

Abstract
Previous experimental and theoretical studies have produced high-resolution descriptions of the native and folding transition states of chymotrypsin inhibitor 2 (CI2). In similar fashion, here we use a combination of NMR experiments and molecular dynamics simulations to examine the conformations populated by CI2 in the denatured state. The denatured state is highly unfolded, but there is some residual native helical structure along with hydrophobic clustering in the center of the chain. The lack of persistent nonnative structure in the denatured state reduces barriers that must be overcome, leading to fast folding through a nucleation–condensation mechanism. With the characterization of the denatured state, we have now completed our description of the folding/unfolding pathway of CI2 at atomic resolution.
Keywords: CI2, nuclear magnetic resonance, molecular dynamics simulations, conformational transitions, nucleation-condensation
Protein folding is a rapid and complex process that is difficult to characterize. To add to this difficulty, the denatured state consists of a large ensemble of conformations interconverting at a rapid rate. The denatured state is often assumed to be devoid of intramolecular interactions, such that the stability of a protein can be explained purely in terms of interactions in the native state. In recent years, it has become apparent that many proteins contain residual structure in the denatured state (ref. 1 and refs. therein). However, detailed characterization of this structure is very challenging, if not impossible in many cases. As such, during folding one follows the transition of a diverse system from an unknown starting point to a well-ordered native state. Further information about the diversity, dynamics, and structure of the denatured state is necessary to characterize and understand better this process.
The simplest folding pathway to define is two state, i.e., involving only the denatured and native states, which are separated by the energetically unfavorable transition state. Chymotrypsin inhibitor 2 (CI2) was the first protein shown to fold by a two-state mechanism, and it has since been the focus of a number of experimental and theoretical studies. It is a 64-residue protein that consists of an α-helix and a three-stranded β-sheet (Fig. 1). The main hydrophobic core is formed by the packing of the α-helix against the β-sheet.
Figure 1.
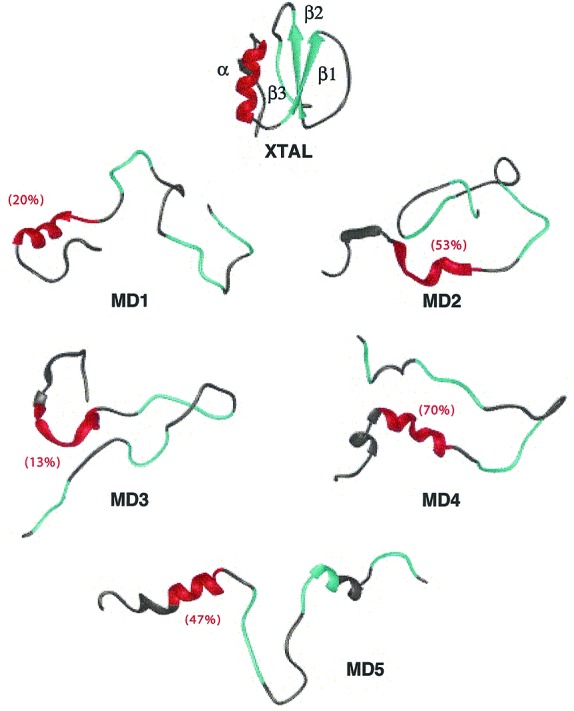
Crystal structure of CI2 (23) and final structures from each of the denaturation simulations (3 ns, MD1–MD4, 20 ns for MD5). The residues (13–23) involved in the native α-helix are colored red. β-Strands 1–3 are colored cyan and are defined as follows: β1, residues 27–34; β2, 46–52; and β3, 59–63. The average helix content of residues 13–23 in each denatured ensemble is given in parentheses (27). Secondary structure, by using the method of Kabsch and Sander (33), is displayed as a ribbon. Fig. 1 was made by using University of California, San Francisco, midasplus (34), and Figs. 3–5 were created with molscript (35) and raster3d (36).
Experimentally, the structure of the transition state has been studied by the protein engineering (Φ-value) method (2). In combination with molecular dynamics (MD) simulations, an atomic-resolution model of the transition state has been proposed (3–7) and verified (8). The rate-limiting step for the folding of CI2 involves the final expulsion of water molecules from the exposed nonpolar side chains and the tight packing of the hydrophobic core. The transition state is similar to an expanded native state with some disruption of the secondary structure.
