

Abstract
The research on secondary metabolites of Aspergillus lentulus afforded eight unusual heterodimeric tetrahydroxanthone derivatives, lentulins A−H (2−9), along with the known compound neosartorin (1). Compounds 1−6 exhibited potent antimicrobial activities especially against methicillin-resistant Staphylococci. Their absolute configurations, particularly the axial chiralities, were unambiguously demonstrated by a combination of electronic circular dichroism (ECD), Rh2(OCOCF3)4-induced ECD experiments, modified Mosher methods, and chemical conversions. Interestingly, compounds 1–4 were the first samples of atropisomers within the dimeric tetrahydroxanthone class. Further investigation of the relationships between their axial chiralities and ECD Cotton effects led to the proposal of a specific CD Exciton Chirality rule to determine the axial chiralities in dimeric tetrahydroxanthones and their derivatives.
Dimeric tetrahydroxanthones and their derivatives like xanthone−chromanone and chromanone dimers, with attractive antibacterial1,2, antifungal3 and antimalarial activities4, are a class of structurally interesting natural products produced by multiple fungal and lichenous species5. More than 70 members with 2,2′-, 4,4′- or 2,4′-axial linkages have been isolated and identified since 19585. Biogenetically, most dimers are formed by two units from the same path6. Two paths derived heterodimers are rare and only four such molecules have been reported, namely eumitrins A1, A2, B, and neosartorin7,8.
All the dimeric tetrahydroxanthone analogues structurally involve axially chiral issues, which, different from the classic central chiralities, are interesting phenomena associated with the hindered rotation of a single bond9. Generally, a rotary energy barrier greater than 23.3 kcal/mol will prevent atropisomeric racemization and lead to stable axially chiral compounds10,11. Due to the presence of axial chiralities and their complicated spatial structures, the absolute configuration determinations of these compounds were quite challenging and mostly unsettled1,4,12,13. Electronic circular dichroism (ECD) calculation was the commonly used method. However, it was time-consuming to find the suitable conformers as well as choose the right calculation method. Sometimes, improper method could even lead to incorrect results14,15.
Recently, eight unusual heterodimeric tetrahydroxanthone derivatives, lentulins A−H (2−9), and the known neosartorin (1), were obtained during our metabolite research of the endophytic fungus Aspergillus lentulus (Fig. 1). Compounds 1–4 were the first samples of dimeric tetrahydroxanthone atropisomers, which along with other derivatives, gave useful samples to reveal the domination of axial chiralities in ECD Cotton effects (CEs). Based on the well determined absolute configurations and theoretical studies, the CD Exciton Chirality method was introduced to their axially chiral demonstration. In addition, these compounds exhibited potent antimicrobial activities especially against methicillin-resistant Staphylococci. Herein, we describe the structural elucidations, axially chiral researches and bioactive studies of compounds 1−9.
Figure 1. Structures and biogenesis relationships of 1–9.

Result and Discussion
Structural elucidation
The crude extract of Aspergillus lentulus, obtained from its rice cultures, was purified through silica gel and ODS column chromatography (CC), as well as preparative HPLC to yield compounds 1–9 (Fig. 1).
Lentulin A (2) was obtained as a yellow powder. Its molecular formula, C34H32O15, was established from the positive HRESIMS ion at m/z 703.1631 (calculated for 703.1633, C34H32O15Na). UV absorption at 337 nm suggested the presence of a long conjugated moiety and IR absorptions at 3451 and 1740 cm−1 indicated the existence of hydroxy and carbonyl groups. The typically doubled NMR data (Supporting Information Tables S1 and S2), especially the presence of two conjugated ketone carbonyls (δC 187.8, 187.2) and two enolic groups (δH 14.11, 1 H, s and δC 178.4, 101.1; δH 14.02, 1 H, s and δC 179.3, 100.2), revealed the dimeric tetrahydroxanthone skeleton of 2. Further comparison of its 1D NMR data and HMBC correlations (Fig. 2) with those of neosartorin (1)16 denoted the identical planar structure between them (Fig. 1), possessing 2,4′-linked blennolide C (unit A)17 and 5-acetyl blennolide A (unit B)18.
Figure 2. Selected HMBC correlations of 2 and 5.

The relative configurations of units A and B in 2 were confirmed the same as those in 1 (Fig. 3), due to the similar NOEs of H-5/OCH3-12, H-5′/OCH3-12′, H-5′/H-6′, OCH3-12′/H-6′, and CH3-14′/CH3-13′16. However, the contrary optical rotations (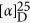 –210.2 for 1;
–210.2 for 1; 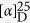 +276.1 for 2) suggested the distinction of their axial chiralities19,20. More importantly, the different long range NOE correlations between units A and B (Fig. 3), especially the observed signal of OH-1/OCH3-12′ and the missing signals of OH-1/CH3-14′ and OCH3-12/H-5′, identified 2 as the atropisomer of 1 with aS* configuration, rather than the aR* in 116.
+276.1 for 2) suggested the distinction of their axial chiralities19,20. More importantly, the different long range NOE correlations between units A and B (Fig. 3), especially the observed signal of OH-1/OCH3-12′ and the missing signals of OH-1/CH3-14′ and OCH3-12/H-5′, identified 2 as the atropisomer of 1 with aS* configuration, rather than the aR* in 116.
Figure 3. Key ROESY correlations of 1–3.

Lentulin B (3), a yellow powder, had the same molecular formula C34H32O15 as neosartorin (1). A comprehensive analysis of their NMR data (Supporting Information Tables S1 and S2) indicated that 3 was an epimer of 1. Unlike neosartorin, the large coupling constants between H-5 and H-6 (3J5–6ax = 12.4 Hz,3J5–6eq = 4.8 Hz)18 suggested H-5 was in the axial position (Fig. 3). The lacked NOE signal between OCH3-12 and H-5 revealed their trans configuration and the absent ROESY correlation of OCH3-12/H-5′ denoted the α-orientation of COOCH3-10. In addition, the similar ROESY cross-peaks of OH-1/CH3-14′, CH3-3/H-3′, and CH3-3/OCH3-12′ (Fig. 3), established the same aR* configuration as neosartorin.
Lentulin C (4) had the same molecular formula C34H32O15 (HRESIMS ion at m/z 703.1637) as 3, and was deduced to be its isomer from their similar 1D NMR data (Supporting Information Tables S1 and S2). The aS* configuration was also demonstrated by the NOE correlations of OH-1/OCH3-12′ and CH3-3/H-3′.
Lentulins D (5) and E (6) were another pair of neosartorin derivatives. NMR data analysis verified their structural similarities with above compounds. However, the lack of enolic signals indicated the absence of olefinic bond (Δ8(9)). And the presence of ester carbonyls (δC 175.6 in 5, δC 175.8 in 6) and the additional methylenes (δH 3.21, 2.97, each 1 H, and δC 39.4 in 5; δH 3.55, 3.07, each 1 H, and δC 40.3 in 6) revealed the γ-lactone moieties18, that were further confirmed by the HMBC correlations from H-5 to C-8, and from H-9 to C-5, C-9a, C-10, and C-11. As derived from 1 and 3 respectively, they were epimers with differently oriented COOCH3-10, which were proved by the present NOE cross-peak of OCH3-12/H-5′ in 5 but absent in 6. Similar to 1, the aR* configurations of 5 and 6 were confirmed by the long ranged NOE signals of OH-1/CH3-14′, CH3-3/H-3′, and CH3-3/OCH3-12′.
