

Abstract
A-kinase anchoring proteins (AKAPs) target the cAMP-regulated protein kinase (PKA) to its physiological substrates. We recently identified a novel anchoring protein, called AKAP-Lbc, which functions as a PKA-targeting protein as well as a guanine nucleotide exchange factor (GEF) for RhoA. We demonstrated that AKAP-Lbc Rho-GEF activity is stimulated by the alpha subunit of the heterotrimeric G protein G12. Here, we identified 14-3-3 as a novel regulatory protein interacting with AKAP-Lbc. Elevation of the cellular concentration of cAMP activates the PKA holoenzyme anchored to AKAP-Lbc, which phosphorylates the anchoring protein on the serine 1565. This phosphorylation event induces the recruitment of 14-3-3, which inhibits the Rho-GEF activity of AKAP-Lbc. AKAP-Lbc mutants that fail to interact with PKA or with 14-3-3 show a higher basal Rho-GEF activity as compared to the wild-type protein. This suggests that, under basal conditions, 14-3-3 maintains AKAP-Lbc in an inactive state. Therefore, while it is known that AKAP-Lbc activity can be stimulated by Gα12, in this study we demonstrated that it is inhibited by the anchoring of both PKA and 14-3-3.
Keywords: A-kinase anchoring protein (AKAP), protein kinase A, Rho, 14-3-3
Introduction
The transduction of signals across the plasma membrane to intracellular targets is a tightly regulated process that underlies diverse physiological functions. It is now evident that specificity of transduction events is controlled at the molecular level by scaffold, anchoring and adaptor proteins, which position signaling enzymes at the proper subcellular localization (Smith and Scott, 2002).
A-kinase anchoring proteins (AKAPs) are prototypic examples of proteins that compartmentalize signaling complexes at precise subcellular sites (Michel and Scott, 2002). Members of this family of proteins bind to regulatory (R) subunit dimers of cAMP-dependent protein kinase (PKA) holoenzyme through an amphipathic helix motif of 14–18 residues (Carr et al, 1991; Newlon et al, 1999). Unique targeting domains on each anchoring protein direct PKA/AKAP complexes to specific subcellular sites, thereby providing a mechanism that positions PKA in close proximity to its physiological substrates (Michel and Scott, 2002). Another important function of AKAPs is to assemble signaling complexes containing multiple kinases, phosphatases and regulatory proteins. By simultaneously interacting with multiple signaling enzymes, AKAPs can integrate diverse transduction pathways that coordinately regulate the function of specific cellular substrates (Michel and Scott, 2002).
Recent findings show that AKAPs can regulate small molecular weight GTPases of the Ras superfamily. This is the case for AKAP-Lbc (Diviani et al, 2001), which functions as a targeting protein for type II PKA as well as guanine nucleotide exchange factor (GEF) for RhoA, a small GTP-binding protein that controls fundamental cell properties such as cell cycle progression, gene expression, actin remodeling and cytokinesis (Etienne-Manneville and Hall, 2002). A truncated form of AKAP-Lbc, called onco-Lbc, was originally isolated as an oncogene from myeloid leukemia patients and shown to represent a constitutively active Rho-GEF (Zheng et al, 1995b). AKAP-Lbc belongs to the Dbl family of GEFs, which all share a Dbl homology (DH) domain and an adjacent pleckstrin homology (PH) domain (Zheng, 2001). The DH domain is responsible for the guanine nucleotide exchange activity, whereas the PH domain controls the subcellular localization of the GEF or contributes to the binding pocket for Rho-GTPases (Etienne-Manneville and Hall, 2002). Interestingly, the Rho-GEF activity of AKAP-Lbc can be strongly enhanced by the α subunit of the heterotrimeric G protein G12 as well as by the activation of G protein-coupled receptors (GPCRs) that couple to G12 (Diviani et al, 2001). This places AKAP-Lbc in the growing family of Rho-specific exchange factors, which includes LARG, PDZ-Rho-GEF and p115 Rho-GEF, which relay signals from heterotrimeric G proteins to Rho (Hart et al, 1998; Fukuhara et al, 1999, 2000; Booden et al, 2002).
It well established that activation of PKA affects cell morphology, inducing loss of stress fibers and focal adhesions and cell rounding. These effects are the consequence of the inhibitory action of PKA on multiple components of the Rho signaling pathway. In fact, recent findings demonstrate that activated PKA directly phosphorylates Rho on serine 188 and inhibits its function (Lang et al, 1996; Ellerbroek et al, 2003). PKA can also phosphorylate the Gα13 subunit and promote its uncoupling from the βγ subunit dimer, thereby inhibiting receptor-mediated activation of RhoA (Manganello et al, 2003). Moreover, Laudanna et al (1997) showed that, in B-lymphocytes, activation of PKA inhibits RhoA GDP/GTP exchange and suggested that PKA might negatively regulate an upstream activator of RhoA. Since AKAP-Lbc can both bind PKA and act as a Rho-GEF, it might represent a signaling complex where the PKA and Rho pathways converge. However, the functional role of anchoring PKA on AKAP-Lbc signaling remains elusive.
In the present study, using a yeast two-hybrid approach, we identified 14-3-3 as a novel protein interacting with AKAP-Lbc. We found that phosphorylation of AKAP-Lbc by the anchored PKA promotes the recruitment of 14-3-3, which, in turn, inhibits AKAP-Lbc Rho-GEF activity. We also demonstrate that binding of 14-3-3 to AKAP-Lbc maintains the anchoring protein in an inactive state under basal conditions. These findings provide a novel mechanistic hypothesis explaining the inhibitory action of PKA on the Rho signaling pathway.
Results
Identification of 14-3-3 as a protein interacting with AKAP-Lbc
We have previously shown that truncation of the N-terminal region of AKAP-Lbc can significantly increase its basal Rho-GEF activity in the absence of external activating stimuli (Diviani et al, 2001). This suggested that inhibitory determinants located in the N-terminal sequence of AKAP-Lbc maintain the protein inactive under basal conditions. To identify novel regulatory proteins interacting with this inhibitory region, a fragment of AKAP-Lbc encompassing residues 1388–1922 (Figure 1A) was used as bait to screen a human heart cDNA library using the yeast two-hybrid system. Five independent positive clones encoding 14-3-3β, six encoding 14-3-3ɛ and one encoding 14-3-3ζ were identified from 6 × 106 cotransformants, as assessed by their ability to grow in the absence of histidine and to produce β-galactosidase. To confirm these interactions, the L40 yeast strain was transformed with the bait plasmid expressing the 1388–1922 fragment of AKAP-Lbc (AKAP-Lbc 1388–1922-pLexA) in combination with either pACT2 vectors containing the 14-3-3 cDNAs (14-3-3-pACT2) or with empty pACT2 vector. As shown in Figure 1A, yeast transformed with the 14-3-3-pACT2 constructs were able to grow in the absence of histidine (upper panel) and to produce β-galactosidase (lower panel), whereas those transformed with the empty pACT2 vector did not.
Figure 1.
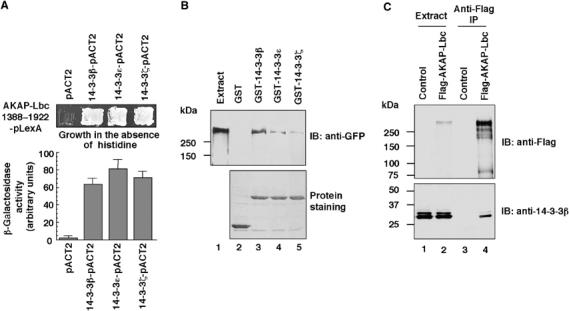
Identification of 14-3-3 as a protein interacting with AKAP-Lbc. (A) Yeast two-hybrid screening. The AKAP-Lbc 1388–1922-pLexA plasmid was transformed into the L40 yeast strain in combination with either the empty pACT2 vector or with pACT2 containing the cDNAs of 14-3-3β, 14-3-3ɛ or 14-3-3ζ. Transformants were plated on His plates to select for histidine prototrophy (upper panel). Quantitative analysis of the interaction between the AKAP-Lbc 1388–1922 fragment and 14-3-3 proteins was performed using the liquid β-galactosidase assay. (B) Extracts from HEK-293 cells overexpressing AKAP-Lbc-GFP were incubated with glutathione-sepharose beads coupled to GST alone, GST-14-3-3β, GST-14-3-3ɛ or GST-14-3-3ζ. Associated AKAP-Lbc-GFP was detected using anti-GFP monoclonal antibodies (upper panel). A control protein staining indicating the expression level of the GST and GST-tagged 14-3-3s used in the pulldown assay is shown (lower panel). (C) Extracts from HEK-293 cells transfected with the empty Flag vector (Control) or with the cDNA encoding Flag-AKAP-Lbc were subjected to immunoprecipitation with anti-Flag monoclonal antibodies. Western blots of the immunoprecipitates (lanes 3 and 4) and of the cell extracts (lanes 1 and 2) were revealed using anti-Flag monoclonal antibodies to detect Flag-AKAP-Lbc (upper panel) or anti-14-3-3β polyclonal antibodies to detect the endogenous 14-3-3β (lower panel).
