

Abstract
Spectroscopic studies have identified a number of proteins that appear to retain significant residual structure under even strongly denaturing conditions. Intrinsic viscosity, hydrodynamic radii, and small-angle x-ray scattering studies, in contrast, indicate that the dimensions of most chemically denatured proteins scale with polypeptide length by means of the power-law relationship expected for random-coil behavior. Here we further explore this discrepancy by expanding the length range of characterized denatured-state radii of gyration (RG) and by reexamining proteins that reportedly do not fit the expected dimensional scaling. We find that only 2 of 28 crosslink-free, prosthetic-group-free, chemically denatured polypeptides deviate significantly from a power-law relationship with polymer length. The RG of the remaining 26 polypeptides, which range from 16 to 549 residues, are well fitted (r2 = 0.988) by a power-law relationship with a best-fit exponent, 0.598 ± 0.028, coinciding closely with the 0.588 predicted for an excluded volume random coil. Therefore, it appears that the mean dimensions of the large majority of chemically denatured proteins are effectively indistinguishable from the mean dimensions of a random-coil ensemble.
Numerous and compelling reports of residual structure in highly denatured proteins have emerged in recent years. For example, NMR studies suggest that significant secondary structure and long-range hydrophobic clusters persist in unfolded proteins even at high concentrations of urea or guanidine hydrochloride (GuHCl) (1–4). Similarly, recent reports have suggested that even the most highly denatured proteins exhibit residual long-range order similar to the native topology (5). This residual structure in the denatured state is widely thought to play a significant role in folding thermodynamics and kinetics (6), and, thus, a better understanding of its magnitude may be key to our understanding of the folding process.
Variations in the population of residual structure in the denatured state will lead to deviations from ideal random-coil behavior when the dimensions of a number of proteins are compared. A hallmark of random-coil behavior is a power-law relationship between polymer length and ensemble average radius of gyration (RG)
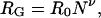 |
[1] |
where N is the number of monomers in the polymer chain, R0 is a constant that is a function of, among other things, the persistence length of the polymer, and ν is an exponential scaling factor. For an ideal (infinitely thin) random-coil chain in a “good” solvent, ν = ½. For an excluded-volume polymer (i.e., a real polymer with nonzero thickness and nontrivial interactions between monomers), Flory (7) has estimated that ν expands to approximately three-fifths, and more precise follow-on estimates (8) stemming from renormalization group models indicate that ν = 0.588. The formation of persistent denatured-state structure, however, should lead to perturbations from ideal random-coil behavior. For example, the formation of hydrophobically stabilized clusters will reduce the excluded-volume effect, producing a smaller RG than that of a polymer lacking such interactions. Similarly, local structure could increase (or decrease) a polymer's mean persistence length, leading to an increase (or decrease) in R0 and the dimensions of the denatured state relative to states lacking such structure. Differences in the magnitude of the residual denatured state structure from one unfolded protein to another would thus be expected to produce significant scatter around any underlying power-law relationship across a diverse set of unfolded proteins.
Although proteins unfolded in water by means of mutation under pressure or at low pH are sometimes relatively compact (9–11), the dimensions of most urea- or GuHCl-denatured proteins obey the theoretically expected random-coil scaling. Studies of this issue date from the 1960s, when Tanford et al. (12) used intrinsic viscosity measurements to determine that, for a set of 12 proteins unfolded in 5–6 M GuHCl, ν = 0.67 ± 0.09 (95% confidence interval). More recent and direct studies have shown that the hydrodynamic radii of sets of 8 and 38 highly denatured, disulfide-free proteins fit power-law relationships with ν = 0.57 ± 0.05 and ≈0.64, respectively, and that the RG of 11 chemically denatured proteins fit a power law with an exponent of 0.58 ± 0.25 (13, 14). We have recently reviewed the small-angle x-ray scattering (SAXS) and small-angle neutron scattering literature and amassed a set of 19 RG of chemically, thermally, or intrinsically unfolded, putatively disulfide-free proteins spanning the range of 52 to 416 residues (11). The RG of approximately two-thirds of these proteins fall roughly on a single power-law curve with an exponent, 0.61 ± 0.06, also within error of the value predicted for excluded volume random-coil behavior. The dimensions of approximately one-third of the proteins appear, however, to represent experimentally significant deviations from this power-law relationship, suggesting that R0 (and perhaps the scaling exponent) can vary widely from protein to protein. Here we critically reevaluate the RG of several previously characterized proteins, including the most significant outliers described in earlier work; tabulate 11 values from the literature; and report SAXS-derived RG for 12 additional chemically denatured proteins and peptides. This study of 28 chemically denatured, prosthetic-group-free and crosslink-free proteins allows us to more exhaustively characterize the extent to which putative residual denatured state structure affects the random-coil-like dimensional scaling of chemically denatured proteins.
