMR (400 MHz, CDCl3) δ 7.07 (td, J = 7.6, 1.3 Hz, 1H), 6.94 (dd, J = 7.5, 1.3 Hz, 1H), 6.73 (td, J = 7.4, 1.0 Hz, 1H), 6.64 (d, J = 7.8 Hz, 1H), 5.68 (s, 1H), 4.99 (s, 1H), 4.82 (s, 1H), 3.62 (s, 3H), 3.56 (dt, J = 8.9, 5.3 Hz, 1H), 3.03-2.85 (m, 1H), 2.67-2.58 (m, 1H), 2.40-2.28 (m, 2H), 2.07 (ddd, J = 12.7, 5.7, 2.4 Hz, 1H), 2.01-1.86 (m, 1H), 1.81 (dd, J = 10.5, 5.4 Hz, 1H), 1.74-1.65 (m, 1H), 1.65-1.46 (m, 2H), 1.31-1.12 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 154.75, 154.15, 149.41, 131.44, 128.48, 123.57, 119.02, 110.94, 109.85, 90.14, 64.42, 51.95, 46.03, 33.40, 32.94, 32.71, 32.16, 24.45; ESI m/z calcd for C18H22N2O2 [M+H]+ 299.1, found 299.2; IR (thin film) 3387, 3305, 2925, 2854, 1689, 1599, 1410, 1354, 1331, 1212, 1108, 1078, 702 cm−1.
Methyl (5aR,10aS)-2-methyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclo-hepta[b]indole-13-carboxylate (11d)
The title compound was synthesized through Method B from 9d. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 70%; 1H NMR (400 MHz, CDCl3) δ 6.87 (dd, J = 7.9, 1.8 Hz, 1H), 6.74 (d, J = 1.8 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 5.57 (s, 1H), 4.98 (s, 1H), 4.82 (s, 1H), 3.61 (s, 3H), 3.55 (ddd, J = 10.2, 6.6, 2.3 Hz, 1H), 2.94 (td, J = 10.6, 7.4 Hz, 1H), 2.61 (dd, J = 14.6, 5.8 Hz, 1H), 2.39-2.29 (m, 3H), 2.15-2.00 (m, 1H), 1.92 (td, J = 14.9, 14.1, 3.4 Hz, 1H), 1.82 (d, J = 13.2 Hz, 1H), 1.77-1.66 (m, 1H), 1.64-1.46 (m, 2H), 1.31-1.10 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 154.71, 154.39, 147.13, 131.60, 128.97, 128.24, 124.13, 110.79, 109.70, 90.46, 64.41, 51.90, 46.00, 33.43, 32.97, 32.64, 32.23, 24.46, 21.02; ESI m/z calcd for C18H21ClN2O2 [M+H]+ 313.2, found 313.2; IR (thin film) 3357, 2944, 2921, 2854, 1678, 1451, 1376, 1346, 1201, 1127, 981, 907, 858, 780 cm−1.
Methyl (5aR,10aS)-2-chloro-10-methylene-7,8,9, 10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclo-hepta[b]indole-13-carboxylate (11e)
The title compound was synthesized through Method B from compound 9e. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 71%; 1H NMR (400 MHz, CDCl3) δ 7.01 (dd, J = 8.3, 2.2 Hz, 1H), 6.88 (d, J = 2.2 Hz, 1H), 6.55 (d, J = 8.3 Hz, 1H), 5.68 (s, 1H), 4.99 (s, 1H), 4.85 (s, 1H), 3.62 (s, 3H), 3.57 (ddd, J = 10.2, 6.5, 2.4 Hz, 1H), 2.95 (td, J = 10.4, 7.7 Hz, 1H), 2.69-2.54 (m, 1H), 2.40-2.27 (m, 2H), 2.16-2.01 (m, 1H), 1.98-1.74 (m, 2H), 1.73-1.47 (m, 3H), 1.31-1.13 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 154.74, 153.42, 147.98, 133.50, 128.41, 123.89, 123.47, 111.62, 110.77, 90.56, 64.50, 52.05, 45.99, 33.40, 32.92, 32.67, 32.07, 24.40; ESI m/z calcd for C18H21ClN2O2 [M+H]+ 333.1, found 333.2; IR (thin film) 3383, 2929, 2884, 2854, 1685, 1447, 1369, 1197, 1164, 978, 899, 814, 780 cm−1.
Ethyl (5aR,10aS)-2-fluoro-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclo-hepta[b]indole-13-carboxylate (11f)
The title compound was synthesized through Method B from compound 9f. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 78%; 1H NMR (400 MHz, CDCl3) δ 6.75 (td, J = 8.8, 2.7 Hz, 1H), 6.65 (dd, J = 8.3, 2.6 Hz, 1H), 6.53 (dd, J = 8.5, 4.2 Hz, 1H), 5.60 (s, 1H), 4.98 (d, J = 1.2 Hz, 1H), 4.84 (d, J = 1.1 Hz, 1H), 4.28-3.91 (m, 2H), 3.59 (ddd, J = 10.3, 7.1, 1.9 Hz, 1H), 3.08-2.85 (m, 1H), 2.61 (ddt, J = 14.7, 6.1, 1.6 Hz, 1H), 2.39-2.22 (m, 2H), 2.08 (ddd, J = 12.7, 5.7, 2.5 Hz, 1H), 1.99-1.76 (m, 2H), 1.75-1.65 (m, 1H), 1.65-1.45 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 158.31, 155.97, 154.38, 153.65, 145.45, 133.20, 133.13, 114.88, 114.65, 111.42, 110.90, 110.66, 110.11, 110.03, 90.76, 64.65, 60.75, 45.91, 33.35, 33.04, 32.64, 32.11, 24.38, 14.69; ESI m/z calcd for C19H23FN2O2 [M+H]+ 331.1, found 331.1; IR (thin film) 3383, 2930, 2877, 2854, 1677, 1491, 1413, 1380, 1350, 1290, 1182, 1111, 1007, 959, 862, 813, 784, 657, 571, 530.
Ethyl (5aR,10aS)-2-fluoro-3-methoxy-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclohepta[b]indole-13-carboxylate (11g)
The title compound was synthesized through Method B from 9g. TLC (hexanes:ethyl acetate, 8:1 v/v) Rf = 0.20; white solid, 71%; 1H NMR (300 MHz, CDCl3) δ 6.65 (d, J = 10.6 Hz, 1H), 6.29 (d, J = 7.0 Hz, 1H), 5.61 (s, 1H), 4.94 (t, J = 1.1 Hz, 1H), 4.80 (d, J = 1.1 Hz, 1H), 4.16-3.95 (m, 2H), 3.82 (s, 3H), 3.64-3.50 (m, 1H), 3.05-2.87 (m, 1H), 2.65-2.52 (m, 1H), 2.36-2.17 (m, 2H), 2.13-2.03 (m, 1H), 1.97-1.76 (m, 2H), 1.73-1.42 (m, 3H), 1.21 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 154.46, 153.87, 148.35, 147.89 (d, J = 12.1 Hz), 145.44 (d, J = 1.8 Hz), 122.27 (d, J = 6.0 Hz), 145.23, 111.30, 111.09, 111.03, 96.02, 90.76, 64.35, 60.74, 56.35, 45.96, 33.31, 33.04, 32.49, 32.07, 24.42, 14.70; ESI m/z calcd for C20H25FN2O3 [M+H]+ 361.2, found 361.2.