HRESIMS (m/z 721.1736 [M + Na]+) gave the molecular formula C34H34O16 for lentulin F (7), with one more H2O unit than 5. An intensive comparison of its NMR data (Supporting Information Tables S1 and S2) with those of 5 confirmed the γ-hydroxy butyric acid moiety in 717, notably through one degree loss of unsaturation and the lack of HMBC correlation from H-5 to C-8. The relative configuration of 7 was confirmed same as 5 from their identical NOE correlations.
8 and 9 were the derivatives of 5 and 6 with the cleavages of γ-lactones and the formations of γ-hydroxy butyric acid methyl esters, which were denoted by the decreases of unsaturation and HMBC correlations from the additional methoxyls to C-8. Similar to 5 and 6, the relative configurations of COOCH3-10 in 8 and 9 were confirmed as β- and α-orientation respectively. Their axial configurations were also assigned same as those of 5 and 6.
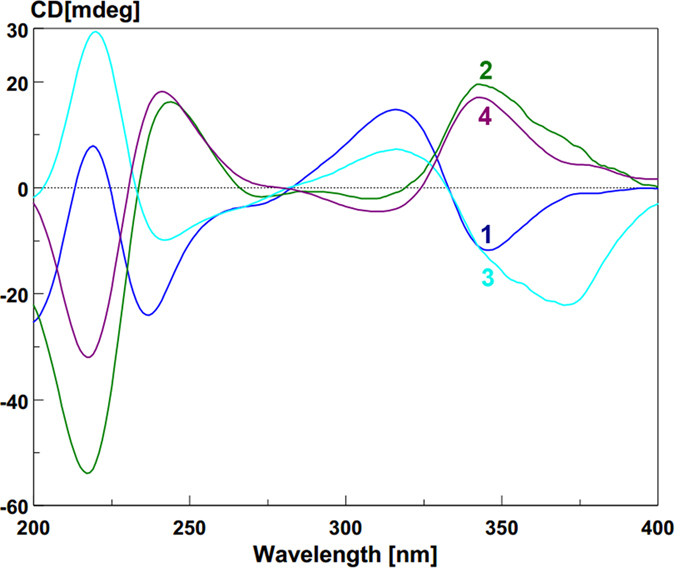
The absolute configuration (aR, 5S, 10R, 5′S, 6′S, 10′R) of neosartorin was determined by ECD calculation, which also predicted the approximate mirror image CEs of its atropisomer16. In the ECD spectrum of 2 (Fig. 4), the CEs well matched the calculation for neosartorin atropisomer with aS, 5S, 10R, 5′S, 6′S, 10′R configuration, proving that their CEs were governed by axial chiralities. The similar mirror images CEs were also found for 3 and 4 (Fig. 4). The identical CEs between 3 and 1 indicated the aR configuration in 3, while 4 was assigned as the atropisomer of 3. A Rh2(OCOCF3)4-induced ECD experiment (Supporting Information Figure S1) suggested the 5S configuration in 4 according to the Bulkiness rule21, confirming its absolute configuration as aS, 5S, 10S, 5′S, 6′S, 10′R. Interestingly, although 3 was the epimer of 1 with different central chiralities at C-10, the signs of their CEs were still the same, also suggesting the domination of axial chiralities in ECD spectra. So were compounds 4 and 2.
Figure 4. ECD spectra of 1–4 (in MeOH).
To avoid any ambiguity and demonstrate the absolute configurations of other derivatives, series of chemical conversions from 1 into 8, 3 into 9, and 7 into 5 were performed (Figs 5 and 6)17,22. Succeeding derivations of 8 and 9 by (R)- and (S)-methoxyphenylacetic acid (MPA) allowed the determination of their absolute configurations22. The negative ΔδH(R-S) values of OCH3-13 and H-7 as well as the positive ΔδH(R-S) values of OCH3-12 and H-9 (Fig. 7) confirmed 5S absolute configurations in both 8 and 9. In combination with above chemical conversions, the 5S configurations in the other compounds were also revealed.
Figure 5. Chemical conversions of 1 and 3.

Figure 6. Chemical Conversion of 7 into 5.

Figure 7. ΔδH(R−S) values of MPA esters for 8 and 9.

A plausible biosynthetic pathway was shown in Fig. 1. The oxidization and reduction of chrysophanol from the two paths gave units A and B6,23. Unit C (epi-blennolide C) was afforded via ring open and closure of unit A and both of them could convert into γ-lactone products through the retro-Dieckmann cyclization, which further yielded other ring cleavage intermediates5,6,17. Interestingly, all the biosynthetic modifications were occured on monomeric unit A, while the derivatization of unit B was limited to 5-acetylation that could prevent the further retro-Dieckmann cyclization. Meanwhile, the dimers were regularly formed by unit B with diverse path A units. These patterned dimerisations together with the specific monomeric modifications, indicated the enzymatic nature of these reactions, rather than the radical coupling24,25.
CD Exciton Chirality method
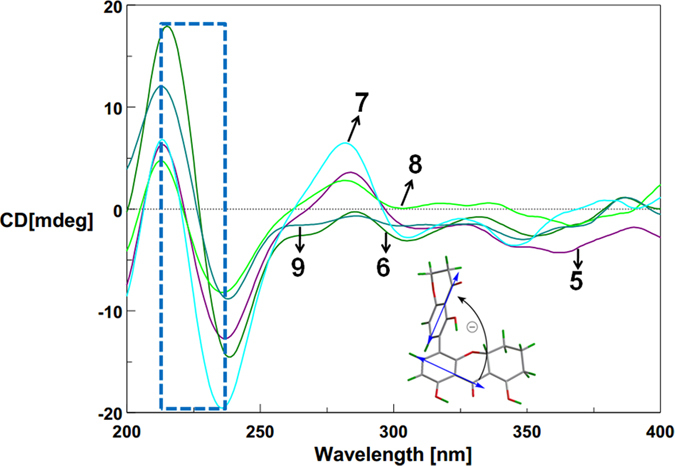
Tetrahydroxanthone monomers possessed two distinct kinds of chromophores (Fig. 8), namely 1-arylpropenone (330 nm) and benzoyl (230 nm). Their corresponding ECD CEs at above wavelengths were not split18. But for the axially linked dimers, their obviously split CEs suggested the two chromophores interacted with each other, and the opposite but not mirror image ECD spectrums (Fig. 4) revealed axial chiralities governed chromophore spatial positions to affect ECD CEs26,27.
Figure 8. CD Exciton Chiralities of 1–4.