To provide biochemical evidence that the full-length AKAP-Lbc can associate with 14-3-3 proteins, pulldown experiments were performed by incubating extracts of HEK-293 cells overexpressing GFP-tagged AKAP-Lbc with purified GST-tagged 14-3-3β, 14-3-3ɛ and 14-3-3ζ. We found that recombinant AKAP-Lbc bound to all three 14-3-3 isoforms. No binding was observed with GST alone (Figure 1B).
To demonstrate that AKAP-Lbc and 14-3-3 interact inside cells, we performed co-immunoprecipitation experiments from HEK-293 cells that were transiently transfected with the empty Flag vector (Control) or the Flag-tagged AKAP-Lbc (Figure 1C). After immunoprecipitating the anchoring protein using anti-Flag antibodies, anti-14-3-3β antibodies were used to immunoblot the immunoprecipitated samples. The Western blots revealed that 14-3-3β endogenously expressed in HEK-293 cells could specifically co-immunoprecipitate with AKAP-Lbc, whereas no bands were immunoprecipitated from cells transfected with the empty Flag vector (Figure 1C, bottom panel, lanes 3 and 4). Similar results were obtained for 14-3-3ɛ and 14-3-3ζ (results not shown). These findings strongly suggest that AKAP-Lbc and 14-3-3 associate inside cells.
Interaction between AKAP-Lbc and 14-3-3 is regulated by PKA
Members of the 14-3-3 family interact with several cellular targets through the association with phosphoserine- or phosphothreonine-containing motifs (Tzivion and Avruch, 2002). Phosphorylation of these motifs is catalyzed predominantly by members of the AGC subfamily of protein kinases such as PKA, PKB and PKC. Since AKAP-Lbc is a PKA-binding protein, we hypothesized that PKA might phosphorylate the anchoring protein and regulate its interaction with 14-3-3. To investigate this hypothesis, we used a solid phase overlay assay to assess whether phosphorylation of the GST-tagged AKAP-Lbc 1388–1922 fragment by PKA could influence its ability to associate with purified S-tagged 14-3-3β. GST or GST-AKAP-Lbc 1388–1922 fragments were expressed in Escherichia coli, purified and preincubated with or without purified catalytic subunit of PKA in the presence of ATP, transferred to nitrocellulose and incubated with 50 nM of S-tagged 14-3-3β. The blot shown in Figure 2A (left panel) revealed that the GST-tagged 1388–1922 fragment of AKAP-Lbc could specifically interact with 14-3-3β only after preincubation with PKA (Figure 2A, lane 2 versus lane 4). No basal interaction was observed in the absence of PKA preincubation since the GST-tagged 1388–1922 fragment of AKAP-Lbc should not undergo PKA-mediated phosphorylation when expressed in bacteria. Similar results were obtained when overlay assays were performed with purified 14-3-3ɛ and 14-3-3ζ (results not shown). This strongly suggests that AKAP-Lbc and 14-3-3 interact directly and that this association is regulated by the phosphorylation of the anchoring protein by PKA.
Figure 2.

Interaction between 14-3-3 and AKAP-Lbc is regulated by PKA. (A) GST or GST-AKAP-Lbc 1388–1922 was incubated for 1 h in the absence (No PKA) or presence (+PKA) of purified PKA catalytic subunit and electroblotted. The membranes were incubated with 50 nM of S-tagged 14-3-3β (left panel) and HRP-conjugated S-protein and revealed by autoradiography. A protein staining showing the amount of GST and GST-AKAP-Lbc 1388–1922 used in the assay is shown (right panel). (B) HeLa-S3 cells were serum starved for 24 h and then treated for 10 min in the absence or presence of 10 μM forskolin (FSK) or 10 μM forskolin+20 μM of myristoylated PKI 5–24 peptide (FSK+PKImyr). Lysates were subjected to immunoprecipitation with either nonimmune IgG or anti-AKAP-Lbc polyclonal antibodies. Immunoblots were revealed using anti-AKAP-Lbc polyclonal antibodies (upper panel), anti-RIIα monoclonal antibodies (middle panel) and anti-14-3-3β monoclonal antibodies. (C) Densitometry of the bands corresponding to 14-3-3β co-immunoprecipitated with AKAP-Lbc from cells that were treated as indicated in (B). The amount of 14-3-3β in the immunoprecipitates was normalized to the 14-3-3β content of the cell extracts. Results are the mean±s.e. of three independent experiments.
To assess whether PKA could modulate the AKAP-Lbc/14-3-3 interaction inside cells, HeLa-S3 cells, which express endogenous AKAP-Lbc, were incubated for 10 min in the absence (untreated) or presence of 10 μM forskolin prior to immunoprecipitation of AKAP-Lbc. As shown in Figure 2B, treatment with forskolin (FSK) induced a 2.4-fold increase in the amount of endogenous 14-3-3 co-immunoprecipitated with AKAP-Lbc when compared to untreated cells (Figure 2B, bottom panel, lanes 3 and 6, and Figure 2C). To demonstrate that the effect induced by forskolin was mediated by PKA, cells were pretreated with the cell-permeable myristoylated form of the PKA inhibitor PKI (PKImyr) before incubation with forskolin (Figure 2B, bottom panel, lane 9, and Figure 2C). PKImyr treatment abolished the interaction between endogenous AKAP-Lbc and 14-3-3β, suggesting that the association between these two proteins requires PKA catalytic activity.
It is well established that 14-3-3 can bind cellular targets both as a monomer and as a dimer. The interaction of monomeric 14-3-3 seems to be independent of the phosphorylation of the target, whereas binding of dimeric 14-3-3 seems to require it (Shen et al, 2003). To determine whether 14-3-3 interacts with AKAP-Lbc as a monomer or as a dimer, we generated a mutant of 14-3-3β that does not undergo dimerization (14-3-3β dm) (see Supplementary Figure 1A). This dimerization-deficient 14-3-3 mutant was originally described for 14-3-3ζ by Shen et al (2003). Our results indicate that binding of wild-type 14-3-3β to AKAP-Lbc is significantly enhanced by PKA phosphorylation, whereas the interaction of the dimerization-deficient mutant (14-3-3β dm) is not (see Supplementary Figure 1B). This suggests that AKAP-Lbc interacts with the dimeric form 14-3-3β in a PKA-dependent manner.
Identification of the binding site for 14-3-3 on AKAP-Lbc
To identify the binding site for 14-3-3 within residues 1388–1922 of AKAP-Lbc, we generated a series of GFP-tagged AKAP-Lbc fragment encompassing residues 1388–1493, 1494–1593, 1594–1693, 1694–1793 and 1794–1922 (see Supplementary Figure 2A), and expressed them in HEK-293 cells. After a 10 min treatment with 10 μM forskolin, cell extracts were prepared and incubated with GST or GST-14-3-3β. Interestingly, only the GFP fusion protein encompassing residues 1494–1593 retained the ability to interact with GST-14-3-3β (see Supplementary Figure 2B). No binding was observed between the GFP-tagged AKAP-Lbc fragments and GST alone.