Methods
We have characterized 17 proteins and peptides under strongly denaturing conditions (Table 1). Fyn SH3, pI3K SH2, and pI3K SH3 were prepared as described in refs. 15–17. Common-type acylphosphatase was expressed from a synthetic gene cloned into the pRSET B plasmid (Invitrogen) and purified as described in ref. 18. A 66-residue, amino-terminally cystine-linked coiled coil derived from the leucine zipper region of the protein GCN4 was synthesized and purified by analogy to Choma et al. (19). Staphylococcal nuclease (Snase), α subunit of tryptophan synthase (α-TS), and outer surface protein A (OspA) were gifts of D. Shortle (The Johns Hopkins University, Baltimore), O. Bilsel (University of Massachusetts Medical School, Worcester), C. R. Matthews (University of Massachusetts Medical School), and S. Koide (University of Chicago, Chicago) (6, 20). Ubiquitin was expressed from the wild-type human expression vector pRSUB (Entrez gi576323), which was mutated (F45W), expressed, and purified as described in ref. 21. Reduced RNase A and creatine kinase of the highest purity available were obtained commercially (Sigma) and used without further purification. Excluding reduced RNase A and the GCN4-p2′ dimer, the cysteine-containing samples were treated with 20 mM iodoacetamide/10 mM tris(2-carboxyethyl)phosphine hydrochloride for 2 h at room temperature before exhaustive dialysis against 10 mM ammonium bicarbonate and lyophilization. The peptides were angiotensin II [sequence DRVYIHPF (Sigma); used without further purification after identity and purity were checked by mass spectroscopy], AK-16 [sequence YGCAKAAAAKACAAKA (United Biochemical Research, Seattle); chemically synthesized and HPLC-purified], a 39-residue fragment of cytochrome c [sequence MIFFMVMPIMIGGFGNWLVPLMIGA PDMAFPRMNNNSFWL (a gift of P. Dawson, The Scripps Research Institute, La Jolla, CA)], and three synthetic peptides of sequence (AAKAA)nGY (acetyl- and amido-terminally capped) where n = 5, 6, and 7.
Table 1. Chemically denatured crosslink-free and prosthetic-group-free proteins.
| Protein | Length | RG,* Å | Conditions | Ref. |
|---|---|---|---|---|
| GroEL | 549 | 82 ± 4 | 4 M urea | 56 |
| yPGK | 416 | 71 ± 1 | 2 M GuHCl | 57 |
| Creatine kinase | 380 | 46.0 ± 1.5 | 6 M GuHCl | This work |
| α-TS | 268 | 48.8 ± 1.0 | 6 M GuHCl | This work |
| Carbonic anhydrase | 260 | 59 ± ≈2 | 6 M GuHCl | 28† |
| OspA | 257 | 49.3 ± 1.9 | 6 M GuHCl | This work |
| DHFR | 167 | 44 ± 2 | 8 M urea | 54 |
| Apomyoglobin | 154 | 40 ± ≈2 | 6 M GuHCl | 28† |
| Snase | 149 | 37.2 ± 1.2 | 6 M GuHCl | This work |
| Lysozyme, reduced | 129 | 35.8 ± 0.5 | 4 M GuHCl | 53 |
| CheY | 129 | 38.0 ± 1.0 | 5-7 M urea | 2 |
| RNase A, reduced | 124 | 33.2 ± 1.0 | 3.25-6 M GuHCl | This work |
| pI3K SH2 | 112 | 29.6 ± 3.3 | 3 M GuHCl | This work |
| pI3K SH3 | 103 | 30.9 ± 0.3 | 2.67 M GuHCl | This work |
| mAcP | 98 | 30.4 ± 1.3 | 6.5-8 M urea | 27 |
| ctACP | 98 | 30.5 ± 0.4 | 5.5-6.8 M urea | This work |
| Protein L | 79 | 26.0 ± 0.6 | 4-5 M GuHCl | 37 |
| Fyn SH3 | 78 | 25.7 ± 0.5 | 6 M GuHCl | This work |
| Ubiquitin | 76 | 25.2 ± 0.2 | 4.9-6 M GuHCl | This work |
| GCN4-p2′ | 66 | 24.1 ± 0.9 | 4.2-6 M GuHCl | This work |
| drK SH3 | 59 | 21.9 ± 0.5 | 2 M GuHCl | 38 |
| Protein G | 52 | 23 ± 1 | 2.3 M GuHCl | 58 |
| N-terminal cytochrome c | 39 | 18.4 ± 1.0 | 4 M urea | This work |
| AK-37 | 37 | 16.9 ± 0.6 | 6 M GuHCl | This work |
| AK-32 | 32 | 14.5 ± 0.6 | 6 M GuHCl | This work |
| AK-27 | 27 | 12.8 ± 0.6 | 6 M GuHCl | This work |
| AK-16 | 16 | 9.8 ± 0.6 | 4 M urea | This work |
| Angiotensin | 8 | 9.1 ± 0.3 | 4 M urea | This work |
Measured or estimated sample standard deviations are indicated. These were derived by using a variety of approaches and widely varying numbers of observations and therefore provide, at best, only a qualitative indicator of experimental precision.
Solvent conditions and approximate sample standard deviations (G. Semisotnov, personal communication).
Lyophilized Fyn SH3, α-TS, creatine kinase, Snase, and OspA were dissolved to concentrations of 10 mg/ml in 6 M GuHCl, 10 mM EDTA, and 20 mM Tris (pH 7) and were equilibrated for >24 h at room temperature before addition of the radical scavenger N-tert-butyl-α-(4-pyridyl)nitrone N′-oxide (5 mM) (Fluka) and subsequent measurements.