When watching parallel to the chiral axes, the two chromophores’ rotary manners were identical to those of the CD Exciton Chirality rule. In particular, the anticlockwise manner of two 1-arylpropenone chromophores16 led to negative exciton couplets centered at around 330 nm like 1 and 3 (Fig. 8a), otherwise positive like 2 and 4 (Fig. 8b). This deduction was simultaneously proved by the split CEs at 230 nm caused by two benzoyl chromophores in the same rotary manners. Interestingly, due to the cleavages of one 1-arylpropenone chromophore in 5–9, their exciton couplets at 330 nm disappeared (Fig. 9), also suggesting the domination of chromophore interactions in ECD CEs. For further verification, different dimeric tetrahydroxanthone analogues were checked, which accurately revealed the same result as determined by X-ray analysis, chemical conversions or ECD calculations (Supporting Information Table S3)15,17,28,29.
Figure 9. CD Exciton Chiralities of 5–9.
The CD Exciton Chirality method has been applied to some axially chiral determinations before, such as those of binaphthyl, biarylic dihydronaphthopyranone30,31, and bis(naphtho-γ-pyrone)32. However, a very few exceptions were reported due to their complicated chromophores and non-negligible intense magnetic transition dipole moments33,34.
Theoretically, the rotational strengths Rα and Rβ of α and β excited states of one exciton couplet can be defined as Equations (1) and (2)33,35 (μ, electric transition dipole moments; m, magnetic transition dipole moments):
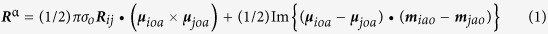 |
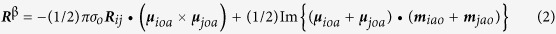 |
The first terms (μμ terms) were the rotational strengths caused by electric transition dipole moments μ and the second terms (μm terms) described the rotational strengths obtained by the combined electric and origin-independent magnetic transition moments. For the common π−π* excitation chromophores such as p-substituted benzoates, naphthoates, and anthroates, their internal magnetic transition moments (miao, mjao) were small and negligible, so were the μm terms35,36. The remaining terms (μμ terms) were the theoretical quantitative parameters of the CD Exciton Chirality rule26. Thus, based on the undisturbed benzoyl and 1-arylpropenone chromophores, the CD Exciton Chirality method could be used for the axial chiralities of dimeric tetrahydroxanthones and their derivatives, such as xanthone−chromanone and chromanone dimers.
Antimicrobial Assays
Using broth microdilution method37, compounds 1–9 were evaluated for their antimicrobial activities against a panel of pathogenic microbes, including multidrug resistant clinical strains. As collated in Table 1, compounds 1–6 showed moderate to significant antibacterial activities against four strains of Gram-positive and three strains of Gram-negative bacteria. 3 was the most potent antibiotic among these compounds, especially against methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-resistant S. epidermidis (MRSE). In view of the weakly active 5 and 6, as well as the inactive ring-opening derivatives (7–9), 8-OH might be important for their antibacterial activities.
Table 1. MIC values (μg/mL) of compounds against a panel of bacteria.
| Compounda | Sa1c | Sa2c | MRSAc | MRSEc | Pac | Kpc | Abc |
|---|---|---|---|---|---|---|---|
| 1 | 6.8 ± 0.6 | >100 | 38.5 ± 4.3 | 31.5 ± 3.7 | >100 | 62.8 ± 4.0 | 63.2 ± 6.3 |
| 2 | 4.7 ± 0.6 | 84.1 ± 9.9 | 65.8 ± 3.9 | 65.7 ± 6.0 | 35.8 ± 4.5 | >100 | >100 |
| 3 | 5.5 ± 0.4 | 3.1 ± 0.3 | 29.3 ± 2.3 | 31.3 ± 3.4 | 8.0 ± 0.6 | 59.1 ± 5.1 | 60.1 ± 4.2 |
| 5 | 13.3 ± 1.5 | >100 | 62.5 ± 5.3 | >100 | >100 | >100 | >100 |
| 6 | 10.6 ± 0.4 | 40.2 ± 0.9 | >100 | >100 | 19.3 ± 0.6 | >100 | >100 |
| penicillinb | 0.5 ± 0.05 | 71.2 ± 6.0 | |||||
| streptomycinb | 0.6 ± 0.03 | ||||||
| tigecyclineb | 0.5 ± 0.03 | 0.7 ± 0.04 | 0.5 ± 0.05 | 0.6 ± 0.03 |
aThe compounds were inactive when MICs > 100 μg/mL, ±SD values were calculated on three individual experiments.
bPositive control.
cSa1, Staphylococcus aureus ATCC 25923; Sa2, S. aureus clinical strain; Pa, Pseudomonas aeruginosa ATCC 9027; Kp, Klebsiella pneumonia clinical strain; Ab, Acinetobacter baumannii clinical strain.
Conclusion
In summary, our study on fungal metabolites afforded nine rare 2,4′-linked heterodimeric tetrahydroxanthone derivatives with attractive antibacterial activities. Compounds 1–4 were first isolated as two pairs of atropisomers, which along with other derivatives, provided great samples to reveal the dominant role of axial chiralities in CEs. The specific CD Exciton Chirality method was therefore proposed to determine the axial chiralities in dimeric tetrahydroxanthones and their derivatives.
Methods
General experimental procedures
Optical rotations were measured on a Jasco P-1020 polarimeter (Jasco, Tokyo, Japan). A UV-2450 spectrophotometer (Shimadzu, Tokyo, Japan) was applied for the measurement of UV spectra. ECD and IR data were collected on Jasco J-810 spectrometer and Bruker Tensor 27 spectrometer (Bruker, Karlsruhe, Germany), respectively. Preparative HPLC was performed on a Shimadzu LC-8A system equipped with Shim-pack RP-C18 column (10 μm, 200 mm × 20 mm i.d., Shimadzu, Tokyo, Japan), using a binary channel UV detector at the wavelengths of 210 and 330 nm, respectively. With TMS as internal standard, NMR data were recorded on a Bruker AVIII-500 NMR instrument (1H NMR, 500 MHz; 13C NMR, 125 MHz). HRESIMS data were obtained using Agilent 6520B Q-TOF mass instrument and Agilent 1100 series LC/MSD-Trap-SL mass analyzer (Agilent Technologies, Santa Clara, USA). The extracts were fractionated through CCs with multiple packing materials such as silica gel (Qingdao Marine Chemical Co. Ltd., Qingdao, China), Sephadex LH-20 (Pharmacia, Stockholm, Sweden) and ODS (40−63 μm, Fuji, Tokyo, Japan). After fluorescence observation under UV light (254 and 356 nm), silica gel GF254 plates were sprayed with vanillin−sulfuric acid to visualize the spots. Optical density (OD) values were determined by a microplate reader (Tecan, Mannedorf, Switzerland).
