

Abstract
GPR40/FFAR1 is a G protein-coupled receptor predominantly expressed in pancreatic β-cells and activated by long-chain free fatty acids, mediating enhancement of glucose-stimulated insulin secretion. A novel series of substituted 3-(4-aryloxyaryl)propanoic acid derivatives were prepared and evaluated for their activities as GPR40 agonists, leading to the identification of compound 5, which is highly potent in in vitro assays and exhibits robust glucose lowering effects during an oral glucose tolerance test in nSTZ Wistar rat model of diabetes (ED50 = 0.8 mg/kg; ED90 = 3.1 mg/kg) with excellent pharmacokinetic profile, and devoid of cytochromes P450 isoform inhibitory activity.
Keywords: GPR40, GPR40 agonist, FFAR1, fatty acids, insulin secretion, type 2 diabetes
Type 2 diabetes, the most prevalent form of diabetes, is a growing epidemic, which is associated with a high degree of morbidity and mortality.1−3 Various available oral therapies include insulin secretagogues, such as sulfonylureas; glucose-lowering effectors, for example, metformin; and activators of PPAR-γ, like pioglitazone.4 However, these available treatments are unable to provide satisfactory glycemic control in many patients.
GPR40 receptor, also known as the long chain free fatty acid 1 receptor (FFAR1), has emerged as one of the potential targets for the effective treatment of type 2 diabetes mellitus (T2DM).5 GPR40 receptor belongs to the class A family of G-protein coupled receptors. GPR40 is mainly expressed in pancreatic β-cells and activated by long-chain free fatty acids. Thereby, resulting in enhancement of glucose-stimulated insulin secretion (GSIS), which is dependent on elevated glucose levels.6 Thus, identification of a small molecule GPR40 agonist, with sufficient pharmacokinetic and pharmacodynamics properties, may offer beneficial treatment for type 2 diabetes and associated complications.7 Additionally, the limited tissue distribution of GPR40 (primarily in pancreatic islets)8 suggests that there may be less possibilities for adverse events associated with GPR40 inhibition in other tissues.
Due to the promising application of GPR40 agonist in type II diabetes treatment, intensive research has been undertaken in the area, and several structurally diverse small molecule modulators of GPR40 have been reported (Figure 1).9−16 However, many of them possessed insufficient potency, lack metabolic stability, or toxicity. Two of these molecules, AMG-83717 and TAK-875,18 were selected for clinical trials as antihyperglycemic agents. The most advanced compound of the two was fasiglifam (TAK-875) from Takeda in phase III. However, in December 2013 Takeda discontinued the development of fasiglifam due to concerns about liver idiopathic toxicity.19
Figure 1.

Selected known GPR40/FFAR1 receptor agonist.
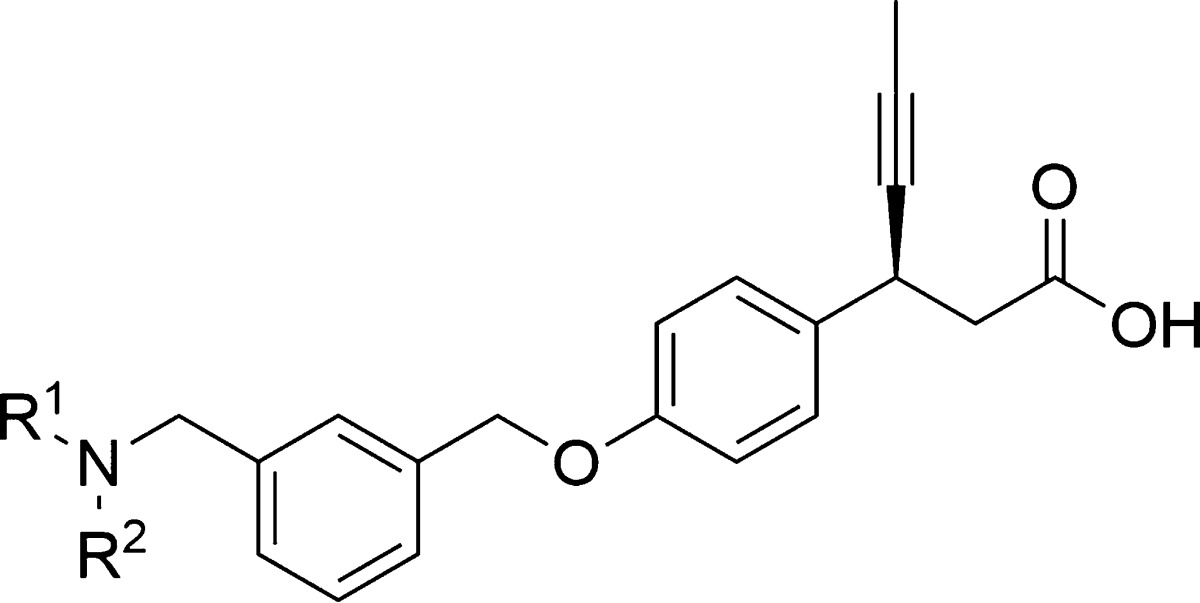
Continuing our research efforts,20 we explored therapies with distinct mechanism of action for type 2 diabetes. This letter describes the identification of (S)-3-(4-((3-((isopropyl(thiophen-3-ylmethyl)amino) methyl) benzyl)oxy)phenyl)hex-4-ynoic acid calcium salt (5) as a novel, potent, and orally efficacious GPR40 agonist. We sought an opportunity in AMG-837 scaffold and focused our efforts in modifying this scaffold. The rational being the introduction of amine substituent in the central phenyl ring of AMG-837, if tolerated, would reduce the lipophilicity and thereby improve the drug-like properties (Figure 2). AMG-837 is very lipophilic (cLog P = 7.6) and likely to have significant CNS exposure.21 Potential drugs with high lipophilicity and poor physiochemical properties are generally correlated with various side effects and have higher risk of failure in clinical trials.22