The remaining samples were treated as follows. The pI3K SH2 and pI3K SH3 domains (10 mg/ml) were dissolved in 20 mM Tris (pH 7) and 2.67 or 3 M GuHCl, respectively, well into their optical denaturation baselines (15) (data not shown). Reduced RNase A was dissolved in 6 M GuHCl and 50 mM Tris (pH 8) at a concentration of ≈40 mg/ml. After ≈30 min, a 30-fold excess of tris(2-carboxyethyl)phosphine hydrochloride was added. After an additional 30 min, the pH was reduced to 2.5 to inhibit reoxidation. This sample was diluted with GuHCl solutions to a final protein concentration of 2 mg/ml and 10 GuHCl concentrations ranging from 3.25 to 6 M in ≈30-fold excess tris(2-carboxyethyl)phosphine hydrochloride and 50 mM Tris (pH 2.5). Ubiquitin (2 mg/ml in 50 mM acetate, pH 5) was equilibrated for ≈1 h before measurements at four GuHCl concentrations ranging from 4.9 to 6 M. Common-type acylphosphatase (2 mg/ml in 50 mM acetate, pH 5.5) was equilibrated for ≈6 h at four urea concentrations ranging from 5.5 to 6.8 M. GCN4-p2′ (2 mg/ml in 50 mM Hepes, pH 7) was unfolded in 4.2, 5, and 6 M GuHCl. The peptides AK-27, AK-32, and AK-37 were equilibrated at 20, 15, and 14 mg/ml, respectively, for 30 min at room temperature in 6 M GuHCl, 1 M NaCl, 3 mM potassium phosphate, and 5 mM tris(2-carboxyethyl)phosphine hydrochloride at a final pH of 4. The remaining peptides (10 mg/ml in 20 mM Tris, pH 7) were dissolved in 4 M urea and 5 mM N-tert-butyl-α-(4-pyridyl)nitrone N′-oxide immediately before measurements.
Scattering Experiments. SAXS experiments were conducted on the BioCAT and BESSRC-CAT beamlines at the Advanced Photon Source and on beamline 4-2 at the Stanford Synchrotron Radiation Laboratory (Stanford University) (Table 2). AK-27, AK-32, and AK-37 were studied at 10 ± 1°C. All other SAXS experiments were conducted at 25 ± 1°C. RG were determined by using the Guinier analysis (22). Forward scattering at zero angle (I0) was monitored in all experiments; no indication of aggregation was observed (data not shown) (23).
Table 2. Experimental conditions.
| Protein/peptide | Cell/beam-line |
|---|---|
| Reduced RNase | Flow/APS/BioCAT |
| Snase | Flow/APS/BESSRC-CAT |
| α-TS | Static/SSRL/4-2 |
| Creatine kinase | Flow/APS/BESSRC-CAT |
| GCN4-p2′ | Flow/APS/BioCAT |
| OspA | Flow/SSRL/4-2 |
| ctAcP | Flow/APS/BioCAT |
| Ubiquitin | Flow/APS/BioCAT |
| Fyn SH3 | Flow/APS/BESSRC-CAT |
| pI3K SH3 | Static/SSRL/4-2 |
| pI3K SH2 | Static/SSRL/4-2 |
| N-terminal cytochrome c | Flow/APS/BESSRC-CAT |
| AK-37 | Flow/APS/BESSRC-CAT |
| AK-32 | Flow/APS/BESSRC-CAT |
| AK-27 | Flow/APS/BESSRC-CAT |
| AK-16 | Flow/APS/BESSRC-CAT |
| Angiotensin | Flow/APS/BESSRC-CAT |
APS, Advanced Photon Source; SSRL, Stanford Synchrotron Radiation Laboratory.
RG Adopted from the Literature. The SAXS-determined RG of 16 disulfide-free, denatured proteins are available from the literature (11, 24–26). Of these, the prosthetic group containing cytochrome c and five highly charged, intrinsically unfolded proteins (unfolded in water) were excluded from our data set (see below). For the few proteins for which multiple denatured-state RG determinations have been reported by SAXS, we adopted data by using the following criteria (in order of preference): data collected over a range of denaturant concentrations (see Table 1) under a single set of conditions and fitted to determine the average unfolded RG value (27) or data collected at the highest GuHCl or urea concentration reported to date (28).
Error Analysis. Reported RG and their estimated confidence intervals represent averages and sample standard deviations derived from at least two independent measurements, except as noted below. The confidence intervals for OspA, Fyn SH3, and the amino-terminal peptide of cytochrome c and the AK-27, AK-32, and AK-37 peptides reflect estimated sample standard deviations for the fitted parameters based on a single experiment. The RG and sample standard deviations for RNase, ubiquitin, common-type acylphosphatase, and GCN4-p2′ reflect the average and estimated standard deviation of measurements taken at 3–10 independent GuHCl concentrations. Estimated sample standard deviations for the data adopted from the literature were either taken as reported (26), or by means of personal communication (G. Semisotnov, Institute for Protein Research, Pushchino, Russia).
Estimates for the parameters in the power-law relationship were obtained by using least-squares regression on the log-transformed data, i.e., log(RG) = log(R0) + νlog(N). For point predictions made by using the fitted linear model, two sources of variability must be taken into account: the variability in the parameter estimated and the naturally occurring variability, i.e., the fact that an actual observation does not precisely equal its predicted value. Closed-form solutions of the predictive intervals (indicated in gray in Fig. 1) exist, assuming a normal distribution for the errors (29). Therefore, data points that have not been used in the model fitting and that fall outside the gray region can be declared outliers at the 95% confidence level used in the calculations for the predictive intervals. This method is primarily used for visualization of the confidence intervals. We have thus also carried out formal hypothesis tests (based on the t distribution) by using the aforementioned formulas.