Analysis of the primary sequence between residues 1493 and 1593 of AKAP-Lbc using the scansite program (http://scansite.mit.edu) identified four motifs at positions 1525, 1559, 1565 and 1584, which could represent potential 14-3-3-binding sites. Based on these predictions, we generated GFP fusions of the 1493–1593 fragment of AKAP-Lbc in which serines 1523 and 1525, 1553 and 1554, 1557 and 1559, 1565, as well as 1582 and 1584 were substituted by alanines (Figure 3A). The GFP fusions were expressed in HEK-293 cells and their ability to interact with GST or GST-14-3-3β assessed using the pulldown assay. As shown in Figure 3B, mutation of serine 1565 completely abolished the interaction between AKAP-Lbc and 14-3-3 (Figure 3B, lane 18), whereas the substitution of the other serines had no effect. This suggests that the binding of 14-3-3 to AKAP-Lbc requires the integrity of serine 1565.
Figure 3.
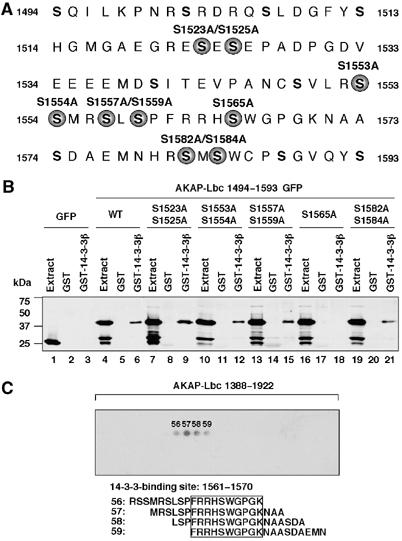
14-3-3 interacts with AKAP-Lbc through a phosphoserine-containing motif. (A) The region of AKAP-Lbc encompassing residues 1494–1593. Serine residues mutated to alanine are indicated by solid circles. (B) Extracts from HEK-293 cells expressing either GFP or GFP-tagged AKAP-Lbc fragments encompassing residues 1494–1593 carrying point mutations of various serines were incubated with glutathione-sepharose beads coupled to GST alone or to GST-tagged 14-3-3β. The GFP-tagged AKAP-Lbc fragments eluted from the beads were detected as indicated in Figure 2C. (C) Peptide array analysis of the 14-3-3-binding site on AKAP-Lbc. An array of overlapping 19-residue peptides spanning the region of AKAP-Lbc included between residues 1388 and 1922 was submitted to PKA phosphorylation and incubated with 50 nM S-tagged 14-3-3β. Solid phase binding was assessed with HRP-conjugated S-protein. The 14-3-3β-binding peptides are numbered. The results are representative of three independent experiments.
To identify the minimal 14-3-3 interaction motif on AKAP-Lbc, we used an S-tagged 14-3-3β fusion protein as a probe to screen a peptide array of overlapping 19-residue peptides (each displaced by three amino acids), spanning the region between residues 1388 and 1922 of AKAP-Lbc (Figure 3C). Prior to the incubation with 50 nM of S-tagged 14-3-3β fusion protein, peptide arrays were treated for 1 h with purified PKA catalytic subunit. Binding of S-tagged 14-3-3β was detected using HRP-conjugated S-protein. Specific binding was detected at one single site between residues 1561 and 1570 of AKAP-Lbc (Figure 3C). No 14-3-3 binding was observed on the array that was not incubated with PKA (results not shown). In conclusion, our mapping analysis revealed that AKAP-Lbc interacts with 14-3-3β in a PKA-dependent manner through a site located between residues 1561 and 1570.
Anchored PKA phosphorylates AKAP-Lbc on serine 1565
Interestingly, serine 1565 within the 14-3-3-binding motif of AKAP-Lbc is predicted to form a phosphorylation site for PKA. Based on our findings that phosphorylation of AKAP-Lbc by PKA is required for the association of 14-3-3, we investigated whether PKA might directly phosphorylate serine 1565. As shown in Figure 4A, the purified GST-AKAP-Lbc 1388–1922 fragment was efficiently phosphorylated by purified PKA (Figure 4A, left panel, lane 2). Phosphorylation was completely abolished by the mutation of serine 1565 to alanine, suggesting that this serine represents the unique PKA phosphorylation site between residues 1388 and 1922 (Figure 4A, left panel, lane 3). No phosphorylation was observed in the presence of the PKA-specific inhibitor PKI (Figure 4A, left panel, lanes 4–6).
Figure 4.
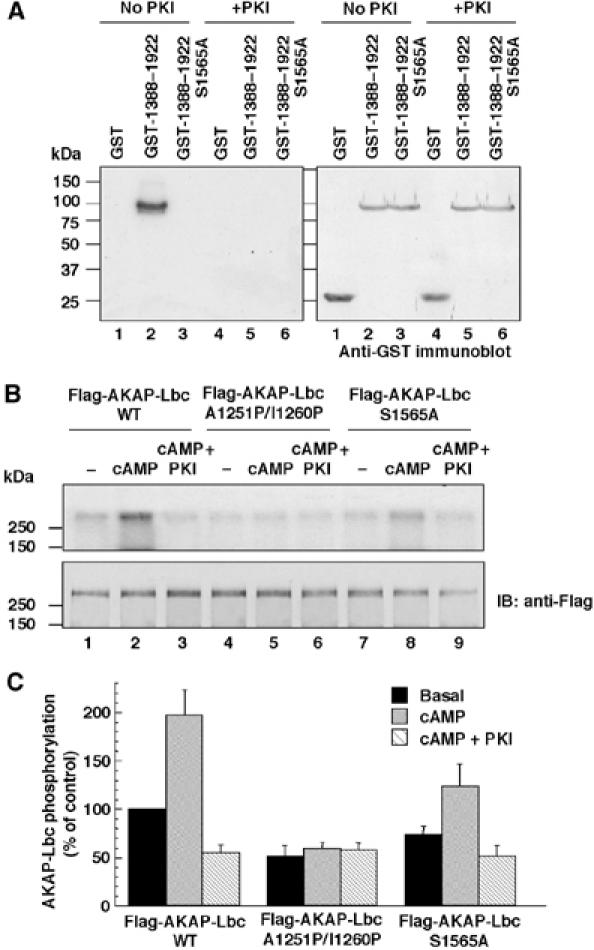
Anchored PKA phosphorylates AKAP-Lbc on serine 1565. (A) GST, GST-AKAP-Lbc 1388–1922 and GST-AKAP-Lbc 1388–1922 S1565A proteins were incubated with purified PKA catalytic subunit and [32P]γ-ATP in the absence or presence of 20 μM PKI, and then electroblotted and subjected to autoradiography (left panel). The relative amount of GST fusion proteins used in the assay was determined by immunoblot using antibodies against GST (right panel). (B) HEK-293 cells were transfected with the cDNAs encoding the Flag-tagged fusions of AKAP-Lbc (lanes 1–3) or of its mutants A1251P/I1260P (lanes 4–6) and S1565A (lanes 7–9). Cell lysates were subjected to immunoprecipitation with anti-Flag monoclonal antibodies. Immunoprecipitates were phosphorylated as described in Materials and methods in the absence or presence of 10 μM cAMP (cAMP) or 10 μM cAMP+20 μM PKI (cAMP+PKI). Proteins were electroblotted and subjected to autoradiography (upper panel). The amount of Flag-AKAP-Lbc present in the immunoprecipitates was determined using anti-Flag monoclonal antibodies (lower panel). (C) Densitometry of the bands corresponding to the phosphorylated Flag-AKAP-Lbc proteins that were treated as indicated in (B). The extent of phosphorylation was normalized to the amount of Flag-AKAP-Lbc present in the immunoprecipitates. Results are the mean±s.e. of three independent experiments.