Methyl (4bS,8aR)-2,4-dimethyl-5-methylene-5,6, 7,8-tetrahydro-9H-8a,4b-(epimino-ethano)carbazole-10-carboxylate (12a)
The title compound was synthesized through Method B from compound 10a. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 90%; 1H NMR (400 MHz, CDCl3) δ 6.35 (d, J = 0.8 Hz, 1H), 6.32 (s, 1H), 5.42 (s, 2H), 4.91 (d, J = 1.4 Hz, 2H), 4.73 (s, 2H), 3.74-3.54 (m, 4H), 3.21-3.08 (m, 1H), 2.64-2.56 (m, 1H), 2.53-2.44 (m, 1H), 2.34-2.28 (m, 1H), 2.26 (s, 3H), 2.20 (s, 3H), 2.17-2.11 (m, 2H), 1.69-1.62 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 154.79, 149.73, 147.90, 138.18, 135.17, 125.11, 122.57, 112.00, 109.11, 89.08, 61.21, 51.85, 46.38, 30.61, 30.16, 30.09, 22.83, 21.42, 18.53; ESI m/z calcd for C19H24N2O2 [M+H]+ 313.2, found 313.0.
Methyl (4bS,8aR)-3-methyl-5-methylene-5,6,7,8-tetrahydro-9H-8a,4b-(epiminoethano)-carbazole-10-carboxylate (12b)
The title compound was synthesized through Method B from compound 10b. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 64%; 1H NMR (400 = 1.7 Hz, 1H), 6.88 (dd, J = 7.7, 1.7 Hz, 1H), 6.57 (d, J = 7.8 Hz, 1H), MHz, CDCl3) δ 6.95 (d, J 5.46 (s, 1H), 4.87 (s, 1H), 4.80 (s, 1H), 3.73-3.51 (m, 4H), 3.17-3.03 (m, 1H), 2.74-2.53 (m, 1H), 2.35-2.12 (m, 7H), 1.72-1.54 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 154.73, 148.74, 146.56, 131.99, 128.66, 128.03, 125.28, 125.24, 112.07, 112.00, 110.60, 89.11, 59.75, 51.84, 46.41, 32.02, 31.67, 31.07, 23.09, 21.09; ESI m/z calcd for C18H22N2O2 [M+H]+ 299.2, found 299.1.
Methyl (4bS,8aR)-3-chloro-5-methylene-5,6,7,8-tetrahydro-9H-8a,4b-(epiminoethano)-carbazole-10-carboxylate (12c)
The title compound was synthesized through Method B from compound 10c. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 2.1 Hz, 1H), 7.03 (dd, J = 8.3, 2.1 Hz, 1H), 6.58 (d, J = 8.3 Hz, 1H), 5.56 (s, 1H), 4.90 (s, 1H), 4.79 (s, 1H), 3.76-3.49 (m, 4H), 3.22-3.01 (m, 1H), 2.77-2.50 (m, 1H), 2.41-2.10 (m, 3H), 1.78-1.47 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 154.74, 147.93, 147.56, 133.85, 128.18, 124.84, 123.46, 112.44, 111.66, 89.23, 59.87, 52.00, 46.39, 31.98, 31.53, 30.93, 22.90; ESI m/z calcd for C17H19ClN2O2 [M+H]+ 319.1, found 319.1.
Ethyl (4bS,8aR)-3-chloro-5-methylene-5,6,7,8-tetrahydro-9H-8a,4b-(epiminoethano)carba-zole-10-carboxylate (12e)
The title compound was synthesized through Method B from compound 10e. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; white solid, 74%; 1H NMR (400 MHz, CDCl3) δ 7.07 (s, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 8.3 Hz, 1H), 5.56 (s, 2H), 4.87 (s, 1H), 4.77 (s, 2H), 4.22-3.93 (m, 2H), 3.64-3.52 (m, 1H), 3.15-3.02 (m, 1H), 2.72-2.52 (m, 1H), 2.33-2.09 (m, 4H), 1.70-1.52 (m, 3H), 1.16 (t, J = 7.1 Hz, 4H); 13C NMR (101 MHz, CDCl3) δ 154.35, 147.96, 147.59, 133.85, 128.09, 124.76, 123.32, 112.28, 111.54, 89.08, 60.67, 59.84, 46.31, 31.92, 31.48, 30.87, 22.82, 14.60; ESI m/z calcd for C18H21ClN2O2 [M+H]+ 333.1, found 333.1.
General protocol for alkylation reaction of indoline (Method C)
To a solution of tetracyclic indoline (11 or 12) in anhydrous CH2Cl2 (0.050 M) was added ROTf (MeOTf or EtOTf, 10 equiv) at 0°C. The resulting mixture was stirred at 0-23 °C for 2-12 h under argon. Then, the mixture is quenched by aqueous NaHCO3, diluted by ethyl acetate, washed with brine, dried over Na2SO4, filtered, concentration in vacuo. The residue is purified by column chromatography on silica gel to give product 13 or 14.
Methyl (5aR,10aS)-1,3,5-trimethyl-10-methyle-ne-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epi-minoethano)cyclohepta[b]indole-13-carboxylate (4)
The title compound was synthesized through Method C from compound 11a and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; white solid, 86%; 1H NMR (500 MHz, CDCl3) δ 6.26 (s, 1H), 6.10 (s, 1H), 5.04 (s, 1H), 5.01 (s, 1H), 3.72-3.41 (m, 5H), 3.05-3.00 (m, 4H), 2.41 (dd, J = 12.3, 5.0 Hz, 1H), 2.28 (s, 3H), 2.26-2.19 (m, 1H), 2.16 (s, 3H), 2.13-2.10 (m, 1H), 1.76-1.69 (m, 3H), 1.57-1.45 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 155.30, 151.86, 151.14, 138.38, 133.88, 123.55, 121.36, 112.73, 104.17, 93.88, 65.48, 51.96, 47.10, 33.39, 32.46, 32.08, 31.49, 31.09, 24.29, 22.85, 21.73, 17.22, 14.30; ESI m/z calcd for C21H28N2O2 [M+H]+ 341.2, found 341.2; IR (thin film) 2925, 2854, 1704, 1596, 1443, 1361, 1190, 1104, 963, 903, 817, 776 cm−1.
Methyl (5aR,10aS)-5-ethyl-1,3-dimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimi-noethano)cyclohepta[b]indole-13-carboxylate (13a)
The title compound was synthesized through Method C from 11a and EtOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil, 25%; 1H NMR (400 MHz, CDCl3) δ 6.20 (s, 1H), 6.08 (s, 3H), 5.01 (s, 2H), 4.96 (s, 4H), 3.71-3.46 (m, 7H), 3.08-3.01 (m, 1H), 2.32-2.19 (m, 5H), 2.17-2.12 (m, 1H), 2.10 (s, 3H), 1.71-1.60 (m, 4H), 1.48-1.41 (m, 1H), 1.32-1.27 (m, 1H), 1.14 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 155.00, 150.73, 150.45, 138.06, 133.86, 124.59, 120.53, 113.36, 104.48, 94.34, 65.72, 51.94, 46.78, 38.16, 33.76, 32.03, 31.81, 23.81, 21.81, 17.54, 12.88; ESI m/z calcd for C22H30N2O2 [M+H]+ 355.2, found 355.0; IR (thin film) 2925, 2858, 1704, 1596, 1443, 1361, 1186, 1104, 1045, 981, 896, 821, 806, 776 cm−1.