Fig. 1.

The RG of the large majority of chemically denatured proteins scale with polymer length, N, by means of the power-law relationship RG = R0Nν. Two statistically significant outliers, creatine kinase and angiotensin II, are indicated. The solid line, which is the least-squares fit ignoring the two potential outliers, produces an exponent, ν = 0.598 ± 0.028 (95% confidence interval), that is indistinguishable from the 0.588 predicted for an excluded-volume random coil. The shaded region represents the 95% confidence intervals for future measurements, assuming that the errors about (log)RG are normally distributed around the fitted relationship. Only the measurements for creatine kinase and angiotensin II fall outside this predictive interval, and, thus, only these measurements can be said to represent unambiguously significant deviations. Error bars indicate the reported experimental (i.e., standard) deviations of the sample. These were derived by using a variety of approaches and widely varying numbers of observations and therefore provide only an approximate indication of experimental precision.
Results
We have collected the RG of 28 chemically denatured, crosslink- and prosthetic-group-free proteins and peptides (Table 1). In preparing this data set we have critically surveyed the literature, measured the RG of 12 new denatured proteins and peptides, and experimentally reevaluated five previously characterized proteins, including the four most statistically significant outliers from the best-fit power law (11).
Denaturation Conditions. Theory suggests that the dimensions of an excluded-volume random-coil polymer will change as the denaturing ability of the solvent improves (30). However, empirical observations suggest that once a protein is unfolded (i.e., at denaturant concentrations well past the denaturation transition midpoint), further increases in solvent quality do not measurably increase RG (11). We have confirmed this observation by monitoring the RG of ubiquitin, reduced RNase A, common-type acylphosphatase, and GCN4-p2′ over wide ranges of denaturant concentration (23). We observe no statistically significant variation in RG for any of these proteins as either urea or GuHCl concentration is increased beyond the end of the unfolding transition (as illustrated, for example, by the small sample standard deviations reported for RG averaged over these ranges; Table 1). Similarly, previous reports indicate that the RG of denatured states produced by high levels of urea are indistinguishable from those produced by high levels of GuHCl (11). Thus, we can reasonably compare RG collected with either denaturant and at any denaturant concentration beyond the end of the unfolding transition.
Redefined RG. We have redetermined the RG of five previously characterized, chemically unfolded proteins. For ubiquitin, we observe an RG that is within error of the previously reported 26.0 ± 1.2 Å (27). The remaining proteins are Snase (31, 32), reduced RNase A (33), creatine kinase (34), and α-TS (35). The RG of reduced, chemically unfolded RNase was previously reported to be significantly more compact than expected for a random coil (11, 33). This is, however, apparently an artifact due to incomplete reduction; measuring the dimensions of RNase in the presence of a strong reductant, we observe that RG remains effectively constant at 33.2 ± 1.0 Å above 3.25 M GuHCl, a value well within error of the expected random-coil dimensions. Similarly, at ≈35 Å and 33 ± 2 Å, the previously reported RG values for Snase denatured in 5 and 8 M urea, respectively, (31, 32) are significantly more compact than the 38.3 Å predicted by the best-fit power law. We find that in 6 M GuHCl, however, this value increases to 37.2 ± 2.4 Å, within error of the expected random-coil value. Gualfetti et al. (35) have previously reported that the RG of α-TS is 34 ± 4 Å in 4–6 M urea. We find that in 6 M GuHCl this value expands to 48.8 ± 1.0 Å, consistent with random-coil predictions. This observation is also consistent with NMR-based reports that at concentrations above 6 M urea the protein undergoes an additional cooperative transition (36). In contrast, we observe an RG of 46 ± 1.5 Å for creatine kinase in 6 M GuHCl, which is within error of previous reports (34) but significantly less than the 67 Å predicted for a 380-residue random coil. It thus appears that, whereas the creatine kinase denatured state in 6 M GuHCl is significantly more compact than an unperturbed random coil, the other previously reported outliers probably reflect the use of less denaturing conditions than those used here.
New RG Measurements. We have also determined the RG of 12 additional chemically denatured polypeptides (Table 1 and Fig. 1). We find that all of the denatured proteins and five of six denatured peptides exhibit RG within error of the best-fit power law. Only the RG of the smallest peptide, the eight-residue angiotensin II, deviates significantly from the expected random-coil value (9.1 ± 0.6 Å observed versus 6.7 Å expected).
Previously Determined RG. We have previously reported the RG of four chemically denatured, disulfide- and prosthetic-group-free proteins (27, 37, 38) (Table 1). SAXS measurements of the chemically denatured-state RG of eight additional disulfide- and prosthetic-group-free proteins have been reported by other groups (Table 1). All 12 RG fit the expected dimensional scaling (Fig. 1).