Figure 2.

Schematic representation of ligand optimization.
A variety of substituted amines were synthesized and assayed for GPR40 activity using Luciferase transactivation assay. The observation revealed in Table 1 indicates that the pyridyl compound 1a, though was significantly less lipophilic when compared to reference AMG-837, however, was also inactive in the in vitro assay. The thiophene substituted secondary amine 1b showed a marked improvement in potency with EC50 value of 118 nM, while corresponding N-methyl derivative 1c had EC50 of 38 nM. Our optimization of the R1 portion began with a brief examination since relatively steep SAR had been found. The results obtained with analogous 1b and 1c suggested that incorporation of the amino-alkyl thiophene substitution may open up new directions for further SAR development.
Table 1. In Vitro Activity of Reference Compounds (TAK-875 & AMG-837) and Compounds 1a–1c.

EC50 values given are expressed as mean ± SEM of three independent experiments
Calculated from Schrödinger 4.4 Software.
Accordingly, we focused our investigation to the substituents on the nitrogen atom, and thus, in our optimization campaign, a series of novel 3-methyl amino thiophene derivatives were synthesized and evaluated as GPR40 agonist (Table 2). Compound 2a, substituted with electron-withdrawing trifluoromethyl group, provided a significant leap in the agonistic activity and found to be extremely potent (EC50 = 0.8 nM), while substitution with electron-donating groups OH and OMe (2b–c) were relatively less potent. However, the unsubstituted branched alkyls (2d–e) were also found to be active. Further, to investigate the effect of cycloalkyls, cyclopropyl was synthesized using our double reductive amination strategy. The smallest, cyclopropyl (2f) was very potent (EC50 = 1.9 nM), while methyl cyclopropyl (2g) was comparatively less potent. The larger, cyclopentyl (2h) and cyclohexyl (2k) also retained the potency. In addition, heterocyclyles (2j–l) were also tolerated, with the notable exception of the N-methylpiperidine (2m).
Table 2. In Vitro Activity of Compounds 2a–2t.


EC50 values given are expressed as mean ± SEM of three independent experiments
Calculated from Schrödinger 4.4 Software.
In an effort to increase the bulk of N-substitution, compound 2n having benzyl subunit was examined and was also found active as GPR40 agonist, including heteroaryls (2o–p). Unlike 1a, the pyridyl compound 2o showed potent GPR40 activity thereby pointing the importance of methyl thiophene substitution. Next, we introduced the amide groups, which were synthesized via reductive amination of commercially available (2-thiophene)methylamine and aldehyde (8), followed by the coupling of resulting amine with corresponding sulfonyl chloride or acids. Notably, sulfonamides 2q and 2r showed significant GPR40 agonistic activity, EC50 of 7 and 10 nM, respectively. By contrast, amide 2s was surprisingly weak (EC50 = 30 nM), while acid (2t) was inactive as GPR40 agonist. In general, a wide variety of substitutions were tolerated, resulting in a series of potent compounds.