Fungal material
The tubers of Pinellia ternata (Araceae) were collected from the suburb of Nanjing, Jiangsu, People’s Republic of China in May, 2014. After surface sterilization with 75% ethanol and 1% NaClO, the tubers were cut into small pieces, which were put on the potato dextrose agar to afford the title strain. From morphological and microscopic characteristics, this fungus was identified as Aspergillus lentulus, which was further reinforced by its internal transcribed spacer (ITS) and 18S rDNA sequences with 100% identity to the reported one (GenBank accession No. HE578064.1). The fungus was cultivated on potato dextrose agar (PDA) at 28 °C for 7 days. Then 16 pieces of the agar were transformed into four 250 mL Erlenmeyer flasks (containing 80 mL potato dextrose liquid medium), which were incubated at 28 °C, and 120 rpm for 6 days to prepare seed culture. A total of 15 Erlenmeyer flasks (500 mL), each containing 80 g of rice and 120 mL of tap water, were used for solid fermentation. After being sterilized at 115 °C for 30 minutes, the flasks were incubated with 20 mL of seed cultures to cultivate at 28 °C for 30 days.
Extraction and isolation
The crude extract (15.2 g), obtained by extracting the rice cultures with EtOAc three times and removing EtOAc under reduced pressure, had obvious antibacterial activity with an MIC value of 162 μg/mL for S. aureus ATCC 25923. With a gradient elution of petroleum ether−EtOAc from 20:1 to 1:2, the extract was fractionated through silica gel CC, giving fractions A−H. Bioactive fraction C (3.5 g, MIC = 78 μg/mL for S. aureus ATCC 25923) was further submitted to ODS CC with MeOH−H2O as the mobile phase to afford 11 subfractions C1−C11 and fraction C9 was purified by Sephadex LH-20 CC (MeOH−CH2Cl2, 1:1) to gave six subfractions. With Shimadzu LC-8A preparative HPLC system using MeOH−H2O (75:25, 0.1% HCOOH) as the mobile phase, compounds 8 (42.3 mg, tR 31.8 min), 3 (55.6 mg, tR 33.7 min), 2 (13.5 mg, tR 35.6 min), 1 (650.3 mg, tR 38.8 min), and 4 (7.8 mg, tR 48.2 min) were isolated from the third subfraction. Fraction C8 was purified likewise by Sephadex LH-20 CC and preparative HPLC using acetonitrile−H2O (61:39, 0.1% HCOOH) to give 7 (14.9 mg, tR 14.7 min), 9 (4.7 mg, tR 26.6 min), 6 (14.6 mg, tR 31.4 min), and 5 (128.5 mg, tR 33.1 min), respectively.
Spectroscopic data
Neosartorin (1): Yellow powder; 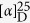 –210.2 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 208 (4.42), 334 (4.43) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−23.1), 219 (+7.9), 237 (−24.1), 316 (+14.7), 346 (−11.8) nm; 1H NMR (500 MHz, CDCl3) δ 13.91 (1 H, s, 8-OH), 13.78 (1 H, s, 8′-OH), 11.54 (1 H, s, 1-OH), 11.37 (1 H, s, 1′-OH), 7.12 (1 H, d, J = 8.5 Hz, H-3′), 6.58 (1 H, d, J = 8.5 Hz, H-2′), 6.47 (1 H, s, H-4), 5.26 (1 H, d, J = 1.3 Hz, H-5′), 4.36 (1 H, dd, J = 4.0, 1.5 Hz, H-5), 3.79 (3 H, s, OCH3-12), 3.65 (3 H, s, OCH3-12′), 2.84 (1 H, ddd, J = 18.6, 11.2, 6.9 Hz, H-7a), 2.39 (1 H, m, H-7b), 2.37 (2 H, m, H-7′), 2.31 (1 H, m, H-6′), 2.16 (1 H, m, H-6a), 2.08 (3 H, s, CH3-3), 1.98 (1 H, m, H-6b), 1.92 (3 H, s, CH3-14′), 0.92 (1 H, d, J = 5.8 Hz, CH3-13′); 13C NMR (125 MHz, CDCl3) δ 187.7 (C-9′a), 187.4 (C-9a), 178.7 (C-8), 177.7 (C-8′), 171.2 (C-11), 170.6 (C-11′), 169.0 (C-15′), 161.7 (C-1′), 159.8 (C-1), 156.7 (C-4a), 155.5 (C-4a′), 148.5 (C-3), 139.9 (C-3′), 118.6 (C-2), 114.6 (C-4′), 110.2 (C-2′), 108.9 (C-4), 106.8 (C-9′b), 104.8 (C-9b), 100.5 (C-9), 100.2 (C-9′), 83.9 (C-10), 82.1 (C-10′), 69.4 (C-5′), 67.0 (C-5), 53.6 (C-12), 53.4 (C-12′), 32.8 (C-7′), 27.8 (C-6′), 24.3 (C-7), 23.1 (C-6), 21.1 (3-CH3), 20.3 (C-14′), 17.0 (C-13′); ESIMS m/z 681.30 [M + H]+.
–210.2 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 208 (4.42), 334 (4.43) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−23.1), 219 (+7.9), 237 (−24.1), 316 (+14.7), 346 (−11.8) nm; 1H NMR (500 MHz, CDCl3) δ 13.91 (1 H, s, 8-OH), 13.78 (1 H, s, 8′-OH), 11.54 (1 H, s, 1-OH), 11.37 (1 H, s, 1′-OH), 7.12 (1 H, d, J = 8.5 Hz, H-3′), 6.58 (1 H, d, J = 8.5 Hz, H-2′), 6.47 (1 H, s, H-4), 5.26 (1 H, d, J = 1.3 Hz, H-5′), 4.36 (1 H, dd, J = 4.0, 1.5 Hz, H-5), 3.79 (3 H, s, OCH3-12), 3.65 (3 H, s, OCH3-12′), 2.84 (1 H, ddd, J = 18.6, 11.2, 6.9 Hz, H-7a), 2.39 (1 H, m, H-7b), 2.37 (2 H, m, H-7′), 2.31 (1 H, m, H-6′), 2.16 (1 H, m, H-6a), 2.08 (3 H, s, CH3-3), 1.98 (1 H, m, H-6b), 1.92 (3 H, s, CH3-14′), 0.92 (1 H, d, J = 5.8 Hz, CH3-13′); 13C NMR (125 MHz, CDCl3) δ 187.7 (C-9′a), 187.4 (C-9a), 178.7 (C-8), 177.7 (C-8′), 171.2 (C-11), 170.6 (C-11′), 169.0 (C-15′), 161.7 (C-1′), 159.8 (C-1), 156.7 (C-4a), 155.5 (C-4a′), 148.5 (C-3), 139.9 (C-3′), 118.6 (C-2), 114.6 (C-4′), 110.2 (C-2′), 108.9 (C-4), 106.8 (C-9′b), 104.8 (C-9b), 100.5 (C-9), 100.2 (C-9′), 83.9 (C-10), 82.1 (C-10′), 69.4 (C-5′), 67.0 (C-5), 53.6 (C-12), 53.4 (C-12′), 32.8 (C-7′), 27.8 (C-6′), 24.3 (C-7), 23.1 (C-6), 21.1 (3-CH3), 20.3 (C-14′), 17.0 (C-13′); ESIMS m/z 681.30 [M + H]+.