In contrast, the denatured state of CI2 appears to be largely unstructured as probed by NMR studies of various fragments (9–11). However, there appears to be some tendency for very weak native helical structure and some weak clustering of hydrophobic residues, particularly near the center of the protein. The pKa values for the acidic residues of denatured CI2 are weakly perturbed from their values in model compounds, 0.3 units lower, suggesting that the denatured state harbors some weak electrostatic interactions (12). For comparison, the pKas of these residues are ≈2 units lower in the native state. So far, the most detailed information has come from studies of fragments, which may or may not reflect the properties of the denatured form of the full-length protein.
Here, a combination of NMR experiments and MD simulations is used to characterize the denatured state of full-length CI2. Our earlier thermal denaturation MD simulations at 498 K have been extended to 3 ns (3–5). Also, the results of a new 20-ns simulation (13) are described. In all cases, CI2 unfolds within ≈1.5 ns at such a high temperature. Accordingly, the analyses here focus on the structures after that time. We also note that recent simulations of the engrailed homeodomain (14) and CI2 (13) demonstrate that the pathway of unfolding is independent of temperature for these proteins and that 498 K simulations provide a good description for unfolding at experimentally attainable temperatures (≤373 K).
Both the simulations and NMR results presented here suggest that denatured CI2 is highly expanded with little residual structure and approaches a random coil, making it more disordered than most other similarly studied proteins. A small amount of residual structure is suggested, however, by chemical shift deviations from random coil values in two segments of the protein, and this structure is observed in the MD-generated denatured ensemble. However, this residual structure is transient and does not affect the relaxation of these residues.
Materials and Methods
NMR Spectroscopy.
A denatured CI2 sample was prepared by dissolving lyophilized protein
in a 0.5-ml 6.4 M guanidium chloride (GdmCl) solution containing 10%
D2O (vol/vol). HNCACB (15) and CBCA(CO)NH
(16) spectra were acquired on a Bruker AMX 500 spectrometer equipped
with an inverse triple-resonance single-axis gradient probe (Karlsruhe,
Germany). 15N relaxation experiments were
performed at 278 K on a Bruker AMX2 600 spectrometer with an inverse
triple-resonance single-axis gradient probe. Standard pulse sequences
were used to measure
{1H-15N} heteronuclear
nuclear Overhauser effect (NOE), 15N longitudinal
(R1) and transverse
(R2) relaxation rates (17, 18). The
R1 relaxation times were set to 100,
200, 300, 400, 500, 600, and 800 ms and 1 s.
R2 experiments were acquired with
relaxation times of 7.26, 29.1, 43.6, 58.1, 72.6, 87.2, 102, 116, 131,
145, 174, and 203 ms. Peak intensities were fit to monoexponential
equations to obtain R1 and
R2, and their associated uncertainties
were estimated from the fitting error. To evaluate
{1H-15N} NOEs,
two-dimensional spectra were recorded with and without NOE
enhancement achieved with a 5-s 1H saturation.
Uncertainties in the
{1H-15N} NOE values
were estimated from the rms noise of the spectra. Correlation
times for the reorientation of individual backbone NH vectors were
estimated by:
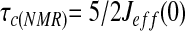 (19), as described previously (1).
(19), as described previously (1).
MD Simulations.
All MD simulations were performed by using the encad program (20) and a previously described force field (21, 22). The high-temperature, 498 K, simulations of CI2 in water began with different experimental starting structures. For MD1, the starting structure was the crystal structure (23). For MD2–MD4, the first three structures from a NMR-derived structural ensemble were used (24). These simulations have been described previously (4), although they were continued for this study. In addition, a 20-ns simulation beginning from the crystal structure was performed, MD5 (13). Structures were saved every 0.2 ps for analysis, resulting in 7,500 structures in each denatured ensemble for MD1–MD4 (1.5–3 ns) and 50,000 (10–20 ns) in MD5, which results in 80,000 structures in the pooled denatured ensemble.