Methyl (5aR,10aS)-5-methyl-10-methylene-7,8,9, 10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclohepta[b]indole-13-carboxylate (13c)
The title compound was synthesized through Method C from compound 11c and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; white solid, 50%; 1H NMR (500 MHz, CDCl3) δ 7.19-7.09 (m, 1H), 6.92-6.83 (m, 1H), 6.68 (t, J = 7.3 Hz, 1H), 6.41 (d, J = 7.8 Hz, 1H), 5.02 (s, 1H), 4.85 (s, 1H), 3.72-3.46 (m, 5H), 3.02-2.92 (m, 4H), 2.37-2.19 (m, 2H), 2.19-2.13 (m, 1H), 1.79-1.61 (m, 2H), 1.64-1.51 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 155.26, 153.65, 151.25, 130.86, 128.70, 122.79, 117.56, 111.09, 105.42, 93.60, 65.35, 52.02, 46.88, 33.63, 33.26, 32.14, 31.45, 29.87, 24.44; ESI m/z calcd for C19H24N2O2 [M+H]+ 313.2, found 313.2; IR (thin film) 2925, 2892, 2858, 1700, 1611, 1436, 1366, 1346, 1197, 1108, 959, 892, 776, 736 cm−1.
Methyl (5aR,10aS)-2,5-dimethyl-10-methylene-7, 8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxylate (13d)
The title compound was synthesized through Method C from compound 11d and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; white solid, 83%; 1H NMR (400 MHz, CDCl3) δ 6.97-6.86 (m, 1H), 6.67 (d, J = 1.8 Hz, 1H), 6.30 (d, J = 7.8 Hz, 1H), 5.00 (s, 1H), 4.83 (s, 1H), 3.76-3.63 (m, 2H), 3.58 (s, 3H), 2.98 (s, 3H), 2.91 (dt, J = 11.4, 5.4 Hz, 1H), 2.39-2.15 (m, 5H), 2.14-2.06 (m, 1H), 1.82-1.47 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 155.27, 153.91, 149.20, 130.95, 128.95, 126.72, 123.56, 110.89, 105.25, 93.86, 65.32, 51.99, 46.91, 33.64, 33.20, 32.21, 31.56, 29.88, 24.46, 20.94; ESI m/z calcd for C20H26N2O2 [M + H]+ 327.2, found 327.2; IR (thin film) 2929, 2880, 2858, 1707, 1503, 1443, 1357, 1339, 1283, 1104, 963, 858, 810, 776 cm−1.
Methyl (5aR,10aS)-2-chloro-5-methyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxylate (13e)
The title compound was synthesized through Method C from compound 11e and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; white solid, 50%; 1H NMR (400 MHz, CDCl3) δ 7.04 (dd, J = 8.3, 2.2 Hz, 1H), 6.79 (d, J = 2.2 Hz, 1H), 6.27 (d, J = 8.3 Hz, 1H), 4.99 (s, 1H), 4.85 (s, 1H); 3.71-3.39 (m, 5H), 2.97-2.88 (m, 4H), 2.29-2.21 (m, 1H), 2.16-2.08 (m, 2H), 1.76-1.67 (m, 2H), 1.53-1.42 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 155.22, 152.91, 149.86, 132.82, 128.46, 123.19, 111.79, 106.20, 93.82, 65.35, 52.11, 46.84, 33.66, 33.21, 32.04, 31.54, 29.86, 24.39; ESI m/z calcd for C19H23ClN2O2 [M+H]+ 347.1, found 347.2; IR (thin film) 2948, 2925, 2884, 2858, 1693, 1491, 1443, 1365, 1343, 1275, 1197, 1115, 974, 903, 810, 791cm−1.
Ethyl (5aR,10aS)-2-fluoro-5-methyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclohepta[b]indole-13-carboxylate (13f)
The title compound was synthesized through Method C from compound 11f and EtOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil, 89%; 1H NMR (500 MHz, CDCl3) δ 6.80 (td, J = 8.9, 2.6 Hz, 1H), 6.61 (dd, J = 8.2, 2.6 Hz, 1H), 6.26 (dd, J = 8.6, 4.0 Hz, 1H), 5.00 (d, J = 1.1 Hz, 1H), 4.86 (s, 1H), 4.02 (s, 0H), 3.72 (t, J = 9.2 Hz, 1H), 3.45 (s, 1H), 3.09-2.83 (m, 4H), 2.26 (td, J = 12.2, 7.7 Hz, 1H), 2.17-2.08 (m, 2H), 1.73 (dddd, J = 30.1, 16.0, 7.1, 3.2 Hz, 1H), 1.56-1.48 (m, 2H), 1.35-1.17 (m, 5H); ESI m/z calcd for C20H25FN2O2 [M+H]+ 345.2, found 345.2.
Ethyl (5aR,10aS)-2-fluoro-3-methoxy-5-methyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxylate (13g)
The title compound was synthesized through Method C from compound 11g and EtOTf. TLC (hex-anes:ethyl acetate, 8:1 v/v) Rf = 0.20; white solid, 93%; 1H NMR (500 MHz, CDCl3) δ 6.61 (d, J = 10.5 Hz, 1H), 6.02 (d, J = 6.8 Hz, 1H), 4.96 (s, 1H), 4.83 (s, 1H), 4.07-4.01 (m, 2H), 3.89 (s, 3H), 3.71 (t, J = 9.2 Hz, 1H), 3.42 (s, 1H), 3.06-2.86 (m, 5H), 2.24 (td, J = 12.2, 7.7 Hz, 1H), 2.10 (dt, J = 11.9, 4.9 Hz, 2H), 1.84-1.66 (m, 2H), 1.59-1.45 (m, 3H), 1.35-1.17 (m, 5H); ESI m/z calcd for C21H27FN2O3 [M+H]+ 375.2, found 375.2.
Methyl (4bS,8aR)-2,4,9-trimethyl-5-methylene-5,6,7,8-tetrahydro-9H-8a,4b-(epimino-ethano)car-bazole-10-carboxylate (14a)
The title compound was synthesized through Method C from compound 12a and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil, 64%; 1H NMR (400 MHz, CDCl3) δ 6.21 (s, 1H), 6.03 (s, 1H), 4.95 (s, 1H), 4.89 (s, 1H), 3.75-3.55 (m, 4H), 3.15 (td, J = 10.3, 6.2 Hz, 1H), 2.99-2.77 (m, 4H), 2.42-2.32 (m, 1H), 2.23 (s, 4H), 2.18 (s, 4H), 1.97 (dt, J = 14.4, 8.2 Hz, 1H), 1.87 (td, J = 14.7, 12.8, 5.6 Hz, 1H), 1.54-1.45 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 155.39, 151.41, 147.76, 138.36, 134.21, 124.07, 120.77, 111.33, 104.36, 92.53, 61.97, 52.06, 46.61, 31.34, 30.45, 29.57, 26.79, 21.74, 20.71, 17.89; ESI m/z calcd for C19H24N2O2 [M+H]+ 327.2, found 327.2; IR (thin film) 2948, 2869, 1704, 1596, 1443, 1369, 1201, 1123, 896, 821, 736 cm −1.
Methyl (4bS,8aR)-3,9-dimethyl-5-methylene-5,6, 7,8-tetrahydro-9H-8a,4b-(epiminoethano)-carbazole-10-carboxylate (14b)
The title compound was synthesized through Method C from compound 12b and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil, 50%; 1H NMR (400 MHz, CDCl3) δ 6.91 (d, J = 7.8 Hz, 1H), 6.79 (s, 1H), 6.28 (d, J = 7.9 Hz, 1H), 4.92 (d, J = 13.0 Hz, 2H), 3.70-3.61 (m, 4H), 3.22-3.15 (m, 1H), 2.94(s, 3H), 2.29-2.14 (m, 7H), 2.08-2.00 (m, 1H), 1.61-1.49 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 155.46, 148.53, 148.21, 131.24, 128.85, 126.37, 123.80, 110.66, 106.07, 92.80, 61.03, 52.04, 47.15, 33.14, 30.69, 30.61, 27.06, 21.22, 20.95; ESI m/z calcd for C19H24N2O2 [M+H]+ 313.2, found 313.2; IR (thin film) 2947, 2921, 2892, 1711, 1502, 1450, 1368, 1216, 1178, 1108, 992, 955, 776 cm−1.
