

Abstract
Germ line DNA directs the development of the next generation and, as such, is profoundly different from somatic cell DNA. Spermatogenic cells obtained from young adult lacI transgenic mice display a lower spontaneous mutant frequency and greater in vitro base excision repair activity than somatic cells and tissues obtained from the same mice. However, spermatogenic cells from old lacI mice display a 10-fold higher mutant frequency. This increased spontaneous mutant frequency occurs coincidentally with decreased in vitro base excision repair activity for germ cell and testicular extracts that in turn corresponds to a decreased abundance of AP endonuclease. To directly test whether a genetic diminution of AP endonuclease results in increased spontaneous mutant frequencies in spermatogenic cell types, AP endonuclease heterozygous (Apex+/−) knockout mice were crossed with lacI transgenic mice. Spontaneous mutant frequencies were significantly elevated (approximately twofold) for liver and spleen obtained from 3-month-old Apex+/− lacI+ mice compared to frequencies from Apex+/+ lacI+ littermates and were additionally elevated for somatic tissues from 9-month-old mice. Spermatogenic cells from 9-month-old Apex+/− lacI+ mice were significantly elevated twofold compared to levels for 9-month-old Apex+/+ lacI+ control mice. These data indicate that diminution of AP endonuclease has a significant effect on spontaneous mutagenesis in somatic and germ line cells.
Maintenance of germ cell genetic integrity is fundamental to the development of healthy offspring. However, DNA is constantly exposed to endogenous sources of damage (15). If left unrepaired, DNA damage can lead to errors during replication, thus generating de novo germ line mutations. Such mutations can result in genetic diseases. It has been suggested that most spontaneous germ line mutations occur in male gametes rather than female gametes (11, 18, 55), and there is a paternal age effect associated with several dominant genetic disorders (8, 11, 14, 19, 36, 52, 55). Studies on germ line mutagenesis were facilitated greatly by the development of mice transgenic for the lacI or lacZ reporter genes. Using these models, a direct assessment of in vivo spontaneous mutant frequencies for male germ cells and somatic cells and tissues is possible. lacI transgenic mice display an approximately 10-fold lower spontaneous mutant frequency (0.6 × 10−5) for male germ cells than for somatic cells and tissues (4.8 × 10−5) obtained from young adults (26, 47, 60). Notably, a 10-fold higher spontaneous mutant frequency was observed for spermatogenic cells obtained from old (28-month-old) mice (2.9 × 10−5 to 4.9 × 10−5) than for young adult (60-day-old) mice (0.4 × 10−5 to 0.8 × 10−5), thereby indicating a paternal age effect in the mouse model (60). The lacI transgenic mice represent the first mouse model used to study mechanisms mediating the paternal age effect.
The results demonstrating a lower spontaneous mutant frequency in the male germ line led to questions about how a lower mutant frequency is realized. Mutant frequency is regulated to a large extent by mechanisms that function to monitor and guard genetic integrity. Among these genetic maintenance mechanisms are DNA repair pathways that recognize DNA damage, remove it, and restore the integrity of the DNA. The base excision repair (BER) pathway restores DNA integrity when challenged by damaged bases (e.g., oxidized or alkylated bases), abasic sites, and some single-strand breaks (29, 35, 39, 41). In this pathway, a damaged base is first recognized and removed by a DNA glycosylase, creating an apurinic/apyrimidinic site (abasic or AP site). The phosphate backbone is nicked, usually by AP endonuclease. A correct base is inserted by a DNA polymerase, the deoxyribose phosphate moiety is removed, and the phosphodiester backbone is sealed by a DNA ligase (29, 39). Several BER genes, including Apex (AP endonuclease), Polβ (DNA polymerase β), Xrcc1, and Lig3 (DNA ligase III), appear to be most abundantly expressed in the testis, or male germ cells specifically, relative to other tissues in the young adult animal (1, 9, 23, 33, 58, 61, 67). In vitro BER assays have revealed a greater activity in testis and spermatogenic cell extracts than in tested somatic cell and tissue extracts (7, 24, 42). These data are consistent with a high level of BER activity contributing to the low spontaneous mutant frequency in the male germ line observed in young adult male mice.
What then causes the increased spontaneous mutant frequency in germ cells with increased age? A significant decrease (50%) in BER activity has been observed in nuclear extracts prepared from spermatogenic cells or testis extracts obtained from old (24- to 26-month-old) mice (7, 25). The decline in in vitro BER activity observed in spermatogenic cells was accompanied by a decline in AP endonuclease (Apex) abundance in one study (25) and by a decline in DNA polymerase β (β-pol) in another (7). Together these data suggest that a decline in BER mediates, at least in part, the increased spontaneous mutant frequency detected in male germ cells obtained from older animals. The purpose of the studies described herein was to more directly test the ability of a diminution in Apex abundance to mediate a decline in BER that would in turn result in an increased spontaneous mutant frequency. Apex heterozygous knockout (Apex+/−) mice (32) were crossed with the lacI transgenic mice, and the spontaneous mutant frequencies were determined for spermatogenic cells and select somatic tissues. The results indicate that inactivation of one Apex allele is sufficient to mediate increased spontaneous mutagenesis in germ line and somatic tissue DNA.
MATERIALS AND METHODS
Generation of heterozygous Apex (Apex+/−) lacI+ transgenic mice.
Mice heterozygous for the Apex (Apex+/−) gene (32) were backcrossed into nontransgenic C57BL/6 mice a minimum of seven to nine generations to obtain an essentially congenic C57BL/6 genetic background. The Apex+/− mice were then crossed with Big Blue mice (Stratagene) that carry a lacI mutation reporter transgene. The lacI transgenic mice were originally made and subsequently maintained in the C57BL/6 genetic background. Progeny resulting from the cross were genotyped by PCR amplification using DNA isolated from ear or tail biopsy specimens (57, 59). The primers used to detect the presence of the inactivated Apex allele were the following neomycin primers: sense neo, 5′-CTGAATGAACTGCAGGACGA-3′, and antisense neo, 5′-CTCTTCGTCCAGATCATCCT-3′. These primers generated a 325-bp PCR fragment spanning positions 162 to 487 of the neomycin resistance gene. Internal control primers detected Apex exon IV that should be present in all mouse DNA samples. The primer set consisted of sense Apex-exon IV, 5′-GTGATTGTGGCTGAATTTGA-3′, and antisense Apxe-exon IV, 5′-GTCTAAAGGAAACCGGAAGT-3′. These primers generate a 624-bp PCR fragment spanning positions 29 to 652 of the murine Apex gene, including a portion of exon IV. To screen for the presence of the lacI gene, the following set of primers were used: sense lacI, 5′-GAAGCGGCGATGGCGGAGCTG-3′, and antisense lacI, 5′-CACCAGTGAGACGGGCAACAG-3′. These primers generated a 857-bp PCR fragment. The PCR conditions used were the following: denaturation at 95°C for 1 min, reannealing at 52°C for 1.5 min, and DNA synthesis at 72°C for 2 min. PCR products were subjected to electrophoresis on 1.2% agarose gels. Male mice heterozygous for the Apex gene and hemizygous for the lacI gene (Apex+/− lacI+) and male siblings wild-type for Apex and hemizygous for lacI (Apex+/+ lacI+) were used to test spontaneous mutant frequencies and apoptosis prevalence at 3 and 9 months of age. Animals were fed a standard rodent chow ad libitum and were maintained under a 12 h-12 h light-dark cycle in an American Association for Accreditation of Laboratory Animal Care-accredited animal facility. All animal manipulations were approved by the Institutional Animal Care and Use Committee.