To investigate whether the PKA holoenzyme anchored to AKAP-Lbc could phosphorylate serine 1565, we overexpressed Flag-tagged forms of AKAP-Lbc and of its mutants A1551P/I1260P (which is impaired in its ability to interact with PKA) and S1565A in HEK-293 cells. The anchoring proteins were immunoprecipitated using anti-Flag antibodies and incubated for 10 min with [32P]γ-ATP in the absence or presence of 10 μM cAMP, or 10 μM cAMP and 20 μM PKI. As shown in Figure 4B, treatment of AKAP-Lbc immunoprecipitates with cAMP increased by two-fold the phosphorylation of the anchoring protein (Figure 4B, upper panel, lane 2, and Figure 4C). This effect was totally abolished by PKI, suggesting that it is entirely mediated by PKA (Figure 4B, upper panel, lane 3, and Figure 4C). The cAMP-induced phosphorylation was abolished in the anchoring-deficient mutant A1551P/I1260P, whereas it was strongly reduced in the mutant S1565A (Figure 4B, upper panel, lanes 5 and 8, and Figure 4C). These results suggest that serine 1565 can be phosphorylated by the PKA holoenzyme anchored to AKAP-Lbc. The increase in phosphorylation of the S1565A mutant of AKAP-Lbc in response to cAMP, which is blocked by PKI, suggests the existence of a second PKA phosphorylation site, which is less phosphorylated as compared to serine 1565. The basal phosphorylation of the S1565A mutant, which is not influenced by cAMP or PKI (Figure 4B, upper panel, lane 4, and Figure 4C), suggests that a kinase other than PKA phosphorylates AKAP-Lbc.
Binding of 14-3-3 to AKAP-Lbc is modulated by phosphorylation of serine 1565 by anchored PKA
To demonstrate that the phosphorylation of serine 1565 of AKAP-Lbc by anchored PKA is required for the association with 14-3-3, serum-starved HEK-293 cells overexpressing the Flag-AKAP-Lbc, Flag-AKAP-Lbc A1551P/I1260P as wells as Flag-AKAP-Lbc S1565A were incubated for 10 min in the absence or presence of 10 μM forskolin or preincubated for 1 h with PKImyr before forskolin treatment. Overexpressed proteins were immunoprecipitated using anti-Flag antibodies and the presence of associated 14-3-3β was assessed using anti-14-3-3β antibodies. Treatment of cells with forskolin (FSK) enhanced the association of 14-3-3β to Flag-AKAP-Lbc by 2.5–fold, whereas pretreatment with PKImyr (FSK+PKImyr) strongly inhibited this interaction (Figure 5A middle panel, lanes 1–3, and Figure 5B). Accordingly, the A1551P/I1260P mutant of AKAP-Lbc was impaired in its ability to associate with 14-3-3 either under basal conditions or after forskolin treatment as compared to the wild-type anchoring protein (Figure 5A, middle panel, lanes 4–6, and Figure 5B), whereas mutation of serine 1565 to alanine abolished the interaction of AKAP-Lbc with 14-3-3 (Figure 5A, middle panel, lanes 7–9, and Figure 5B). These results strongly suggest that phosphorylation of serine 1565 by the PKA holoenzyme associated with AKAP-Lbc is required for the binding of 14-3-3 to the anchoring protein.
Figure 5.
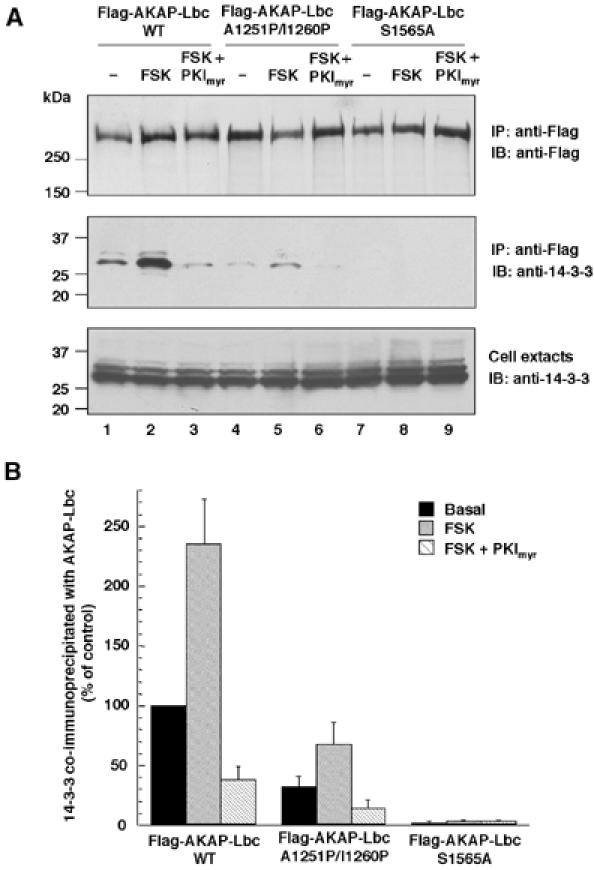
Anchored PKA is required for the association of 14-3-3 with AKAP-Lbc. (A) HEK-293 cells expressing the Flag-tagged fusions of AKAP-Lbc (lanes 1–3) or of its mutants A1251P/I1260P (lanes 4–6) and S1565A (lanes 7–9) were serum starved for 24 h, and then treated for 10 min in the absence or presence of 10 μM forskolin (FSK) or 10 μM forskolin+20 μM of myristoylated PKI 5–24 peptide (FSK+PKImyr). Cell lysates were subjected to immunoprecipitation with anti-Flag antibodies. Western blots of the immunoprecipitates and of the cell extracts were revealed using anti-Flag monoclonal antibodies to detect Flag-AKAP-Lbc (upper panel) or anti-14-3-3β rabbit polyclonal antibodies to detect the endogenous 14-3-3β (middle and lower panels). (B) Densitometry of the bands corresponding to 14-3-3β co-immunoprecipitated with AKAP-Lbc from cells that were treated as indicated in (A). The amount of 14-3-3β in the immunoprecipitates was normalized to the 14-3-3β content of the cell extracts. Results are the mean±s.e. of three independent experiments.
14-3-3 inhibits AKAP-Lbc Rho-GEF activity in vitro
Members of the 14-3-3 family regulate the activity of a variety of signaling proteins (Tzivion and Avruch, 2002). To determine whether 14-3-3 could modulate the signaling properties of the AKAP-Lbc complex, we used an in vitro GDP/GTP exchange assay to measure the effect of increasing concentrations of purified His6-tagged 14-3-3β on the Rho-GEF activity of AKAP-Lbc. Flag-AKAP-Lbc was immunoprecipitated from HEK-293 cells and submitted to PKA phosphorylation prior to the beginning of the exchange reaction. As shown in Figure 6A, Flag-AKAP-Lbc was able to promote the incorporation of [35S]GTPγS into purified RhoA. Addition of His6-14-3-3β to the exchange reaction resulted in the inhibition of AKAP-Lbc Rho-GEF activity with a maximal inhibition observed at a concentration of 200 nM. Addition of similar concentrations of a control protein (His-tagged regulatory subunit IIα of PKA) did not induce any effect (results not shown). These results suggest that 14-3-3 can directly inhibit AKAP-Lbc Rho-GEF activity.
Figure 6.