Methyl (4bS,8aR)-3-chloro-9-methyl-5-methylene-5,6,7,8-tetrahydro-9H-8a,4b-(epimino-ethano)carbazole-10-carboxylate (14c)
The title compound was synthesized through Method C from compound 12c and MeOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil, 65%; 1H NMR (400 MHz, CDCl3) δ7.03 (dd, J = 8.3, 2.2 Hz, 1H), 6.90 (d, J = 2.2 Hz, 1H), 6.25 (d, J = 8.2 Hz, 1H), 4.92 (d, J = 13.0 Hz, 2H), 3.70-3.61 (m, 4H), 3.22-3.15 (m, 1H), 2.94(s, 3H), 2.29-2.14 (m, 4H), 2.08-2.00 (m, 1H), 1.61-1.49 (m, 2H); ESI m/z calcd for C18H21ClN2O2 [M+H]+ 333.1, found 333.1.
Ethyl (4bS,8aR)-3,9-dimethyl-5-methylene-5,6,7, 8-tetrahydro-9H-8a,4b-(epiminoethano)-carbazole-10-carboxylate (14d)
The title compound was prepared through deprotecting methyl carbamate of compound 14b, ethyl carbonylation reaction with ClCOOEt. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil; 1H NMR (400 MHz, CDCl3) δ 6.91 (d, J = 7.8 Hz, 1H), 6.79 (s, 1H), 6.28 (d, J = 7.9 Hz, 1H), 4.94 (s, 1H), 4.90 (s, 1H), 4.22-4.01 (m, 2H), 374-3.58 (m, 1H), 3.22-3.10 (m, 1H), 2.93 (s, 3H), 2.29-2.13 (m, 7H), 2.07-2.00 (m, 1H), 1.53-1.47 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 155.12, 148.60, 148.26, 131.35, 128.85, 126.38, 123.81, 110.62, 106.08, 92.79, 61.09, 60.69, 47.19, 33.17, 30.76, 30.68, 27.10, 21.24, 20.98, 14.80; ESI m/z calcd for C20H26N2O2 [M+H]+ 327.2, found 327.2; IR (thin film) 2933, 2865, 1704, 1502, 1406, 1380, 1335, 1212, 1115, 1070, 951, 802, 771 cm−1.
Ethyl (4bS,8aR)-3-chloro-9-methyl-5-methyl-ene-5,6,7,8-tetrahydro-9H-8a,4b-(epimino-ethano)-carbazole-10-carboxylate (14e)
The title compound was synthesized through Method C from compound 12e and EtOTf. TLC (hexanes:ethyl acetate, 12:1 v/v) Rf = 0.20; colorless oil, 68%; 1H NMR (400 MHz, CDCl3) δ 7.03 (dd, J = 8.3, 2.2 Hz, 1H), 6.90 (d, J = 2.2 Hz, 1H), 6.25 (d, J = 8.2 Hz, 1H), 4.92 (d, J = 3.8 Hz, 2H), 417-4.01 (m, 2H), 3.72-3.60 (m, 1H), 3.20-3.14 (m, 1H), 2.93 (s, 3H), 2.67-2.64 (m, 1H), 2.29-2.09 (m, 4H), 2.05-1.98 (m, 1H), 1.57-11.41 (m, 2H), 1.23-1.18 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 154.97, 148.96, 147.77, 132.99, 128.35, 123.31, 121.61, 111.17, 106.83, 92.64, 60.81, 46.99, 33.07, 30.51, 30.42, 27.13, 20.88, 14.74; ESI m/z calcd for C19H23ClN2O2 [M+H]+ 347.1, found 347.1; IR (thin film) 2981, 2940, 2873, 1700, 1607, 1481, 1406, 1380, 1335, 1212, 1115, 1085, 944, 806, 776, 735 cm−1.
(5aS,10aS)-1,3,5-Trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclohepta[b]indole (5)
A solution of Kf18 in toluene (0.10 M) was added Me3SiI (2.0 equiv) at room temperature. Then mixture was heated to 70 °C for 5 h, until Kf18 disappeared. The mixture was cooled to 0 °C in ice bath, followed by addition of aqueous NaHCO3. The mixture was diluted with ethyl acetate, washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel to give product. TLC (CH2Cl2:MeOH, 16:1 v/v) Rf = 0.20; colorless oil, 96%; 1H NMR (400 MHz, CDCl3) δ 6.20 (s, 1H), 6.05 (s, 1H), 5.16 (s, 1H), 5.06 (s, 1H), 3.27 (s, 1H), 3.08 (dd, J = 11.2, 6.1 Hz, 1H), 2.80 (s, 3H), 2.58 (ddd, J = 12.3, 11.2, 4.4 Hz, 1H), 2.37 (td, J = 12.3, 6.3 Hz, 1H), 2.25 (s, 3H), 2.24-2.18 (m, 1H), 2.15 (dd, J = 12.1, 4.3 Hz, 1H), 2.09 (s, 3H), 2.07-2.03 (m, 1H), 1.73-1.55 (m, 1H), 1.52-1.43 (m, 1H), 1.37-1.18 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 152.22, 151.07, 138.16, 133.89, 125.48, 120.34, 112.36, 102.82, 94.75, 64.51, 45.47, 37.17, 34.72, 34.64, 31.79, 28.30, 24.06, 21.74, 17.28; ESI m/z calcd for C19H26O2 [M+H]+ 283.2, found 283.2; IR (thin film) 3312, 3085, 2925, 2854, 1596, 1480, 1443, 1365, 1305, 1227, 1074, 1030, 892, 817 cm−1.
General procedure for the preparation of compound 6a, 6b and 6g (Method D)
To a solution of compound 5 (1.0 equiv), N,N-diisopropylethylamine (DIPEA, 6.0 equiv), and 4-dimethylaminopyridine (DMAP, 0.2 equiv) in DMF (0.030 M) at 0 °C was added the alkyl chloroformate or 4-chloroben-zenesulfonyl chloride (3.0 equiv). Then the mixture was stirred at room temperature overnight. The mixture was then quenched by aqueous NaHCO3, diluted by ethyl acetate, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give product.
Ethyl (5aR,10aS)-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxylate (6a)
The title compound was prepared through Method D from compound 5 and ethyl chloroformate. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; yellow oil, 90%; 1H NMR (400 MHz, CDCl3) δ 6.23 (s, 1H), 6.06 (s, 1H), 5.01 (s, 1H), 4.98 (s, 1H), 4.01 (s, 2H), 3.69 (t, J = 9.1 Hz, 1H), 3.38 (s, 1H); 3.03-2.96 (m, 4H), 2.37 (dd, J = 12.3, 4.8 Hz, 1H), 2.24 (s, 3H), 2.21-2.15 (m, 1H), 2.12-2.06 (m, 4H), 1.73-1.64 (m, 2H), 1.58-1.40 (m, 4H), 1.19 (bs, 3H); 13C NMR (101 MHz, CDCl3) δ 154.90, 151.90, 151.16, 138.35, 133.90, 123.58, 121.29, 112.69, 104.12, 93.79, 65.46, 60.61, 47.07, 33.39, 32.47, 31.57, 31.03, 24.30, 21.75, 17.24, 14.77; ESI m/z calcd for C22H30N2O2 [M+H]+ 355.2, found 355.2; IR (thin film) 2925, 2858, 1704, 1596, 1380, 1335, 1190, 1104, 1130, 959, 896, 862, 826 cm−1.