Results
NMR experiments were performed on the GdmCl-denatured state of CI2, for which chemical shift deviations from random coil and relaxation rates were obtained. Five MD simulations were performed of CI2 in explicit water at high temperature (498 K) and neutral pH (acidic groups unprotonated). Four of these simulations have been presented up to, and just beyond, the transition state (MD1–MD4) (3, 4). For this study, these simulations were continued to 3 ns, and an additional 20-ns unfolding simulation (MD5) was performed, along with a 3-ns quench (Q) simulation, where an unfolded structure (1.5 ns of MD1) was simulated at a lower, quasinative temperature (335 K). Taken together, these simulations total 35 ns. As a control, a 5.3-ns 298 K native simulation has been described (25), in addition to a newer 50-ns simulation (13).
NMR Studies of Denatured CI2.
The structure of denatured CI2 was monitored by comparing the chemical shift values in 6.4 M GdmCl with those derived from model peptides (26). Most of the nuclei of denatured CI2 had chemical shift values similar to those derived from model peptides, suggesting that the denatured state of CI2 resembles a random coil (Fig. 2). However, significant chemical shift deviations were found for 10 residues: V19-L21 and I30-T36.
Figure 2.

Structural and dynamic results of NMR experiments of denatured CI2. The deviation of the chemical shifts from random coil values for denatured CI2: (A) 13C, (B) 15N, and (C) 1H. Relaxation rates and correlation times for denatured CI2: (D) R1, (E) R2 and (F) {1H-15N} NOE values. Plots of the correlation times (τc) for denatured CI2 parallel those of the R2 values.
The dynamic behavior of CI2 denatured in 6.4 M GdmCl was monitored by 15N relaxation of backbone resonances. Longitudinal (R1), transverse (R2) 15N relaxation rates and heteronuclear {1H-15N} NOEs for denatured CI2 were measured at 278 K (Fig. 2). The average R1, R2 and {1H-15N} NOEs values were 1.75 ± 0.19, 7.12 ± 1.68, and −0.69 ± 0.12, respectively. The heteronuclear NOE values, which are indicative of picosecond mobility, were fairly constant across the primary sequence, with the exception of large negative values near the termini. This “end effect” is also reflected in the R2 values (Fig. 2).
A generalized correlation time τc(NMR), the characteristic time required for the autocorrelation function to decay to zero, for individual NH vectors was estimated by a reduced spectral density mapping approach (19). The average τc(NMR) value was 4.4 ± 1.2 ns. The “end effect” was also reflected in the values of τc(NMR) of denatured CI2 at 278 K, which ranged from ≈1 ns at the termini to ≈5–6 ns for residues in the middle of the polypeptide chain. This “end effect” of dynamics behavior has also been observed in other denatured proteins (ref. 1 and refs. therein).
MD Simulations.
The Cα rms deviation from the native state for the denatured region of the simulations varied between 9 and 16 Å. The denatured structures were expanded with little persistent secondary structure and few tertiary contacts (Fig. 1). In fact, the radius of gyration was 36–80% greater in the denatured state compared with the crystal structure, and the solvent-accessible surface area increased by 47–52% with a 58–69% increase in the nonpolar exposure. There was some dynamic residual native helical structure in three of the five simulations, but the β-sheet was lost in all simulations. There were also some dynamic hydrophobic clusters in the denatured ensemble, of which the more persistent were in the center of the protein. Overall, the protein was mobile, and the N and C termini had a 1.5 times larger Cα rms fluctuation around the mean structure in the denatured state compared with residues in the middle of the chain.
The helical structure in denatured CI2 resided between residues 17–21 (Figs. 1 and 3A), whereas the native helix spans residues 13–23. The residual helical structure is not just a result of limited sampling such that it lacks the necessary time to unfold. Instead, the helix is very dynamic and completely unfolded and refolded over time. On average, the helix refolded two or more turns (>63% helix) from a completely unfolded conformation (0% helix), as assessed by (Φ, Ψ) angles (27), 35 times per nanosecond. Also, a simulation starting with the 1.5-ns structure from MD1 (≈11 Å rms deviation from the native state) was performed at 335 K to address this point further (Q simulation). This starting structure contained no helical structure and was highly unfolded, but over the initial 2.5 ns, helical structure formed between residues 17–21 (Fig. 3B).
Figure 3.
