

Abstract
PAM (Protein Associated with Myc) is an almost ubiquitously expressed protein that is one of the most potent inhibitors of adenylyl cyclase activity known so far. Here we show that PAM is localized at the endoplasmic reticulum in HeLa cells and that upon serum treatment PAM is recruited to the plasma membrane, causing an inhibition of adenylyl cyclase activity. We purified the serum factor that induced PAM translocation and identified it as sphingosine-1-phosphate (S1P). Within 15 min after incubation with S1P, PAM appeared at the plasma membrane and was detectable for up to 120 min. Sphingosine-1-phosphate induced adenylyl cyclase inhibition in two phases: an initial (1–10 min) and a late (20–240 min) phase. The initial adenylyl cyclase inhibition was Gi-mediated and PAM independent. In the late phase, adenylyl cyclase inhibition was PAM dependent and attenuated cyclic AMP (cAMP) signaling by various cAMP-elevating signals. This makes PAM the longest lasting nontranscriptional regulator of adenylyl cyclase activity known to date and presents a novel mechanism for the temporal regulation of cAMP signaling.
Keywords: adenylyl cyclase, cAMP, inhibitory G protein, PAM, sphingosine-1-phosphate
Introduction
PAM (Protein Associated with Myc) is an extremely large protein of 510 kDa that was originally identified by its ability to bind specifically to the N-terminus of myc (Guo et al, 1998). While PAM mRNA is expressed in almost every human tissue tested so far, a high PAM expression was found in pancreas, skeletal muscle, ovary, and human aortic endothelial cells. Moreover, its expression is exceptionally high in brain, spinal cord, dorsal root ganglia, and thymus (Guo et al, 1998; Scholich et al, 2001; Yang et al, 2002; Ehnert et al, 2004). Several physiological functions of PAM have recently been reported, including spinal nociceptive processing, synaptogenesis, senescence, and cell differentiation (Schaefer et al, 2000; Wan et al, 2000; Zhen et al, 2000; DiAntonio et al, 2001; Jiao et al, 2002; Semov et al, 2002; Burgess et al, 2004; Ehnert et al, 2004). Especially well documented is the involvement of mammalian PAM and its homologues in Drosophila (highwire) and Caenorhabditis elegans (rpm-1) in presynaptic terminal organization (Zhen et al, 2000; Burgess et al, 2004), the regulation of synaptic growth (Wan et al, 2000; DiAntonio et al, 2001; Burgess et al, 2004), and neurite growth and targeting (Schaefer et al, 2000; Burgess et al, 2004). Chromosomal deletions that impair PAM expression in mice were lethal at birth and PAM has been proposed to be a candidate gene for respiratory distress and ventilatory disorders that arise from defective neuronal control of breathing (Peterson et al, 2002; Burgess et al, 2004).
It becomes increasingly evident that PAM, through different domains, can associate with a variety of proteins, ultimately participating in the aforementioned diverse cellular functions. Using pull-down assays, Guo et al (1998) showed that a centrally located region of human PAM (residues 2413–2672) interacts with myc. Later, employing the yeast two-hybrid assay and co-immunoprecipitation, an interaction between the C-terminus of mammalian PAM (residues 4492–4641) and tuberin was demonstrated (Murthy et al, 2004). Tuberin is a protein that is involved in the development of an autosomal dominant disorder, tuberous sclerosis complex, which affects several organs including the brain, resulting in mental retardation and seizures. Finally, using the yeast two-hybrid system with a recombinant functional domain of adenylyl cyclase type V (AC; E.C.4.6.1.1) (Scholich et al, 1997; 1999; Wittpoth et al, 1999) as bait, as well as employing functional assays, we demonstrated that PAM interacts with AC through its N-terminal RCC1-like domain (residues 499–1066) (Scholich et al, 2001). While the functional consequences of the PAM–myc and the PAM–tuberin interactions remain unclear, PAM proved to be a potent regulator of cyclic AMP (cAMP) signaling that inhibits the enzymatic activity of several adenylyl cyclase isoforms at pico- and nanomolar concentrations (Scholich et al, 2001; Ehnert et al, 2004). The IC50 values for AC inhibition by PAM are therefore significantly lower than those observed with P-site inhibitors (Dessauer and Gilman, 1997; Tesmer et al, 2000) or α and βγ subunits of the inhibitory G protein, Gi (Taussig et al, 1994; Wittpoth et al, 1999).
Until now, nothing is known about signals or signaling pathways leading to activation of PAM. Identification of such signals will advance the understanding of the physiological functions of PAM in different tissues and cells. Therefore, in this study, we aimed to identify pathways that lead to the activation of the AC-inhibitory properties of PAM. We found that the bioactive sphingolipid metabolite sphingosine-1-phosphate (S1P) activates the AC-inhibitory actions of PAM in HeLa cells. PAM mediates a sustained inhibition of receptor- as well as forskolin-stimulated AC enzyme activity, making it the longest lasting nontranscriptional regulator of AC activity known to date. Thus, AC inhibition by PAM is able to bridge the gap in the temporal regulation of ACs between short-term (e.g. phosphorylation) and long-term (e.g. transcriptional) regulation.
Results
Serum treatment induces PAM translocation and PAM-dependent AC inhibition
Previously, our laboratory generated two antibodies that were raised against peptides corresponding to amino-acid residues 135–153 and 4601–4614 of human PAM (Ehnert et al, 2004). Signals generated in immunohistochemical experiments using both antibodies on tissue sections of adult rat brain and spinal cords were identical to those observed by in situ hybridization (Yang et al, 2002; Ehnert et al, 2004), demonstrating the specificity of these antibodies and allowing their use in immunohistochemical investigations.
Using these antibodies, we found in serum-starved HeLa cells an apparent colocalization of PAM with calnexin, a marker for the endoplasmic reticulum (ER) (Figure 1A). In contrast to a previous report stating a primarily nuclear localization of PAM (Guo et al, 1998), both of our antibodies exhibited only little nuclear immunostaining. However, the antibody described by Guo et al (1998) was raised against a portion of the C-terminus of PAM that includes several common protein motifs such as C2H2 and ring zinc-fingers, as well as putative B-box motifs (Guo et al, 1998). Therefore, cross-recognition with other proteins might occur and limit the value of this antibody for immunohistological experiments.
Figure 1.