Benzyl (5aR,10aS)-1,3,5-Trimethyl-10-methyl-ene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epi-minoethano)cyclohepta[b]indole-13-carboxylate (6b)
The title compound was prepared through Method D from compound 5 and benzyl chloroformate. TLC (hexanes:ethyl acetate, 10:1 v/v) Rf = 0.20; yellow oil, 54%; 1H NMR (400 MHz, CDCl3) δ 7.44-7.28 (m, 5H), 6.25 (s, 1H), 6.09 (s, 1H), 5.20-4.95 (m, 5H), 3.77 (dd, J = 10.6, 7.3 Hz, 1H), 3.41 (s, 1H), 3.07 (ddd, J = 12.3, 10.5, 5.2 Hz, 1H), 2.99 (s, 3H), 2.40 (dd, J = 12.3, 5.1 Hz, 1H), 2.31-2.20 (m, 4H), 2.14 (s, 4H), 1.80-1.45 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 154.55, 151.87, 151.09, 138.41, 137.12, 135.28, 133.91, 128.72, 128.68, 128.56, 128.48, 127.93, 127.77, 123.51, 121.37, 112.74, 104.20, 94.06, 69.88, 66.38, 65.44, 47.15, 33.37, 32.44, 31.56, 31.07, 29.74, 24.27, 21.75, 17.22; ESI m/z calcd for C27H32N2O2 [M+H]+ 417.2, found 417.2; IR (thin film) 2929, 2858, 1704, 1598, 1458, 1477, 1443, 1395, 1354, 1339, 1272, 1190, 959, 896, 736, 702 cm−1.
(5aR,10aS)-13-((4-Chlorophenyl)sulfonyl)-1,3,5-trimethyl-10-methylene-7,8,9,10-tetra-hydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole (6g)
The title compound was prepared through Method D from compound 5 and 4-chlorobenzenesulfonyl chloride. TLC (hexanes:ethyl acetate, 8:1 v/v) Rf = 0.20; yellow oil, 40%; 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 8.6 Hz, 1H), 7.42 (d, J = 8.6 Hz, 2H), 6.26 (s, 1H), 6.11 (s, 1H), 5.01 (s, 1H), 4.95 (s, 1H), 3.31 (dd, J = 10.0, 6.7 Hz, 1H), 3.23 (s, 1H), 3.07-2.93 (m, 4H), 2.26 (s, 4H), 2.17-2.10 (m, 1H), 2.08 (s, 3H), 1.80-1.63 (m, 4H), 1.45 (p, J = 12.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 151.52, 150.09, 140.06, 138.71, 138.58, 133.66, 129.31, 129.23, 129.09, 128.58, 123.34, 121.85, 113.52, 104.44, 99.31, 66.84, 48.50, 33.52, 32.05, 31.98, 30.74, 24.27, 21.72, 17.24; ESI m/z calcd for C25H29ClN2O2S [M+H]+ 457.2, found 457.1; IR (thin film) 2925, 2858, 1598, 1480, 1335, 1160, 1093, 884, 825, 736 cm−1.
Tert-butyl (5aR,10aS)-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxylate (6c)
To a solution of triphosgene (9.5 mg, 1.0 equiv) in anhydrous CH2Cl2 (0.8 mL) at -78 °C was added pyridine (21 μL). The mixture was stirred for 10 min at -78 °C. A solution of substrate 5 (9.0 mg, 1.0 equiv) in CH2Cl2 (0.4 mL) was added to the above solution slowly. The resulting solution was stirred at -78 °C for 30 min before warmed to 0 °C over 1 h and stirred for another 30 min at 0°C. A solution of °BuOH (22 mg, 10 equiv) and Et3N (44 μL, 10 equiv) in anhydrous CH2Cl2 (0.8 mL) was added to the above solution at 0 °C and stirred for 2 h before quenched by aqueous NaHCO3, diluted by ethyl acetate, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give product 6c. TLC (hexanes:ethyl acetate, 3:1 v/v) Rf = 0.20; yellow oil, 46%; 1H NMR (400 MHz, CDCl3) δ 6.17 (s, 1H), 6.05 (s, 1H), 5.19 (s, 1H), 5.07 (s, 1H), 3.01 (dd, J = 11.3, 6.2 Hz, 1H), 2.85 (s, 3H), 2.59-2.52 (m, 1H), 2.46 (td, J = 11.6, 4.3 Hz, 1H), 2.28-2.15 (m, 5H), 2.06 (s, 3H), 1.98 (dd, J = 11.8, 4.2 Hz, 1H), 1.81-1.72 (m, 2H), 1.68-1.62 (m, 1H), 1.59-1.53 (m, 1H), 1.47 (s, 9H), 1.32-1.27 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 174.67, 153.57, 151.09, 137.92, 133.45, 125.92, 120.16, 112.84, 103.59, 93.96, 81.09, 67.37, 53.13, 46.17, 37.54, 34.60, 30.34, 28.24, 27.93, 21.78, 17.38; ESIm/z calcd for C24H34N2O2 [M+H]+ 383.2, found 383.2; IR (thin film) 2929, 2858, 1730, 1596, 1458, 1369, 1141, 896, 821, 736 cm−1.
Acetylation of 5 using anhydrides to afford analogues 6d and 6e (Method E)
To a solution of 5 (1.0 equiv), Et3N (6.0 equiv) and DMAP (0.2 equiv) in CH2Cl2 (0.03 M) at 0 °C was added anhydride (Ac2O or (CF3CO)2O, 3.0 equiv). The resulting mixture was slowly warmed to room temperature and stirred overnight before quenched by aqueous NaHCO3, diluted by ethyl acetate, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give product 6d or 6e.
1-((5aR,10aS)-1,3,5-Trimethyl-10-methylene-7,8, 9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cy-clohepta[b]indol-13-yl)ethan-1-one (6d)
The title compound was prepared through Method E. TLC (hexanes:ethyl acetate, 5:1 v/v) Rf = 0.20; colorless oil, 90%; 1H NMR (400 MHz, CDCl3) δ 6.25 (s, 1H), 6.10 (s, 1H), 5.01 (s, 1H), 5.00 (s, 1H), 3.53 (dd, J = 10.0, 7.3 Hz, 2H), 3.17 (ddd, J = 12.4, 9.8, 5.2 Hz, 1H), 3.02 (s, 3H), 2.48 (dd, J = 12.4, 5.1 Hz, 1H), 2.29-2.27 (m, 4H), 2.15 (s, 3H), 2.12-2.04 (m, 1H), 1.98 (s, 3H), 1.82-1.64 (m, 3H), 1.63-1.43 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 170.15, 152.08, 151.11, 138.60, 133.94, 123.06, 121.29, 112.66, 104.22, 95.09, 65.22, 48.95, 33.26, 32.48, 32.41, 31.24, 29.46, 25.79, 24.32, 21.77, 17.20; ESI m/z calcd for C21H28N2O [M+H]+ 325.2, found 325.2; IR (thin film) 2925, 2858, 1655, 1598, 1395, 1275, 1194, 1032, 896, 825 cm−1.
2,2,2-Trifluoro-1-((5aR,10aS)-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indol-13-yl)ethan-1-one (6e)
The title compound was prepared through Method E. TLC (hexanes:ethyl acetate, 6:1 v/v) Rf = 0.20; colorless oil, 78%; 1H NMR (500 MHz, CDCl3) δ 6.80 (s, 1H), 6.75 (s, 2H), 5.03 (s, 1H), 4.71 (s, 1H), 3.13 (s, 3H), 2.99 (dq, J = 13.7, 6.9 Hz, 1H), 2.93-2.78 (m, 3H), 2.68 (dq, J = 18.8, 7.1 Hz, 2H), 2.61-2.48 (m, 3H), 2.38 (s, 3H), 2.32 (s, 5H), 2.10-2.01 (m, 1H), 1.81-1.54 (m, 3H);13C NMR (101 MHz, CDCl3) δ 175.32, 146.58, 143.95, 139.52, 135.49, 128.71, 128.04, 114.21, 109.42, 99.84, 62.89, 37.98, 36.33, 34.98, 31.68, 31.38, 24.32, 21.58, 18.88; ESI m/z calcd for C21H22F3N2O [M+H]+ 379.2, found 379.1; IR (thin film) 3257, 3093, 2929, 2858, 1707, 1491, 1383, 1175, 1160, 1130, 1056, 1037, 914, 840, 732 cm−1.