With multiple examples of highly potent GPR40 agonists in hand, we next evaluated them against the cytochrome P450 (CYP450) enzymes using recombinant CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4.23 The number of active compounds tested against CYP450 isoforms were unfortunately found to show strong CYP450 inhibition and unable to pass this hurdle in spite of low nanomolar potency against GPR40 receptor, including most potent compound 2a. Interestingly, as an exception, compound 2d (EC50 = 8.6 nM) with N-isopropyl substituent showed no significant inhibitory activity against CYP450 isoforms (Supporting Information). Compound 2d was then selected for early stage in vivo studies and additional profiling activities.
Synthesis of compound 2d is depicted in Scheme 1. The reductive amination24 of commercially available 3-thiophene-aldehyde (3) and isopropyl amine using sodium triacetoxyborohydride resulted in secondary amine intermediate 4. Compound 4 on further reductive amination under similar conditions with aldehyde intermediate, (S)-3-(4-((3-formylbenzyl)oxy)phenyl)hex-4-ynoic acid (8), afforded 2d in high yields. The aldehyde intermediate 8 was obtained from (S)-3-(4-hydroxyphenyl)hex-4-ynoic acid (6) as shown in Scheme 2. Phenol 6 was synthesized via five-step reported procedure using commercially available 4-hydroxybenzaldehyde and Meldrum’s acid.25 Treatment of 6 with 40% aqueous tetrabutylphosphonium hydroxide (n-Bu4POH) in THF, followed by addition of 3-formyl benzyl bromide (7), afforded aldehyde intermediate 8. In order to improve the stability and aqueous solubility, 2d was converted to its corresponding calcium salt (5) via two-step sequence of formation of sodium salt followed by its conversion to calcium salt, with excellent chemical purity for the further pharmacological studies.26 It should be emphasized that calcium salt was preferred as it offered advantages of crystallinity, API stability, API scale up, and hygroscopicity.
Scheme 1. Synthesis of Compounds 2d and 5.

Reagent and conditions: (a) CH(CH3)2NH2, NaB(OAc)3H, CH3COOH, dry THF, 0 °C to r.t., 16 h; (b) 8, NaB(OAc)3H, CH3COOH, dry THF, 0 °C to r.t., 16 h; (c) NaOH, MeCN/H2O, r.t., 3 h; (d) CaCl2, MeOH/H2O, r.t., 16 h.
Scheme 2. Synthesis of (S)-3-(4-((3-Formylbenzyl)oxy)phenyl)hex-4-ynoic Acid (8).

Reagent and conditions: (a) n-Bu4POH, THF/H2O, 0 °C to r.t., 16 h.
(S)-3-(4-((3-((Isopropyl(thiophen-3-ylmethyl)amino)methyl)benzyl)oxy)phenyl)hex-4-ynoic acid calcium salt (5) was next evaluated in cell-based functional IP1 ELISA assay and calcium flux assay. To our delight, 5 was found to be highly potent in both the assays with EC50 of 10.5 and 11.6 nM, respectively. Furthermore, 5 was also evaluated in insulin assay in RINm cells and showed dose-dependent insulin secretion with EC50 of 20 μM, while TAK-875 showed EC50 of 27 μM in the similar assay. All these in vitro assays indicate excellent penetration. The compound 5, with high in vitro potency, was next evaluated for physiochemical properties and metabolic stability assays in addition to in vivo pharmacokinetic profile. Notably, 5 have good aqueous solubility and acceptable lipophilicity with cLogP in the middle of the ideal range. The compound is stable toward rat liver microsomes and also demonstrated good Caco2 permeability (Supporting Information). These drug-like properties of compound 5 further enabled its potential for in vivo validation. The in vivo pharmacokinetic investigation in fasted SD rats revealed that 5 exhibited good oral absorption having Cmax of 2.78 μg/mL with an AUC of 6.6 μg.h/mL and terminal half-life of 2 h, at an oral dose of 3 mg/kg (Table 3). Pharmacokinetic studies in rats showed a fast oral absorption, higher plasma concentration, and an acceptable half-life and demonstrated an excellent oral bioavailability (100%). The desirable pharmacokinetic profile of compound 5 allowed for the progression of the compound into in vivo efficacy studies.