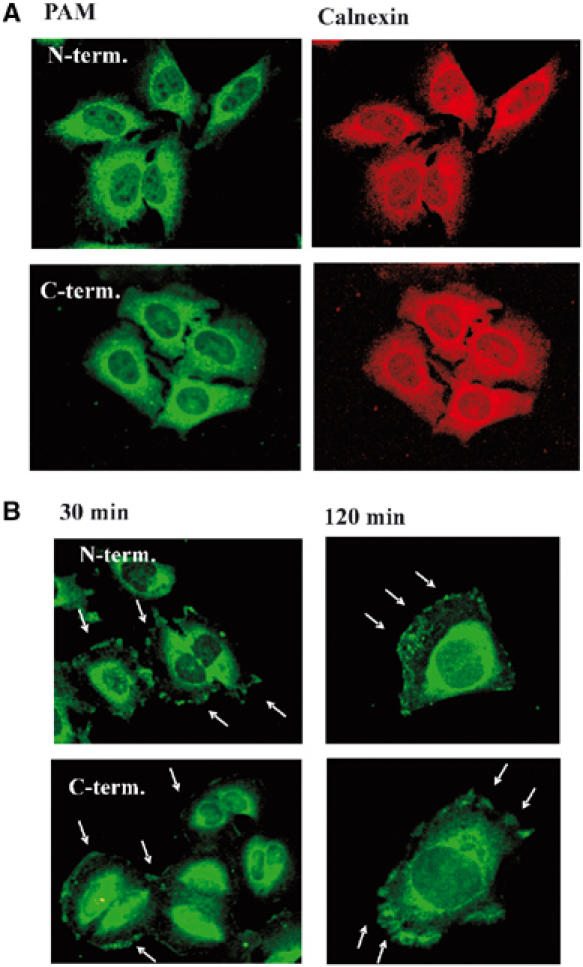
PAM is recruited from the ER to the plasma membrane in HeLa cells upon serum stimulation. (A) Serum-starved HeLa cells (24 h) were fixed and stained with anti-PAM antibodies raised against peptides corresponding to the N- or C-terminus of PAM (green) and anti-calnexin antibody (red) as described in Materials and methods. (B) HeLa cells were serum starved for 24 h, then treated with 10% fetal bovine serum for 30 or 120 min and stained using the N- and C-terminal anti-PAM antibodies to monitor the subcellular localization. The Arrows indicate membrane localization of PAM.
After a 20–30 min incubation of serum-starved HeLa cells with 10% fetal bovine serum, a partial translocation of PAM to the plasma membrane was observed, which was visible for up to 2 h. Similar results were obtained with both antibodies (Figure 1B). Although the majority of cells showed a regular distribution of the translocated PAM along the membrane, in some cells PAM was found in ruffle-like structures (Figure 1B). Since PAM is a potent inhibitor of AC enzyme activity in HeLa, we investigated if translocation of PAM to the plasma membrane was associated with an inhibition of AC activity. Indeed, serum treatment of HeLa cells reduced basal intracellular cAMP accumulation (Figure 2A) as well as Gαs- and forskolin-stimulated AC activity in isolated HeLa membranes (Figure 2B). To determine if this serum-induced decrease of AC activity was mediated by PAM, we lowered the amount of endogenous PAM employing antisense oligonucleotides (ODNs) against PAM as previously described (Scholich et al, 2001). As shown in Figure 2C, in HeLa cells treated with antisense ODN the amount of PAM, as determined by Western analysis, was decreased by 60% as compared to cells treated with sense or mutant antisense ODNs. Reprobing the same membrane with anti-Hsp70 antibody showed equal loading of protein (Figure 2C). The treatment of HeLa cells with PAM-antisense ODNs abolished the serum-induced inhibition of Gαs- and forskolin-stimulated AC activity (Figure 2D). Importantly, HeLa cells transfected with sense or mutated antisense ODNs still exhibited the serum-induced inhibition of Gαs- and forskolin-stimulated AC activity to the same extent as untransfected cells (compare Figure 2B and D). These data suggest that endogenous PAM exerts an inhibitory influence on AC activity after stimulation of HeLa cells with serum.
Figure 2.
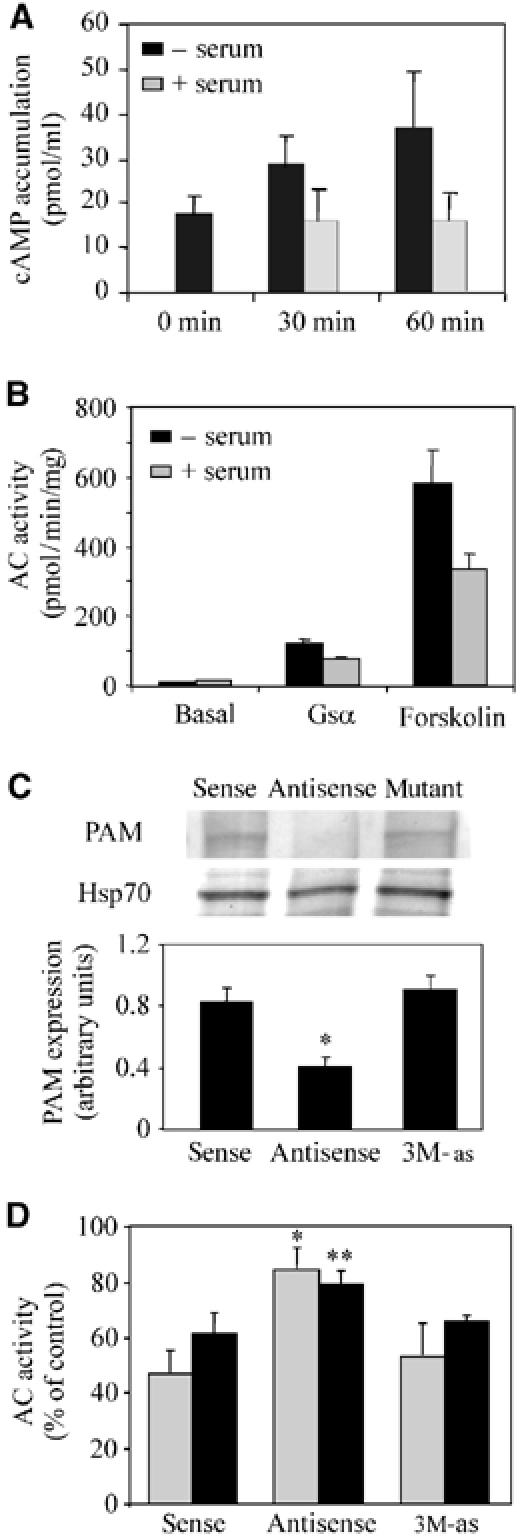
Inhibition of AC enzyme activity after serum treatment of HeLa cells is PAM dependent. (A) Basal cAMP accumulation in HeLa cells was measured in the presence of 100 μM IBMX and in the absence and presence of 10% serum either directly (0 min) or after 30 and 60 min incubation, as described in Materials and methods. The mean of five independent experiments±s.e.m. is shown. (B) Adenylyl cyclase activity determined in the presence of 80 nM Gαs* or 100 μM forskolin in membrane preparations of HeLa cells incubated for 1 h with (gray bars) or without serum (black bars). The mean±s.e. of 2–5 experiments, each performed in triplicates, is shown. (C) Serum-starved HeLa cells were transfected with 3 μM each of antisense, sense, and antisense ODNs harboring three point mutations (3M-as) as described in Materials and methods. Cells were harvested and subjected to Western blot analysis using a 7% SDS–PAGE (30 μg protein/lane). Even protein loading was checked by reprobing the membrane with an anti-Hsp70 antibody. The lower panel shows the densitometric evaluation of PAM expression of three independent experiments. Student's t-test for expression compared to cells transfected with sense ODNs: *P⩽0.05. (D) Serum-starved HeLa cells were treated with sense, antisense, and mutated (3M-as) ODNs. After 24 h, the cells were incubated for 1 h with 10% FBS. Cells were harvested and AC activity in the presence of Gαs* (80 nM; gray bars) and forskolin (100 μM; black bars) was determined. The data are presented as % AC activity of the transfected, serum-treated cells compared to transfected, untreated cells (100%). Data from at least three experiments measured in triplicate are presented as the mean±s.e. Student's t-test for activities compared to cells transfected with sense ODNs: *P⩽0.05, **P⩽0.01. Forskolin-stimulated AC activities were 45.3±10 pmol/mg/min (sense), 68.7±12 pmol/mg/min (antisense), and 59.6±13 pmol/mg/min (3M-as).