Random-Coil Dimensional Scaling. Do the dimensions of chemically unfolded proteins differ significantly from the expectations of a random-coil model? We find no evidence that this is true when an ordinary least-squares regression is applied to the data excluding the candidate outliers angiotensis II (eight residues) and creatine kinase (380 residues). For this regression, the log10 of RG was used as the dependent variable, and the log10 of the polypeptide length (in residues) was used as the independent variable. The least-squares estimate for the parameters of this relationship yields R0 = 1.330 [95% confidence intervals: (1.256, 1.408)] and ν = 0.598 ± 0.028 (95% confidence intervals). The latter is clearly consistent with the theoretically predicted value of 0.588. We note, however, that this analysis makes the erroneous assumption that all data points are equally precisely measured. In an attempt to correct this, we have also performed a weighted least-squares regression by using the inverse of the reported sample standard deviations as case weights. The result of this weighted fitting (r2 = 0.989), ν = 0.605 ± 0.027, is effectively identical to that obtained from the unweighted fit.
Only creatine kinase and angiotensin II fall outside of the calculated 95% confidence interval. To determine whether these measurements reflect statistically significant deviations from the theoretically predicted relationship, we calculated 95% predictive intervals for future observations, i.e., prediction intervals for RG given any arbitrary N (Fig. 1, gray). The RG of both proteins fall well outside this region, strongly suggesting that they do not obey the expected power-law relationship. Formal hypothesis testing, assuming the data points were observations stemming from the model, yields P values (the probability that an outlier of equal or greater magnitude would be observed by chance) of 10–6 and 7 × 10–5. Thus, the RG of only 2 of 28 chemically denatured, crosslink-free, prosthetic-group-free peptides and proteins deviate significantly from random-coil behavior.
Proteins Excluded from the Data Set. We have excluded from consideration the RG of six previously characterized, crosslink-free, unfolded proteins. One is the prosthetic group containing cytochrome c, which, despite containing a very electron-dense iron (leading to an anomalous reduction in RG), falls quite close to the best-fit line (27) (observed, 30.2 ± 0.2 Å; expected, 30.9 Å). We have also excluded the intrinsically unfolded proteins prothymosin-α, the carboxyl-terminal domain of caldesmon, α-synuclein, β-synuclein, and γ-synuclein (39, 24, 25). In water, all five are expanded relative to the chemically denatured states of normally well folded proteins (observed/expected RG: 37.8 ± 0.9/32.0, 40.8 ± 0.8/36.1, 40 ± 1/36.9, 49 ± 1/35.8, and 61 ± 1/34.7 Å, respectively). Notably, however, all five of these proteins are unusually highly charged at pH 7; prothymosin-α, for example, is composed of 49% aspartate or glutamate residues. Because of their anomalously high charges, the physics of these proteins may differ significantly from that of the chemically denatured states of more typical, globular proteins.
Discussion
Twenty-six of the 28 noncrosslinked, prosthetic-group-free, chemically denatured polypeptides examined to date exhibit the dimensional scaling expected for a set of excluded-volume random coils. Only the eight-residue angiotensin II and the 380-residue creatine kinase represent unambiguously significant outliers from this relationship.
The presence of only two outliers suggests that, although the dimensions of the large majority of chemically denatured polypeptides are indistinguishable from random coil, a small minority of chemically denatured polypeptides exhibit residual structure of sufficient magnitude to measurably perturb R0 and thus produce significant deviations from random-coil dimensions. For the eight-residue peptide angiotensin II, the only member of the data set exhibiting a significantly larger than expected RG, the discrepancy may reflect the persistence length of the polypeptide chain; chain stiffness will cause a very short polymer to expand relative to the dimensions predicted by naively extrapolating the behavior of longer polymers (7). Creatine kinase, in contrast, is alone significantly more compact than expected for a random-coil ensemble. We presume that this compaction reflects residual structure that is persistent even in 6 M GuHCl. We note, however, that whereas numerous optical probes suggest that the chemically unfolded state of creatine kinase exhibits residual structure at low denaturant concentrations, at high denaturant concentrations the spectroscopic signals indicative of both secondary and tertiary structure are lost, and the protein produces a Kratky scattering profile consistent with a random-coil ensemble (34, 40). Similarly, although the RG of GuHCl-denatured creatine kinase is that of a polymer half its length, the protein migrates in SDS/PAGE as expected for an unfolded, 380-residue chain (J.E.K. and K.W.P., unpublished data). The origins of this significant compaction, and the reason only 1 of 28 polypeptides might exhibit it, thus, remain open to question.
Although the observation of random-coil dimensional scaling across the large majority of chemically denatured proteins is consistent with the results of previous scattering studies, it is seemingly inconsistent with compelling spectroscopic studies that suggest that many denatured proteins populate significant residual structure. For example, proteins unfolded at high concentrations of denaturant produce Kratky scattering profiles exhibiting the monotonic increase indicative of an expanded, coil-like conformation (11), and the x-ray and neutron scattering profiles of denatured yeast phosphoglycerate kinase and neocarzinostatin closely match those calculated for excluded-volume random coils (41, 42). The observation of random-coil dimensional scaling is seemingly inconsistent, however, with previous spectroscopic studies, which indicate significant residual structure for even highly denatured proteins. NMR dipolar-coupling analysis of gel-aligned Snase suggests, for example, that the unfolded protein retains a native-like topology even in 8 M urea (5, 43), an observation that has recently been expanded to other polypeptides (44, 45) (D. Shortle, personal communication). And although dihydrofolate reductase and reduced lysozyme fall on the best-fit power law (Fig. 1), NMR relaxation studies and the observation of residual, native-like 1H-15N heteronuclear single quantum correlation (HSQC) peaks suggest that the chemically denatured states of both proteins contain significant, sequence-distant hydrophobic clusters (3, 4). Even in the absence of long-range order or sequence-distant hydrophobic interactions, NMR coupling experiments and molecular dynamics simulations suggest that the main-chain torsion angles of small, unfolded peptides populate values similar to those observed in native proteins (46, 47). Consistent with these results, simulations of polypeptides suggest that excluded-volume effects extend beyond sequential residue pairs and may thus significantly constrain the overall structure of the chemically denatured ensemble (48, 49). Thus, there appears to be a potentially serious discrepancy between simulations and spectroscopy, which indicate the denatured state is relatively ordered, and scattering studies that, for the large majority of proteins, produce no significant deviations from random-coil behavior.