Preparation of mixed spermatogenic cells and somatic tissue collection.
Apex+/− lacI+ and control Apex+/+ lacI+ mice were humanely euthanized with isofluorane, and then testis, liver, and spleen were collected immediately. Liver and spleen were sliced into small pieces, frozen in liquid nitrogen, and stored at −80°C until use for DNA isolation. Preparation of the mixed spermatogenic cells was conducted as described previously (4, 5). Briefly, each pair of testes was removed, decapsulated, placed in enriched Krebs ringer bicarbonate medium (EKRB), and kept on ice. Collagenase (0.5 mg/ml) and trypsin (0.5 mg/ml) treatments were performed by incubation at 33°C in 5% CO2-enriched air for 15 min, with shaking. DNase (1 μg/ml) treatment was performed subsequently. The resulting mixed spermatogenic cell suspension was passed through a pipette repeatedly to disaggregate cells, and then the cell suspension was filtered through a 100-μm-pore-size nylon cell strainer (Millipore) and centrifuged at 1,200 × g for 12 min at 4°C. The cell pellet was washed in EKRB and resuspended in fresh EKRB with gentle passage through a pipette. A minimum of 7.5 × 106 cells/ml was obtained per cell suspension. Mixed spermatogenic cells were pelleted by centrifugation, frozen by immersion in liquid nitrogen, and stored at −80°C until used for DNA isolation.
Mutant frequency determinations.
High-molecular-weight genomic DNA was isolated from the cells and tissues of interest by using the RecoverEase DNA isolation kit (Stratagene) per the manufacturer's recommendations. The λ shuttle vector transgene carrying the lacI mutation reporter gene was rescued from genomic DNA samples by using Transpack packaging extracts as recommended by the manufacturer (Stratagene). Ten NZY bottom agar-containing assay trays were prepared for each sample. Each packaging reaction mixture was added to a culture of SCS-8 cells prior to being combined with agarose containing the chromogenic substrate 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). Plates were incubated overnight at 37°C to allow plaque formation. Putative mutant plaques were identified by their blue appearance and were marked for isolation. All identified putative mutant plaques were cored and replated on fresh X-Gal/NZY plates to confirm that the lacI mutations were likely mouse derived (20, 21, 40, 43, 54). Mutant frequency was calculated by dividing the number of confirmed mutant plaques by the total number of PFU for that sample.
Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays and histology.
Following euthanasia, animal body weights and testis weights were recorded for 9-month-old Apex+/+ and Apex+/− animals. Testis, spleen, and liver were removed, labeled, and rapidly submerged in formalin solution. Tissue blocks were prepared by personnel in the San Antonio Cancer Institute-sponsored histology facility housed within the Department of Pathology. One slide per animal and tissue of interest (i.e., testis, spleen, and liver) was stained with hematoxylin and eosin (H&E).
Paraffin-embedded testis cross-sections corresponding to Apex+/− and wild-type Apex+/+ mice were subjected to the terminal deoxynucleotidyl end-labeling procedure as originally described (17). Sections were deparaffinized and proteinase K treated (40 μg/ml) for 15 min at room temperature. Three percent H2O2-MeOH was used to block internal peroxidases. Slides were rinsed first in double-distilled H2O and then in terminal deoxynucleotidyl transferase (TdT) buffer (30 mM Trizma base [pH 7.2], 140 mM sodium cacodylate, 1 mM CoCl2). Slides were incubated with 195 U of TdT (Gibco, BRL)/μl and 1 U of biotinylated dUTP (Boehringer Mannheim)/μl at 37°C in a moist chamber for 2 h. After incubation, slides were placed in TB buffer (300 mM NaCl, 30 mM sodium citrate) for 5 min at room temperature and then were washed with double-distilled H2O. Two percent bovine serum albumin was applied to slides for 10 min followed by rinses in distilled H2O and phosphate-buffered saline (PBS) buffer. Incubation (37°C, 1 h) was performed with streptavidin horseradish peroxidase (Dako) diluted 1:200 in streptavidin peroxidase diluent (BioGenex). After a PBS rinse, DAB (3,3-diaminobenzidine tetrahydrochloride; Polysciences Inc.) was added (10 mg/10 ml) to automation buffer (100 mM Tris-HCl, 100 mM Tris-base [pH 7.5], 0.75% Brij 35) in conjunction with 1 μl of 30% H2O2/ml. Subsequent to rinses in distilled H2O, samples were treated with 0.2% OsO4 in PBS. The sections were counterstained with methyl green (1% in double-distilled H2O) following rinses in PBS. Sections were dehydrated prior to mounting coverslips with permount solution.
Slides thus prepared were scored with a Zeiss Photomicroscope III. The percentage of tubules containing apoptotic cells, with apoptosis defined as a TUNEL-positive signal, was determined by dividing the number of seminiferous tubule cross-sections containing at least one TUNEL-positive cell by the total number of scored tubules. TUNEL-positive cell types were identified to determine the numbers of spermatogonia and primary spermatocytes yielding a positive signal. Spermatogenic cell types were determined by cell morphology and position within the seminiferous tubule.
Statistical analysis.
Body weights and testis weights were compared by using the nonparametric Wilcoxon rank sum test. Statistical analyses were performed in a manner similar to that of previously described analyses for lacI mutant frequencies (45, 60). The numbers of mutants were described by Poisson random variables, and the mutant frequencies were compared by using the conditional binomial test. The TUNEL data were analyzed by using analysis of variance, and comparisons among mean values were Bonferroni adjusted.
RESULTS
Comparisons of weights and histology.
Comparisons of body weights, testis weights, and histology of selected tissues were made to ascertain if there were gross differences in the Apex+/− and Apex+/+ mice. The body weights of 9-month-old Apex+/− mice, 37.1 ± 1.0 g, were similar to those of Apex+/+ mice, 35.5 ± 2.1 g. Testis weights were similar between Apex+/− mice, 0.11 ± 0.003 g, and Apex+/+ mice, 0.11 ± 0.003 g. Histological differences were not observed in H&E-stained cross-sections of the testis and spleen from 3- and 9-month-old Apex+/− mice compared to those of aged-matched Apex+/+ mice (Fig. 1). Similarly, no differences were observed in livers of each genotype at 3 months (Fig. 2). However, the presence of centrilobular fatty changes surrounding the central vein in the liver was common (5 of 10 mice examined) in 9-month-old Apex+/− mice relative to that in livers from 9-month-old Apex+/+ mice (Fig. 2).