Sphingosine-1-phosphate induces PAM-mediated inhibition of AC enzyme activity
The serum component responsible for PAM translocation was purified using a series of hydrophobic, anionic exchange, and gel-filtration columns as detailed in Figure 3A. Several important observations were made concerning the physical properties of the substance by determining its binding abilities to the different resins. First, since heat inactivation of the serum or protease cleavage of compound-enriched fractions was not interfering with serum-induced PAM translocation, proteins or peptides were excluded as the active factors (data not shown). Second, its binding to a phenyl-sepharose column suggested the presence of a hydrophobic group. Third, its strong interaction with anionic exchangers points towards a strong negative charge in the substance. Finally, comparison of its elution profile with molecular weight standards using a Superdex 30pg gel-filtration column allowed us to estimate a molecular weight for the serum factor of 350–450 (Figure 3B). Based on the physical properties stated above, we hypothesized that the substance might be a phospholipid. Therefore, we used the HPLC analysis and labeling protocol for the detection of phospholipids, as described by Caligan et al (2000), and detected two substances, A and B, with retention times of 6 and 29 min, respectively (Figure 3C). Repeated freeze–thawing decreased the amount of substance B and increased the amount of substance A, suggesting that substance A is a degradation product of substance B. Accordingly, fractions containing only substance A did not induce PAM translocation, while fractions containing substance B were able to induce PAM translocation. The retention time of substance B was found to be the same as for S1P (Figure 3C).
Figure 3.
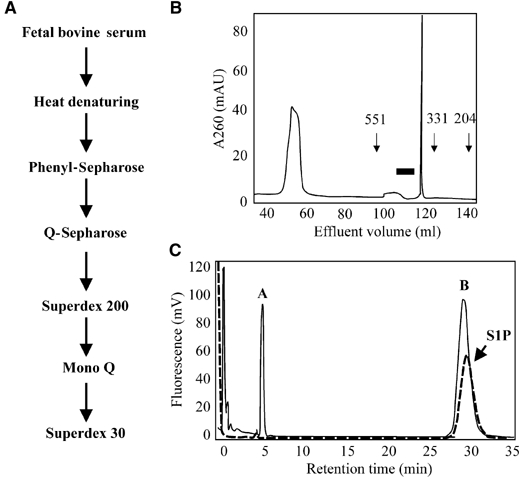
Purification of the PAM-activating serum factor. (A) Schematic representation of the purification strategy for the serum factor responsible for PAM translocation. (B) Gel-filtration analysis of the PAM-activating factor using a Superdex 30pg column as described in Materials and methods. Elution of substances was monitored using UV detection at 260 nm. The arrows indicate the elution times for size standards. The black bar indicates fractions that induced PAM translocation and AC inhibition in HeLa cells. (C) HPLC analysis of the pooled positive fractions (solid line) after the last purification step in comparison to an S1P standard (dotted line). The samples were subjected to phthaldialdehyde labeling for detection of sphingosine and its derivates, as described by Caligan et al (2000).
Commercially available S1P exhibited basically the same properties towards PAM activation and translocation as fetal bovine serum. Similar to that observed after serum treatment, HeLa cells that were treated for 20 min with 0.5 μM S1P exhibited a partial translocation of PAM to the plasma membrane (Figure 4A). Membrane localization of PAM was demonstrated by colocalization with the multi-drug resistance transporter 1 (Mdr1; Figure 4B). Incubation with increasing amounts of S1P (0.01–10 μM) showed dose-dependent PAM translocation (Figure 4C). S1P concentrations that induced PAM translocation ranged from 0.1 to 3 μM (Figure 4C). Human plasma concentrations of S1P were determined to be around 0.2 μM (Igarashi and Yatomi, 1998). Since in various tissues the S1P concentrations can reach locally much higher values (Igarashi and Yatomi, 1998; Edsall and Spiegel, 1999), the S1P concentrations that induce PAM translocation in HeLa cells are within the physiological range. In cells incubated with 0.5 μM S1P, PAM was detected at the plasma membrane as early as 10 min after the beginning of treatment and reached a maximum after 30 min, with 70–80% of the cells showing PAM translocation (Figure 4C). The recruitment of PAM to the plasma membrane was specific for S1P, since neither sphingosine nor the structure-related lysophosphatidic acid was able to induce translocation of PAM at concentrations between 0.01 and 10 μM (data not shown).
Figure 4.
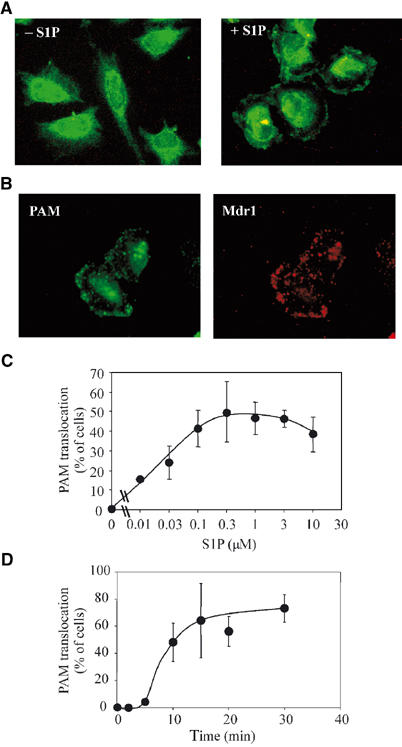
S1P induces the translocation of PAM to the plasma membrane. (A) Serum-starved HeLa cells were treated with 0.5 μM S1P for 30 min and stained with anti-PAM antibodies to monitor the subcellular localization of PAM. (B) Same as (A), except that membrane localization of PAM (green) is shown by costaining with Mdr1 (red). (C, D) HeLa cells were treated with increasing concentrations of S1P for 20 min (C) or with 0.5 μM S1P for varying times (D) and stained with anti-PAM antibodies to monitor the subcellular localization. The mean percentage of cells exhibiting translocation±variance of 2–5 experiments is shown.
Next, we investigated the actions of S1P on the AC activity in HeLa cells. Gαs*- and forskolin-stimulated AC activity in membrane preparations from cells treated with 0.5 μM S1P for different times presented similar temporal inhibition patterns (Figure 5A). An initial phase of inhibition was observed as early as 1 min after S1P incubation, followed by a recovery of AC enzyme activity to almost initial levels after 10–15 min of S1P treatment. Afterwards, a second phase of AC inhibition was seen following 20–60 min of S1P treatment. Notably, the time of appearance of the second inhibition phase coincides with the translocation of PAM to the membrane.
Figure 5.