Lentulin A (2): Yellow powder; 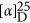 +276.1 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 205 (4.61), 337 (4.54) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−22.1), 217 (−53.9), 246 (+15.7), 306 (−2.0), 342 (+19.5) nm; IR (KBr) νmax 3451, 1740, 1622, 1453, 1222 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.20 [M + H]+; HRESIMS m/z 703.1631 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
+276.1 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 205 (4.61), 337 (4.54) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−22.1), 217 (−53.9), 246 (+15.7), 306 (−2.0), 342 (+19.5) nm; IR (KBr) νmax 3451, 1740, 1622, 1453, 1222 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.20 [M + H]+; HRESIMS m/z 703.1631 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
Lentulin B (3): Yellow powder; 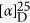 −319.4 (c 0.14, MeOH); UV (MeOH) λmax (log ε) 205 (5.04), 334 (5.03) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−1.8), 220 (+29.5), 242 (−9.9), 316 (+7.3), 370 (−22.2) nm; IR (KBr) νmax 3451, 2955, 1743, 1622, 1452, 1366, 1218 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.20 [M + H]+; HRESIMS m/z 703.1632 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
−319.4 (c 0.14, MeOH); UV (MeOH) λmax (log ε) 205 (5.04), 334 (5.03) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−1.8), 220 (+29.5), 242 (−9.9), 316 (+7.3), 370 (−22.2) nm; IR (KBr) νmax 3451, 2955, 1743, 1622, 1452, 1366, 1218 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.20 [M + H]+; HRESIMS m/z 703.1632 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
Lentulin C (4): Yellow powder; 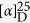 +69.0 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 203 (4.80), 335 (4.69) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−2.9), 217 (−32.0), 241 (+18.2), 310 (−4.5), 343 (+17.1) nm; IR (KBr) νmax 3458, 1735, 1625, 1458, 1384, 1220 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.11 [M + H]+; HRESIMS m/z 703.1637 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
+69.0 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 203 (4.80), 335 (4.69) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−2.9), 217 (−32.0), 241 (+18.2), 310 (−4.5), 343 (+17.1) nm; IR (KBr) νmax 3458, 1735, 1625, 1458, 1384, 1220 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.11 [M + H]+; HRESIMS m/z 703.1637 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
Lentulin D (5): Yellow powder; 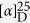 −114.3 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 205 (4.92), 278 (4.57), 336 (4.55) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−7.4), 213 (+6.3), 237 (−12.7), 284 (+3.6), 363 (−4.3) nm; IR (KBr) νmax 3455, 2959, 1791, 1746, 1628, 1453, 1368, 1218 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.30 [M + H]+; HRESIMS m/z 703.1636 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
−114.3 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 205 (4.92), 278 (4.57), 336 (4.55) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−7.4), 213 (+6.3), 237 (−12.7), 284 (+3.6), 363 (−4.3) nm; IR (KBr) νmax 3455, 2959, 1791, 1746, 1628, 1453, 1368, 1218 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 681.30 [M + H]+; HRESIMS m/z 703.1636 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
Lentulin E (6): Yellow powder; 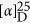 −211.4 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 205 (5.00), 278 (4.65), 338 (4.67) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−0.3), 215 (+18.0), 238 (−14.5), 352 (−2.6), 387 (+1.1) nm; IR (KBr) νmax 3451, 2962, 1791, 1750, 1630, 1592, 1479, 1385, 1220 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 698.09 [M + NH4]+; HRESIMS m/z 703.1627 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
−211.4 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 205 (5.00), 278 (4.65), 338 (4.67) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−0.3), 215 (+18.0), 238 (−14.5), 352 (−2.6), 387 (+1.1) nm; IR (KBr) νmax 3451, 2962, 1791, 1750, 1630, 1592, 1479, 1385, 1220 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 698.09 [M + NH4]+; HRESIMS m/z 703.1627 [M + Na]+ (calcd for 703.1633, C34H32O15Na).
Lentulin F (7): Yellow powder; 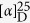 −111.7 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 205 (5.05), 278 (4.70), 335 (4.72) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−8.5), 213 (+6.8), 236 (−19.5), 282 (+6.5), 345 (−3.6), 379 (+0.9) nm; IR (KBr) νmax 3455, 1743, 1631, 1478, 1385, 1220 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 699.04 [M + H]+; HRESIMS m/z 721.1736 [M + Na]+ (calcd for 721.1739, C34H34O16Na).
−111.7 (c 0.13, MeOH); UV (MeOH) λmax (log ε) 205 (5.05), 278 (4.70), 335 (4.72) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−8.5), 213 (+6.8), 236 (−19.5), 282 (+6.5), 345 (−3.6), 379 (+0.9) nm; IR (KBr) νmax 3455, 1743, 1631, 1478, 1385, 1220 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 699.04 [M + H]+; HRESIMS m/z 721.1736 [M + Na]+ (calcd for 721.1739, C34H34O16Na).
Lentulin G (8): Yellow powder; 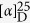 −159.5 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 205 (4.82), 276 (4.45), 346 (4.38) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−4.1), 213 (+4.8), 236 (−8.2), 282 (+2.8), 368 (−1.6) nm; IR (KBr) νmax 3461, 2957, 1741, 1625, 1418, 1367, 1217 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 713.20 [M + H]+; HRESIMS m/z 735.1893 [M + Na]+ (calcd for 735.1896, C35H36O16Na).
−159.5 (c 0.16, MeOH); UV (MeOH) λmax (log ε) 205 (4.82), 276 (4.45), 346 (4.38) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 200 (−4.1), 213 (+4.8), 236 (−8.2), 282 (+2.8), 368 (−1.6) nm; IR (KBr) νmax 3461, 2957, 1741, 1625, 1418, 1367, 1217 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 713.20 [M + H]+; HRESIMS m/z 735.1893 [M + Na]+ (calcd for 735.1896, C35H36O16Na).
Lentulin H (9): Yellow powder; 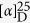 −161.5 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 205 (4.88), 277 (4.52), 347 (4.51) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 213 (+12.1), 238 (−8.8), 286 (−0.7), 349 (−3.0), 387 (+1.1) nm; IR (KBr) νmax 3446, 2956, 1741, 1625, 1588, 1452, 1368, 1217 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 713.08 [M + H]+; HRESIMS m/z 735.1899 [M + Na]+ (calcd for 735.1896, C35H36O16Na).
−161.5 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 205 (4.88), 277 (4.52), 347 (4.51) nm; ECD (3.9 × 10−4 M, MeOH) λ (Δε) 213 (+12.1), 238 (−8.8), 286 (−0.7), 349 (−3.0), 387 (+1.1) nm; IR (KBr) νmax 3446, 2956, 1741, 1625, 1588, 1452, 1368, 1217 cm−1; 1H NMR and 13C NMR data, see Supporting Information
Tables S1 and S2; ESIMS m/z 713.08 [M + H]+; HRESIMS m/z 735.1899 [M + Na]+ (calcd for 735.1896, C35H36O16Na).