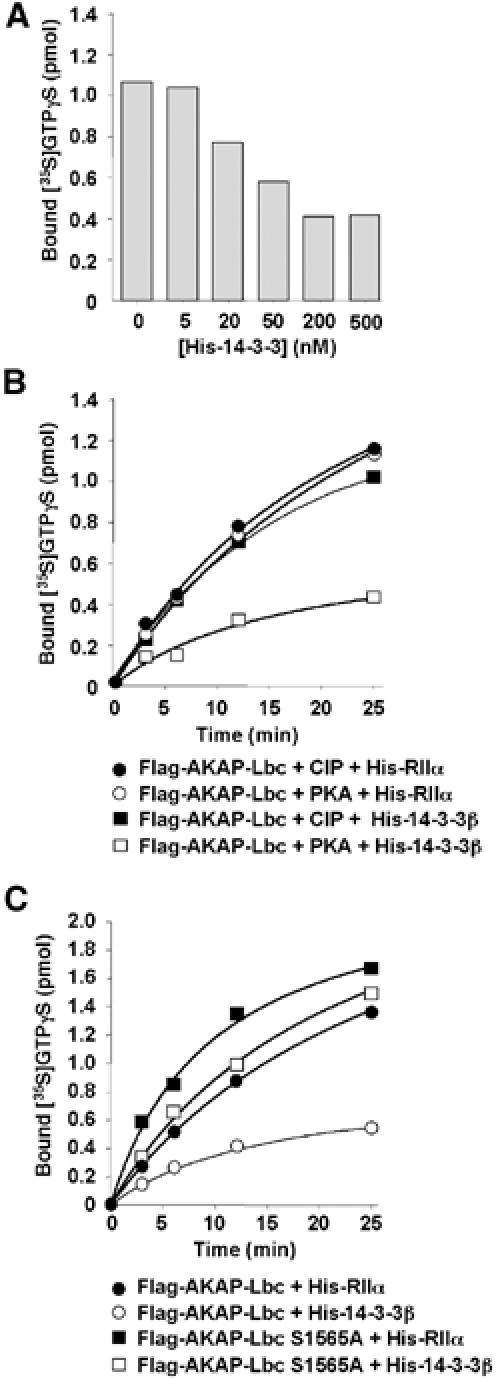
14-3-3 inhibits AKAP-Lbc Rho-GEF activity in vitro. (A) HEK-293 cells expressing Flag-AKAP-Lbc were serum starved for 24 h. Lysates were then subjected to immunoprecipitation with anti-Flag monoclonal antibodies. Immunoprecipitates were incubated with purified PKA and then incubated with GDP-loaded RhoA, [35S]GTPγS and concentrations of His-tagged 14-3-3β ranging from 5 to 500 nM. Exchange reactions were terminated after 25 min. (B) Immunoprecipitates of Flag-AKAP-Lbc were incubated with CIP (closed symbols) or with purified PKA (open symbols) and then incubated with GDP-loaded RhoA, [35S]GTPγS and 200 nM of His-tagged purified RIIα (circles) or 14-3-3β (squares). Exchange reactions were terminated after 25 min. (C) Immunoprecipitates of Flag-tagged AKAP-Lbc (circles) or AKAP-Lbc S1565A (squares) were incubated for 1 h with purified PKA and then incubated with GDP-loaded RhoA, [35S]GTPγS and 200 nM of His-tagged RIIα (closed symbols) or 14-3-3β (open symbols). Exchange reactions were terminated after 25 min. Results are representative of three independent experiments.
To determine whether the inhibitory effect induced by 14-3-3 requires the phosphorylation of AKAP-Lbc by PKA, we pretreated AKAP-Lbc immunoprecipitates with calf intestine phosphatase (CIP) or with PKA before incubation with 200 nM of His-14-3-3β or His-RIIα. As shown in Figure 6B, His-14-3-3β could exert his inhibitory effect only when AKAP-Lbc was preincubated with PKA (open squares). No inhibition of Rho-GEF activity was observed after pretreatment with CIP (closed squares) or when AKAP-Lbc was incubated with PKA and His-RIIα (open circles), suggesting that PKA and His-14-3-3 act coordinately to regulate AKAP-Lbc function negatively.
To establish whether 14-3-3 inhibits AKAP-Lbc Rho-GEF activity by interacting directly with the phosphoserine-containing motif located between residues 1561 and 1570, we measured the effect of mutating serine 1565 on the inhibitory function of 14-3-3β. As shown in Figure 6C, the Rho-GEF activity of the S1565A mutant of AKAP-Lbc was unaffected by His-14-3-3β (open squares). Altogether these findings suggest that 14-3-3 can inhibit AKAP-Lbc function by associating with the serine 1565-containing motif in a PKA-dependent manner.
14-3-3 inhibits AKAP-Lbc Rho-GEF activity inside cells
To determine whether 14-3-3 can inhibit AKAP-Lbc function inside cells, we overexpressed AKAP-Lbc as well as its mutants S1565A and A1251P/I1260P in HEK-293 cells and assessed their ability to activate RhoA using the Rhotekin pulldown assay.
We previously showed that AKAP-Lbc displays a low basal activity state in serum-starved cells and that this activity can be enhanced by treatment of cells with serum or lysophosphatidic acid (LPA) (Diviani et al, 2001). Accordingly, treatment of cells with 10% serum (S) strongly stimulated the Rho-activating effect of AKAP-Lbc as compared to untreated cells (Figure 7A, upper panel, lanes 4–5, and Figure 7B). Interestingly, forskolin treatment (S+FSK) inhibited by 95% the stimulatory effect of serum on AKAP-Lbc Rho-GEF activity (Figure 7A, upper panel, lane 6, and Figure 7B) suggesting that stimulation of the PKA pathway can inhibit the activation of AKAP-Lbc induced by serum.
Figure 7.
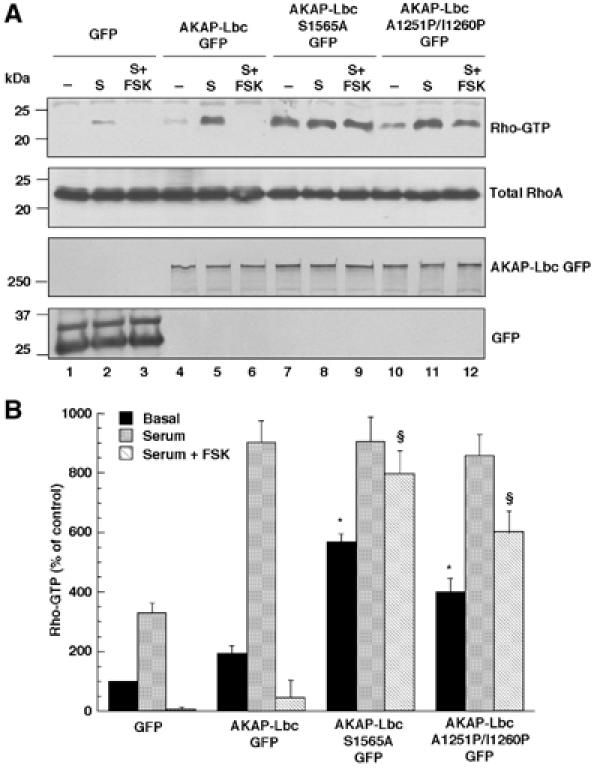
Anchoring of both PKA and 14-3-3 to AKAP-Lbc inhibits its Rho-GEF activity inside cells. (A) HEK-293 cells expressing GFP (lanes 1–3) or the GFP-tagged forms of AKAP-Lbc (lanes 4–6) or of its mutants A1251P/I1260P (lanes 7–9) and S1565A (lanes 10–12) were serum starved for 24 h, and then treated for 1 h in the absence or presence of 10% fetal calf serum (S) or 10% fetal calf serum+10 μM forskolin (S+FSK). Cell lysates were incubated with GST-RBD beads. The bound RhoA was detected with a monoclonal anti-RhoA antibody (upper panel). The amounts of total RhoA, GFP and AKAP-Lbc GFP in the cell lysates were assessed using monoclonal antibodies against RhoA (middle panel) and GFP (lower panels). (B) Quantitative analysis of the GTP-RhoA associated with RBD beads was obtained by densitometry. The RhoA bound to RBD (upper panel) was normalized to the RhoA content of cell extracts (middle panel). Results are expressed as mean±s.e. of four independent experiments. Statistical significance was analyzed by paired Student's test. *P<0.05 as compared with Rho-GTP levels measured in untreated cells expressing AKAP-Lbc GFP. §P<0.05 as compared with Rho-GTP levels measured in forskolin-treated cells expressing AKAP-Lbc GFP.
Remarkably, both the S1565A and A1251P/I1260P mutants of AKAP-Lbc displayed basal Rho-GEF activities significantly higher than wild-type AKAP-Lbc (Figure 7A, upper panel, lanes 7 and 10, and Figure 7B) and showed an increased resistance to forskolin-mediated inhibition (Figure 7A, upper panel, lanes 9 and 12, and Figure 7B). These results suggest that anchoring of PKA and binding of 14-3-3 to AKAP-Lbc are both required to maintain the anchoring protein in a low-activity state under basal unstimulated conditions. They also provide evidence that the inhibitory effect of forskolin on AKAP-Lbc Rho-GEF activity is mediated by the anchoring of both PKA and 14-3-3 to AKAP-Lbc.