Reconciliation of short-range, sequence-local order with near random-coil behavior may lie in the observation that R0 can be surprisingly independent of the detailed, sequence-local structure of the unfolded chain (11, 50, 51). As an extreme example, in the accompanying paper Fitzkee and Rose (52) report that random-coil-like RG may be observed even if the large majority of the polypeptide chain is participating in native-like local structure. This occurs despite the two-fifths-power dependence of R0 on persistence length (7), which seems likely to vary with local structure content. This, in turn, suggests that the formation of local structure involves compensatory changes in both the persistence length and excluded-volume terms that contribute to R0 such that it and, thus, RG are effectively independent of such structure.
It is perhaps more difficult to reconcile random-coil dimensional scaling with spectroscopic evidence for sequence-distant interactions in even the most highly denatured proteins, because the formation of such structure should significantly reduce RG. For example, the RG of chemically denatured reduced Rnase A and lysozyme expand by 30% and 60%, respectively, upon reduction of their disulfide bonds (this work and refs. 33 and 53), and breaking a single (nonnatural) crosslink increases the RG of chemically denatured dihydrofolate reductase by 45% (54). We presume that the almost complete absence of similarly large deviations from ideal random-coil scaling in noncrosslinked, chemically denatured proteins indicates that the pinning together of sequence-distant residues in specific, well populated hydrophobic clusters is rare. More generally, Miller and Goebel (50) have calculated that the formation of “irregular knots of compact structure” can be consistent with random-coil-like dimensional scaling but only if the fraction of residues that participates in collapsed structure is the same in all proteins. Variation in the size of these putative compact structures, which seems likely given the widely varying sequences used here, would produce deviations from random-coil scaling that are of approximately the same magnitude as the fraction of residues involved in the clusters (50). In this light, our results suggest that, at any given instant, significantly less than 3% of the residues in a typical chemically denatured protein are participating in compact, nonregular structures.
A possible reconciliation between the widespread spectroscopic evidence for denatured-state structure and the relative paucity of significant deviations from random-coil dimensional scaling reported here lies in the observation that the relevant spectroscopic probes do not provide firm estimates of the population of such structure (C. Dobson and C. Frieden, personal communication). The extent to which SAXS and spectroscopy produce very differently weighted ensemble averages might also contribute; whereas scattering measurements produce a root mean squared average RG, thus slightly overweighing more expanded configurations, nuclear Overhauser effect signals scale inversely with the mean sixth power of distance, thus significantly overweighing potentially rare compact structures. Lastly, it has been suggested that residual denatured-state structure may reflect the observation that the “mean” structure of the unfolded state, at least under native conditions, can be native-like even if any single conformation contains few native-like structural elements (55). If this mean structure hypothesis holds for the chemically denatured state, the native-like spectral signals observed for chemically unfolded proteins may arise from an effectively random-coil ensemble in which native-like structure is rarely adopted but accurately approximates the ensemble average. The questions of whether this mean structure hypothesis holds for the chemically denatured state and whether a native-like conformational average can generate native-like spectroscopic signals without perturbing random-coil dimensional scaling remain unanswered.
Acknowledgments
Osman Bilsel, Philip Dawson, Shohei Koide, David Shortle, and Bob Matthews generously provided protein samples. We thank Ken Dill, Chris Dobson, Carl Freiden, Phil Pincus, and George Rose for critical commentary and clarifying discussions. This work was supported by the National Institutes of Health (K.W.P. and V.S.P.), the Packard Foundation Interdisciplinary Science Program (T.R.S. and P.T.), and a faculty innovation award from The Johns Hopkins University (to I.R.). The Stanford Synchrotron Radiation Laboratory is supported by the U.S. Department of Energy and the National Institutes of Health. The Advanced Photon Source is supported by the U.S. Department of Energy, Basic Energy Sciences, and the Office of Science, and BioCAT is a National Institutes of Health-supported research center.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GuHCl, guanidine hydrochloride; SAXS, small-angle x-ray scattering; Snase, staphylococcal nuclease; α-TS, α subunit of tryptophan synthase; OspA, outer surface protein A.
References
- 1.Kazmirski, S. L., Wong, K. B., Freund, S. M. V., Tan, Y. J., Fersht, A. R. & Daggett, V. (2001) Proc. Natl. Acad. Sci. USA 98, 4349–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia, P., Serrano, L., Durand, D., Rico, M. & Bruix, M. (2001) Protein Sci. 10, 1100–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodsdon, M. E. & Frieden, C. (2001) Biochemistry, 40, 732–742. [DOI] [PubMed] [Google Scholar]
- 4.Klein-Seetharaman, J., Oikawa, M., Grimshaw, S. B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L. J., Dobson, C. M. & Schwalbe, H. (2002) Science 295, 1719–1722. [DOI] [PubMed] [Google Scholar]
- 5.Shortle, D. & Ackerman, M. S. (2001) Science 293, 487–489. [DOI] [PubMed] [Google Scholar]
- 6.Shortle, D. (1996) FASEB J. 10, 27–34. [DOI] [PubMed] [Google Scholar]
- 7.Flory, P. J. (1953) in Principles of Polymer Chemistry (Cornell Univ. Press, Ithaca, NY).