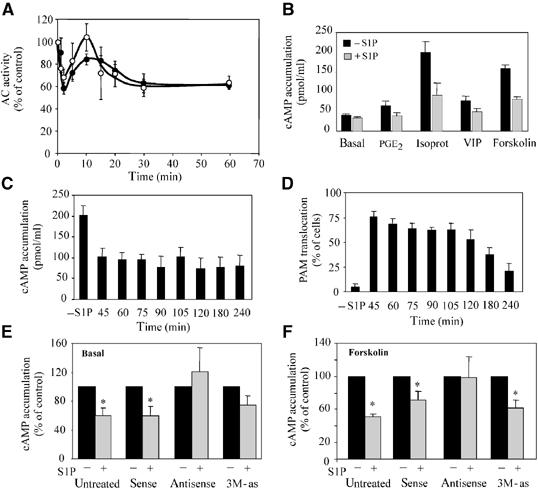
S1P induces PAM-dependent inhibition of AC enzyme activity in HeLa cells. (A) Serum-starved HeLa cells were treated for varying times with 0.5 μM S1P. Cells were harvested and AC activity in the presence of 80 nM Gαs* (black circles) or 100 μM forskolin (white circles) was determined as described in Materials and methods. Data from at least three experiments each measured in triplicate are presented as the mean±s.e.m. The data are shown as percentage of activity of untreated cells to allow better comparison. (B) After serum starving Hela cells for 24 h, fresh medium was added and the cells were treated for 15 min with 0.5 μM S1P. Then the medium was changed again and the cells were incubated for 5 min with 100 μM IBMX, followed by 15 min of incubation with either 300 nM PGE2, 100 nM VIP, 1 μM isoproterenol, or 5 μM forskolin in the presence of IBMX. The mean of 3–6 experiments±s.e.m. is shown. (C) Serum-starved Hela cells were given fresh serum-free medium and, except for the control, treated for 15 min with 0.5 μM S1P. Afterwards, the medium was exchanged against fresh serum-free medium. Then, after varying times, the cells were incubated for 5 min with 100 μM IBMX, followed by 15 min with 5 μM forskolin in the presence of IBMX. The total times after the beginning of S1P treatment are indicated. The mean of 2–7 experiments±s.e. is shown. (D) Same as (C), except that all cells were subjected to immunohistological treatment to determine the percentage of cells exhibiting PAM transloction. (E) Basal cAMP accumulation was measured in the absence and presence after 30 min of continuous incubation with 0.5 μM S1P and 100 μM IBMX. Prior to the S1P incubation, the cells were transfected with sense, antisense, and 3M-as ODNs as described in Materials and methods. Data of 6–7 experiments measured in triplicate are presented as the mean±s.e.m. The data are presented as % AC activity of transfected, S1P-treated cells (gray bars) compared to transfected, untreated cells (100%; black bars). Student's t-test to compare cells in the presence or absence of S1P: *P⩽0.05, mutated antisense ODN P⩽0.054. (F) The experimental setting was the same as in (B), except that the cells were transfected with sense, antisense, and 3M-as ODNs 24 h prior to incubation with S1P and forskolin. Data of five experiments measured in triplicate are presented as the mean±s.e.m. The data are presented as % AC activity of transfected, S1P-treated cells (gray bars) compared to transfected, untreated cells (100%; black bars). Student's t-test to compare cells in the presence or absence of S1P: *P⩽0.05.
Then we examined the consequences of the second-phase inhibition for stimulated cAMP accumulation. HeLa cells were pretreated with 0.5 μM S1P for 15 min, then fresh medium was added and the cells were incubated with IBMX along with the respective cAMP-elevating agent. The stimulated cAMP accumulation after treatment with prostaglandin E2 (PGE2), vasoactive intestinal peptide (VIP), isoproterenol or forskolin, a direct activator of enzyme activity of all AC isoforms, was decreased by 60–80% by S1P pretreatment (Figure 5B). Furthermore, we found that S1P preincubation attenuated forskolin-stimulated cAMP accumulation over a time period of up to 4 h (Figure 5C). During this time period, the degree of inhibition remained constant at 50% as compared to the cAMP accumulation in untreated cells. In all, 60–70% of the HeLa cells exhibited PAM translocation for nearly 2 h before the number of cells showing PAM translocation decreased gradually (Figure 5D).
To investigate the involvement of PAM in the late phase of S1P-mediated AC inhibition, we decreased the amount of endogenous PAM employing antisense ODNs as described above. Treatment of HeLa cells with 0.5 μM S1P reduced basal and forskolin-stimulated cAMP concentration in the cells after 30 min incubation by 40% (Figure 5E and F). Pretreatment of these cells with antisense ODN attenuated the S1P-induced decrease of basal and forskolin-stimulated cAMP accumulation (Figure 5E, F). In contrast, transfection of HeLa cells with sense ODNs had no influence on the S1P-induced decrease of cAMP accumulation (Figure 5E and F). PAM antisense ODNs carrying three mutations that are less effective than unaltered antisense ODNs (Scholich et al, 2001) were also unsuccessful in suppressing the S1P-induced decrease of cAMP accumulation (Figure 5E and F). Taken together, these data demonstrate that PAM mediates the inhibitory actions of S1P on AC enzyme activity during the late-phase inhibition. Previously, we reported that addition of exogenous PAM to HeLa membranes did not alter forskolin-stimulated AC enzyme activity (Scholich et al, 2001). However, because of technical restraints due to the size of PAM (510 kDa), a maximal PAM concentration of 30 nM was used in these experiments. Hence, inhibition of forskolin-stimulated AC enzyme activity in HeLa cells may require PAM concentrations above these relative low concentrations.
PAM activation by S1P is PTX-sensitive and requires PKC and PLC activity
Finally, we addressed the question whether PAM activation is achieved through one of the known S1P-activated signaling pathways or whether PAM activation by S1P represents a novel pathway. The occurrence of both, sustained inhibition of cAMP accumulation and PAM translocation, was independent of the time of incubation with S1P. The S1P-induced inhibition of cAMP accumulation and PAM translocation were the same if cells were pretreated with S1P for only 2.5 min or constantly incubated with S1P over 35 min (Figure 6A and B). This finding indicates the rapid initiation of a signal transduction pathway by S1P, which resulted in PAM translocation and finally in inhibition of AC enzyme activity.
Figure 6.