FIG. 1.

Histological appearance of testis and spleen. Standard H&E staining was performed on tissue sections from 3- and 9-month-old Apex+/+ and Apex+/− male mice. No significant differences were observed between ages or genotypes. (a) Testis from a 3-month-old Apex+/+ mouse. (b) Testis from a 3-month-old Apex+/− mouse. (c) Testis from a 9-month-old Apex+/+ mouse. (d) Testis from a 9-month old Apex+/− mouse. (e) Spleen from a 3-month-old Apex+/+ mouse. (f) Spleen from a 3-month-old Apex+/− mouse. (g) Spleen from a 9-month-old Apex+/+ mouse. (h) Spleen from a 9-month-old Apex+/− mouse. Magnification of testis, ×16. Magnification of spleen, ×6.3.
FIG. 2.

Histological appearance of liver. H&E staining was performed on liver sections obtained from a 3-month-old Apex+/+ mouse (a), a 3-month-old Apex+/− mouse (b), a 9-month-old Apex+/+ mouse (c), and 9-month-old Apex+/− mouse (d). Note the fatty changes designated by arrowheads.
Spontaneous mutant frequencies for mixed spermatogenic cells.
The spontaneous mutant frequencies for mixed spermatogenic cells obtained from 3-month-old Apex+/− lacI+ and Apex+/+ lacI+ mice were not significantly different from each other at 1.6 × 10−5 ± 0.3 × 10−5 and 1.0 × 10−5 ± 0.2 × 10−5, respectively (Fig. 3, Table 1). Thus, a significantly increased mutant frequency was not observed in spermatogenic cells obtained from 3-month-old Apex+/− lacI+ mice. However, the spontaneous mutant frequencies were more than twofold higher and were significantly different for mixed spermatogenic cells obtained from 9-month-old Apex+/− lacI+ mice (3.3 × 10−5 ± 0.5 × 10−5) than for 9-month-old Apex+/+ lacI+ mice (1.4 × 10−5 ± 0.3 × 10−5) (Fig. 3, Table 1). The mutant frequency for spermatogenic cells obtained from 9-month-old Apex+/− lacI+ mice was twofold higher than that for spermatogenic cells obtained from 3-month-old Apex+/− lacI+ mice (Fig. 3, Table 1).
FIG. 3.

Age-dependent spontaneous mutant frequencies of mixed spermatogenic cells obtained from Apex+/+ lacI+ and Apex+/− lacI+ mice. A significant increase in spontaneous mutant frequency was detected for the lacI gene recovered from mixed spermatogenic cells obtained from 9-month-old Apex+/− lacI+ mice. Results are presented as means ± standard errors of the means. An asterisk indicates the significant increase in spontaneous mutant frequency of spermatogenic cells obtained from 9-month-old Apex+/− lacI+ mice compared to those of 9-month-old Apex+/+ lacI+ control mice. The letter “a” indicates a significant increase in spontaneous mutant frequency detected for samples obtained from 9-month-old Apex+/− lacI+ mice compared to those of 3-month-old Apex+/− lacI+ mice. Black bars, 3-month-old mice; white bars, 9-month-old mice. P values of <0.05 were considered significant.
TABLE 1.
Spontaneous mutant frequencies for a lacI transgene recovered from select tissues of Apex+/+ lacI+ and Apex+/− lacI+ mice
| Cell or tissue type | Donor mouse age (mo) | No. of mice | Genotype | PFU | No. of confirmed mutants | Mutant frequency (10−5) | SEM (10−5) |
|---|---|---|---|---|---|---|---|
| Mixed spermatogenic cells | 3 | 5 | Apex+/+lacI+ | 1,878,569 | 19 | 1.0 | 0.2 |
| Mixed spermatogenic cells | 3 | 6 | Apex+/−lacI+ | 1,399,046 | 22 | 1.6 | 0.3 |
| Mixed spermatogenic cells | 9 | 5 | Apex+/+lacI+ | 1,170,125 | 16 | 1.4 | 0.3 |
| Mixed spermatogenic cells | 9 | 5 | Apex+/−lacI+ | 1,526,500 | 51 | 3.3a,b | 0.5 |
| Liver | 3 | 7 | Apex+/+lacI+ | 1,744,500 | 45 | 2.6 | 0.4 |
| Liver | 3 | 4 | Apex+/−lacI+ | 754,500 | 52 | 6.9a | 1.0 |
| Liver | 9 | 5 | Apex+/+lacI+ | 962,034 | 64 | 6.6b | 0.8 |
| Liver | 9 | 5 | Apex+/−lacI+ | 1,098,797 | 124 | 11.3a,b | 1.0 |
| Spleen | 3 | 6 | Apex+/+lacI+ | 2,056,500 | 69 | 3.4 | 0.4 |
| Spleen | 3 | 4 | Apex+/−lacI+ | 1,525,250 | 115 | 7.5a | 0.7 |
| Spleen | 9 | 5 | Apex+/+lacI+ | 1,441,674 | 73 | 5.1b | 0.6 |
| Spleen | 9 | 6 | Apex+/−lacI+ | 1,666,317 | 240 | 14.4a,b | 0.9 |
Apex+/− mice are significantly different from Apex+/+ mice of the same age.
Nine-month-old mice are significantly different from 3-month-old mice of the same genotype.
Liver spontaneous mutant frequencies.
A significantly higher (approximately twofold) mutant frequency was observed for liver samples obtained from 3-month-old Apex+/− lacI+ mice (6.9 × 10−5 ± 1.0 × 10−5) compared to that of Apex+/+ lacI+ mice (2.6 × 10−5 ± 0.4 × 10−5) (Fig. 4, Table 1). An additional 1.5-fold increase in spontaneous mutant frequency was detected in samples obtained from 9-month-old Apex+/− lacI+ mice (11.3 × 10−5 ± 1.0 × 10−5) (Fig. 4, Table 1). This mutant frequency is significantly higher than that obtained for samples from 3-month-old mice of the same genotype and from that of age-matched Apex+/− lacI+ mice (6.6 × 10−5 ± 0.8 × 10−5) (Fig. 4, Table 1). The mutant frequency for liver samples obtained from 9-month-old Ape +/+ lacI+ mice is significantly higher (approximately 2.5-fold) than the mutant frequency for liver samples prepared from 3-month-old mice of the same genotype (Fig. 4, Table 1). Thus, both genotypes displayed an age-related increase in spontaneous mutant frequencies for liver samples, with the Apex+/− lacI+ mice consistently displaying higher mutant frequencies.
FIG. 4.