Table 3. Pharmacokinetics Properties of 5 in SD Rats.
| parameters | compd 5 |
|---|---|
| PO dose (mg/kg) | 3 |
| Tmax (h) | 1 (0.25–1) |
| Cmax (μg/mL) | 2.78 ± 0.68 |
| AUC(O-t) (μg·h/mL) | 6.61 ± 1.00 |
| T1/2, po (h) | 2.01 ± 0.19 |
| MRT (h) | 2.69 ± 0.55 |
| IV dose (mg/kg) | 1 |
| CO (μg/mL) | 1.30 ± 0.35 |
| AUC(0-t) (μg·h/mL) | 1.10 ± 0.29 |
| Vss (L/kg) | 1.05 ± 0.09 |
| CL (mL/min/kg) | 13.83 ± 1.56 |
| T1/2, iv (h) | 1.78 ± 0.16 |
| MRT (h) | 1.27 ± 0.05 |
| %F | 100 |
Single dose PK study of compound 5 in fasted female SD rat by oral route. Formulation detail: 5% Tween80 + 5%PEG400 + 90% Na-CMC (0.5%), oral.
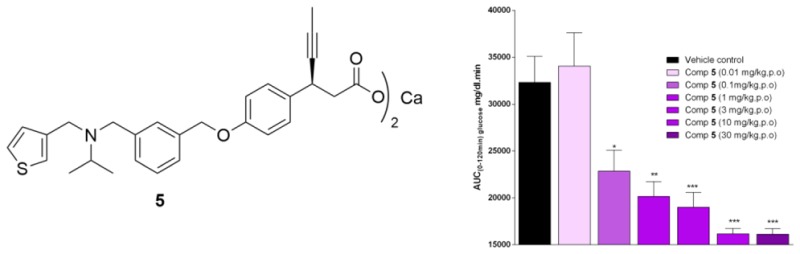
In vivo efficacy of the lead compound 5 was evaluated by an oral glucose tolerance test (OGTT) in nSTZ Wistar rat model.27,28 A single oral administration of 5 at 0.1 mg/kg dose decreases the glucose levels by 30% w.r.t. vehicle control (Figure 3), indicating that even at this low dose, compound 5 markedly reduced the glucose excursion compared to the control. The glucose-lowering potency of 5 at 0.1 mg/kg was found to be similar to that of TAK-875. Single oral doses of 5 robustly lowered the blood glucose excursion during an oral glucose tolerance test in a dose-proportional manner from 0.01 to 30 mg/kg when the compound was administered 1 h before the oral glucose challenge, in nSTZ Wistar rats. It should be noted that compound 5 exhibited a significant blood glucose-lowering effect at a much lower dose with an ED50 = 0.8 mg/kg and ED90 = 3.1 mg/kg. These results can be at least in part rationalized by the good oral exposure of 5. Moreover, 5 also showed around 14% AUC-glucose reduction in db/db mice model at 10 mg/kg dose (Supporting Information). It is interesting to note that TAK-875 does not show any measurable decrease in glucose in this mice model.
Figure 3.
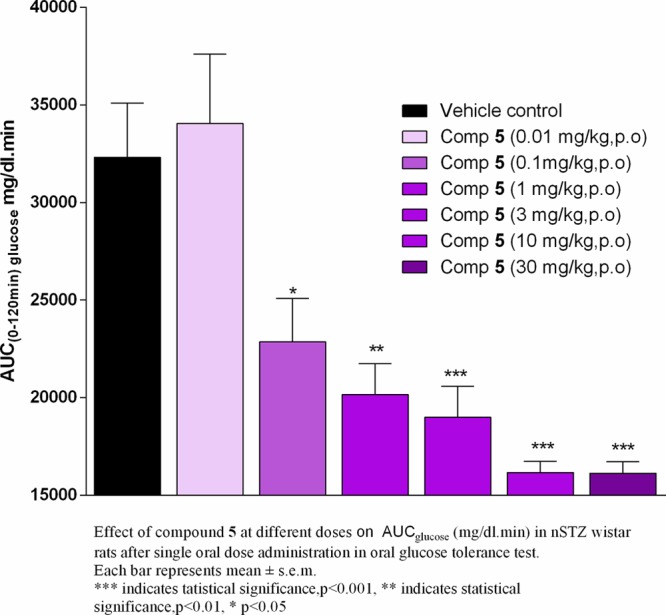
In vivo efficacy studies of 5 in nSTZ Wistar rats.