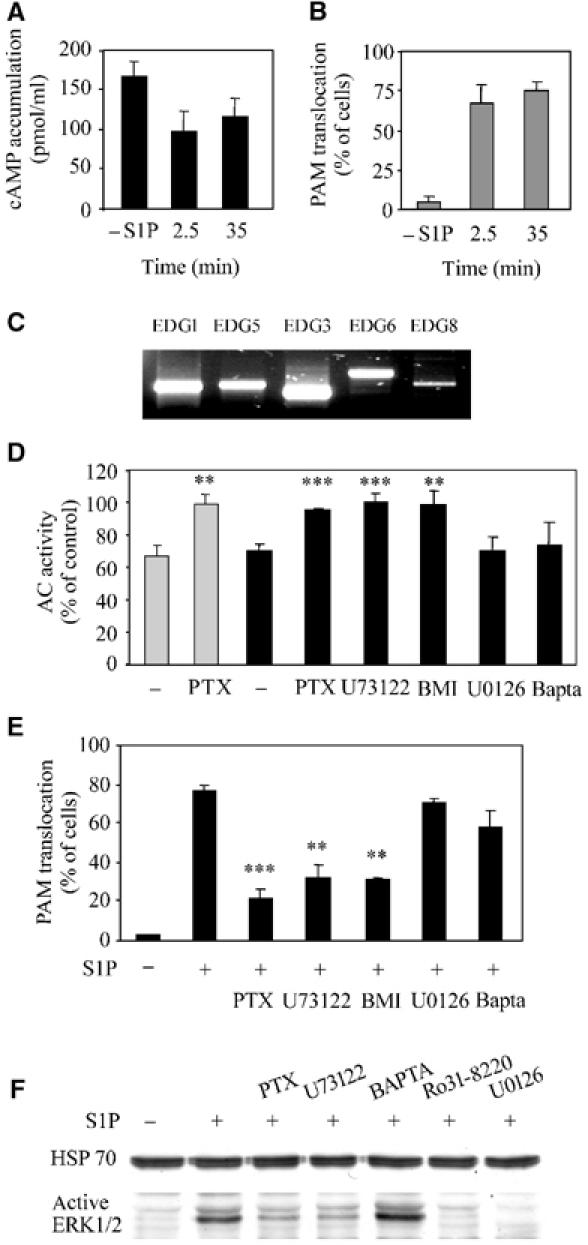
PAM-dependent inhibition of AC enzyme activity is mediated by PLC and PKC signaling. (A) Serum-starved Hela cells were given fresh serum-free medium and treated for 2.5 min or constantly for 35 min with 0.5 μM S1P. After the 2.5 min treatment, the medium was exchanged against fresh serum-free medium. At 15 min after beginning the respective S1P treatments, the cells were incubated for 5 min with 100 μM IBMX, followed by 15 min of incubation with 5 μM forskolin in the presence of IBMX. The mean±s.e.m. is shown. (B) Same as (A), except that all cells were subjected to immunohistological treatment to determine the percentage of cells exhibiting PAM transloction. (C) RT–PCR analysis of the S1P receptor isoform expression in serum-starved HeLa cells. (D) Serum-starved HeLa cells were treated for 3 min (gray bars) or 60 min (black bars) with 0.5 μM S1P. Prior to incubation with S1P, the cells were incubated for 24 h with 1 μg/ml PTX or 15 min with 10 μM U73122, BAPTA-AM, U0126, or 1 μM BMI. AC activity was determined with membrane preparations in the presence of 80 nM Gαs*. Data from 2–6 experiments, each measured in triplicate, are presented as the mean±s.e. Student's t-test: **P⩽0.025, ***P⩽0.005. (E) PAM translocation is mediated by PLC and PKC signaling. HeLa cells were treated with 0.5 μM S1P for 20 min and stained with anti-PAM antibodies to monitor the subcellular localization. Prior to S1P –treatment, the cells were incubated for 24 h with 1 μg/ml PTX or for 15 min with 1 μM BMI, 10 μM U73122, BAPTA-AM, Ro31-8220, or U0126. Data are presented as the mean±s.e. Student's t-test: **P⩽0.025, ***P⩽0.005. (F) Western blot analysis of HeLa cells treated with 0.5 μM S1P for 5 min. Prior to incubation with S1P, the cells were incubated for 24 h with 1 μg/ml PTX, 10 μM U73122, BAPTA-AM, Ro31-8220, or U0126. Cells were harvested in boiling Laemmli buffer containing 100 mM sodium phosphate buffer (pH 7.4) and 0.2 mM sodium orthovanadate. Samples were subjected to Western blot analysis using antiactive ERK1/2 antibody. Anti-Hsp70 antibody was used to control for even loading.
S1P is a natural ligand for a family of five G-protein-coupled receptors. The mRNAs for all the five known S1P-receptor isoforms were detected by semiquantitative RT–PCR in HeLa cells (Figure 6C). Four different G proteins, Gi, Gq, G12, and G13, can be activated by these receptors (Hla et al, 2001; Siehler and Manning, 2002; Spiegel and Milstien, 2002). Out of these four G proteins, only Gi is inhibited by pertussis toxin (PTX). Pretreatment of HeLa cells with PTX eliminated early and delayed inhibitory actions of S1P on Gαs*-stimulated AC activity (Figure 6D) as well as PAM translocation to the plasma membrane (Figure 6E), suggesting that PAM activation is mediated by Gi. Notably, neither PTX treatment nor treatment with any of the other inhibitors used in this study interfered by themselves with forskolin- or Gαs*-activated AC activity or induced PAM translocation (data not shown).
Gi activation by S1P receptors can stimulate phospholipase C (PLC) activity in several cell types (Kluk and Hla, 2002; Siehler and Manning, 2002; Spiegel and Milstien, 2002). Pretreatment of HeLa cells with the PLC inhibitor U73122 eliminated PAM translocation and delayed AC inhibition (Figure 6D and E). PLC converts phosphatidylinositol 4,5-biphosphate to inositol 1,4,5-triphophate (IP3), a calcium-mobilizing second messenger, and 1,2-diacylglycerol, an activator of protein kinase C (PKC). Although an increase of intracellular calcium was detected by calcium imaging after S1P stimulation (data not shown), BAPTA-AM, a chelator of intracellular calcium, did not interfere with PAM translocation or delayed AC inhibition (Figure 6E). In contrast, the PKC inhibitors bisindoylmaleimide I (BMI) and Ro31-8220 eliminated S1P-induced PAM translocation as well as AC inhibition, suggesting the involvement of PKC in PAM translocation (Figure 6D and E).
PKC activation by S1P can stimulate the extracellular signal-regulated kinase 1/2 (ERK1/2) in astrocytes (Sato et al, 1999), C6-glioma cells (Sato et al, 1999), and T24 cells (Shu et al, 2002). In HeLa cells, S1P incubation induced ERK1/2 activation, as detected by antiactive ERK (Figure 6F) and anti-phospho-Tyr183 ERK antibodies (data not shown). ERK1/2 phosphorylation was prevented by preincubation with PTX, U73122, Ro31-8220, and the MEK1/2 inhibitor U0126, but not by BAPTA-AM (Figure 6F), indicating that ERK1/2 activation is downstream of Gi, PLC, and PKC. However, ERK1/2 activation was not necessary for PAM activation since U0126 did not alter PAM translocation or S1P-induced inhibition of AC activity (Figure 6D, E). Taken together, the findings demonstrate that S1P induces PAM translocation and inhibition of AC enzyme activity through a signaling cascade that includes Gi, PLC, and PKC, but is independent of ERK1/2 activation and increased intracellular calcium concentrations.
Discussion
Cyclic AMP is a ubiquitous secondary messenger in eukaryotes and controls numerous physiological responses such as growth, apoptosis, migration, and differentiation. To address the specific needs of a cell, a complex network of regulators exists, which controls the spatial and temporal aspects of cAMP signaling (Patel et al, 2001; Neves and Iyengar, 2002; Neves et al, 2002). This regulatory network comprises factors that interact directly with adenylyl cyclases, control the activation level of G proteins, are involved in receptor desensitization, or exercise feedback regulation (Patel et al, 2001). Some of these regulators, for example, G proteins or calcium, are fast acting but last only for a short time in the order of milliseconds to seconds (Schleicher et al, 1993; Gorbunova and Spitzer, 2002). Other regulators, such as protein kinases, show a delayed regulation of AC activity, which normally peaks after several minutes (Choi and Toscano, 1988; Raymond, 1991). A third regulatory level targets the transcriptional control that acts between hours and days. However, no regulator of AC enzyme activity is known that bridges the regulatory time gap between minutes (e.g. phosphorylation effects) and long-term (e.g. transcriptional) actions. Here we show that the sphingolipid S1P activates the AC-inhibitory actions of PAM in HeLa cells to allow a sustained suppression of cAMP signaling, thus filling the already stated gap in the temporal regulation.