Age-dependent spontaneous mutant frequencies for liver and spleen obtained from Apex+/+ lacI+ and Apex+/− lacI+ mice. Liver and spleen samples obtained from 3-month-old Apex+/− lacI+ mice displayed a significantly higher spontaneous mutant frequency than tissues from 3-month-old Apex+/+ lacI+ mice (as indicated by asterisks). Each tissue displayed a significant increase in mutant frequency when obtained from 9-month-old mice compared to that from 3-month-old mice of the same genotype (indicated by the letter “a”). Samples from 9-month-old Apex+/− lacI+ mice displayed even higher spontaneous mutant frequencies than 9-month-old Apex+/+ lacI+ mice (indicated by asterisks). Results are presented as means ± standard errors of the means. Black bars, 3-month-old Apex+/+ lacI+ mice; white bars, 3-month-old Apex+/− lacI+ mice; striped bars, 9-month-old Apex+/+ lacI+ mice; cross-hatched bars, 9-month-old Apex+/− mice. P values of <0.05 were considered significant.
Spleen spontaneous mutant frequencies.
A twofold significantly elevated spontaneous mutant frequency was detected in spleen samples prepared from 3-month-old Apex+/− lacI+ mice (7.5 × 10−5 ± 0.7 × 10−5) compared to that for Apex+/+ lacI+ mice (3.4 × 10−5 ± 0.4 × 10−5) (Fig. 4, Table 1). The mutant frequency in the Apex+/− lacI+ mice increased an additional twofold in samples from 9-month-old mice (14.4 × 10−5 ± 0.9 × 10−5) (Fig. 4, Table 1). The mutant frequency obtained for spleen samples from 9-month-old Apex+/− lacI+ mice was significantly higher than that for 3-month-old mice of the same genotype and for 9-month-old Apex+/+ lacI+ mice (5.0 × 10−5 ± 0.6 × 10−5) (Fig. 4, Table 1). The mutant frequency for 9-month-old Apex+/− lacI+ mice was approximately twofold higher than that for 3-month-old Apex+/− lacI+ mice and was threefold higher than that for 3-month-old Apex+/+ lacI+ mice. In comparison, the mutant frequency for 9-month-old Apex+/+ lacI+ mice was more modestly, yet significantly, increased (approximately 1.5-fold) relative to the mutant frequency for 3-month-old mice of the same genotype.
Apoptotic prevalence.
The percentage of seminiferous tubules with TUNEL-positive cells was similar between 3-month-old Apex+/− and Apex+/+ mice (Fig. 5). However, a significant difference was detected between 9-month-old Apex+/− and Apex+/+ mice. The number of seminiferous tubule cross-sections displaying TUNEL-positive cells was 32.8% for Apex+/− mice and 19.3% for Apex+/+ mice (P < 0.05) (Fig. 5, Table 2). Analysis of apoptotic cell types revealed a higher proportion of TUNEL-positive spermatogonia for 9-month-old Apex+/− mice than for Apex+/+ mice (39.1% versus 20.2%) (Fig. 6; Table 2) and a greater prevalence of TUNEL-positive primary spermatocytes for Apex+/− mice than for Apex+/+ control mice (54.4% versus 17.3%) at 9 months of age. There were no significant differences in the proportions of apoptotic spermatogonia and primary spermatocytes between genotypes at 3 months.
FIG. 5.
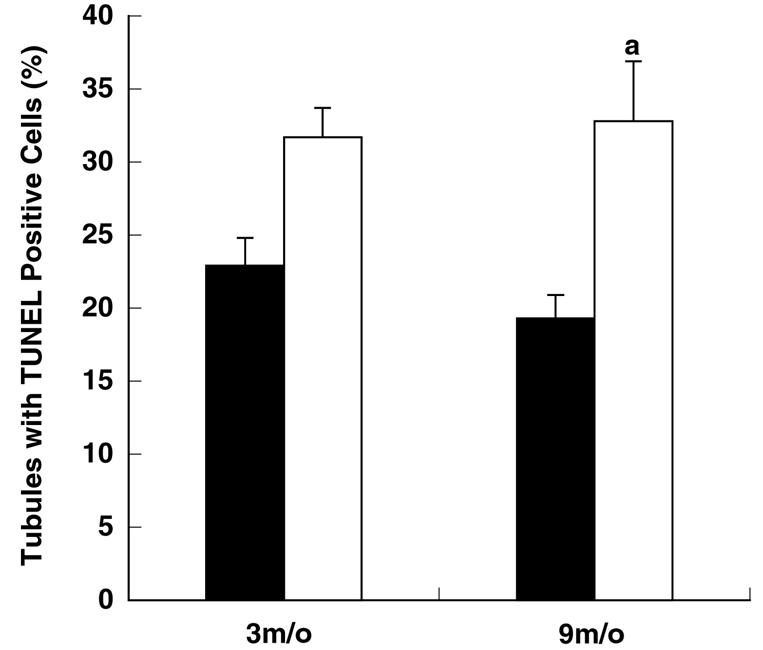
Prevalence of seminiferous tubules displaying TUNEL-positive cells. Cross-sections of seminiferous tubules were examined for TUNEL-positive cells. Significantly more seminiferous tubules with TUNEL-positive cells were detected for 9-month-old Apex+/− mice than for 9-month-old Apex+/+ mice (indicated by the letter “a”). Results are presented as means ± standard errors of the means. Black bars, Apex+/+ mice; white bars, Apex+/− mice. P values of <0.05 were considered significant.
TABLE 2.
Apoptosis prevalence in seminiferous tubule cross-sections from Apex+/+ and Apex+/− mice
| Age (mo) | Genotype | No. of mice | % Affected tubules ± SEM | No. of apoptotic spermatogonia/tubule ± SEM | No. of apoptotic primary spermatocytes/tubule ± SEM | No. of undetermined apoptotic cell type/tubule ± SEM | No. of tubule cross-sections scored |
|---|---|---|---|---|---|---|---|
| 3 | Apex+/+ | 4 | 22.9 ± 1.9 | 0.23 ± 0.06 | 0.28 ± 0.07 | 0.05 ± 0.03 | 628 |
| 3 | Apex+/− | 6 | 31.7 ± 2.0 | 0.37 ± 0.05 | 0.37 ± 0.05 | 0 | 685 |
| 9 | Apex+/+ | 6 | 19.3 ± 1.6 | 0.20 ± 0.02 | 0.17 ± 0.03 | 0 | 617 |
| 9 | Apex+/− | 10 | 32.8 ± 4.1a | 0.39 ± 0.05a | 0.54 ± 0.09a | 0.05 ± 0.02 | 628 |
Values are significantly different from mice of the same age but with different genotypes.
FIG. 6.