Absolute configuration of 5-secondary alcohol in 4
To a CH2Cl2 solution of 4 (0.5 mg/mL), 0.8 mg of Rh2(OCOCF3)4 was added and the first induced ECD spectrum was recorded immediately. The following spectra were measured every 5 minutes until reaching a stationary state. The absolute configuration of 5-sencondary alcohol was determined by the induced CE at around 350 nm.
Conversion of neosartorin (1) and lentulin B (3) into lentulin D (5) and lentulin E (6)
To solutions of 1 and 3 (each 1.5 mg, 0.0022 mmol) in 2 mL CH2Cl2, triethylamine (10 μL, 0.072 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl, 2.0 mg, 0.010 mmol) were added separately. The solutions were stirred at room temperature for 24 h. The reactions were quenched by 2 mL of 0.2 M HCl and the mixtures were extracted with EtOAc three times. Then, the EtOAc layers were washed by water and brine. After being evaporated under gas flow, the residues were purified over Shimadzu LC-8A preparative HPLC system with acetonitrile−H2O (61:39, 0.1% HCOOH) as the mobile phase. Synthetic 5 and 6 were obtained at tR 33.1 min and tR 31.4 min, respectively. All the spectral data of the synthetic products were the same as the natural products including their optical rotations {synthetic 5, 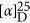 −108.6 (c 0.11, MeOH); natural 5,
−108.6 (c 0.11, MeOH); natural 5, 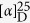 −114.9 (c 0.13, MeOH); synthetic 6,
−114.9 (c 0.13, MeOH); synthetic 6, 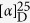 −207.0 (c 0.12, MeOH); natural 6,
−207.0 (c 0.12, MeOH); natural 6, 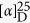 −211.4 (c 0.10, MeOH)}.
−211.4 (c 0.10, MeOH)}.
Methanolysis of lentulin D (5) and lentulin E (6)
Compounds 5 (2.0 mg, 0.0029 mmol) and 6 (1.5 mg, 0.0022 mmol) were dissolved in 0.5% HCl−MeOH (1.0 mL) and were stirred at room temperature for 6 h, respectively22. The residues were obtained by evaporating the solutions under gas flow, and were chromatographied by preparative HPLC to give synthetic 8 (1.5 mg, 0.0021 mmol, 72%, tR 31.8 min in 75% MeOH with 0.1% HCOOH) and synthetic 9 (1.2 mg, 0.0017 mmol, 76%, tR 26.6 min in 61% acetonitrile with 0.1% HCOOH), respectively. All the spectral data of the synthetic 8 and 9 were the same as the natural products {synthetic 8, 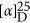 −148.3 (c 0.08, MeOH); natural 8,
−148.3 (c 0.08, MeOH); natural 8, 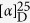 −159.5 (c 0.16, MeOH); synthetic 9,
−159.5 (c 0.16, MeOH); synthetic 9, 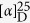 −174.5 (c 0.07, MeOH); natural 9,
−174.5 (c 0.07, MeOH); natural 9, 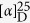 −161.5 (c 0.07, MeOH)}.
−161.5 (c 0.07, MeOH)}.
Mosher esterfication of lentulins G (8) and H (9)
To 2 mL of CH2Cl2 solution of 8 (2.1 mg, 0.0029 mmol), triethylamine (10 μL, 0.072 mmol), DMAP (0.2 mg, 0.0016 mmol), EDC·HCl (4.6 mg, 0.024 mmol), and (R)-MPA (3.5 mg, 0.021 mmol) were added respectively22. The mixture was poured into 2 mL of 0.2 M HCl after being stirred for 24 h at room temperature and was extracted with EtOAc three times. After washing by water and brine, the EtOAc layer was evaporated to give a residue, which was finally purified using preparative HPLC with MeOH−H2O (83:17, 0.1% HCOOH) to yield 8a (1.0 mg, 0.0012 mmol, 40%, tR 22.1 min). In the same manner, 8b (0.8 mg, 0.00093 mmol, 39%, tR 23.7 min) was obtained by preparative HPLC (83% MeOH with 0.1% HCOOH) from 1.7 mg of 8. Subsequently, 9a (0.4 mg, 0.00046 mmol, 36%, tR 25.2 min) and 9b (0.5 mg, 0.00058 mmol, 38%, tR 26.3 min) were yielded from 1.0 mg of 9 using preparative HPLC (81% MeOH with 0.1% HCOOH).
Data for lentulin G (R)-MPA ester 8a: δH 13.820 (1 H, s, 8′-OH), 11.720 (1 H, s, 1-OH), 11.387 (1 H, s, 1′-OH), 7.317−7.438 (5 H, aromatic), 7.120 (1 H, d, J = 7.7 Hz, H-3′), 6.591 (1 H, d, J = 8.1 Hz, H-2′), 6.433 (1 H, s, H-4), 5.506 (1 H, dd, J = 9.3, 2.3 Hz, H-5), 5.269 (1 H, br d, H-5′), 4.777 (1 H, s, H-MPA), 3.803 (3 H, s, OCH3-12), 3.643 (3 H, s, OCH3-12′), 3.616 (3 H, s, OCH3-13), 3.420 (3 H, s, OCH3-MPA), 3.158 (1 H, d, J = 16.9 Hz, H-9a), 3.023 (1 H, d, J = 17.0 Hz, H-9b), 2.353 (2 H, m, H-7′), 2.312 (1 H, m, H-6′), 2.175 (1 H, d, J = 5.4 Hz, H-7a), 2.162 (1 H, d, J = 7.6 Hz, H-7b), 2.071 (3 H, s, CH3-3), 1.936 (3 H, s, CH3-14′), 1.923−1.907 (2 H, overlap, H-6), 0.923 (1 H, d, J = 6.6 Hz, CH3-13′); ESI-MS: m/z 878.09 [M + NH4]+, m/z 895.11 [M + Cl]−.
Data for lentulin G (S)-MPA ester 8b: δH 13.792 (1 H, s, 8′-OH), 11.637 (1 H, s, 1-OH), 11.385 (1 H, s, 1′-OH), 7.320−7.385 (5 H, aromatic), 7.131 (1 H, d, J = 7.7 Hz, H-3′), 6.603 (1 H, d, J = 8.2 Hz, H-2′), 6.275 (1 H, s, H-4), 5.520 (1 H, dd, J = 9.6, 2.3 Hz, H-5), 5.245 (1 H, br d, H-5′), 4.797 (1 H, s, H-MPA), 3.703 (3 H, s, OCH3-12), 3.696 (3 H, s, OCH3-13), 3.641 (3 H, s, OCH3-12′), 3.425 (3 H, s, OCH3-MPA), 2.837 (1 H, d, J = 16.8 Hz, H-9a), 2.594 (1 H, d, J = 17.0 Hz, H-9b), 2.371 (1 H, d, J = 7.2 Hz, H-7a), 2.350 (2 H, m, H-7′), 2.349 (1 H, d, J = 6.6 Hz, H-7b), 2.308 (1 H, m, H-6′), 2.058 (3 H, s, CH3-3), 1.882 (3 H, s, CH3-14′), 2.047−1.994 (2 H, overlap, H-6), 0.908 (1 H, d, J = 6.0 Hz, CH3-13′); ESI-MS: m/z 878.09 [M + NH4]+, m/z 895.03 [M + Cl]−.