For the S1P-induced inhibition of AC activity in HeLa cells, two phases can be distinguished: First, an immediate inhibition of AC enzyme activity (1–10 min of S1P treatment) that is most likely due to an inhibitory G protein since this initial inhibition is PTX sensitive. Also, it has been demonstrated in several cell lines and animal models that Gi mediates S1P-dependent AC inhibition (Hla et al, 2001; Kluk and Hla, 2002; Siehler and Manning, 2002; Spiegel and Milstien, 2002). An involvement of PAM in mediating this initial inhibition is unlikely since PAM cannot be detected at the plasma membrane during this time period. After the initial inhibition phase, a delayed, sustained AC inhibition (up to 4 h after the beginning of S1P treatment) occurs. This sustained AC inhibition is PAM dependent since the reduction of endogenous PAM with antisense ODNs abolished the S1P-induced inhibition of cAMP accumulation. Since PAM is downstream of Gi in its signaling pathway, a role of Gi, other than its part in the signaling cascade leading to PAM activation, in the sustained AC inhibition can be ruled out. Taken together, the findings suggest that PAM activation leads to a delayed but sustained AC inhibition. This sustained AC inhibition is the longest known nontranscriptional regulatory mechanism for AC enzyme activity, and may bridge the time between short-term inhibition (e.g. G proteins or calcium) and transcriptional changes. Since the sustained inhibitory actions of PAM diminished the stimulatory effect of a variety of cAMP-elevating agents (forskolin, isoproterenol, VIP, and PGE2), we propose that PAM activation by S1P causes a general inhibition of cAMP signaling. This inhibition might play an important role in suppressing cAMP-elevating signals during the time that is needed for relative slow transcriptional or cellular (e.g. cytoskeletal rearrangements) changes to occur in the cell.
The mechanism by which PAM achieves the sustained inhibition of AC enzyme activity is unclear. One possible mechanism of action could be a targeted degradation of ACs by PAM. This possibility is supported by recent reports indicating that PAM might act as a ubiquitin ligase (Grossberger et al, 1999; DiAntonio et al, 2001; Murthy et al, 2004). The degradation of ACs after ubiquitination would also explain why forskolin-, VIP-, PGE2- and isoproterenol-elicited cAMP accumulation is all inhibited to a similar extent and why the inhibition is ongoing although PAM gradually disappears from the plasma membrane after 2 h. On the other hand, PAM was found at the plasma membrane from 20 to 120 min after S1P incubation in similar percentages of cells. This indicates the necessity of PAM to stay in close proximity of the ACs to allow ongoing inhibition. However, we cannot rule out the possibility that PAM fulfills other functions at the membrane, except AC regulation, during this time. In addition, we demonstrated previously that the RCC1 domain of PAM is sufficient to inhibit AC of type V enzyme activity and that RCC1 and AC type V physically interact with each other. Furthermore, PAM and its RCC1 domain inhibited Gαs-stimulated AC type V activity with equivalent potency (Scholich et al, 2001). Yet, the RCC1 domain does not contain any of the ubiquitin ligase motifs that are found in the PAM sequence. Therefore, it seems that PAM and its RCC1 domain inhibit AC activity through the same mechanism, which makes it unlikely that PAM inhibits AC enzyme activity through a ubiquitination process.
S1P regulates multiple cellular functions, ranging from suppression of ceramide-induced apoptosis (Cuvillier et al, 1996), regulation of neuronal cell survival and differentiation (Edsall et al, 1997; Van Brocklyn et al, 1999), tumor cell invasiveness (Sadahira et al, 1992), cytoskeletal rearrangements and migration (Lee et al, 1999), angiogenesis, and vascular maturation (Liu et al, 2000) to T-cell proliferation and inhibition of neutrophil motility (Sadahira et al, 1992; Graeler et al, 2002; Jin et al, 2003). The signal transduction pathways that are activated by S1P have been the focus of intensive research over the past few years and it is now widely accepted that regulation of cAMP signaling plays a central role in the signal transduction pathways activated by S1P (Kluk and Hla, 2002; Spiegel and Milstien, 2003). Here, we show that the inhibition of AC activity after S1P treatment is, at least in HeLa cells, not solely due to the direct inhibition of AC by Gi, but also depends on the activation of PAM through a signal transduction pathway that comprises the activation of Gi, PLC, and PKC. This pathway is known to be initiated in many cells and tissues by S1P, and PAM activation may also be involved in this common pathway in other cells. Since PAM is found in a great variety of tissues, PAM is also potentially involved in transducing the actions of S1P in these tissues and might represent a previously unknown common mechanism for long-term regulation of AC signaling.
Materials and methods
Materials
S1P, PTX, U0126, and U73122 were purchased from Tocris (Ellisville, MO), and the anti-Hsp70 and the anti-calnexin antibodies were from Calbiochem (San Diego, CA). HeLa cells (ACC 75) were acquired from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany. The anti-active ERK1/2 antibody was obtained from Promega (Madison, WI). Forskolin, Ro31-8220, BAPTA-AM, and BMI (GF109203X) were supplied by Sigma (St Louis, MO). DMEM medium and fetal bovine serum were from Gibco (Karlsruhe, Germany). All other chemicals were purchased from Sigma unless otherwise specified.
Immunofluorescence staining
Antibodies against PAM were generated by injection in rabbits of peptides corresponding to amino-acid residues 135–153 and 4601–4614 of human PAM (Ehnert et al, 2004). HeLa cells (100 000 cells/35 mm dish) were grown on glass coverslips in DMEM with 10% FBS and 1% penicillin/streptomycin. To visualize translocation of PAM, HeLa cells were serum-starved overnight and then treated with 10% serum or S1P for the indicated times. Wherever indicated, cells were preincubated before serum or S1P treatment for 15 min with the indicated concentrations of inhibitors. Cells were fixed in 4% paraformaldehyde in PBS for 10 min and then permeabilized in 0.1% Triton X-100 for 5 min. The coverslips were incubated for 1 h in 3% BSA in PBS, for 1 h with anti-PAM antibodies, and, where indicated, with anti-calnexin antibody (BD Biosciences, Heidelberg, Germany), followed by incubation with FITC-labeled goat anti-rabbit antibody (Sigma, St Louis, MO) in PBS containing 1% BSA. The coverslips were then washed with PBS and mounted. For the analysis, confocal (Bio-Rad, Hercules, CA) and regular fluorescence microscopes (Nikon, Duesseldorf, Germany) were used.
For the assessment of PAM–translocation, the following procedure was applied: Pictures of ∼100 cells from each experimental condition were taken. Translocation occurrence was assessed by generating line densitometric profiles of the cells using the ImageJ 1.32 (NIH, USA) software. Cells were judged translocation positive if the PAM signal strength at the plasma membrane was increased by at least 50% as compared to the signal of the cytosolic region between ER and plasma membrane. The background was determined in a region of the picture that contained no cells and was subtracted from the two other values.
cAMP accumulation
HeLa cells were plated at 200 000 cells per 35 mm dishes and serum-starved the next day for 24 h. Then fresh serum-free medium was given and 5 min prior to the respective treatment (e.g. forskolin or VIP) 100 μM IBMX was added. After the incubation with S1P or serum for the indicated times, the cells were incubated for 20 min in 1 M HCl, scraped off the dish, and centrifuged for 5 min at 10 000 g. The supernatant was dried and used for cAMP measurements. cAMP accumulation in the cells was determined by employing the cAMP detection Kit (Assay Design Inc., Ann Arbor, MI) according to the manufacturer's instructions.