In conclusion, a series of novel 3-(4-aryloxyaryl)propanoic acid derivatives were prepared and evaluated for their activities as GPR40 agonists. Compound 5 was identified as a structurally distinct GPR40/FFAR1 agonist possessing potent activity in vitro, and excellent plasma glucose lowering efficacy in animal models of diabetes with desired pharmacokinetic profile.
Acknowledgments
Authors thank the management of Zydus Research Centre, Cadila Healthcare Ltd. for support and encouragement. ZRC Communication No. 496.
Glossary
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- CMC
carboxymethylcellulose
- NMP
N-methyl-2-pyrrolidone
- T2D
type II diabetes
- SAR
structure–activity relationship
- GSIS
glucose-stimulated insulin secretion
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00331.
Experimental procedures and analytical data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- IBD Diabetes Atlas, 6th ed.; International Diabetes Federation: Brussells, Belgium, 2013; http://www.diabetesatlas.org. [Google Scholar]
- Defronzo R. A. From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 2009, 58, 773–795. 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kles K. A.; Vinik A. I. Curr. Pathophysiology and treatment of diabetic peripheral neuropathy: the case for diabetic neurovascular function as an essential component. Curr. Diabetes Rev. 2006, 2, 131–145. 10.2174/157339906776818569. [DOI] [PubMed] [Google Scholar]
- Ashiya M.; Smith R. E. T. Non-insulin therapies for type 2 diabetes. Nat. Rev. Drug Discovery 2007, 6, 777–778. 10.1038/nrd2420. [DOI] [PubMed] [Google Scholar]
- Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat. Rev. Drug Discovery 2009, 8, 369. 10.1038/nrd2782. [DOI] [PubMed] [Google Scholar]
- Itoh Y.; Kawamata Y.; Harada M.; Kobayashi M.; Fujii R.; Fukusumi S.; Ogi K.; Hosoya M.; Tanaka Y.; Uejima H.; Tanaka H.; Maruyama M.; Satoh R.; Okubo S.; Kizawa H.; Komatsu H.; Matsumura F.; Noguchi Y.; Shinohara T.; Hinuma S.; Fujisawa Y.; Fujino M. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- Tan C. P.; Feng Y.; Zhou Y. P.; Eiermann G. J.; Petrov A.; Zhou C.; Lin S.; Salituro G.; Meinke P.; Mosley R.; Akiyama T. E.; Einstein M.; Kumar S.; Berger J. P.; Mills S. G.; Thornberry N. A.; Yang L.; Howard A. D. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes 2008, 57, 2211–2219. 10.2337/db08-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayasam G. V.; Tulasi V. K.; Davis J. A.; Bansal V. S. Fatty acid receptors as new therapeutic targets for diabetes. Expert Opin. Ther. Targets 2007, 11, 661–671. 10.1517/14728222.11.5.661. [DOI] [PubMed] [Google Scholar]
- Defossa E.; Wagner M. Recent developments in the discovery of FFA1 receptor agonists as novel oral treatment for type 2 diabetes mellitus. Bioorg. Med. Chem. Lett. 2014, 24, 2991–3000. 10.1016/j.bmcl.2014.05.019. [DOI] [PubMed] [Google Scholar]