4-Oxo-4-((5aR,10aS)-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indol-13-yl)butanoic acid (6f)
To a solution of compound 5 (1.0 equiv), N,N-diiso-propylethylamine (DIPEA, 8.0 equiv) and 4-dimethyl-aminopyridine (DMAP, 0.2 equiv) in anhydrous CH2Cl2 (0.03M) at 0 °C was added succinic anhydride (5.0 equiv). The mixture was stirred at room temperature overnight before quenched by aqueous HCl (1.0 M), diluted by ethyl acetate, washed with brine, dried over Na2SO4, filtered, concentrated to afford compound 6f. 1H NMR (400 MHz, CDCl3) δ 6.26 (s, 1H), 6.09 (s, 1H), 5.00 (s, 2H), 3.54 (dd, J = 9.8, 7.4 Hz, 1H), 3.46 (dd, J = 14.0, 6.5 Hz, 1H), 3.12 (ddd, J = 12.5, 9.9, 5.3 Hz, 1H), 2.98 (s, 3H), 2.67-2.41 (m, 6H), 2.25 (s, 3H), 2.13 (s, 3H), 2.10-2.02 (m, 1H), 1.78-1.64 (m, 2H), 1.63-1.43 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 175.10, 172.07, 151.72, 150.69, 138.82, 134.02, 122.63, 121.73, 112.98, 104.55, 96.31, 64.97, 47.97, 33.09, 32.47, 32.40, 31.75, 31.21, 30.50, 29.34, 24.22, 21.76, 17.18; ESI m/z calcd for C23H30N2O3 [M+H]+ 383.2, found 383.2.
Amino((5a R,10aS)-1,3-dimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indol-13-yl)methaniminium 2,2,2-tri-fluoroacetate (6k)
To a solution of 5 (1.0 equiv), Et3N (4.0 equiv) in ClCH2CH2Cl (0.070 M), at 0 °C was added N,N′-di-Boc-N″-trifluoromethanesulfonylguanidine (6.0 equiv) under argon. The mixture was stirred at room temperature for 3 days before diluted with ethyl acetate, washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vauo. The residue was purified by column chromatography on silica gel to give product Boc-6k.TLC (hexanes:ethyl acetate, 2:1 v/v) Rf = 0.20; yellow oil, 38%; 1H NMR (400 MHz, CDCl3) δ 6.44 (s, 1H), 6.36 (s, 1H), 5.21 (d, J = 11.4 Hz, 1H), 5.05 (d, J = 8.8 Hz, 2H), 4.95 (d, J = 11.4 Hz, 1H), 3.72 (dd, J = 11.6, 8.3 Hz, 1H), 3.02 (td, J = 11.8, 6.5 Hz, 1H), 2.69 (dd, J = 13.0, 6.5 Hz, 1H), 2.40-2.34 (m, 1H), 2.33-2.27 (m, 1H), 2.23 (s, 3H), 2.18 (s, 3H), 2.07 (d, J = 6.3 Hz, 1H), 2.05-2.02 (m, 1H), 1.84 (d, J = 13.6 Hz, 1H),1.78-1.68 (m, 2H), 1.48 (s, 12H), 1.41 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 151.57, 150.23, 148.76, 139.16, 135.85, 125.33, 123.85, 120.96, 117.77, 114.31, 108.77, 90.12, 64.26, 58.55, 47.50, 32.87, 32.65, 32.20, 30.09, 28.45, 28.29, 28.10, 27.95, 21.59, 17.20; IR (thin film) 3301, 2985, 2933, 1789, 1737, 1559, 1328, 1257, 1190, 1130, 1104, 821, 788, 728 cm−1. To a solution of the Boc-6k (16 mg) in CH2Cl2 (1.2 mL) at 0 °C was added trifluoroacetic acid (1.2 mL). The mixture was stirred for 12 h before concentrated in vacuo to afford 6k: white solid, 95%. 1H NMR (500 MHz, Methanol-d4) δ 6.64 (s, 1H), 6.57 (s, 1H), 5.24-5.17 (m, 2H), 5.14 (s, 1H), 4.70 (d, J = 13.4 Hz, 1H), 3.73 (t, J = 9.3 Hz, 1H), 3.07-2.96 (m, 1H), 2.90 (ddd, J = 11.7, 9.7, 7.2 Hz, 1H), 2.57 (ddd, J = 13.3, 11.6, 8.9 Hz, 1H), 2.29 (s, 3H), 2.27 (s, 4H), 2.20-2.09 (m, 3H), 1.95-1.87 (m, 2H), 1.82-1.73 (m, 2H); 13C NMR (101 MHz, Methanol-d4) δ 159.85, 153.15, 150.13, 147.58, 139.09, 136.06, 125.62, 113.99, 108.70, 90.20, 62.76, 50.91, 46.48, 32.29, 32.07, 31.18, 29.74, 21.89, 20.04, 15.85.
(5aR,10aS)-N-Benzyl-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxamide (6h)
To a solution of compound 5 (1.0 equiv), Et3N (4.0 equiv) in CH3CN (0.030 M) at 0 °C was added benzyl isocyanate (6.0 equiv). The mixture was stirred at room temperature for 2 h before diluted with ethyl acetate, washed with H2O and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give 6h. TLC (hexanes:ethyl acetate, 5:1 v/v) Rf = 0.20; yellow oil, 91%; 1H NMR (400 MHz, CDCl3) δ 7.58-7.03 (m, 5H), 6.21 (s, 1H), 6.08 (s, 1H), 4.98 (s, 1H), 4.42 (t, J = 5.5 Hz, 1H), 4.38-4.20 (m, 2H), 3.79-3.55 (m, 1H), 3.35 (td, J = 7.8, 1.2 Hz, 1H), 3.07-3.00 (m, 4H), 2.51-2.40 (m, 1H), 2.36-2.27 (m, 1H), 2.25 (s, 3H), 2.13 (s, 3H), 2.10-2.04 (m, 1H), 1.70 (d, J = 6.3 Hz, 2H), 1.57-1.45 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 156.78, 152.07, 151.22, 139.70, 138.50, 133.92, 128.70, 127.81, 127.33, 123.37, 121.08, 112.62, 104.18, 94.92, 65.19, 46.51, 44.66, 33.36, 32.52, 32.07, 31.10, 31.05, 24.38, 21.77, 17.24; ESI m/z calcd for C27H33N3O [M+H]+ 416.2, found 416.2; IR (thin film) 3376, 2948, 2918, 2858, 1640, 1536, 1477, 1343, 1238, 1201, 1030, 974, 903, 821, 732 cm−1.
(5aR,10aS)-N-Ethyl-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole-13-carboxamide (6i)
TLC (hexanes:ethyl acetate, 5:1 v/v) Rf = 0.20; yellow oil, 50%; 1H NMR (300 MHz, CDCl3) δ 6.23 (s, 1H), 6.08 (s, 1H), 4.99 (d, J = 1.3 Hz, 2H), 4.08 (t, J = 5.4 Hz, 1H), 3.63 (dd, J = 15.2, 6.9 Hz, 1H), 3.33 (td, J = 7.7, 1.3 Hz, 1H), 3.17 (qd, J = 7.2, 5.4 Hz, 2H), 3.08-2.97 (m, 4H), 2.47 (ddd, J = 12.2, 5.3, 1.2 Hz, 1H), 2.32 (dd, J = 12.0, 7.7 Hz, 1H), 2.28 (s, 3H), 2.15 (s, 3H), 2.12-2.03 (m, 1H), 1.77-1.45 (m, 6H), 1.08 (t, J = 7.2 Hz, 3H); ESI m/z calcd for C22H31N3O [M+H]+ 354.2, found 354.2.