Adenylyl cyclase activity assays
HeLa cells were plated at 200 000 cells per 35 mm dishes and serum-starved the next day for 24 h. The cells were pretreated with inhibitors as described above when indicated, prior to the incubation with 10% serum or 0.5 μM S1P. Cell membranes were prepared following the protocol described by Kassis and Fishman (1982) with the following modification: At the end of the incubation with S1P or serum, the cells were immediately scraped off the plates in ice-cold 50 mM Tris, pH 7.4, 0.5 mM EDTA, and snap frozen. Aliquots of the purified membranes were stored at −80°C until use. AC activity assays were performed in a volume of 100 μl for 15 min at room temperature in the presence of 100 μM MgCl2 as previously described (Chen et al, 1995). Gαs* (80 nM) or forskolin (100 μM) was used to stimulate adenylyl cyclase enzyme activity of the HeLa membranes (7.5 μg protein/assay).
Expression and purification of recombinant Gsa
The hexahistidyl-tagged, constitutively active, Q213L mutant of Gαs (Gαs*) was expressed and purified as described in our previous publication (Scholich et al, 1999). To ensure maximal activation of Gαs*, the G protein was incubated with 1 μM GTPγS in the presence of MgCl2 (25 mM) for 30 min prior to use in AC activity assays.
Antisense oligodeoxynucleotides
The sequences of the oligodeoxynucleotides (ODNs) were chosen as published previously (Scholich et al, 2001). HeLa cells were plated on 35 mm dishes (300 000 cells) and grown in DMEM medium containing 10% FBS and 1% penicillin/streptomycin for 24 h. The ODNs (3 μM each) were introduced into the cells by transfection using Tfx20 reagent (Promega, Madison, WI) in 1 ml serum-free medium according to the manufacturer's instructions. After 2 h, 1 ml DMEM containing 10% FBS was added. After 6 h, the medium was exchanged against serum-free medium and the cells were incubated for another 16 h before being used for the experiments. Western blots for PAM were performed as described previously (Ehnert et al, 2004). For the densitometric evaluation of the Western blots, the ImageJ (NIH, USA) software was used.
Purification of the PAM-activating serum factor
The PAM-activating serum factor was purified following the flow scheme shown in Figure 3A. After each purification step, the flow-through and the eluted fractions were tested for their ability to induce PAM translocation in HeLa cells. Positive fractions were pooled and submitted to the next purification step. Fetal bovine serum (188 ml) (batch 40G121OK; Gibco, Karlsruhe, Germany) was heated for 20 min at 100°C and then centrifuged three times at 18 000 g for 20 min. The supernatant was adjusted to a final concentration of 1 M NaCl and was loaded on a phenyl-Sepharose 15/10 column (Amersham, Pharmacia, Piscataway, NJ). The active serum factor was eluted by a step gradient with 0.3 M NaCl. Next, it was loaded on a Q-Sepharose 15/10 column (Amersham, Pharmacia, Piscataway, NJ). The column was washed with 400 mM NaCl in TE (50 mM Tris–HCl (pH 7.4), 0.5 mM EDTA) and eluted with 1 M NaCl in TE. Positive fractions were eluted at a salt concentration of 700 mM NaCl. The eluate was loaded on a Superdex 200pg gel-filtration column (Amersham, Pharmacia, Piscataway, NJ). Positive fractions were pooled, loaded on a MonoQ 5/5 FPLC column (Amersham, Pharmacia, Piscataway, NJ) and washed with 400 mM NaCl in TE. The protein was eluted with a gradient from 400 to 1000 mM NaCl in TED. Positive fractions eluted with 650–750 mM NaCl were pooled and loaded on a Superdex 30pg gel-filtration column (Amersham, Pharmacia, Piscataway, NJ). The protein was eluted with TE and the fractions were analyzed for their ability to induce translocation of PAM in HeLa cells. Positive fractions were pooled, stored at −80°C and used within 2 weeks. S1P was detected using phthaldialdehyde labeling followed by HPLC separation as described by Caligan et al (2000).
RT–PCR
Total RNA was isolated by guanidinium isothiocyanate/phenol/chloroform extraction (Chomczynski and Sacchi, 1987). In all, 2 μg of total RNA was annealed with 0.6 μM of each oligo (dT) primer and reverse-transcribed using reverse transcriptases (Promega, Madison, WI) for 30 min at 37°C. The cDNA was then immediately used for amplification (one-step PCR). ODN primers used for the amplification of the various EDG receptor isoforms were chosen and used as described by Hornuss et al (2001).
ERK1/2 Western blot analysis
HeLa cells were plated at 200 000 cells per 35 mm dishes and serum-starved the next day for 24 h. The cells were pretreated with inhibitors when indicated prior to stimulation with 10% serum or 0.5 μM S1P. The cells were then prepared for Western blots by adding boiling 1 × Laemmli buffer containing 100 mM sodium phosphate and 0.2 mM sodium orthovanadate. Antiactive ERK1/2 and anti-Phospho-Tyr106 (both Promega, Madison, WI) were employed to detect phosphorylated ERK1/2 following the manufacturer's instructions.
Acknowledgments
We are grateful to Dr AG Gilman, University of Texas Southwestern Medical School, and Dr C Kleuss, Freie Universität Berlin, for supplying the Gsα cDNA constructs. We also thank A Schmidt and M Seegel for their help in the identification of S1P and Dr J Calderwood for revising the manuscript. This work was supported by an unrestricted grant from Aventis, Frankfurt, Germany, and in part by the DFG grant SCHO817 1-1.