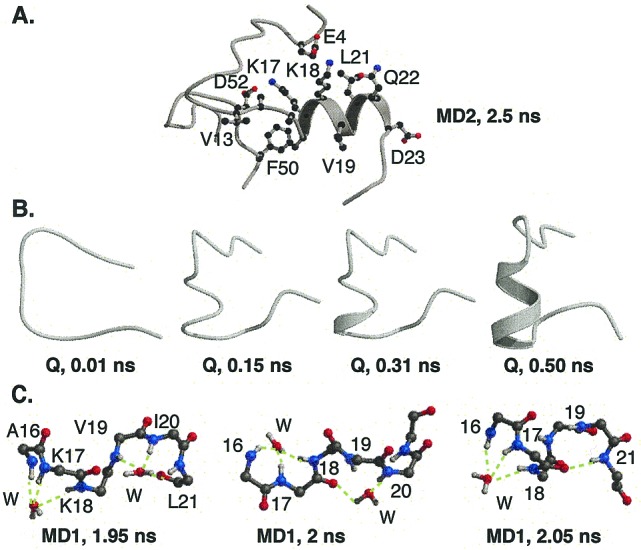
Residual helical structure in the denatured state. (A) Residual helix in the 2.5-ns structure from the MD2 simulation. The hydrophobic side chains of the helix are protected from solvent by the rest of the denatured protein. The more hydrophilic side chains of Lys-17 and -18 form salt bridges with acidic side chains allowing their hydrophobic methylene side chains to cluster away from solvent. (B) Residues 11 (Top) to 26 from the Q simulation. Native helix formed between residues 17–22. (C) Mechanism of the formation of the initial helical turn in the denatured state. In MD1, two water molecules form hydrogen bonds with the main chain amide hydrogens of residues 17–21. One water molecule initiated a turn-like structure between residues 16–18, whereas the other led to a turn-like structure between residues 19–21. The two water molecules ordered the region for helical formation. At 2.05 ns, the carbonyl oxygens of residues 16 and 17 formed i → i + 4 hydrogen bonds with residues 20 and 21, expelling the water from the region.
Helical formation was also observed in the last third of the MD1 simulation (Fig. 3C). In both the Q and MD1 simulations, the first turn of the helix formed via an i → i + 4 hydrogen bond between the amide hydrogen of Leu-21 and the carbonyl oxygen of Lys-17 (MD1, 2.05 ns; Q, 0.2 ns). Before the formation of this hydrogen bond, a water molecule induced a turn-like structure in residues 19–21 by hydrogen bonding with their amide hydrogens (Fig. 3C). In MD1, there was also another water molecule that formed a similar turn-like structure with residues 16–18 (Fig. 3C), whereas in the Q simulation, residues 17 and 18 hydrogen bonded to the main chain of the protein. Thus, in both simulations, the converting segments were fairly ordered before the formation of the initial helical turn. The first helical hydrogen bond formed from these two ordered segments acting as a nucleus, the water was expelled, and the helix extended in both directions.
Although the native helix was populated in the simulations, it fluctuated and was only marginally stable (Fig. 1). As a result, the helical structure was effectively averaged out when considering the average (Φ, Ψ) values of this region over the full ensemble. The 3JNH-CαH coupling constants were calculated from the simulations for the denatured state ensemble from the average Φ angles (28). Essentially all of the residues had coupling constants between 5.5–8.5 Hz, as determined both by MD and NMR, with average values of 7.0 and 7.2 Hz, respectively. Neither simulation nor experiment showed a decrease in the coupling constants in the areas of residual helical structure.
Besides the secondary structure, experimental studies also suggest there is hydrophobic clustering between residues Ile-29 and Ile-37. This region of CI2 is highly hydrophobic with the sequence IIVLPVGTI. The number of NOEs and various chemical shift values suggest there is a hydrophobic cluster in this region in CI2 fragments (29) and in the full-length protein, as illustrated in Fig. 2. In the native protein, this region encompasses the C-terminal half of β-strand 1 and part of the active site loop. In the simulations, the 29–37 region makes a number of hydrophobic contacts within itself as well as with other hydrophobic regions in the molecule (Fig. 4).
Figure 4.