Quantitative assessment of spermatogenic cells undergoing apoptosis. Cell types identified as TUNEL positive were also identified according to spermatogenic cell types. Two major populations of spermatogenic cell types were found to display TUNEL-positive signals, namely spermatogonia and primary spermatocytes. Frequencies were calculated by dividing the number of spermatogonia or primary spermatocytes displaying a TUNEL-positive signal by the number of seminiferous tubule cross-sections scored. Significantly greater numbers of spermatogonia and primary spermatocytes were found to display a TUNEL-positive signal from 9-month-old Apex+/− mice than from age-matched Apex+/+ mice (indicated by the letter “a”). Results are presented as means ± standard errors of the means. Black bars, 3-month-old Apex+/+ mice; white bars, 3-month-old Apex+/− mice; striped bars, 9-month-old Apex+/+ mice; cross-hatched bars, 9-month-old Apex+/− mice. P values of <0.05 were considered significant.
DISCUSSION
Among the most frequent spontaneous lesions in DNA, apurinic/apyrimidinic (AP or abasic) sites occur minimally 9,000 to 10,000 times per cell per day (30, 31, 38). These data suggest that AP sites are a major source of DNA damage and potentially a major source of spontaneous mutagenesis (62). A member of the BER pathway, AP endonuclease (Apex) is the predominant DNA repair enzyme capable of recognizing and initiating repair of AP lesions. Apex is a multifunctional protein, possessing 3′ to 5′ exonuclease, 3′ phosphatase, 3′ phosphodiesterase, and RNase H activities (13). Apex also functions as a strong p53 activator (28) and stimulates DNA binding of multiple transcription factors (56, 63-65). The interaction of Apex with transcription factors is related largely to redox activity. Thus, Apex may provide molecular cross-talk among DNA repair, transcription, and apoptosis pathways, all of which are involved in maintaining genomic integrity. The purpose of the study was to determine directly if inactivation of an Apex allele would be sufficient to mediate increased spontaneous genomic instability.
Gross measures of animal health, including body weights, testis weights, and histological analyses of spleen, liver, and testis, revealed no detectable manifestation of abnormal development or disease, with the exception of centrilobular fatty changes in the livers of several 9-month-old Apex+/− mice. Based on these assessments, the Apex+/− mice were considered normal young adults. However, examination of mutant frequencies revealed approximately twofold higher spontaneous mutant frequencies for spleen and liver of young adult (3-month-old) Apex+/− mice compared to those of Apex+/+ controls of the same age. The effects of an inactivated Apex allele were exacerbated with increased age, such that the spontaneous mutant frequencies in spleen and liver of 9-month-old Apex+/− mice were approximately doubled for each tissue. These data indicate that inactivation of one Apex allele results in cells with reduced protection against mutagenesis, possibly emanating from AP sites.
In stark contrast to somatic tissue from 3-month-old mice, spermatogenic cells obtained from 3-month-old Apex+/− lacI+ mice did not display an increased mutant frequency compared to that of Apex+/+ lacI+ mice of the same age. There are several possible explanations. (i) The abundance of BER proteins and in vitro BER activity is lower in tested somatic tissue compared to that of testis or spermatogenic cell types (7, 24, 42). Reduced Apex in somatic tissues may then compromise the inherently lower BER activity in somatic tissues. Indeed, while this study was under review, Raffoul et al. (48) reported decreased BER activity for liver nuclear extracts obtained from independently generated Apex+/− mice. Thus, the increased mutant frequency in somatic tissues is consistent with reduced in vitro BER activity for Apex+/− mice. (ii) AP sites do not occur as frequently in spermatogenic cells as in somatic cells. (iii) Other repair proteins and/or repair pathways compensate for the inactivated Apex allele in spermatogenic cells. (iv) The greater inherent expression of Apex in male germ cells is sufficient to accommodate the need for Apex in its various capacities even when one Apex allele is inactivated. Notably, Apex+/− mice display reduced in vitro BER activity in spermatogenic cell nuclear extracts obtained from young adult mice (25), indicating this is not likely. (v) Expression from the intact Apex allele may be enhanced in spermatogenic cells; however, the reduced BER activity in nuclear extracts prepared from Apex+/− mice (25) indicates this is unlikely. (vi) A higher frequency of apoptosis may occur in the germ cells and remove aberrant spermatogenic cells, thereby precluding a higher mutant frequency. However, the results of apoptosis prevalence in the testis of 3-month-old mice in the present study were not consistent with this possible explanation. (vii) Embryonic spermatogonia enter a mitotic arrest and reenter a mitotic cycle after birth (22, 44). In contrast, liver and spleen cells undergo continuous cell divisions until after birth. Both organs (liver and spleen) in the newborn continue to exhibit high cell division rates which slow down as the animals mature. The mitotic arrest experienced by spermatogenic cells may preclude an increased mutant frequency at 3 months of age. However, by 9 months of age the spermatogenic cells have undergone many more rounds of replication and therefore have had more opportunity to fix mutations.
Despite the mechanism that precluded a higher spontaneous mutant frequency in spermatogenic cells obtained from 3-month-old Apex+/− lacI+ mice, the mutant frequency was elevated in spermatogenic cells from 9-month-old mice. The increase was approximately twofold compared to that of Apex+/+ lacI+ mice of the same age. These data provide strong evidence that inactivation of an Apex allele is sufficient to result in an increased spontaneous mutant frequency in the male germ line, and they support the hypothesis that reduced Apex abundance in spermatogenic cells from old mice contributes to the reduced BER activity observed for nuclear extracts prepared from these cell types (25) and for the increased spontaneous mutant frequency at old ages (60). These data are also consistent with increased spontaneous mutagenesis observed in yeast after inactivation of the major AP endonuclease, APN1 (49).
The increased frequencies of mutants for somatic tissues obtained from Apex+/− mice and reported herein are consistent with reduced Apex mediating genomic instability. As reported by Raffoul et al. (48), Apex+/− mice display reduced BER activity in nuclear extracts prepared from liver. However, the data from a previous study (25) and the present study are incongruent with the testis results presented by Raffoul et al. (48). Their data show an increased abundance of DNA polymerase β and increased BER in testis nuclear extracts prepared from Apex+/− mice compared to levels for wild-type mice. Notably, however, the BER activity reported for testis of Apex+/− and wild-type mice was lower than the level of liver BER activity (48). Apex abundance was similar in testis of wild-type mice and liver in one set of presented data. Thus, the testis data reported by Raffoul et al. (48) are inconsistent with results from previous reports (7, 24, 42). These differences between studies might have been caused by different experimental approaches. For example, the nuclear extracts prepared by Raffoul et al. (48) were prepared from frozen tissue. The extracts prepared for the studies reported by Intano et al. (24, 25) were prepared from fresh tissue or cell preparations. It seems possible that the content of the nuclear extracts was different in the various studies due to the different preparation methods.