- 8.LeGuillou, J. C. & Zinn-Justin, J. (1977) Phys. Rev. Lett. 39, 95–98. [Google Scholar]
- 9.Doniach, S. (2001) Chem. Rev. 101, 1763–1778. [DOI] [PubMed] [Google Scholar]
- 10.Woenckhaus, J., Kohling, R., Thiyagarajan, P., Littrell, K. C., Royer, C. A. & Winter, R. (2001) Biophys. J. 80, 1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millett, I., Doniach, S. & Plaxco, K. W. (2002) Adv. Protein Chem. 62, 241–262. [DOI] [PubMed] [Google Scholar]
- 12.Tanford, C., Kawahara, K. & Lapanjes, S. (1966) J. Biol. Chem. 241, 1921–1923. [PubMed] [Google Scholar]
- 13.Wilkins, D. K., Grimshaw, S. B., Receveur, V., Dobson, C. M., Jones, J. A. & Smith, L. J. (1999) Biochemistry 38, 16424–16431. [DOI] [PubMed] [Google Scholar]
- 14.Tcherkasskaya, O. & Uversky, V. N. (2001) Proteins Struct. Funct. Genet. 44, 244–254. [DOI] [PubMed] [Google Scholar]
- 15.Booker, G. W., Breeze, A. L., Downing, A. K., Panayotou, G., Gout, I., Waterfield, M. D. & Campbell, I. D. (1992) Nature 358, 684–687. [DOI] [PubMed] [Google Scholar]
- 16.Guijarro, J. I., Morton, C. J., Plaxco, K. W., Campbell, I. D. & Dobson, C. M. (1998) J. Mol. Biol. 276, 657–667. [DOI] [PubMed] [Google Scholar]
- 17.Plaxco, K. W., Guijarro, J. I., Morton, C. J., Pitkeathly, M., Campbell, I. D. & Dobson, C. M. (1998) Biochemistry 37, 2529–2537. [DOI] [PubMed] [Google Scholar]
- 18.Taddei, N., Chiti, F., Fiaschi, T., Bucciantini, M., Capanni, C., Stefani, M., Serrano, L., Dobson, C. M. & Ramponi, G. (2000) J. Mol. Biol. 300, 633–647. [DOI] [PubMed] [Google Scholar]
- 19.Choma, C. T., Lear, J. D., Nelson, M. J., Dutton, L. P., Robertson, D. E. & DeGrado, W. F. (1994) J. Am. Chem. Soc. 116, 856–865. [Google Scholar]
- 20.Koide, S., Bu, Z. M., Risal, D., Pham, T. N., Nakagawa, T., Tamura, A. & Engelman, D. M. (1999) Biochemistry 38, 4757–4767. [DOI] [PubMed] [Google Scholar]
- 21.Larsen, C. N., Krantz, B. A. & Wilkinson, K. D. (1998) Biochemistry 37, 3358–3368. [DOI] [PubMed] [Google Scholar]
- 22.Guinier, A. & Fournet, G. (1955) Small-Angle Scattering of X-Rays (Wiley, New York).
- 23.Jacob, J., Krantz, B., Dothager, R. S., Thiyagarajan, P. & Sosnick, T. R. (2004) J. Mol. Biol. 338, 369–382. [DOI] [PubMed] [Google Scholar]
- 24.Permyakov, S. E., Millett, I. S., Doniach, S., Permyakov, E. A. & Uversky, V. N. (2003) Protein Struct. Funct. Genet. 53, 855–862. [DOI] [PubMed] [Google Scholar]
- 25.Uversky, V. N., Li, J., Souillac, P., Millett, I. S., Doniach, S., Jakes, R., Goedert, M. & Fink, A. L. (2002) J. Biol. Chem. 277, 11970–11978. [DOI] [PubMed] [Google Scholar]
- 26.Arai, M., Inobe, T., Maki, K., Ikura, T., Kihara, H., Amemiya, Y. & Kuwajima, K. (2003) Protein Sci. 12, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millet, I. S., Townsley, L., Chiti, F., Doniach, S. & Plaxco, K. W. (2002) Biochemistry 41, 321–325. [DOI] [PubMed] [Google Scholar]
- 28.Semisotnov, G. V., Kihara, H., Kotova, N. V., Kimura, K., Amemiya, Y., Wakabayashi, K., Serdyuk, I. N., Timchenko, A. A., Chiba, K., Nikaido, K., et al. (1996) J. Mol. Biol. 262, 559–574. [DOI] [PubMed] [Google Scholar]
- 29.Weisberg, S. (1985) Applied Linear Regression (Wiley, New York), 2nd Ed.