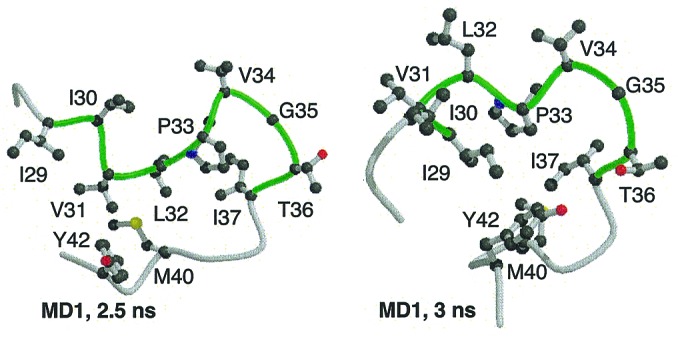
Hydrophobic clustering near the center of the sequence. The main chain of residues 27–43 is presented with residues 29–37 in green.
A recent fluorescence resonance energy transfer study on the denatured state of CI2 tracked the distance between residues 1 and 40 (30). Correspondingly, this distance (N of Met-1 and S of Met-40) was calculated over the denatured MD ensembles. The average pooled value for the 3-ns simulations (MD1–MD4) was 22 ± 5 Å. Similar distances were obtained for MD5 up to ≈5 ns, after which time this distance increased dramatically. The average value from 10–20 ns was 43 ± 7 Å, which is in good agreement with the experimental value of ≈45 Å.
In previous experimental and simulation studies, a nucleation site for the folding of CI2 was suggested to include regions around Ala-16, Leu-49, and Ile-57. The distances between these residues increased drastically over the individual simulations after passing through the transition state. Snapshots from MD1 reveal how closely these three residues of the nucleation site cluster in the crystal structure and MD transition state model (Fig. 5). After the disruption of this cluster, the structure quickly unfolded, as seen in the 0.5-ns structure, where the loop between β-strands 2 and 3 moved away from the helix to expose the hydrophobic core. By 2 ns, the structure was completely unfolded with little residual structure (Fig. 5).
Figure 5.

The folding nucleus comes together only in the transition state. (A) Snapshots from MD1 highlighting the nucleation-site region, Ala-16, Leu-49, and Ile-57. These residues are tightly packed in the native structure and retain some contacts through, but not beyond, the transition state. (B) Distances between the three residues as a function of simulation time.
Discussion
An Unfolded Protein.
The overall view from the NMR experiments and the MD simulations is that the denatured state of CI2 is highly unfolded. The chemical shift deviations from random coil suggest that there are only two small regions (residues 19–21 and 30–36) with residual structure (Fig. 2). The MD simulations also reveal only two areas of residual structure. In three of the five simulations, residual native helical structure in the denatured state was localized between residues 17–21 (Figs. 1 and 3). Moreover, NMR experiments of an isolated 40-residue peptide of CI2 suggest that there is nascent native helical structure (9). The other area of residual structure in the MD simulations was the turn stabilized by hydrophobic interactions around residues 29–37 (Fig. 4). Also, NMR studies of CI2 peptides reveal a high number of NOEs in this region of the sequence (9). The residual structure was transient and weakly populated such that it was not evident in the coupling constants, average Φ values, or relaxation results. For example, the experimental R2 values and correlation times, τc, were relatively constant along the sequence, with the exception of low values at the termini (Fig. 2). Correspondingly, the simulations show an end effect of increased mobility at the termini.
In fact, CI2 appears to have a much more unfolded denatured state than many other small proteins. For example, one ideal protein for comparison is barnase, whose entire folding pathway has been well characterized by experiment and simulation (ref. 1 and refs. therein). Unlike CI2, the structural parameters (e.g., chemical shift deviation, backbone dynamics) of denatured barnase show significant nonrandom-coil behavior (1). For example, the correlation times, τc, of urea-denatured barnase exhibit a large variation across the primary sequence (1). Slower dynamics are found in two regions of denatured barnase residues corresponding to (i) α1 and α2 (≈residues 15–30) and (ii) β-strands 3 and 4 (≈85–100). The presence of residual structure in these regions is also supported by chemical shift data and by MD simulations.