References
- Burgess RW, Peterson KA, Johnson MJ, Roix JJ, Welsh IC, O'Brien TP (2004) Evidence for a conserved function in synapse formation reveals phr1 as a candidate gene for respiratory failure in newborn mice. Mol Cell Biol 24: 1096–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligan TB, Peters K, Ou J, Wang E, Saba J, Merrill AH Jr (2000) A high-performance liquid chromatographic method to measure sphingosine 1-phosphate and related compounds from sphingosine kinase assays and other biological samples. Anal Biochem 281: 36–44 [DOI] [PubMed] [Google Scholar]
- Chen Z, Nield HS, Sun H, Barbier A, Patel TB (1995) Expression of type V adenylyl cyclase is required for epidermal growth factor-mediated stimulation of cAMP accumulation. J Biol Chem 270: 27525–27530 [DOI] [PubMed] [Google Scholar]
- Choi EJ, Toscano WA Jr. (1988) Modulation of adenylate cyclase in human keratinocytes by protein kinase C. J Biol Chem 263: 17167–17172 [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381: 800–803 [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Gilman AG (1997) The catalytic mechanism of mammalian adenylyl cyclase. Equilibrium binding and kinetic analysis of P-site inhibition. J Biol Chem 272: 27787–27795 [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412: 449–452 [DOI] [PubMed] [Google Scholar]
- Edsall LC, Pirianov GG, Spiegel S (1997) Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J Neurosci 17: 6952–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edsall LC, Spiegel S (1999) Enzymatic measurement of sphingosine 1-phosphate. Anal Biochem 272: 80–86 [DOI] [PubMed] [Google Scholar]
- Ehnert C, Tegeder I, Pierre S, Birod K, Nguyen HV, Schmidtko A, Geisslinger G, Scholich K (2004) Protein associated with Myc (PAM) is involved in spinal nociceptive processing. J Neurochem 88: 948–957 [DOI] [PubMed] [Google Scholar]
- Gorbunova YV, Spitzer NC (2002) Dynamic interactions of cyclic AMP transients and spontaneous Ca(2+) spikes. Nature 418: 93–96 [DOI] [PubMed] [Google Scholar]
- Graeler M, Shankar G, Goetzl EJ (2002) Cutting edge: suppression of T cell chemotaxis by sphingosine 1-phosphate. J Immunol 169: 4084–4087 [DOI] [PubMed] [Google Scholar]
- Grossberger R, Gieffers C, Zachariae W, Podtelejnikov AV, Schleiffer A, Nasmyth K, Mann M, Peters JM (1999) Characterization of the DOC1/APC10 subunit of the yeast and the human anaphase-promoting complex. J Biol Chem 274: 14500–14507 [DOI] [PubMed] [Google Scholar]
- Guo Q, Xie J, Dang CV, Liu ET, Bishop JM (1998) Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc Natl Acad Sci USA 95: 9172–9177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hla T, Lee MJ, Ancellin N, Paik JH, Kluk MJ (2001) Lysophospholipids—receptor revelations. Science 294: 1875–1878 [DOI] [PubMed] [Google Scholar]
- Hornuss C, Hammermann R, Fuhrmann M, Juergens UR, Racke K (2001) Human and rat alveolar macrophages express multiple EDG receptors. Eur J Pharmacol 429: 303–308 [DOI] [PubMed] [Google Scholar]
- Igarashi Y, Yatomi Y (1998) Sphingosine 1-phosphate is a blood constituent released from activated platelets, possibly playing a variety of physiological and pathophysiological roles. Acta Biochim Pol 45: 299–309 [PubMed] [Google Scholar]
- Jiao X, Trifillis P, Kiledjian M (2002) Identification of target messenger RNA substrates for the murine deleted in azoospermia-like RNA-binding protein. Biol Reprod 66: 475–485 [DOI] [PubMed] [Google Scholar]
- Jin Y, Knudsen E, Wang L, Bryceson Y, Damaj B, Gessani S, Maghazachi AA (2003) Sphingosine 1-phosphate is a novel inhibitor of T-cell proliferation. Blood 101: 4909–4915 [DOI] [PubMed] [Google Scholar]
- Kassis S, Fishman PH (1982) Different mechanisms of desensitization of adenylate cyclase by isoproterenol and prostaglandin E1 in human fibroblasts. Role of regulatory components in desensitization. J Biol Chem 257: 5312–5318 [PubMed] [Google Scholar]
- Kluk MJ, Hla T (2002) Signaling of sphingosine-1-phosphate via the S1P/EDG-family of G-protein-coupled receptors. Biochim Biophys Acta 1582: 72–80 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99: 301–312 [DOI] [PubMed] [Google Scholar]
- Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 106: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Han S, Beauchamp RL, Smith N, Haddad LA, Ito N, Ramesh V (2004) Pam and its ortholog highwire interact with and may negatively regulate the TSC1.TSC2 complex. J Biol Chem 279: 1351–1358 [DOI] [PubMed] [Google Scholar]
- Neves SR, Iyengar R (2002) Modeling of signaling networks. BioEssays 24: 1110–1117 [DOI] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R (2002) G protein pathways. Science 296: 1636–1639 [DOI] [PubMed] [Google Scholar]
- Patel TB, Du Z, Pierre S, Cartin L, Scholich K (2001) Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene 269: 13–25 [DOI] [PubMed] [Google Scholar]
- Peterson KA, King BL, Hagge-Greenberg A, Roix JJ, Bult CJ, O'Brien TP (2002) Functional and comparative genomic analysis of the piebald deletion region of mouse chromosome 14. Genomics 80: 172–184 [DOI] [PubMed] [Google Scholar]
- Raymond JR (1991) Protein kinase C induces phosphorylation and desensitization of the human 5-HT1A receptor. J Biol Chem 266: 14747–14753 [PubMed] [Google Scholar]
- Sadahira Y, Ruan F, Hakomori S, Igarashi Y (1992) Sphingosine 1-phosphate, a specific endogenous signaling molecule controlling cell motility and tumor cell invasiveness. Proc Natl Acad Sci USA 89: 9686–9690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Tomura H, Igarashi Y, Ui M, Okajima F (1999) Possible involvement of cell surface receptors in sphingosine 1-phosphate-induced activation of extracellular signal-regulated kinase in C6 glioma cells. Mol Pharmacol 55: 126–133 [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Hadwiger GD, Nonet ML (2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26: 345–356 [DOI] [PubMed] [Google Scholar]
- Schleicher S, Boekhoff I, Arriza J, Lefkowitz RJ, Breer H (1993) A beta-adrenergic receptor kinase-like enzyme is involved in olfactory signal termination. Proc Natl Acad Sci USA 90: 1420–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholich K, Barbier AJ, Mullenix JB, Patel TB (1997) Characterization of soluble forms of nonchimeric type V adenylyl cyclases. Proc Natl Acad Sci USA 94: 2915–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholich K, Mullenix JB, Wittpoth C, Poppleton HM, Pierre SC, Lindorfer MA, Garrison JC, Patel TB (1999) Facilitation of signal onset and termination by adenylyl cyclase. Science 283: 1328–1331 [DOI] [PubMed] [Google Scholar]
- Scholich K, Pierre S, Patel TB (2001) Protein associated with Myc (PAM) is a potent inhibitor of adenylyl cyclases. J Biol Chem 276: 47583–47589 [DOI] [PubMed] [Google Scholar]
- Semov A, Marcotte R, Semova N, Ye X, Wang E (2002) Microarray analysis of E-box binding-related gene expression in young and replicatively senescent human fibroblasts. Anal Biochem 302: 38–51 [DOI] [PubMed] [Google Scholar]
- Shu X, Wu W, Mosteller RD, Broek D (2002) Sphingosine kinase mediates vascular endothelial growth factor-induced activation of ras and mitogen-activated protein kinases. Mol Cell Biol 22: 7758–7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siehler S, Manning DR (2002) Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochim Biophys Acta 1582: 94–99 [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S (2002) Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem 277: 25851–25854 [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4: 397–407 [DOI] [PubMed] [Google Scholar]
- Taussig R, Tang WJ, Hepler JR, Gilman AG (1994) Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J Biol Chem 269: 6093–6100 [PubMed] [Google Scholar]
- Tesmer JJ, Dessauer CW, Sunahara RK, Murray LD, Johnson RA, Gilman AG, Sprang SR (2000) Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry 39: 14464–14471 [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Tu Z, Edsall LC, Schmidt RR, Spiegel S (1999) Sphingosine 1-phosphate-induced cell rounding and neurite retraction are mediated by the G protein-coupled receptor H218. J Biol Chem 274: 4626–4632 [DOI] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26: 313–329 [DOI] [PubMed] [Google Scholar]
- Wittpoth C, Scholich K, Yigzaw Y, Stringfield TM, Patel TB (1999) Regions on adenylyl cyclase that are necessary for inhibition of activity by beta gamma and G(ialpha) subunits of heterotrimeric G proteins. Proc Natl Acad Sci USA 96: 9551–9556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Scholich K, Poser S, Storm DR, Patel TB, Goldowitz D (2002) Developmental expression of PAM (protein associated with MYC) in the rodent brain. Brain Res Dev Brain Res 136: 35–42 [DOI] [PubMed] [Google Scholar]
- Zhen M, Huang X, Bamber B, Jin Y (2000) Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26: 331–343 [DOI] [PubMed] [Google Scholar]