To further demonstrate that endogenous 14-3-3 can inhibit AKAP-Lbc function inside cells, we determined whether the overexpression of a dominant-negative mutant of 14-3-3 (in which arginines 56 and 60 were mutated to alanine) could antagonize the effect of endogenous 14-3-3 on AKAP-Lbc function. This mutant, which was originally described for 14-3-3η, is impaired in its ability to interact with cellular targets (Thorson et al, 1998). Overexpression of Flag-14-3-3β R56A/R60A increased significantly the basal Rho-GEF activity of AKAP-Lbc and strongly reduced the inhibitory effect of forskolin on AKAP-Lbc function (see Supplementary Figure 3). Overexpression of dominant-negative 14-3-3β did not induce any effects in cells that were not transfected with AKAP-Lbc (see Supplementary Figure 3). These results reinforce the notion that binding of 14-3-3 to AKAP-Lbc maintains the anchoring protein in an inhibited conformation. In order to determine whether the inhibition of AKAP-Lbc Rho-GEF activity observed upon binding of 14-3-3 could be attributed to the release of RhoA from AKAP-Lbc, we determined the ability of RhoA to co-immunoprecipitate with wild-type AKAP-Lbc or AKAP-Lbc S1565A. Interestingly, the 14-3-3-binding-deficient mutant of AKAP-Lbc displayed a stronger association with RhoA either under basal unstimulated conditions or in the presence of forskolin as compared to wild-type AKAP-Lbc (see Supplementary Figure 4A). These results suggest that binding of 14-3-3 to AKAP-Lbc inhibits the association of RhoA.
In order to determine the cellular consequences of abolishing the 14-3-3-binding site on AKAP-Lbc, we measured the ability of AKAP-Lbc and AKAP-Lbc S1565A to induce the formation of stress fibers in NIH3T3 fibroblasts. We could show that, in serum-starved cells, the S1565A mutant induces a higher density of stress fibers as compared to the wild-type protein (see Supplementary Figure 4B). This suggests that the higher Rho-GEF activity of the S1565A mutant correlates with an increased ability to induce stress fibers formation.
Discussion
GEFs of the Dbl family are multifunctional molecules, which play a crucial role in the transduction of signals leading to the activation of Rho. Because of their implication in diverse physiological processes such as growth and development, skeletal muscle formation and neuronal axon guidance, the mechanisms involved in their regulation has been intensively investigated (Zheng, 2001). Our present findings indicate that the RhoA-selective exchange factor AKAP-Lbc is regulated in a bidirectional manner by signals that activate or deactivate its Rho-GEF activity. Stimulation of Gα12 by serum or LPA is known to enhance the Rho-GEF activity of AKAP-Lbc (Figure 8). Here, we demonstrated that elevation of the cellular concentration of cAMP activates the PKA holoenzyme anchored to AKAP-Lbc, which, in turn, phosphorylates the anchoring protein on serine 1565. This induces the recruitment of 14-3-3, which inhibits the Rho-GEF activity of AKAP-Lbc (Figure 8). Therefore, AKAP-Lbc represents a molecular platform where the cAMP/PKA pathway and Rho signaling pathways converge.
Figure 8.
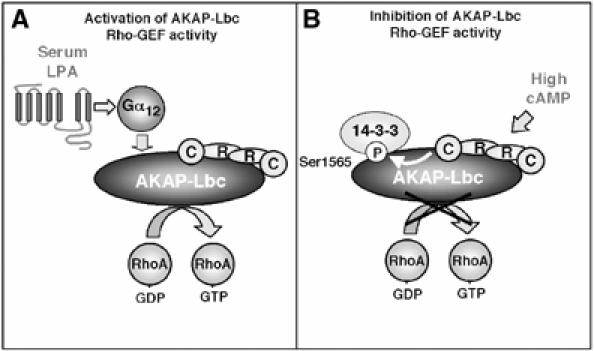
Model for the 14-3-3-mediated inhibition of AKAP-Lbc. (A) AKAP-Lbc is activated in response to serum or LPA stimulation through a Gα12-mediated signaling pathway. (B) Elevation of the cellular concentration of cAMP activates the PKA holoenzyme anchored to AKAP-Lbc. The catalytic subunit released from the PKA holoenzyme can phosphorylate AKAP-Lbc on serine 1565. This induces the recruitment of 14-3-3, which inhibits the Rho-GEF activity of AKAP-Lbc.
It is well established that PKA has an antagonistic effect on the Rho signaling pathway. This is in part mediated by the inhibitory action of PKA on RhoA (Lang et al, 1996; Ellerbroek et al, 2003). However, recent studies indicate that PKA can also regulate the activity of upstream activators of RhoA such as Gα13 (Manganello et al, 2003). Our findings now indicate that PKA can phosphorylate and promote the inhibition of the Rho-GEF AKAP-Lbc, an effect that requires the anchoring of PKA to AKAP-Lbc. This suggests that PKA needs to be activated locally within the AKAP-Lbc complex in order to promote efficient phosphorylation AKAP-Lbc. We also tested the hypothesis whether AKAP-Lbc could modulate the PKA-mediated phosphorylation of RhoA. However, the overexpression of AKAP-Lbc or a PKA-anchoring-deficient mutant of AKAP-Lbc did not induce any modulation of the phosphorylation of RhoA by PKA (results not shown), suggesting that RhoA is regulated by another AKAP. Recent studies have shown that AKAP110, a testis-specific anchoring protein, associates directly with Gα13 (Niu et al, 2001). However, it remains to be established whether this AKAP regulates the phosphorylation of Gα13.
Members of the 14-3-3 family bind their cellular targets primarily through the association with the phosphoserine-containing motifs RSXpSXP and RXXXpSXP, where pS denotes both phosphoserine and threonine (Muslin et al, 1996; Yaffe et al, 1997). Our mapping analysis identified a minimal 14-3-3-binding site (FRRHSWGPGK), which diverges from these canonical 14-3-3-binding sites. In fact, while it contains an arginine three positions upstream of the phosphorylated serine (position 0), it does not contain a serine in the −2 position. In addition, the proline residue, which is normally observed in the +2 position, is located at position +3. This 14-3-3-binding motif is strikingly similar to that identified in the tyrosine phosphatase PTPH1 by Zhang et al (1997), which also lacks a serine residue two positions upstream of the phosphorylated serine and a proline two positions downstream. These findings suggest that the canonical consensus motifs, although strongly favored, do not represent an absolute requirement for high-affinity binding of 14-3-3.
The recent structural analysis of 14-3-3 revealed its dimeric structure (Liu et al, 1995; Xiao et al, 1995). Although both monomeric and dimeric forms of 14-3-3 can interact with cellular proteins, the interaction of monomeric 14-3-3 seems to be independent of the phosphorylation of the target, whereas binding of dimeric 14-3-3 seems to require it (Shen et al, 2003). Our results indicate that AKAP-Lbc interacts with the dimeric form of 14-3-3 in a PKA-dependent manner (see Supplementary Figure 1). However, our mapping studies identified only a single 14-3-3-binding site between residues 1561 and 1570 of AKAP-Lbc. Interestingly, proteins such as cofilin (Gohla and Bokoch, 2002), the ribosomal S6 kinase (Cavet et al, 2003) and the Yes-associated protein YAP (Basu et al, 2003) also interact with 14-3-3 through a single phosphoserine-containing motif. These results could be explained by a recent model, which proposes that some cellular proteins could interact with 14-3-3 through a single dominant site (Yaffe, 2002). If this site is deleted or not phosphorylated, then the secondary site might be too weak to promote a stable 14-3-3 interaction. Once the primary site is phosphorylated and bound to one-half of the 14-3-3 dimer, then the secondary sites could interact with the second half of the dimer by virtue of the high local concentration induced by its proximity. Therefore, we cannot exclude the possibility that AKAP-Lbc might contain a secondary low-affinity 14-3-3-binding site, which alone might not interact with 14-3-3. We have shown that AKAP-Lbc contains a second PKA phosphorylation site in addition to serine 1565 (Figure 4A). One can raise the hypothesis whether this site might represent a second 14-3-3-binding motif that is functionally subordinated to the dominant site containing serine 1565.