The radius of gyration and tertiary contacts from the denatured regions of the MD simulations further reflect how much more unfolded CI2 is compared with barnase. With respect to both properties, CI2 is more expanded and has fewer interactions than barnase. For example, the ratio of the radius of gyration over the square root of the number of residues is 2.2 ± 0.3 for the pooled denatured state of CI2, whereas it is 1.6 ± 0.1 for the pooled denatured state of barnase. This information, combined with the NMR results, suggests that the denatured state of CI2 is highly unfolded compared with other well-characterized denatured proteins.
Although the denatured state of CI2 is highly unfolded, it is not an ideal random coil. A recent theoretical study of polyalanine peptides by Pappu et al. (31) suggests that the Flory isolated-pair hypothesis is incorrect for polypeptides and that proteins do not fold from random coils, in line with certain other experimental and simulation studies. The investigators find that their peptide conformations segregate into helices or extended coils with few partially helical conformations. Further, the conversion from helix → coil occurs in an all-or-none transition, as with globular proteins. However, such cooperativity is generally not actually observed for peptides and also contrasts with our experiments and simulations on the folding of CI2 and the structures of its fragments. One possible explanation for the discrepancies between our results and those of Pappu et al. is their exceptionally simple representation of the system, the neglect of solvent, and the only stabilizing interactions considered were intrachain hydrogen bonds. In reality, there are stabilizing solvent–protein interactions and hydrophobic clustering, as observed in the simulations presented here. In addition, entropic considerations can favor partially helical states, which is the preferred state for small helical peptides. When CI2 unfolds, the α-helix is not confined to two states; instead, a range of structures are observed for residues 13–23.
A fluorescence resonance energy transfer study monitoring the distance distribution between dyes attached to residues 1 and 40 suggests that these residues are ≈45 Å apart in the denatured state (30). This distance is approaching the theoretical distance of 50 Å for a 40-residue random coil. The corresponding average distance in the 3-ns simulations (MD1–MD4) was ≈22 Å. However, the 20-ns simulation had an average distance of ≈43 Å. Although the protein underwent dramatic expansion over time, which was underrepresented in the shorter simulations, the average local residual structure, both in terms of helical structure and hydrophobic clustering, was independent of simulation time after reaching the denatured state
The α-Helix and the Role of Solvent in Helix Folding.
Three of the five simulations adopted residual helical structure. With the maintenance of the helix, the protein better sequesters hydrophobic residues from solvent including the hydrophobic portion of the two lysines (Fig. 3A). That the helical structure was transient and not observed in all of the simulations suggests that it was only marginally stable and may not be observed experimentally by such methods as circular dichroism spectroscopy.
The residual helical structure is not just a result of short simulations that are not long enough to allow helix unfolding to occur. The helix unfolded and refolded at various times in the simulations (Figs. 3 and 5). Also, to test the propensity of this portion of the sequence to form dynamic helical structure, a 335 K quenched simulation of a snapshot of the MD1 simulation devoid of helix was performed. In this case, the unstructured protein refolded to form an α-helix between residues 17–21. Thus, this region is inclined to form helical structure. Water played a major role in the reformation of this helix. The first turn of the helix formed via an i → i + 4 hydrogen bond between residues 17 and 21. Before the formation of this hydrogen bond, a solvent molecule induced turn-like structures in residues 16–18 and 19–21 by forming hydrogen bonds with their amide hydrogens, thereby creating a conformation poised for helix formation. The large hydrophobic side chains of residues 19–21 helped to sterically trap the water near the amide hydrogens, thus inducing the turn-like structure. These results reflect the importance of including an explicit water model when studying protein folding, as the individual water molecules can play a critical role in the mechanism of conformational change.
Summary of the Folding Pathway of CI2.
Both experimental and theoretical studies have been undertaken in an attempt to characterize all steps along the pathway of folding/unfolding of chymotrypsin inhibitor 2. Overall past studies by using the two methods produce quite similar results. Both methods indicate that there is partial structure formation in the transition state: the Φ values are intermediate between values indicative of fully formed or fully disrupted structure. The α-helix is the most structured region of the protein, although it is very mobile and its termini are distorted. The β-sheet is disrupted substantially, with essentially complete loss of structure at the periphery away from the core (Fig. 5).