While the increased mutant frequency observed in Apex+/− lacI+ mice is consistent with the scenario just presented, it is important to consider that Apex is a multifunctional protein. It is not, therefore, clear that the increased spontaneous mutant frequency detected in Apex+/− lacI+ mice is due exclusively to decreased BER. Indeed, changes in apoptosis prevalence might have a significant effect on mutant frequency. Apoptosis is thought to be activated when cells realize intolerable levels of DNA damage (6). Accordingly, it seemed possible that there would be elevated levels of apoptosis in Apex+/− mice, because AP sites can mediate a cytotoxic effect (12, 13, 51). The fraction of seminiferous tubule cross-sections with TUNEL-positive cells was significantly larger in samples from 9-month-old Apex+/− mice than in 9-month-old Apex+/+ mice. This increased prevalence of apoptotic cells may reflect a response to increased levels of DNA damage due to inactivation of one Apex allele. However, the increased prevalence of apoptosis is not sufficient to prevent an increased mutant frequency. Apex activation of p53 can lead to apoptosis in somatic cells (16). With reduced Apex, such as that in Apex+/− mice (34, 48), one would expect a reduction in p53-mediated apoptosis (16). At present, it is not clear that the observed apoptosis in pachytene spermatocytes of Apex+/− mice is mediated by p53. However, among spermatogenic cell types, p53 expression appears most robust in pachytene spermatocytes (53) and p53 is involved in ionizing radiation-induced apoptosis in spermatogenic cells (53).
Essentially all cell types scored TUNEL positive in the testis were spermatogonia or primary spermatocytes, and this is in agreement with other studies (2, 37). The relative proportions of apoptotic spermatogonia and primary spermatocytes were assessed to determine if one cell type was more prone to apoptosis in the Apex+/− mice. A larger fraction of the apoptotic cells were primary spermatocytes (54%) in the seminiferous tubule cross-sections prepared from 9-month-old Apex+/− mice, whereas apoptotic spermatogonia were less abundant (39%). No differences in the relative proportions of spermatogonia (20%) and primary spermatocytes (17%) was observed for seminiferous tubule cross-sections prepared from 9-month-old Apex+/+ mice. These results suggest that primary spermatocytes are more susceptible to apoptosis in Apex+/− mice. Interestingly, primary spermatocytes are more robustly affected by a decreased abundance of apoptosis in p53−/− mice (66). The role of p53 in mediating apoptosis in spermatogenic cells in the environment of reduced Apex is not known.
Apoptosis in spermatogenic cells can be mediated by intrinsic or extrinsic signals (46). The process of apoptosis is essential to successful spermatogenesis. The essentiality of apoptosis has been demonstrated in mouse models in which apoptosis is defective (50). Spermatogenesis is severely impaired in these models. The amount of apoptosis varies in the process of spermatogenesis, with peaks of apoptosis occurring embryonically and during the first wave of spermatogenesis after birth (10, 37). Fluctuations in apoptosis prevalence vary in the adult animal (3, 27), although the cause for such fluctuations is not known.
Raffoul et al. (48) suggest that the effects they detected in Apex+/− mice are due to the redox function of Apex in part because addition of 100 ng of purified recombinant human AP endonuclease (APE) did not restore the BER activity in 50 μg of liver nuclear extracts prepared from Apex+/− mice. However, a previous study showed that addition of purified recombinant APE, in amounts of 1.25, 2.5, 5, 10, and 20 ng, did restore BER activity in freshly prepared mixed spermatogenic cell nuclear extracts obtained from Apex+/− mice (25). Once again, differences in experimental conditions may account for the differences in results. However, in neither case was the redox function of Apex tested directly; rather, the effects of changes in Apex abundance were tested. Multiple Apex functions possibly contribute to the various phenotypic effects observed in Apex+/− mice.
In summary, the data presented here support the hypothesis that inactivation of an Apex allele results in increased spontaneous mutant frequencies in somatic tissues and spermatogenic cells. In somatic tissues the genomic instability effect mediated by inactivation of one Apex allele is realized at a very young age, i.e., by 3 months of age. Genetic instability is greater in somatic tissues by 9 months of age. In comparison, genetic instability is delayed in spermatogenic cells but is observed at the still relatively young age of 9 months. Apoptosis is enhanced among spermatogenic cells in 9-month-old Apex+/− mice, particularly among primary spermatocytes, but the amount of apoptosis is insufficient to prevent an increased mutant frequency. The contributions of various Apex functions to genomic stability remain to be determined directly, and it will also be important to determine if reduced BER activity in and of itself is sufficient to mediate genomic instability in the male germ line.
Acknowledgments
This work was supported by grants ES09136 and AG21163, AG19316, and CA 86936 from the NIH, the Environmental Hazards Center of the South Texas Veteran's Health Care System (STVHCS), and a merit award from the VHCS.
The views presented are the responsibility of the authors and are not necessarily those of the NIH or VHCS.
REFERENCES
- 1.Alcivar, A. A., L. E. Hake, and N. B. Hecht. 1992. DNA polymerase-β and poly(ADP)ribose polymerase mRNAs are differentially expressed during the development of male germinal cells. Biol. Reprod. 46:201-207. [DOI] [PubMed] [Google Scholar]
- 2.Allan, D. J., B. V. Harmon, and J. F. R. Kerr. 1987. Cell death in spermatogenesis, p. 229-259. In C. S. Potter (ed.), Perspectives on cell death. Oxford University Press, New York, N.Y.
- 3.Barnes, C. J., B. W. Covington, IV, I. L. Cameron, and M. Lee. 1998. Effect of aging on spontaneous and induced mouse testicular germ cell apoptosis. Aging-Clin. Exp. Res. 10:497-501. [DOI] [PubMed] [Google Scholar]
- 4.Bellve, A. R. 1993. Purification, culture, and fractionation of spermatogenic cells. Methods Enzymol. 225:84-113. [DOI] [PubMed] [Google Scholar]
- 5.Bellve, A. R., J. C. Cavicchia, C. F. Millette, D. A. O'Brien, Y. M. Bhatnagar, and M. Dym. 1977. Spermatogenic cells of the prepuberal mouse. J. Cell Biol. 74:68-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein, C., H. Bernstein, C. M. Payne, and H. Garewal. 2002. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat. Res. 511:145-178. [DOI] [PubMed] [Google Scholar]
- 7.Cabelof, D. C., J. J. Raffoul, S. Yanamadala, C. Ganir, Z. M. Guo, and A. R. Heydari. 2002. Attenuation of DNA polymerase β-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat. Res. 500:135-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson, K. M., J. Bracamontes, C. E. Jackson, R. Clark, A. Lacroix, S. A. Wells, Jr., and P. J. Goodfellow. 1994. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am. J. Hum. Genet. 55:1076-1082. [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, J., A. E. Tomkinson, W. Ramos, Z. B. Mackey, S. Danehower, C. A. Walter, R. A. Shultz, J. M. Besterman, and I. Husain. 1995. Mammalian DNA ligase III: molecular cloning, chromosomal location, and expression in spermatocytes undergoing meiotic recombination. Mol. Cell. Biol. 15:5412-5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coucouvanis, E. C., S. W. Sherwood, Carswell-C. Crumpton, E. G. Spack, and P. P. Jones. 1993. Evidence that the mechanism of prenatal germ cell death in the mouse is apoptosis. Exp. Cell Res. 209:238-247. [DOI] [PubMed] [Google Scholar]
- 11.Crow, J. F. 2000. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 1:40-46. [DOI] [PubMed] [Google Scholar]
- 12.Demple, B., and L. Harrison. 1994. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63:915-948. [DOI] [PubMed] [Google Scholar]
- 13.Evans, A. R., M. Limp-Foster, and M. R. Kelley. 2000. Going APE over ref-1. Mutat. Res. 461:83-108. [DOI] [PubMed] [Google Scholar]
- 14.Francke, U., J. Felsenstein, S. M. Gartler, B. R. Migeon, J. Dancis, J. E. Seegmiller, F. Bakay, and W. L. Nyhan. 1976. The occurrence of new mutants in the X-linked recessive Lesch-Nyhan disease. Am. J. Hum. Genet. 28:123-137. [PMC free article] [PubMed] [Google Scholar]
- 15.Friedberg, E. C., G. C. Walker, and W. Siede. 1995. DNA repair and mutagenesis. ASM Press, Washington, D.C.