Data for lentulin H (R)-MPA ester 9a: δH 13.938 (1 H, s, 8′-OH), 11.674 (1 H, s, 1-OH), 11.402 (1 H, s, 1′-OH), 7.340−7.408 (5 H, aromatic), 7.155 (1 H, d, J = 7.7 Hz, H-3′), 6.594 (1 H, d, J = 8.8 Hz, H-2′), 6.461 (1 H, s, H-4), 5.425 (1 H, dd, J = 8.4, 2.9 Hz, H-5), 5.297 (1 H, br d, H-5′), 4.856 (1 H, s, H-MPA), 3.687 (3 H, s, OCH3-12), 3.636 (3 H, s, OCH3-12′), 3.612 (3 H, s, OCH3-13), 3.432 (3 H, s, OCH3-MPA), 3.421 (1 H, d, J = 16.4 Hz, H-9a), 3.304 (1 H, d, J = 16.4 Hz, H-9b), 3.093 (2 H, m, H-7), 2.390 (2 H, m, H-7′), 2.342 (1 H, m, H-6′), 2.053 (3 H, s, CH3-3), 2.014 (3 H, s, CH3-14′), 1.956−1.815 (2 H, overlap, H-6), 0.938 (1 H, d, J = 6.0 Hz, CH3-13′); ESI-MS: m/z 861.19 [M + H]+.
Data for lentulin H (S)-MPA ester 9b: δH 13.919 (1 H, s, 8′-OH), 11.613 (1 H, s, 1-OH), 11.392 (1 H, s, 1′-OH), 7.340−7.537 (5 H, aromatic), 7.131 (1 H, d, J = 9.3 Hz, H-3′), 6.579 (1 H, d, J = 8.5 Hz, H-2′), 6.294 (1 H, s, H-4), 5.427 (1 H, dd, J = 9.8, 3.9 Hz, H-5), 5.295 (1 H, br d, H-5′), 4.815 (1 H, s, H-MPA), 3.701 (3 H, s, OCH3-13), 3.636 (3 H, s, OCH3-12′), 3.515 (3 H, s, OCH3-12), 3.494 (2 H, m, H-7), 3.443 (3 H, s, OCH3-MPA), 2.890 (1 H, d, J = 15.7 Hz, H-9a), 2.784 (1 H, d, J = 15.7 Hz, H-9b), 2.381 (2 H, m, H-7′), 2.360 (1 H, m, H-6′), 2.022 (3 H, s, CH3-3), 1.978 (3 H, s, CH3-14′), 1.964−1.879 (2 H, overlap, H-6), 0.946 (1 H, d, J = 6.0 Hz, CH3-13′); ESI-MS: m/z 861.20 [M + H]+.
Conversion of lentulin F (7) into lentulin D (5)
A sample of 7 (1.8 mg, 0.0026 mmol) was dissolved in 2 mL of CH2Cl2, followed by addition of triethylamine (10 μL, 0.072 mmol) and EDC·HCl (2.0 mg, 0.010 mmol)17. The other experimental details were the same as above. Using Shimadzu LC-8A preparative HPLC system with acetonitrile−H2O (61:39, 0.1% HCOOH) as the mobile phase, synthetic 5 (1.4 mg, 0.0021 mmol, 80%) was obtained at 33.1 min. All the spectral data of the product were the same as natural 5 including their optical rotation {synthetic 5, 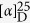 −104.7 (c 0.14, MeOH); natural 5,
−104.7 (c 0.14, MeOH); natural 5, 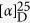 −114.9 (c 0.13, MeOH)}.
−114.9 (c 0.13, MeOH)}.
Antimicrobial assays
Using broth microdilution method, the antimicrobial activities were measured in sterile 96-well plates (Thermo, USA) against five strains of Gram-positive bacteria (S. aureus ATCC 25923, S. aureus clinical strain, MRSA, MRSE, Bacillus subtilis ATCC 6633), four strains of Gram-negative bacteria (Pseudomonas aeruginosa ATCC 9027, Escherichia coli ATCC 25922, Klebsiella pneumonia clinical strain, Acinetobacter baumannii clinical strain), as well as one fungal strain (Candida albicans ATCC 24433). MIC values were determined by comparing OD values with the growth control through a microplate reader, using penicillin G, streptomycin, tigecycline, and fluconazole as the positive controls.
Additional Information
How to cite this article: Li, T.-X. et al. Unusual dimeric tetrahydroxanthone derivatives from Aspergillus lentulus and the determination of their axial chiralities. Sci. Rep. 6, 38958; doi: 10.1038/srep38958 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This research work was funded by the National Natural Science Foundation of China (81503218), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R63), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Fundamental Research Funds for the Central Universities (JKQZ2013016).
Footnotes
Author Contributions T.-X. Li, M.-H. Yang, and Y. Wang carried out the experimental work. X.-B. Wang and J. Luo assisted the ECD research. J. Luo, J.-G. Luo, and L.-Y. Kong provided oversight. T.-X. Li, M.-H. Yang, and L.-Y. Kong conceived the experiments and wrote the manuscript. All authors approved the final version of the manuscript.