The results from the NMR experiments and MD simulations here indicate that CI2 has a highly unfolded denatured state with only two small regions of dynamic residual structure. This structure includes a partially populated portion of the native helix and a small amount of hydrophobic clustering near the center of the sequence. These regions of structure may bias the unfolded protein toward the native state during folding to help guide the protein along the folding pathway, thereby overcoming the Levinthal Paradox.
Although the denatured state of CI2 does not have the dimensions of a random coil, it is extremely expanded, much more so than the denatured state of barnase, for example. The lack of persistent nonnative structure in the denatured state reduces barriers that the protein must overcome during its folding. Considering the simulations from the folding direction, particular patches on the α-helix and the β-sheet are important in nucleating the core, and structure is looser radiating away from this area (Fig. 5). These patches of structure do not exist in the denatured state (Figs. 1 and 5), and fragment studies show that there is little drive for such structure (10, 32). Instead, the nucleation site remains embryonic until sufficient long-range contacts are made (the nucleation-condensation mechanism). These patches of structure come together only in or near the transition state (Fig. 5). Given the nonlocal nature of these tertiary contacts, they are necessarily linked to secondary structure for productive folding to occur. After coming together, collapse and condensation of structure around these patches are rapid (Fig. 5). With the agreement between experiment and simulation of all three “discrete” states of CI2 (native, transition, and denatured), the simulated transitions between these states also appear to be plausible, thereby providing an atomic-level description of the full folding/unfolding pathway.
CI2 and barnase are two complementary paradigms for experimental and simulation studies on folding; CI2 displays two-state kinetics and folds from a highly disordered denatured state, whereas barnase folds via multistate kinetics from a somewhat structured denatured state. The simulations not only provide detailed structural information, but they also fill in the experimentally inaccessible parts of the pathway. For example, two-state kinetics may mask the presence of a high-energy intermediate that is kinetically silent. Simulations detect the presence of the folding intermediates in barnase, but similar evidence is not observed for CI2, suggesting that there is not a high-energy intermediate.
Acknowledgments
We thank Dr. A. Deniz and P. G. Schultz (The Scripps Research Institute, La Jolla, CA) and S. Weiss (Lawrence Berkeley National Laboratory, Berkeley, CA) for kindly providing the fluorescence resonance energy transfer data. We also thank the European Community for access to the SON (Netherlands Foundation for Chemical Research) NMR Large Scale Facility, Utrecht, The Netherlands. Helpful discussions with members of the group of Professor R. Kaptein, in particular R. Boelens, M. Czisch, and R. Wechselberger, are appreciated. S.L.K. received support from a National Institutes of Health training grant (GM 08268) and a Hitchings–Elion Fellowship from the Burroughs–Wellcome Fund. V.D. is grateful for support by National Institutes of Health (GM 50789).
Abbreviations
- CI2
chymotrypsin inhibitor 2
- MD
molecular dynamics
- NOE
nuclear Overhauser effect
- Gdm
guanidium
- Q
quench
References
- 1.Wong K B, Clarke J, Bond C J, Neira J L, Freund S M V, Fersht A R F, Daggett V. J Mol Biol. 2000;296:1257–1282. doi: 10.1006/jmbi.2000.3523. [DOI] [PubMed] [Google Scholar]
- 2.Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 3.Li A, Daggett V. Proc Natl Acad Sci USA. 1994;91:10430–10434. doi: 10.1073/pnas.91.22.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li A, Daggett V. J Mol Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 5.Daggett V, Li A, Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1996;257:430–440. doi: 10.1006/jmbi.1996.0173. [DOI] [PubMed] [Google Scholar]
- 6.Otzen D E, Itzhaki L S, elMasry N F, Jackson S E, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazaridis T, Karplus M. Science. 1997;278:1928–1931. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 8.Ladurner A G, Itzhaki L S, Daggett V, Fersht A R. Proc Natl Acad Sci USA. 1998;95:8473–8478. doi: 10.1073/pnas.95.15.8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Prat Gay G, Ruiz-Sanz J, Davis B, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10943–10946. doi: 10.1073/pnas.91.23.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neira J L, Itzhaki L S, Ladurner A G, Davis B, de Prat Gay G, Fersht A R. J Mol Biol. 1997;268:185–197. doi: 10.1006/jmbi.1997.0932. [DOI] [PubMed] [Google Scholar]
- 11.Tan Y-J. Ph.D. thesis. Cambridge, U.K: Univ. of Cambridge; 1995. [Google Scholar]
- 12.Tan Y J, Oliveberg M, Davis B, Fersht A R. J Mol Biol. 1995;254:980–992. doi: 10.1006/jmbi.1995.0670. [DOI] [PubMed] [Google Scholar]
- 13.Alonso, D. O. V., Pan, Y. P., Ham, S., Bennion, B. & Daggett, V. (2001) J. Mol. Biol., in press.