- 16.Gaiddon, C., N. C. Moorthy, and C. Prives. 1999. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 18:5609-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gravrieli, Y., Y. Sherman, and S. A. Ben-Sasson. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119:493-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haldane, J. B. S. 1935. The rate of spontaneous mutation of a human gene. J. Genet. 31:317-326. [DOI] [PubMed] [Google Scholar]
- 19.Haldane, J. B. S. 1947. The mutation rate of the gene for haemophilia, and its segregation ratios in males and females. Ann. Eugen. 13:262-271. [DOI] [PubMed] [Google Scholar]
- 20.Hill, K. A., V. L. Buettner, B. W. Glickman, and S. S. Sommer. 1999. Spontaneous mutations in the Big Blue transgenic system are primarily mouse derived. Mutat. Res. 436:11-19. [DOI] [PubMed] [Google Scholar]
- 21.Hill, K. A., H. Nishino, V. L. Buettner, A. Halangoda, S. Li, and S. S. Sommer. 1999. The Big Blue transgenic mouse mutation detection assay: the mutation pattern of sectored mutant plaques. Mutat. Res. 425:47-54. [DOI] [PubMed] [Google Scholar]
- 22.Hilscher, B., W. Hilscher, B. Bulthoff-Phnolz, U. Krämer, A. Birke, H. Pelzer, and G. Gauss. 1974. Kinetics of gametogenesis. I. Comparative histological and autoradiographic studies of oocytes and transitional prospermatogonia during oogenesis and prespermatogenesis. Cell Tissue Res. 154:443-470. [DOI] [PubMed] [Google Scholar]
- 23.Hirose, F., Y. Hotta, M. Yamaguchi, and A. Matsukage. 1989. Difference in the expression level of DNA polymerase β among mouse tissues: high expression in the pachytene spermatocytes. Exp. Cell Res. 181:169-180. [DOI] [PubMed] [Google Scholar]
- 24.Intano, G. W., C. A. McMahan, R. B. Walter, J. R. McCarrey, and C. A. Walter. 2001. Mixed spermatogenic germ cell nuclear extracts exhibit high base excision repair. Nucleic Acids Res. 29:1366-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Intano, G. W., C. A. McMahan, J. R. McCarrey, R. B. Walter, A. E. McKenna, Y. Matusmoto, M. A. MacInnes, D. J. Chen, and C. A. Walter. 2002. Base excision repair is limited by different proteins in male germ cell nuclear extracts prepared from young and old mice. Mol. Cell. Biol. 22:2410-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohler, S. W., G. S. Provost, A. Fieck, P. L. Kretz, W. O. Bullock, J. A. Sorge, D. L. Putman, and J. M. Short. 1991. Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice. Proc. Natl. Acad. Sci. USA 88:7958-7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kojima, S., H. Masahiko, S. Okada, T. Dukuda, Y. Toyama, S. Yuasa, H. Ito, and T. Tokuhisa. 2001. Testicular germ cell apoptosis in Bcl6-deficient mice. Development 128:57-65. [DOI] [PubMed] [Google Scholar]
- 28.Jayaraman, L. J., K. G. K. Murthy, C. Zhu, T. Curran, S. Xanthoudakis, and C. Prives. 1997. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 11:558-570. [DOI] [PubMed] [Google Scholar]
- 29.Lindahl, T. 2000. Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat. Res. 462:129-135. [DOI] [PubMed] [Google Scholar]
- 30.Lindahl, T., and B. Nyberg. 1972. Rate of depurination of native deoxyribonucleic acid. Biochemistry 11:3610-3618. [DOI] [PubMed] [Google Scholar]
- 31.Loeb, L. A., and B. D. Preston. 1986. Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 20:201-230. [DOI] [PubMed] [Google Scholar]
- 32.Ludwig, D. L., M. A. MacInnes, Y. Takiguchi, P. E. Purtymun, M. Henrie, M. Flanner, J. Meneses, R. A. Pedersen, and D. J. Chen. 1998. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat. Res. 409:17-29. [DOI] [PubMed] [Google Scholar]
- 33.Mackey, Z. B., W. Ramos, D. S. Levin, C. A. Walter, J. R. McCarrey, and A. E. Tomkinson. 1997. An alternative splicing event which occurs in mouse pachytene spermatocytes generates a form of DNA ligase III with distinct biochemical properties that may function in meiotic recombination. Mol. Cell. Biol. 17:989-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meira, L. B., S. Devaraj, G. E. Kisby, D. K. Burns, R. L. Daniel, R. E. Hammer, S. Grundy, I. Jialal, and E. C. Friedberg. 2001. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 61:5552-5557. [PubMed] [Google Scholar]
- 35.Memisoglu, A., and L. Samson. 2000. Base excision repair in yeast and mammals. Mutat. Res. 451:39-51. [DOI] [PubMed] [Google Scholar]
- 36.Moloney, D. M., S. F. Slaney, M. Oldridge, S. A. Wall, P. Sahlin, G. Stenman, and A. O. M. Wilkie. 1996. Exclusive paternal origin of new mutations in Apert syndrome. Nat. Genet. 13:48-53. [DOI] [PubMed] [Google Scholar]
- 37.Mori, C., N. Nakamura, D. J. Dix, M. Fujioka, S. Nakagawa, K. Shiota, and E. M. Eddy. 1997. Morphological analysis of germ cell apoptosis during postnatal testis development in normal and Hsp70-2 knockout mice. Dev. Dyn. 208:125-136. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura, J., and J. A. Swenberg. 1999. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 59:2522-2526. [PubMed] [Google Scholar]
- 39.Nilsen, H., and H. E. Krokan. 2001. Base excision repair in a network of defense and tolerance. Carcinogenesis 22:987-998. [DOI] [PubMed] [Google Scholar]
- 40.Nishino, H., V. L. Buettenr, and S. S. Sommer. 1996. Towards validation of the Big Blue transgenic mouse mutagenesis assay: the mutation spectrum of ex vivo pinpoint mutant plaques. Mutat. Res. 372:97-105. [DOI] [PubMed] [Google Scholar]
- 41.Norbury, C. J., and I. A. Hickson. 2001. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 41:367-401. [DOI] [PubMed] [Google Scholar]