(5aR,10aS)-1,3,5-Trimethyl-10-methylene-7,8,9, 10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclohepta[b]indole-13-carboxamide (6j)
To a solution of compound 5 (1.0 equiv), N,N-diisopropylethylamine (DIPEA, 6.0 equiv) in CH2Cl2 (0.030 M) at -78 °C was added trichloroacetyl isocyanate (6.0 equiv). The mixture was warmed slowly to room temperature over 1 h, and stirred at room temperature for another 1.5 h before diluted with CH2Cl2, washed with H2O and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by column chromatography on silica gel to give 6j. TLC (hexanes:ethyl acetate, 2:3 v/v) Rf = 0.20; yellow oil, 40%; 1H NMR (500 MHz, CDCl3) δ 6.26 (s, 1H), 6.11 (s, 1H), 5.01 (s, 1H), 4.26 (s, 2H), 3.58 (d, J = 14.5 Hz, 1H), 3.39 (t, J = 7.9 Hz, 1H), 3.11 (ddd, J = 11.8, 8.2, 5.3 Hz, 1H), 3.04 (s, 3H), 2.51 (dd, J = 12.3, 5.2 Hz, 1H), 2.34 (td, J = 11.4, 10.7, 6.9 Hz, 1H), 2.28 (s, 3H), 2.17 (s, 3H), 2.10 (d, J = 13.7 Hz, 2H), 1.79-1.61 (m, 2H), 1.58-1.48 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 154.03, 152.02, 151.13, 138.57, 133.95, 123.20, 121.21, 112.68, 104.29, 94.81, 68.05, 65.24, 47.48, 33.29, 32.52, 31.99, 31.06, 30.60, 24.35, 21.76, 17.23; ESI m/z calcd for C20H27N3O [M+H]+ 326.2, found 326.2; IR (thin film) 3413, 3339, 2925, 2858, 1722, 1652, 1592, 1417, 1361, 1235, 1201, 1022, 959, 896, 825 cm−1.
Reduction of 6a or 6d to afford compound 6l and 6m (Method F)
To a solution of substrate (6a or 6d) in THF (0.025 M), at 0 °C was added LiAlH4 (1.0 M in THF, 5.0 equiv). The mixture was stirred at 65 °C for 1.0 h before cooled to 0 °C and quenched with ethyl acetate followed by aqueous NaHCO3. The mixture was then diluted with ethyl acetate, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give product 6l or 6m.
(5aS,10aS)-1,3,5,13-Tetramethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epimino-ethano)cyclohepta[b]indole (6l)
The title compound was prepared through Method F from 6a. TLC (CH2Cl2: MeOH, 9:1 v/v) Rf = 0.20; yellow oil, 98%; 1H NMR (400 MHz, CDCl3) δ 6.24 (s, 1H), 6.03 (s, 1H), 5.20 (s, 1H), 5.10 (s, 1H), 3.10 (s, 1H), 2.93 (s, 3H), 2.61 (td, J = 12.1, 5.6 Hz, 1H), 2.54-2.47 (m, 1H), 2.43 (s, 3H), 2.38-2.30 (m, 1H), 2.25 (s, 3H), 2.20 (dt, J = 12.4, 1.9 Hz, 1H), 2.07 (s, 4H), 1.83-1.50 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 152.28, 149.74, 138.41, 133.80, 125.74, 121.15, 113.82, 107.73, 102.64, 65.48, 53.42, 35.67, 34.75, 32.60, 32.12, 31.78, 23.60, 21.72, 17.24, 14.28; ESI m/z calcd for C20H28N2 [M+H]+ 297.2, found 297.2; IR (thin film) 2925, 2854, 1596, 1462, 1443, 1235, 1030, 896, 817 cm−1.
(5aS,10aS)-13-Ethyl-1,3,5-trimethyl-10-methylene-7,8,9,10-tetrahydro-5H,6H-5a,10a-(epiminoethano)cyclohepta[b]indole (6m)
The title compound was prepared through Method F from compound 6d. TLC (CH2Cl2:MeOH, 9:1 v/v) Rf = 0.20; yellow oil, 87%; 1H NMR (500 MHz, CD2Cl2) δ 6.15 (s, 1H), 5.94 (s, 1H), 5.16 (s, 1H), 5.08 (s, 1H), 3.03 (dd, J = 8.4, 6.2 Hz, 1H), 2.90 (dd, J = 11.6, 7.4 Hz, 1H), 2.86 (s, 3H), 2.39 (td, J = 11.7, 6.2 Hz, 1H), 2.33-2.25 (m, 2H), 2.24 (s, 4H), 2.22-2.15 (m, 2H), 2.07 (s, 3H), 1.96 (dd, J = 11.6, 4.3 Hz, 1H), 1.70-1.61 (m, 2H), 1.51-1.41 (m, 3H), 1.07 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CD2Cl2) δ 153.01, 151.66, 137.39, 133.09, 126.50, 119.26, 112.02, 101.26, 65.20, 48.82, 42.73, 34.85, 32.66, 32.36, 32.02, 31.63, 29.65, 23.52, 21.21, 16.95, 14.54; ESI m/z calcd for C21H30N2 [M+H]+ 311.2, found 311.2; IR (thin film) 2925, 2854, 1596, 1484, 1458, 1227, 1160, 963, 896, 817 cm−1.
Supplementary Material
A novel class of tetracyclic indolines was discovered in a β-lactam potentiator screen in MRSA
The compound selectively potentiates β-lactams but not any other classes of antibiotics
This novel class of compounds does not have antibacterial activity on its own, nor do they inhibit the β-lactamase activity directly
Structure-activity relationship (SAR) study identified a more potent analogue with reduced mammalian cell toxicity
Acknowledgments
We are grateful to Dr. Ann Eakin (Concept Acceleration Program, National Institute of Allergy and Infectious Diseases) for helpful discussions. The following reagents were provided by the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for distribution by BEI Resources, NIAID, NIH: Staphylococcus aureus strains NR-45898, NR-46231, and NR-46070. Funding for this project was provided by Colorado Bioscience Discovery Evaluation Grant Program and National Institute of Health under grant number R21-AI121581.