- 14.Mayor U, Johnson C M, Daggett V, Fersht A R. Proc Natl Acad Sci USA. 2000;97:13518–13522. doi: 10.1073/pnas.250473497. . (First Published November 21, 2000; 10.1073/pnas.250473497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wittekind M, Mueller L. J Magn Reson B. 1993;101:201–205. [Google Scholar]
- 16.Muhandiram D R, Kay L E. J Magn Reson B. 1994;103:203–216. [Google Scholar]
- 17.Wagner G. Curr Opin Struct Biol. 1993;3:748–754. [Google Scholar]
- 18.Palmer A G., 3rd Curr Opin Struct Biol. 1997;7:732–737. doi: 10.1016/s0959-440x(97)80085-1. [DOI] [PubMed] [Google Scholar]
- 19.Peng J W, Wagner G. J Magn Reson. 1992;98:308–332. [Google Scholar]
- 20.Levitt M. ENCAD–Energy Calculations and Dynamics. Rehovot, Israel: Yeda; 1990. [Google Scholar]
- 21.Levitt M, Hirshberg M, Sharon R, Daggett V. Comput Phys Commun. 1995;91:215–231. [Google Scholar]
- 22.Levitt M, Hirshberg M, Sharon R, Laidig K E, Daggett V. J Phys Chem B. 1997;101:5051–5061. [Google Scholar]
- 23.Harpaz Y, Elmasry N, Fersht A R, Henrick K. Proc Natl Acad Sci USA. 1994;91:311–315. doi: 10.1073/pnas.91.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludvigsen S, Shen H Y, Kjaer M, Madsen J C, Poulsen F M. J Mol Biol. 1991;222:621–635. doi: 10.1016/0022-2836(91)90500-6. [DOI] [PubMed] [Google Scholar]
- 25.Li A, Daggett V. Protein Eng. 1995;8:1117–1128. doi: 10.1093/protein/8.11.1117. [DOI] [PubMed] [Google Scholar]
- 26.Wishart D S, Sykes B D. Methods Enzymol. 1994;239:363–392. doi: 10.1016/s0076-6879(94)39014-2. [DOI] [PubMed] [Google Scholar]
- 27.Daggett V, Kollman P A, Kuntz I D. Biopolymers. 1991;31:1115–1134. doi: 10.1002/bip.360310911. [DOI] [PubMed] [Google Scholar]
- 28.Karplus M. J Chem Phys. 1959;30:11–15. [Google Scholar]
- 29.de Prat Gay G, Ruiz-Sanz J, Neira J L, Corrales F J, Otzen D E, Ladurner A G, Fersht A R. J Mol Biol. 1995;254:968–979. doi: 10.1006/jmbi.1995.0669. [DOI] [PubMed] [Google Scholar]
- 30.Deniz A A, Lawrence T A, Beligere G S, Dahan M, Martin A B, Chemla D S, Dawson P E, Schultz P G, Weiss S. Proc Natl Acad Sci USA. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. . (First Published May 2, 2000; 10.1073/ pnas.090104997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pappu R V, Srinivasan R, Rose G D. Proc Natl Acad Sci USA. 2000;97:12565–12570. doi: 10.1073/pnas.97.23.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itzhaki L S, Neira J L, Ruiz-Sanz J, de Prat Gay G, Fersht A R. J Mol Biol. 1995;254:289–304. doi: 10.1006/jmbi.1995.0617. [DOI] [PubMed] [Google Scholar]
- 33.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 34.Ferrin T E, Huang C C, Jarvis L E, Langridge R. J Mol Graphics. 1988;6:13–27. [Google Scholar]
- 35.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 36.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]