- 30.Dill, K. A. & Shortle, D. (1991) Annu. Rev. Biochem. 60, 795–825. [DOI] [PubMed] [Google Scholar]
- 31.Flanagan, J. M., Kataoka, M., Shortle, D. & Engelman, D. M. (1992) Proc. Natl. Acad. Sci. USA 89, 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flanagan, J. M., Kataoka, M., Fujisawa, T. & Engelman, D. M. (1993) Biochemistry 32, 10359–10370. [DOI] [PubMed] [Google Scholar]
- 33.Sosnick, T. R. & Trewhella, J. (1992) Biochemistry 31, 8329–8335. [DOI] [PubMed] [Google Scholar]
- 34.Zhou, J. M., Fan, Y. X., Kihara, H., Kimura, K. & Amemiya, Y. (1997) FEBS Lett. 415, 183–185. [DOI] [PubMed] [Google Scholar]
- 35.Gualfetti, P. J., Iwakura, M., Lee, J. C., Kihara, H., Bilsel, O., Zitzewitz, J. A. & Matthews, C. R. (1999) Biochemistry, 38, 13367–13378. [DOI] [PubMed] [Google Scholar]
- 36.Zitzewitz, J. A., Gualfetti, P. J., Perkons, I. A., Wasta, S. A. & Matthews, C. R. (1999) Protein Sci. 8, 1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plaxco, K. W., Millett, I. S., Segel, D. J., Doniach, S. & Baker, D. (1999) Nat. Struct. Biol. 6, 554–556. [DOI] [PubMed] [Google Scholar]
- 38.Choy, W. Y., Mulder, F. A. A., Crowhurst, K. A., Muhandiram, D. R., Millett, I. S., Doniach, S., Forman-Kay, J. D. & Kay, L. E. (2002) J. Mol. Biol. 316, 101–112. [DOI] [PubMed] [Google Scholar]
- 39.Uversky, V. N., Gillespie, J. R., Millett, I. S., Khodyakova, A. V., Vasiliev, A. M., Chernovskaya, T. V., Vasilenko, R. N., Kozovskaya, G. D., Dolgikh, D. A., Fink, A. L., et al. (1999) Biochemistry 38, 15009–15016. [DOI] [PubMed] [Google Scholar]
- 40.Couthon, F., Clottes, E., Ebel, C. & Vial, C. (1995) Eur. J. Biochem. 234, 160–170. [DOI] [PubMed] [Google Scholar]
- 41.Calmettes, P., Durand, D., Desmadril, M., Minard, P., Receveur, V. & Smith, J. C. (1994) Biophys. Chem. 53, 105–114. [DOI] [PubMed] [Google Scholar]
- 42.Petrescu, A. J., Receveur, V., Calmettes, P., Durand, D. & Smith, J. C. (1998) Protein Sci. 7, 1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ackerman, M. S. & Shortle, D. (2002) Biochemistry 41, 3089–3095. [DOI] [PubMed] [Google Scholar]
- 44.Ackerman, M. S. & Shortle, D. (2002) Biochemistry 41, 13791–13797. [DOI] [PubMed] [Google Scholar]
- 45.Ohnishi, S. & Shortle, D. (2003) Protein Struct. Funct. Genet. 50, 546–551. [DOI] [PubMed] [Google Scholar]
- 46.Fiebig, K. M., Schwalbe, H., Buck, M., Smith, L. J. & Dobson, C. M. (1996) J. Am. Chem. Soc. 100, 2661–2666. [Google Scholar]
- 47.Peti, W., Hennig, M., Smith, L. J. & Schwalbe, H. (2000) J. Am. Chem. Soc. 122, 12017–12018. [Google Scholar]
- 48.Pappu, R. V., Srinivasan, R. & Rose, G. D. (2000) Proc. Natl. Acad. Sci. USA 97, 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Gunsteren, W. F., Burgi, P., Peter, C. & Daura, X. (2001) Angew. Chem. Int. Ed. 40, 351–355. [PubMed] [Google Scholar]
- 50.Miller, W. G. & Goebel, C. V. (1968) Biochemistry 7, 3925–3935. [DOI] [PubMed] [Google Scholar]
- 51.Segel, D. J., Fink, A. L., Hodgson, K. O. & Doniach, S. (1998) Biochemistry 37, 12443–12451. [DOI] [PubMed] [Google Scholar]
- 52.Fitzkee, N. C. & Rose, G. D. (2004) Proc. Natl. Acad. Sci. USA 101, 12497–12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoshino, M., Hagihara, Y., Hamada, D., Kataoka, M. & Goto, Y. (1997) FEBS Lett. 416, 72–76. [DOI] [PubMed] [Google Scholar]
- 54.Arai, M., Kataoka, M., Kuwajima, K., Matthews, C. R. & Iwakura, M. (2003) J. Mol. Biol. 329, 779–791. [DOI] [PubMed] [Google Scholar]
- 55.Zagrovic, B., Snow, C. D., Khaliq, S., Shirts, M. R. & Pande, V. S. (2002) J. Mol. Biol. 323, 153–164. [DOI] [PubMed] [Google Scholar]
- 56.Arai, S. & Hirai, M. (1999) Biophys. J. 76, 2192–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Receveur, V., Garcia, P., Durand, D., Vachette, P. & Desmadril, M. (2000) Proteins 38, 226–238. [DOI] [PubMed] [Google Scholar]
- 58.Smith, C. K., Bu, Z. M., Anderson, K. S., Sturtevant, J. M., Engelman, D. M. & Regan, L. (1996) Protein Sci. 5, 2009–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]