We also tested the possibility that the 14-3-3 dimer might link two distinct AKAP-Lbc molecules. However, our results rule out this hypothesis since we could not co-immunoprecipitate fragments of AKAP-Lbc containing the 14-3-3-binding site neither in the absence nor in the presence of overexpressed 14-3-3 (results not shown).
Members of the 14-3-3 family regulate a variety of signal transduction enzymes either by affecting their catalytic activity or by altering their subcellular localization (Tzivion and Avruch, 2002). Here, we show that 14-3-3 binding to AKAP-Lbc negatively regulates its Rho-GEF activity. This inhibitory effect reflects a direct inhibition of AKAP-Lbc activity rather than an alteration of its intracellular localization since it can be observed in vitro in the presence of purified 14-3-3 and AKAP-Lbc. Moreover, we could show that mutation of the 14-3-3-binding site on AKAP-Lbc does not affect its subcellular distribution. In fact, both wild-type and 14-3-3-binding-deficient AKAP-Lbc showed an identical cytoplasmic localization (results not shown). Importantly, our results indicate that binding of 14-3-3 to AKAP-Lbc decreases the amount of RhoA associated with the anchoring protein. This suggests that 14-3-3 inhibits AKAP-Lbc Rho-GEF activity by interfering with the interaction between AKAP-Lbc and RhoA. A possible model is that 14-3-3 maintains AKAP-Lbc in an inactive conformation that cannot interact with Rho.
Interestingly, it has been shown that 14-3-3ɛ and 14-3-3μ directly associate with a RhoA-specific exchange factor called p190RhoGEF (Zhai et al, 2001). However, whether this interaction affects the signaling properties of p190RhoGEF remains to be elucidated. Very recent results demonstrate that another Rho-GEF, called Rho-GEF-H1, interacts with 14-3-3 following PAK1-mediated phosphorylation (Zenke et al, 2004). Interestingly, the 14-3-3-binding site of GEF-H1 is located on a region of the molecule that exerts a negative regulation on the Rho-GEF activity. However, whether 14-3-3 binding directly affects the Rho-GEF activity of GEF-H1 remains to be established. Therefore, to our knowledge, our findings represent the first direct evidence that 14-3-3 family members can regulate the Rho-GEF activity of GEFs of the Dbl family.
Several members of the Dbl family exist in an inactive basal state prior to their activation (Zheng, 2001). Upstream signals, such as G protein α or βγ subunits, protein kinases, adaptor or scaffolding proteins, as well as phosphoinositol lipids have been shown to contribute to GEF activation processes (Zheng, 2001). We have previously shown that inhibitory determinants present in the amino-terminal sequence of AKPA-Lbc maintain the protein in a low activity state (Diviani et al, 2001). Here, we show that this basal inhibited state is controlled by the binding of 14-3-3 to a site located in the N-terminal regulatory region of AKAP-Lbc. This is demonstrated by the fact that mutation of the 14-3-3-binding site on AKAP-Lbc or overexpression of dominant-negative mutants of 14-3-3β significantly increased the basal Rho-GEF activity of AKAP-Lbc. Anchoring of PKA to AKAP-Lbc plays an important role in maintaining the anchoring protein inactive under basal conditions. In fact, PKA-binding-deficient mutants of AKAP-Lbc display a higher basal Rho-GEF activity. This implies that the tonic activity of anchored PKA is sufficient to support basal interaction between 14-3-3 and AKAP-Lbc. Recent evidence suggests that additional members of Dbl family can be maintained at a basal state by regulatory protein. For example, binding of the tumor suppressor nm23H1 to Tiam1 decreases the GEF activity of Tiam1 toward Rac1 (Otsuki et al, 2001), whereas interaction of a yet unidentified protein with p115RhoGEF inhibits its ability to activate RhoA (Wells et al, 2001). Therefore, it appears that exchange factors of the Dbl family can form macromolecular complexes that can be activated or inactivated by associated regulatory proteins.
In conclusion, the implications of our findings are two-fold. Firstly, they demonstrate the functional role of anchoring PKA on AKAP-Lbc signaling. Whereas it is known that AKAP-Lbc signaling can be potentiated by alpha subunit of G12, in this study we demonstrated that it is inhibited by the anchoring of both PKA and 14-3-3. Secondly, they provide a novel hypothesis explaining the inhibitory action of PKA on Rho signaling, suggesting that AKAP-Lbc might play a role in integrating the PKA and Rho pathway.
Materials and methods
Standard methods discussing the generation of expression constructs, the purification of recombinant proteins in bacteria, cell culture and transfection, GST pulldown and immunoprecipitation experiments, SDS–PAGE and Western blotting, and the solid phase overlay assay are described in Supplementary material.
Yeast two-hybrid screening
The yeast strain L40 was transformed with the AKAP-Lbc 1388–1922-pLexA plasmid encoding a fragment of AKAP-Lbc encompassing residues 1388–1922 fused to LexA. Clones were selected and subsequently transformed with 250 μg of a human heart Matchmaker cDNA library in the pACT2 vector (Clontech). Of six million double transformants, 52 exhibited moderate to strong growth on histidine-deficient plates. The library plasmids isolated from positive clones were cotransformed in the L40 strain with either the original bait vector or the empty pLexA plasmid, and the specificity of the interactions was confirmed by growth on histidine-deficient plates as well as by β-galactosidase activity (Yeast Protocols Handbook, Clontech).
In vitro phosphorylation experiments
Purified GST-tagged AKAP-Lbc 1388–1922 or AKAP-Lbc 1388–1922 S1565A fragments (2 μg) were incubated with the catalytic subunit of PKA (5 U) and [32P]γ-ATP (10 μCi), in 50 mM Tris pH 7.4, 5 mM MgCl2 and 1 mM ATP-Na2 for 1 h at 30°C. Reactions were ended by the addition of SDS–PAGE sample buffer and loaded on SDS–PAGE gels.
For the phosphorylation of full-length Flag-AKAP-Lbc constructs, proteins were immunoprecipitated from HEK-293 cells using an anti-Flag affinity resin as indicated above. After immunoprecipitation, beads were washed five times in kinase buffer and then incubated for 1 h in kinase buffer containing 1 mM ATP in the absence or presence of 10 U of purified PKA. Phosphorylation reactions were terminated by washing beads five times with a buffer containing 50 mM Tris and 100 mM NaCl. Phosphorylated AKAP proteins were then processed for the GDP/GTP exchange assay as indicated below.
Auto-spot peptide synthesis
Peptide arrays were synthesized on cellulose paper using an Auto-Spot Robot ASP 222 (AbiMed, Langenfeld, Germany). After synthesis, the amino termini were acetylated with 2% acetic acid anhydride in dimethyl formamide. The peptides were then deprotected by 1 h treatment with dichloromethane:trifluoroacetic acid (1:1), containing 3% triisopropylsilane and 2% water. After extensive washes in TBS-Tween, arrays were incubated for 4 h at 30°C in 10 ml of 50 mM Tris pH 7.4 and 5 mM MgCl2 in the absence or presence of 100 U of purified PKA catalytic subunit (Sigma). Arrays were subsequently washed five times in TBS-Tween and used for the overlay assay.
GDP/GTP exchange assay
The exchange assays were performed as previously described (Zheng et al, 1995a). A 2 μg portion of the recombinant RhoA was incubated for 5 min in 60 μl of loading buffer (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 2 mM EDTA, 0.2 mM DTT, 100 μM AMP-PNP and 10 μM GDP) at room temperature. MgCl2 was then added to a final concentration of 5 mM and the incubation was continued for an additional 15 min. To initiate the exchange reaction, protein aliquots (20 μl) of GDP-loaded GTPases were mixed at room temperature with 80 μl reaction buffer (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10 mM MgCl2, 100 μM AMP-PNP, 0.5 mg/ml bovine serum albumin and 5 μM [35S]GTPγS (11 000 cpm/pmol)) containing immunoprecipitated Flag-AKAP-Lbc or Flag-AKAP-Lbc S1565A with or without 200 nM of purified His-tagged 14-3-3. Aliquots (15 μl) of samples were taken at various time points and added to 10 ml of ice-cold PBS. Bound and free nucleotides were separated by filtration through BA85 nitrocellulose filters.