References
- Stewart M. et al. Rugulotrosins A and B: two new antibacterial metabolites from an Australian isolate of a Penicillium sp. J Nat Prod 67, 728–730 (2004). [DOI] [PubMed] [Google Scholar]
- Rezanka T. & Sigler K. Hirtusneanoside, an unsymmetrical dimeric tetrahydroxanthone from the lichen Usnea hirta. J Nat Prod 70, 1487–1491 (2007). [DOI] [PubMed] [Google Scholar]
- Chutrakul C. et al. Ascherxanthone B from Aschersonia luteola, a new antifungal compound active against rice blast pathogen Magnaporthe grisea. J Appl Microbial 107, 1624–1631 (2009). [DOI] [PubMed] [Google Scholar]
- Isaka M., Palasarn S., Kocharin K. & Saenboonrueng J. A cytotoxic xanthone dimer from the entomopathogenic fungus Aschersonia sp. BCC 8401. J Nat Prod 68, 945–946 (2005). [DOI] [PubMed] [Google Scholar]
- Masters K.-S. & Bräse S. Xanthones from fungi, lichens, and bacteria: the natural products and their synthesis. Chem Rev 112, 3717–3776 (2012). [DOI] [PubMed] [Google Scholar]
- Tabata N., Tomoda H., Matsuzaki K. & Omura S. Structure and biosynthesis of xanthoquinodins, anticoccidial antibiotics. J Am Chem Soc 115, 8558–8564 (1993). [Google Scholar]
- Yang D.-M., Takeda N., Iitaka Y., Sankawa V. & Shibata S. The structures of eumitrins A 1, A 2 and B: the yellow pigments of the lichen, Usnea bayleyi (Stirt.) Zahlbr. Tetrahedron 29, 519–528 (1973). [Google Scholar]
- Proksa B., Uhrin D., Liptaj T. & Šturdíková M. Neosartorin, an ergochrome biosynthesized by Neosartorya fischeri. Phytochemistry 48, 1161–1164 (1998). [Google Scholar]
- Smyth J. E., Butler N. M. & Keller P. A. A twist of nature–the significance of atropisomers in biological systems. Nat Prod Rep 32, 1562–1583 (2015). [DOI] [PubMed] [Google Scholar]
- Bringmann G., Gulder T., Gulder T. A. & Breuning M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem Rev 111, 563–639 (2010). [DOI] [PubMed] [Google Scholar]
- LaPlante S. R. et al. Assessing atropisomer axial chirality in drug discovery and development. J Med Chem 54, 7005–7022 (2011). [DOI] [PubMed] [Google Scholar]
- Wagenaar M. M. & Clardy J. Dicerandrols, new antibiotic and cytotoxic dimers produced by the fungus Phomopsis longicolla isolated from an endangered mint. J Nat Prod 64, 1006–1009 (2001). [DOI] [PubMed] [Google Scholar]
- Isaka M. et al. Phomoxanthones A and B, novel xanthone dimers from the endophytic fungus Phomopsis species. J Nat Prod 64, 1015–1018 (2001). [DOI] [PubMed] [Google Scholar]
- Elsässer B. et al. X‐ray structure determination, absolute configuration and biological activity of phomoxanthone A. Eur J Org Chem 2005, 4563–4570 (2005). [Google Scholar]
- Rönsberg D. et al. Pro-apoptotic and immunostimulatory tetrahydroxanthone dimers from the endophytic fungus Phomopsis longicolla. J Org Chem 78, 12409–12425 (2013). [DOI] [PubMed] [Google Scholar]
- Ola A. R. et al. Absolute configuration and antibiotic activity of neosartorin from the endophytic fungus Aspergillus fumigatiaffinis. Tetrahedron Lett 55, 1020–1023 (2014). [Google Scholar]
- Kikuchi H., Isobe M., Kurata S., Katou Y. & Oshima Y. New dimeric and monomeric chromanones, gonytolides D–G, isolated from the fungus Gonytrichum sp. Tetrahedron 68, 6218–6223 (2012). [DOI] [PubMed] [Google Scholar]
- Zhang W. et al. New mono‐and dimeric members of the secalonic acid family: blennolides A–G isolated from the fungus Blennoria sp. Chem Eur J 14, 4913–4923 (2008). [DOI] [PubMed] [Google Scholar]
- Qin T. et al. Atropselective syntheses of (−) and (+) rugulotrosin A utilizing point-to-axial chirality transfer. Nat Chem 7, 234–240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bara R. et al. Atropisomeric dihydroanthracenones as inhibitors of multiresistant Staphylococcus aureus. J Med Chem 56, 3257–3272 (2013). [DOI] [PubMed] [Google Scholar]
- Gerards M. & Snatzke G. Circular dichroism, XCIII determination of the absolute configuration of alcohols, olefins, epoxides, and ethers from the CD of their “in situ” complexes with [Rh2(COOCF3)4]. Tetrahedron: Asymmetry 1, 221–236 (1990). [Google Scholar]
- Kikuchi H. et al. Structures of the dimeric and monomeric chromanones, gonytolides A–C, isolated from the fungus Gonytrichum sp. and their promoting activities of innate immune responses. Org Lett 13, 4624–4627 (2011). [DOI] [PubMed] [Google Scholar]
- Krick A. et al. Potential cancer chemopreventive in vitro activities of monomeric xanthone derivatives from the marine algicolous fungus Monodictys putredinis. J Nat Prod 70, 353–360 (2007). [DOI] [PubMed] [Google Scholar]
- Wezeman T., Bräse S. & Masters K. S. Xanthone dimers: a compound family which is both common and privileged. Nat Prod Rep 32, 6–28 (2015). [DOI] [PubMed] [Google Scholar]
- Bringmann G., Günther C., Ochse M., Schupp O. & Tasler S. Prog Chem Org Nat Prod 1–249 (Springer, 2001). [DOI] [PubMed] [Google Scholar]
- Harada N. & Nakanishi K. Exciton chirality method and its application to configurational and conformational studies of natural products. Accounts Chem Res 5, 257–263 (1972). [Google Scholar]
- Zhang H.-J., Zhang Y.-M., Luo J.-G., Luo J. & Kong L.-Y. Anti-inflammatory diterpene dimers from the root barks of Aphanamixis grandifolia. Org Biomol Chem 13, 7452–7458 (2015). [DOI] [PubMed] [Google Scholar]
- Wu G.-W. et al. Versixanthones A–F, cytotoxic xanthone–chromanone dimers from the marine-derived fungus Aspergillus versicolor HDN1009. J Nat Prod 78, 2691–2698 (2015). [DOI] [PubMed] [Google Scholar]
- Shim S. H., Baltrusaitis J., Gloer J. B. & Wicklow D. T. Phomalevones A−C: dimeric and pseudodimeric polyketides from a fungicolous Hawaiian isolate of Phoma sp.(Cucurbitariaceae). J Nat Prod 74, 395–401 (2011). [DOI] [PubMed] [Google Scholar]
- Bode S. E., Drochner D. & Müller M. Synthesis, biosynthesis, and absolute configuration of vioxanthin. Angew Chem 119, 6020–6024 (2007). [DOI] [PubMed] [Google Scholar]
- Isaka M., Yangchum A., Rachtawee P., Komwijit S. & Lutthisungneon A. Hopane-type triterpenes and binaphthopyrones from the scale insect pathogenic fungus Aschersonia paraphysata BCC 11964. J Nat Prod 73, 688–692 (2010). [DOI] [PubMed] [Google Scholar]
- Ohkawa Y. et al. Antiangiogenic metabolites from a marine-derived fungus, Hypocrea vinosa. J Nat Prod 73, 579–582 (2010). [DOI] [PubMed] [Google Scholar]
- Bruhn T. et al. Axially chiral BODIPY DYEmers: an apparent exception to the exciton chirality rule. Angew Chem. 53, 14592–14595 (2014). [DOI] [PubMed] [Google Scholar]
- Jurinovich S., Guido C. A., Bruhn T., Pescitelli G. & Mennucci B. The role of magnetic–electric coupling in exciton-coupled ECD spectra: the case of bis-phenanthrenes. Chem Commun 51, 10498–10501 (2015). [DOI] [PubMed] [Google Scholar]
- Harada N., Nakanishi K. & Berova N. Electronic CD exciton chirality method: principles and applications. Compr Chiropt Spectrosc 2, 115–166 (2012). [Google Scholar]
- Berova N., Di Bari L. & Pescitelli G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem Soc Rev 36, 914–931 (2007). [DOI] [PubMed] [Google Scholar]
- Li T.-X., Yang M.-H., Wang X.-B., Wang Y. & Kong L.-Y. Synergistic antifungal meroterpenes and dioxolanone derivatives from the endophytic Fungus Guignardia sp. J Nat Prod 78, 2511–2520 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.