- Negoro N.; Sasaki S.; Mikami S.; Ito M.; Suzuki M.; Tsujihata Y.; Ito R.; Harada A.; Takeuchi K.; Suzuki N.; Miyazaki J.; Santou T.; Odani T.; Kanzaki N.; Funami M.; Tanaka T.; Kogame A.; Matsunaga S.; Yasuma T.; Momose Y. Discovery of TAK-875: A Potent, Selective, and Orally Bioavailable GPR40 Agonist. ACS Med. Chem. Lett. 2010, 1 (6), 290–294. 10.1021/ml1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen E.; Hansen S. V.; Urban C.; Hudson B. D.; Wargent E. T.; Grundmann M.; Jenkins L.; Zaibi M.; Stocker C. J.; Ullrich S.; Kostenis E.; Kassack M. U.; Milligan G.; Cawthorne M. A.; Ulven T. Discovery of TUG-770: A Highly Potent Free Fatty Acid Receptor 1 (FFA1/GPR40) Agonist for Treatment of Type 2 Diabetes. ACS Med. Chem. Lett. 2013, 4, 441–445. 10.1021/ml4000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houze J. B.; Zhu L.; Sun Y.; Akerman M.; Qiu W.; Zhang A. J.; Sharma R.; Schmitt M.; Wang Y.; Liu J.; Liu J.; Medina J. C.; Reagan J. D.; Luo J.; Tonn G.; Zhang J.; Lu J. Y. L.; Chen M.; Lopez E.; Nguyen K.; Yang L.; Tang L.; Tian H.; Shuttleworth S. J.; Lin D. C. -H. AMG 837: A potent, orally bioavailable GPR40 agonist. Bioorg. Med. Chem. Lett. 2012, 22, 1267–1270. 10.1016/j.bmcl.2011.10.118. [DOI] [PubMed] [Google Scholar]
- Garrido D. M.; Corbett D. F.; Dwornik K. A.; Goetz A. S.; Littleton T. R.; McKeown S. C.; Mills W. Y.; Smalley T. L.; Briscoe C. P.; Peat A. J. Synthesis and activity of small molecule GPR40 agonists. Bioorg. Med. Chem. Lett. 2006, 16, 1840–1845. 10.1016/j.bmcl.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Tang C.; Chang E.; Ge M.; Lin S.; Cline E.; Tan C. P.; Feng Y.; Zhou Y. P.; Eiermann G. J.; Petrov A.; Salituro G.; Meinke P.; Mosley R.; Akiyama T. E.; Einstein M.; Kumar S.; Berger J.; Howard A. D.; Thornberry N.; Mills S. G.; Yang L. Discovery of 5-aryloxy-2,4-thiazolidinediones as potent GPR40 agonists. Bioorg. Med. Chem. Lett. 2007, 20, 1298–1301. 10.1016/j.bmcl.2009.10.052. [DOI] [PubMed] [Google Scholar]
- Hansen S. V. F.; Christiansen E.; Urban C.; Hudson B. D.; Stocker C. J.; Due-Hansen M. E.; Wargent E. T.; Shimpukade B.; Almeida R.; Ejsing C. S.; Cawthorne M. A.; Kassack M. U.; Milligan G.; Ulven T. Discovery of a Potent Free Fatty Acid 1 Receptor Agonist with Low Lipophilicity, Low Polar Surface Area, and Robust in Vivo Efficacy. J. Med. Chem. 2016, 59, 2841–2846. 10.1021/acs.jmedchem.5b01962. [DOI] [PubMed] [Google Scholar]
- Takano R.; Yoshida M.; Inoue M.; Honda T.; Nakashima R.; Matsumoto K.; Yano T.; Ogata T.; Watanabe N.; Hirouchi M.; Yoneyama T.; Ito S.; Toda N. Discovery of DS-1558: A Potent and Orally Bioavailable GPR40 Agonist. ACS Med. Chem. Lett. 2015, 6 (3), 266–270. 10.1021/ml500391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D. C.; Zhang J.; Zhuang R.; Li F.; Nguyen K.; Chen M.; Tran T.; Lopez E.; Lu J. Y. L.; Li X. N.; Tang L.; Tonn G. R.; Swaminath G.; Reagan J. D.; Chen J.-L.; Tian H.; Lin Y.-J.; Houze J. B.; Luo J. AMG 837: A novel GPR40/FFA1 agonist that enhances insulin secretion and lowers glucose levels in rodents. PLoS One 2011, 6, e27270. 10.1371/journal.pone.0027270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik H.; Vakilynejad M.; Wu J.; Viswanathan P.; Dote N.; Higuchi T.; Leifke E. Safety, tolerability, pharmacokinetics, and pharmacodynamic properties of the GPR40 agonist TAK-875: Results from a double-blind, placebo-controlled single oral dose rising study in healthy volunteers. J. Clin. Pharmacol. 2012, 52, 1007–1016. 10.1177/0091270011409230. [DOI] [PubMed] [Google Scholar]