Rhotekin Rho-binding domain pulldown assay
HEK-293 cells grown in 100 mm dishes were transfected with 24 μg of the AKAP-Lbc/pEGFP, AKAP-Lbc A1251P/I1260P-pEGFP or AKAP-Lbc S1565A-pEGFP constructs. At 24 h after transfection, cells were incubated in DMEM without serum for an additional 24 h. Cells were then treated for 1 h with 10% fetal calf serum in the absence or presence of 50 μM forskolin (Sigma) and lysed in Rho-binding domain (RBD) lysis buffer (50 mM Tris pH 7.2, 150 mM NaCl, 1% (w/v) Triton X-100, 30 mM MgCl2, 1 mM DTT, 10% glycerol, 1 mM benzamidine, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 1 mM PMSF). Lysates were subjected to centrifugation at 38 000 g for 10 min at 4°C and incubated with 30 μg of RDB beads for 1 h at 4°C. Beads were then washed three times with RBD buffer without sodium deoxycholate, resuspended in SDS sample buffer and analyzed by SDS–PAGE.
Supplementary Material
Supplementary Material
Acknowledgments
We acknowledge Monique Nenniger-Tosato for excellent technical assistance, Dr Olivier Staub for critical reading of the manuscript and Dr Hitoshi Kurose (Fukuoka, Japan) for providing the Rhotekin-RBD construct. We thank Jacquelyn Soderling in the laboratory of John Scott (Vollum Institute, Portland, Oregon) for constructing the pLexA and pGEX vectors containing the AKAP-Lbc 1388–1922 cDNA. This work was supported by grants 3100-067955 (to DD) and 3100AO-100703 (to SC) of the Fonds National Suisse de la Recherche Scientifique.
References
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J (2003) Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell 11: 11–23 [DOI] [PubMed] [Google Scholar]
- Booden MA, Siderovski DP, Der CJ (2002) Leukemia-associated Rho guanine nucleotide exchange factor promotes G alpha q-coupled activation of RhoA. Mol Cell Biol 22: 4053–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser IDC, Bishop SM, Acott TS, Brennan RG, Scott JD (1991) Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem 266: 14188–14192 [PubMed] [Google Scholar]
- Cavet ME, Lehoux S, Berk BC (2003) 14-3-3beta is a p90 ribosomal S6 kinase (RSK) isoform 1-binding protein that negatively regulates RSK kinase activity. J Biol Chem 278: 18376–18383 [DOI] [PubMed] [Google Scholar]
- Diviani D, Soderling J, Scott JD (2001) AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J Biol Chem 276: 44247–44257 [DOI] [PubMed] [Google Scholar]
- Ellerbroek SM, Wennerberg K, Burridge K (2003) Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 278: 19023–19031 [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629–635 [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Chikumi H, Gutkind JS (2000) Leukemia-associated Rho guanine nucleotide exchange factor (LARG) links heterotrimeric G proteins of the G(12) family to Rho. FEBS Lett 485: 183–188 [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS (1999) A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem 274: 5868–5879 [DOI] [PubMed] [Google Scholar]
- Gohla A, Bokoch GM (2002) 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr Biol 12: 1704–1710 [DOI] [PubMed] [Google Scholar]
- Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Galpha13. Science 280: 2112–2114 [DOI] [PubMed] [Google Scholar]
- Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J (1996) Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 15: 510–519 [PMC free article] [PubMed] [Google Scholar]
- Laudanna C, Campbell JJ, Butcher EC (1997) Elevation of intracellular cAMP inhibits RhoA activation and integrin-dependent leukocyte adhesion induced by chemoattractants. J Biol Chem 272: 24141–24144 [DOI] [PubMed] [Google Scholar]
- Liu D, Bienkowska J, Petosa C, Collier RJ, Fu H, Liddington R (1995) Crystal structure of the zeta isoform of the 14-3-3 protein. Nature 376: 191–194 [DOI] [PubMed] [Google Scholar]
- Manganello JM, Huang JS, Kozasa T, Voyno-Yasenetskaya TA, Le Breton GC (2003) Protein kinase A-mediated phosphorylation of the Galpha13 switch I region alters the Galphabetagamma13-G protein-coupled receptor complex and inhibits Rho activation. J Biol Chem 278: 124–130 [DOI] [PubMed] [Google Scholar]
- Michel JJ, Scott JD (2002) AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol 42: 235–257 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897 [DOI] [PubMed] [Google Scholar]
- Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA (1999) The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat Struct Biol 6: 222–227 [DOI] [PubMed] [Google Scholar]
- Niu J, Vaiskunaite R, Suzuki N, Kozasa T, Carr DW, Dulin N, Voyno-Yasenetskaya TA (2001) Interaction of heterotrimeric G13 protein with an A-kinase-anchoring protein 110 (AKAP110) mediates cAMP-independent PKA activation. Curr Biol 11: 1686–1690 [DOI] [PubMed] [Google Scholar]
- Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K, Sugimura H (2001) Tumor metastasis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proc Natl Acad Sci USA 98: 4385–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YH, Godlewski J, Bronisz A, Zhu J, Comb MJ, Avruch J, Tzivion G (2003) Significance of 14-3-3 self-dimerization for phosphorylation-dependent target binding. Mol Biol Cell 14: 4721–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Scott JD (2002) Signaling complexes: junctions on the intracellular information super highway. Curr Biol 12: R32–R40 [DOI] [PubMed] [Google Scholar]
- Thorson JA, Yu LW, Hsu AL, Shih NY, Graves PR, Tanner JW, Allen PM, Piwnica-Worms H, Shaw AS (1998) 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol Cell Biol 18: 5229–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion G, Avruch J (2002) 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J Biol Chem 277: 3061–3064 [DOI] [PubMed] [Google Scholar]
- Wells CD, Gutowski S, Bollag G, Sternweis PC (2001) Identification of potential mechanisms for regulation of p115 RhoGEF through analysis of endogenous and mutant forms of the exchange factor. J Biol Chem 276: 28897–28905 [DOI] [PubMed] [Google Scholar]
- Xiao B, Smerdon SJ, Jones DH, Dodson GG, Soneji Y, Aitken A, Gamblin SJ (1995) Structure of a 14-3-3 protein and implications for coordination of multiple signaling pathways. Nature 376: 188–191 [DOI] [PubMed] [Google Scholar]
- Yaffe MB (2002) How do 14-3-3 proteins work?—Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett 513: 53–57 [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91: 961–971 [DOI] [PubMed] [Google Scholar]
- Zenke FT, Krendel M, DerMardirossian C, King CC, Bohl BP, Bokoch GM (2004) p21-activated kinase 1 phosphorylates and regulates 14-3-3 binding to GEF-H1, a microtubule-localized Rho exchange factor. J Biol Chem 279: 18392–18400 [DOI] [PubMed] [Google Scholar]
- Zhai J, Lin H, Shamim M, Schlaepfer WW, Canete-Soler R (2001) Identification of a novel interaction of 14-3-3 with p190RhoGEF. J Biol Chem 276: 41318–41324 [DOI] [PubMed] [Google Scholar]
- Zhang SH, Kobayashi R, Graves PR, Piwnica-Worms H, Tonks NK (1997) Serine phosphorylation-dependent association of the band 4.1-related protein-tyrosine phosphatase PTPH1 with 14-3-3beta protein. J Biol Chem 272: 27281–27287 [DOI] [PubMed] [Google Scholar]
- Zheng Y (2001) Dbl family guanine nucleotide exchange factors. Trends Biochem Sci 26: 724–732 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Hart MJ, Cerione RA (1995a) Guanine nucleotide exchange catalyzed by dbl oncogene product. Methods Enzymol 256: 77–84 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Olson MF, Hall A, Cerione RA, Toksoz D (1995b) Direct involvement of the small GTP-binding protein Rho in lbc oncogene function. J Biol Chem 270: 9031–9034 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material