ABBREVIATIONS
- RMA
resistance-modifying agent
- CLSI
Clinical Laboratory Standards Institutes
- MRSA
methicillin-resistant Staphylococcus aureus
- MIC
minimum inhibitory concentration
- MRC
minimum re-sensitizing concentration
- GI50
half-growth inhibitory concentration
- HA-MRSA
hospital-acquired MRSA
- CA-MRSA
community-acquired MRSA
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alharbi SA, Wainwright M, Alahmadi TA, Salleeh HB, Faden AA, Chinnathambi A. What if Fleming had not discovered penicillin? Saudi J Biol Sci. 2014;21:289–293. doi: 10.1016/j.sjbs.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleming A. PA Norstedt & Söner. 1947. Nobel lecture on penicillin. [Google Scholar]
- 3.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 4.Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest. 2003;111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stryjewski ME, Corey GR. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clin Infect Dis. 2014;58:10–19. doi: 10.1093/cid/cit613. [DOI] [PubMed] [Google Scholar]
- 6.Center for Disease Control and Prevention. Antibiotic Resistance Threats. CDC; Atlanta, GA: 2013. [Google Scholar]
- 7.Coates ARM, Halls G, Hu Y. Novel classes of antibiotics or more of the same? Br J Pharmacol. 2011;163:184–194. doi: 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 9.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 10.Abreu AC, McBain AJ, Simoes M. Plants as Sources of new antimicrobials and resistance-modifying agents. Nat Prod Rep. 2012;29:1007–1021. doi: 10.1039/c2np20035j. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo, She X, Kales S, Liang L, Liu J, Wu J, Maria JS, Su J, Pan J, Hailey J, Mcguiness D, Tan CM, Flattery A, Walker S, Black T, Roemer T. Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem Biol. 2013;20:272–284. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 13.Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 14.Pasquina L, Santa Maria JP, Jr, Wood BM, Moussa SH, Matano LM, Santiago M, Martin SE, Lee W, Meredith TC, Walker S. A synthetic lethal approach for compound and target identification in Staphylococcus aureus. Nat Chem Bio. 2016;12:40–45. doi: 10.1038/nchembio.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lupoli TJ, Lebar MD, Markovski M, Bernhardt T, Kahne D, Walker S. Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J Am Chem Soc. 2013;136:52–55. doi: 10.1021/ja410813j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasquina LW, Santa Maria JP, Walker S. Teichoic acid biosynthesis as an antibiotic target. Curr Opin Microbiol. 2013;16:531–537. doi: 10.1016/j.mib.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J. Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem Biol. 2013;20:272–284. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown S, Xia G, Luhachack LG, Campbell J, Meredith TC, Chen C, Winstel V, Gekeler C, Irazoqui JE, Peschel A, Walker S. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc Natl Acad Sci U S A. 2012;109:18909–18914. doi: 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki T, Swoboda JG, Campbell J, Walker S, Gilmore MS. In vitro antimicrobial activity of wall teichoic acid biosynthesis inhibitors against Staphylococcus aureus isolates. Antimicrob Agents Chemother. 2011;55:767–774. doi: 10.1128/AAC.00879-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalia VC. Quorum sensing inhibitors: an overview. Biotechnol Adv. 2013;31:224–245. doi: 10.1016/j.biotechadv.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 21.O’Loughlin CT, Miller LC, Siraporn A, Drescher K, Semmelhack MF, Bassler BL. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A. 2013;110:17981–17986. doi: 10.1073/pnas.1316981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huigens RW, III, Richards JJ, Parise G, Ballard TE, Zeng W, Deora R, Melander C. Inhibition of Pseudomonas aeruginosa biofilm formation with Bromoageliferin analogues. J Am Chem Soc. 2007;129:6966–6967. doi: 10.1021/ja069017t. [DOI] [PubMed] [Google Scholar]
- 23.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King AM, Reid-Yu SA, Wang W, King DT, DePascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature. 2014;510:503–506. doi: 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilke KE, Francis S, Carlson EE. Inactivation of Multiple Bacterial Histidine Kinases by Targeting the ATP-Binding Domain. ACS Chem Biol. 2015;10:328–335. doi: 10.1021/cb5008019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris TL, Worthington RJ, Hittle LE, Zurawski DV, Ernst RK, Melander C. Small molecule downregulation of PmrAB reverses lipid A modification and breaks colistin resistance. ACS Chem Biol. 2014;9:122–127. doi: 10.1021/cb400490k. [DOI] [PubMed] [Google Scholar]
- 27.Beesley T, Gascoyne N, Knott-Hunziker V, Petursson S, Waley SG, Jaurin B, Grundström T. The inhibition of class C beta-lactamases by boronic acids. Biochem J. 1983;209:229–233. doi: 10.1042/bj2090229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Defoirdt T, Boon N, Bossier P. Can bacteria evolve resistance to quorum sensing disruption? PLoS Pathog. 2010;6:e1000989. doi: 10.1371/journal.ppat.1000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geddes AM, Klugman KP, Rolinson GN. Introduction: historical perspective and development of amoxicillin/clavulanate. Int J Antimicrob Agents. 2007;30:109–112. doi: 10.1016/j.ijantimicag.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Rossolini GM. Acquired Metallo-β-Lactamases: An Increasing Clinical Threat. Clin Infect Dis. 2005;41:1557–1558. doi: 10.1016/j.ijantimicag.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 31.Therien AG, Huber JL, Wilson KE, Beaulieu P, Caron A, Claveau D, Deschamps K, Donald RG, Galgoci AM, Gallant M, Gu X. Broadening the spectrum of β-lactam antibiotics through inhibition of signal peptidase type I. Antimicrob Agents Chemother. 2012;56:4662–4670. doi: 10.1128/AAC.00726-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worthington RJ, Melander C. Overcoming resistance to β-lactam antibiotics. J Org Chem. 2013;78:4207–4213. doi: 10.1021/jo400236f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Podoll JD, Liu Y, Chang L, Walls S, Wang W, Wang X. Bio-inspired synthesis yields a tricyclic indoline that selectively resensitizes methicillin-resistant Staphylococcus aureus (MRSA) to β-lactam Antibiotics. Proc Natl Acad Sci U S A. 2013;110:15573–15578. doi: 10.1073/pnas.1310459110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Connor SE, Maresh JJ. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/B512615K. [DOI] [PubMed] [Google Scholar]
- 35.Xu W, Wang W, Wang X. Gold-catalyzed cyclization leads to a bridged tetracyclic indolenine that represses β-lactam resistance. Angew Chem Int Ed Engl. 2015;54:9546–9549. doi: 10.1002/anie.201503736. [DOI] [PubMed] [Google Scholar]
- 36.Barbour PM, Podoll JD, Marholz LJ, Wang X. Discovery and initial structure– activity relationships of N-benzyl tricyclic indolines as antibacterials for methicillin-resistant Staphylococcus aureus. Bioorg Med Chem Lett. 2014;24:5602–5605. doi: 10.1016/j.bmcl.2014.10.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griffiths B, Burl J, Wang X. Bio-inspired discovery of chemical reactions and biological probes. Synlett. 2016 doi: 10.1055/s-0035-1561638. [DOI] [Google Scholar]
- 38.Chang L, Podoll JD, Wang W, Walls S, O’Rourke CP, Wang X. Structure–activity relationship studies of the tricyclic indoline resistance-modifying agent. J Med Chem. 2014;57:3803–3817. doi: 10.1021/jm500146g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barbour PM, Wang W, Chang L, Pickard KL, Rais R, Slusher BS, Wang X. Property-guided synthesis of aza-tricyclic indolines: development of gold catalysis en route. Adv Synth Catal. 2016;358:1482–1490. doi: 10.1002/adsc.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility testing for bacteria that grew aerobically. Clinical and Laboratory Standards Institute; Wayne, PA: 2009. Approved Standard M7-A10. [Google Scholar]
- 41.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattor M, Ogasawara N, Hayashi H, Hiramatsu K. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. The Lancet. 2001;357:1225–1240. doi: 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 42.Roberts JC, Krueger RL, Peak KK, Veguilla W, Cannons AC, Amuso PT, Cattani J. Community associated methicillin-resistant Staphylococcus aureus epidemic clone USA300 in isolates from Florida and Washington. J. Clin. Microbiol. 2006;44:225–226. doi: 10.1128/JCM.44.1.225-226.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sopirala MM, Mangino JE, Gebreyes WA, Biller B, Bannerman T, Balada-Llasat JM, Pancholi P. Synergy testing by Etest, microdilution checkerboard, and time-kill methods for pan-drug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother. 2010;54:4678–4683. doi: 10.1128/AAC.00497-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llarrul LI, Toth M, Champion MM, Mobashery S. Activation of BlaR1 protein of methicillin-resistant Staphylococcus aureus, its proteolytic processing, and recovery from induction of resistance. J Biol Chem. 2011;286:38148–38158. doi: 10.1074/jbc.M111.288985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.