- 42.Olsen, A.-K., H. Bjortuft, R. Wiger, J. A. Holme, E. C. Seeberg, M. Bjoras, and G. Brunborg. 2001. Highly efficient base excision repair (BER) in human and rate male germ cells. Nucleic Acids Res. 29:1781-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paashuis-Lew, Y., X. B. Zhang, and J. A. Heddle. 1997. On the origin of spontaneous mutations and sectored plaques detected in transgenic mice. Mutat. Res. 373:277-284. [DOI] [PubMed] [Google Scholar]
- 44.Peters, H. 1970. Migration of gonocytes into the mammalian gonad and their differentiation. Phil. Trans. R. Soc. Lond. B. 259:91-101. [DOI] [PubMed] [Google Scholar]
- 45.Piegorsch, W. W., B. H. Margolin, M. D. Shelby, A. Johnson, J. E. French, R. W. Tennant, and K. R. Tindall. 1995. Study design and sample sizes for a lacI transgenic mouse mutation assay. Environ. Mol. Mutagen. 25:231-245. [DOI] [PubMed] [Google Scholar]
- 46.Print, C. G., and K. L. Loveland. 2000. Germ cell suicide: new insights into apoptosis during spermatogenesis. BioEssays 22:423-430. [DOI] [PubMed] [Google Scholar]
- 47.Provost, G. S., and J. M. Short. 1994. Characterization of mutations induced by ethylnitrosourea in seminiferous tubule germ cells of transgenic B6D3F1 mice. Proc. Natl. Acad. Sci. USA 91:6564-6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raffoul, J. J., D. C. Cabelof, J. Nakamura, L. B. Meira, E. C. Friedberg, and A. R. Heydari. 2004. Apurinic/apyrimidinic endonuclease (APE/REF-1) haploinsufficient mice display tissue-specific differences in DNA polymerase β-dependent base excision repair. J. Biol. Chem. 279:18425-18433. [DOI] [PubMed] [Google Scholar]
- 49.Ramotar, D., S. C. Popoff, E. B. Gralla, and B. Demple. 1991. Cellular role of yeast Apn 1 apurinic endonuclease/3′-diesterase: repair of oxidative and alkylation DNA damage and control of spontaneous mutation. Mol. Cell. Biol. 11:4537-4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguez, I., C. Ody, K. Araki, I. Garcia, and P. Vassalli. 1997. An early and massive of germinal cell apoptosis is required for the development of normal spermatogenesis. EMBO J. 16:2262-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaaper, R. M., T. A. Kunkel, and L. A. Loeb. 1983. Infidelity of DNA synthesis associated with bypass of apurinic sites. Proc. Natl. Acad. Sci. USA 80:487-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuffenecker, I., N. Ginet, D. Goldgar, C. Eng, B. Chambe, A. Boneu, C. Houdent, D. Pallo, M. Schlumberger, C. Thivolet, and G. M. Lenoir. 1997. Prevalence and paternal origin of de novo RET mutations in multiple endocrine neoplasia type 2A and familial medullary thyroid carcinoma. Am. J. Hum. Genet. 60:233-237. [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz, D., N. Goldfinger, and V. Rotter. 1993. Expression of p53 protein in spermatogenesis is confined to the tetraploid pachytene primary spermatocytes. Oncogene 8:1487-1494. [PubMed] [Google Scholar]
- 54.Stuart, G. R., N. J. Gorelick, J. L. Andrews, J. B. DeBoer, and B. W. Glickman. 1996. The genetic analysis of lacI mutations in sectored plaques from Big Blue transgenic mice. Environ. Mol. Mutagen. 28:385-392. [DOI] [PubMed] [Google Scholar]
- 55.Vogel, F., and R. Rathenberg. 1975. Spontaneous mutation in man. Adv. Hum. Genet. 4:223-318. [DOI] [PubMed] [Google Scholar]
- 56.Walker, L. J., C. N. Robson, E. Black, D. Gillespie, and I. D. Hickson. 1993. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol. 13:5370-5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter, C. A., D. Nasr-Schirf, and V. J. Luna. 1989. Identification of transgenic mice carrying the CAT gene with PCR amplification. BioTechniques 7:1065-1070. [PubMed] [Google Scholar]
- 58.Walter, C. A., D. A. Trolian, M. B. McFarland, K. A. Street, G. R. Gurram, and J. R. McCarrey. 1996. Xrcc-1 expression during male meiosis in the mouse. Biol. Reprod. 55:630-635. [DOI] [PubMed] [Google Scholar]
- 59.Walter, C. A. 1999. Transgenic manipulation of the mouse genome, p. 387-415. In B. P. Yu (ed.), Methods in aging research. CRC Press, New York, N.Y.
- 60.Walter, C. A., G. W. Intano, J. R. McCarrey, C. A. McMahan, and R. B. Walter. 1998. Mutation frequency declines during spermatogenesis in young mice but increases in old mice. Proc. Natl. Acad. Sci. USA 95:10015-10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walter, C. A., J. Lu, M. Bhakta, Z.-Q. Zhou, L. H. Thompson, and J. R. McCarrey. 1994. Testis and somatic Xrcc-1 DNA repair gene expression. Som. Cell Mol. Genet. 20:451-461. [DOI] [PubMed] [Google Scholar]
- 62.Wilson, D. M., III, and D. Barsky. 2001. The major human abasic endonuclease: formation, consequences, and repair of abasic lesions in DNA. Mutat. Res. 485:283-307. [DOI] [PubMed] [Google Scholar]
- 63.Xanthoudakis, S., and T. Curran. 1992. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA binding activity. EMBO J. 11:653-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xanthoudakis, S., G. Miao, F. Wang, Y. C. Pan, and T. Curran. 1992. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 11:3323-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xanthoudakis, S., G. G. Miao, and T. Curran. 1994. The redox and DNA repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA 91:23-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yin, Y., B. C. Stahl, W. C. DeWolf, and A. Morgentaler. 1998. p53-mediated germ cell quality control in spermatogenesis. Dev. Biol. 204:165-171. [DOI] [PubMed] [Google Scholar]
- 67.Zhou, Z.-Q., and C. A. Walter. 1995. Expression of the DNA repair gene XRCC1 in baboon tissues. Mutat. Res. 348:111-116. [DOI] [PubMed] [Google Scholar]