- Kaku K.; Enya K.; Nakaya R.; Ohira T.; Matsuno R. Efficacy and safety of fasiglifam (TAK-875), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebocontrolled, phase III trial. Diabetes. Diabetes, Obes. Metab. 2015, 17, 675–681. 10.1111/dom.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S.; Patil A.; Aware U.; Deshmukh P.; Darji B.; Sasane S.; Sairam K. V. V. M; Priyadarsiny P.; Giri P.; Patel H.; Giri S.; Jain M.; Desai R. C. Discovery of a Potent and Orally Efficacious TGR5 Receptor Agonist. ACS Med. Chem. Lett. 2016, 7 (1), 51–55. 10.1021/acsmedchemlett.5b00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Wang Y.; Ma Z.; Schmitt M.; Zhu L.; Brown S. P.; Dransfield P. J.; Sun Y.; Sharma R.; Guo Q.; Zhuang R.; Zhang J.; Luo J.; Tonn G. R.; Wong S.; Swaminath G.; Medina J. C.; Lin D. C.-H.; Houze J. B. Optimization of GPR40 Agonists for Type 2 Diabetes. ACS Med. Chem. Lett. 2014, 5, 517–521. 10.1021/ml400501x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring M. J.; Arrowsmith J.; Leach A. R.; Leeson P. D.; Mandrell S.; Owen R. M.; Pairaudeau G.; Pennie W. D.; Pickett S. D.; Wang J.; Wallace O.; Weir A. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discovery 2015, 14, 475–486. 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Kumar G. N.; Surapaneni S. Role of drug metabolism in drug discovery and development. Med. Res. Rev. 2001, 21 (5), 397. 10.1002/med.1016. [DOI] [PubMed] [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures. J. Org. Chem. 1996, 61 (11), 3849–3862. 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- Walker S. D.; Borths C. J.; DiVirgilio E.; Huang L.; Liu P.; Morrison H.; Sugi K.; Tanaka M.; Woo J. C. S.; Faul M. M. Development of a Scalable Synthesis of a GPR40 Receptor Agonist. Org. Process Res. Dev. 2011, 15, 570–580. 10.1021/op1003055. [DOI] [Google Scholar]
- Spectral data of calcium (S)-3-(4-((3-((isopropyl(thiophen-3-ylmethyl)amino)methyl) benzyl)oxy) phenyl) hex-4-ynoate (5): White powder; mp: 124.5 °C. IR (KBr): ν = 3435, 2960, 2918, 2868, 2818, 1608, 1550, 1508, 1440, 1383, 1359, 1240 cm–1. 1H NMR (400 MHz, DMSO-d6): δ = 7.43–7.42 (m, 2H), 7.28–7.24 (m, 6H), 7.04 (d, J = 4.4 Hz, 1H), 6.89 (d, J = 8.4 Hz, 2H), 5.02 (s, 2H), 4.02 (s, 1H), 3.50 (d, J = 7.2 Hz, 4H), 2.84–2.77 (sept, J = 6.4 Hz, 1H), 2.43 (dd, J1 = 6.8 Hz, J2 = 7.2 Hz, 1H), 2.28 (dd, J1 = 6.8 Hz, J2 = 7.2 Hz, 1H), 1.73 (s, 3H), 0.99 (d, J = 6.4 Hz, 6H). 13C NMR and DEPT (100 MHz, DMSO-d6): δ = 177.78 (C), 157.23 (C), 142.11 (C), 141.4 (C), 137.46 (C), 135.81 (C), 128.83 (CH), 128.62 (CH), 128.40 (CH), 127.94 (CH), 127.69 (CH), 126.26 (CH), 122.18 (CH), 114.77 (CH), 83.18 (C), 77.32 (C), 69.66 (CH2), 52.89 (CH2), 48.59 (CH), 48.48 (CH2), 46.86 (CH2), 33.52 (CH), 17.88 (CH3), 3.78 (CH3). MS (EI): m/z (%) = 462.05 (100) (M+H)+. HPLC (% Purity) = 99.38%; Calcium Content (C56H60CaN2O6S2) Calcd.: 4.17%. Found: 3.99%.
- Shafrir E. Animal models of non insulin dependent diabetes. Diabetes/Metab. Rev. 1992, 8, 179–208. 10.1002/dmr.5610080302. [DOI] [PubMed] [Google Scholar]
- Ito R.; Tsujihata Y.; Suzuki M.; Miyawaki K.; Matsuda K.; Takeuchi K. Fasiglifam/TAK-875, a Selective GPR40 Agonist, Improves Hyperglycemia in Rats Unresponsive to Sulfonylureas and Acts Additively with Sulfonylureas. J. Pharmacol. Exp. Ther. 2016, 357 (1), 217–27. 10.1124/jpet.115.230730. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.