

Abstract
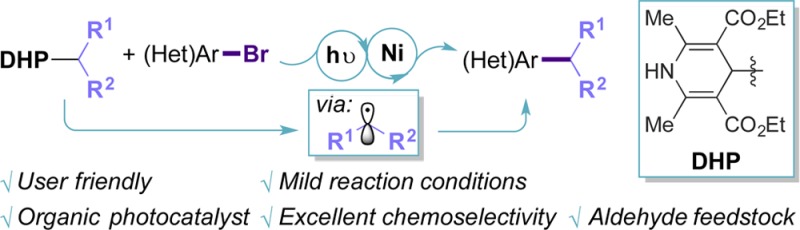
A Ni/photoredox dual catalytic cross-coupling is disclosed in which a diverse range of (hetero)aryl bromides are used as electrophiles, with 1,4-dihydropyridines serving as precursors to Csp3-centered alkyl radical coupling partners. The reported method is characterized by its extremely mild reaction conditions, enabling access to underexplored cores.
Keywords: 1,4-dihydropyridines; nickel catalysis; photocatalysis; nickel/photoredox dual catalysis; oxidative deformylation
In the last decades, cross-coupling reactions have become among the most highly used means to construct new C–C bonds.1 However, despite their numerous advantages, cross-coupling reactions, especially in the context of Csp2–Csp3 bond formation, suffer from some limitations that hinder their more widespread utilization. In particular, the high activation energy barrier associated with the transmetalation step in the coupling of many Csp3-hybridized nucleophiles results in a need to use unstable and functional group-intolerant organometallic reagents, negatively impacting the generality and operational simplicity of these methods.2 In this regard, the recently developed nickel/photoredox dual catalytic process has proved to be an excellent alternative and complementary approach to overcome such limitations.3 In such protocols, a facile transmetalation-like event is triggered, being initiated by single-electron oxidation of an organometallic reagent. The odd-electron nature of this reactivity paradigm proceeds most rapidly with Csp3 coupling partners, effectively inverting the reactivity hierarchy observed in more conventional cross-coupling processes. As a result, reactive organometallic nucleophiles can be replaced by a variety of functional group-tolerant radical precursors.
Among alkyl radical precursors, different partners have been previously reported such as alkyltrifluoroborates,4 alkylsilicates,5 carboxylic acids,6 halides,7 and activated C–H bonds.8 However, it remains of utmost importance to introduce new feedstock functional groups amenable to oxidative fragmentation, forming suitable alkyl radicals.9 Within this context, aldehydes represent an attractive option because aliphatic aldehydes are abundant in nature and readily available from commercial sources. Although nature has long ago developed very effective means to promote oxidative deformylation reactions,10 such transformations still pose significant challenges for the scientific community, which rely on two distinct approaches (Scheme 1). First, an acyl radical can be formed through a hydrogen-atom transfer (HAT) process, usually involving thiyl radicals. Subsequently, decarbonylation delivers the targeted alkyl radical (route a).11 This latter step is usually slow, and acylated byproducts are often observed. Alternatively, superoxometal complexes have been employed (route b);12 however, strong, stoichiometric oxidants are required, thus hampering the applicability and breadth of substrates employed.
Scheme 1. Different Approaches toward the Oxidative Deformylation of Aldehydes.
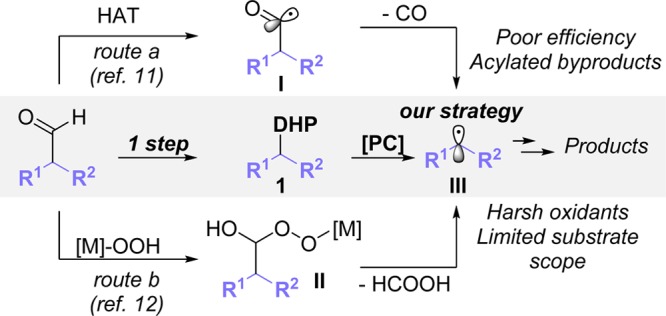
Being aware of the inherent difficulties associated with the formation of alkyl radicals directly from aldehydes,13 we explored different aldehyde derivatives able to undergo photochemical homolysis. 1,4-Dihydropyridines (DHPs) can be easily prepared from aldehydes in one step, even with high functionalization levels,14 and their photochemical oxidation delivers hydrogen (H2) with concomitant pyridine formation. However, in the presence of alkyl substituents in position 4, the oxidation has been shown to generate carbon-centered alkyl radicals.15 Importantly, the Nishibayashi group16 has successfully applied such an approach in aromatic substitution reactions. More recently, Ma and Cheng coupled a variety of DHPs with activated tertiary alkyl bromides under photocatalytic conditions.17
To ensure effective oxidative cleavage of the 1,4-DHP (Eox = +1.05 V vs SCE),18−20 the organic dye 4CzIPN (excited state Ered = +1.35 V vs SCE) was employed as a photocatalyst,21 and different nickel(II) sources were tested (Table 1, entries 1–5).22 As shown, dtbbpy outperformed other ligands tested, with dMeObpy and bpy showing lower yields (entries 2–4 in Table 1). Interestingly, the preformed [Ni(dtbbpy)(H2O)4]Cl2 afforded higher yields with no deleterious effects observed from the water. Other photocatalysts were also explored (entries 6–8 in Table 1). Not surprisingly, [Ru(bpy)3](PF6)2 only delivered traces of product, likely because of its low reduction potential (*Ered = +0.77 V vs SCE) whereas an Ir-photocatalyst (entry 6 in Table 1) delivered the product in moderate yields (*Ered = +1.32 V vs SCE). The highly oxidizing organic photocatalyst mesityl acridinium (*Ered = +2.06 V vs SCE)23 was next explored, but afforded no product, likely because of its inability to reduce the putative Ni(I) intermediate.4j Consequently, 4CzIPN was chosen because of its lower cost ($6.01/g), more straightforward preparation, and higher activity.21 Finally, although other common solvents for photoredox cross-coupling reactions were examined (entries 9 and 10 in Table 1), acetone outperformed both CH3CN and DMF. As anticipated, control experiments showed that all parameters were essential for the reaction to proceed.18 Importantly, we observed complete consumption of 1a in the presence of the photocatalyst, delivering the pyridine byproduct. Therefore, in some cases, we decided to boost the reactivity by increasing the amount of Ni-catalyst and DHP, thus obtaining higher yields. It should be noted that the disclosed reaction conditions are particularly user-friendly, allowing the construction of complex structural motifs in a “dump and stir” fashion.
Table 1. Optimization of the Reaction Conditionsa.
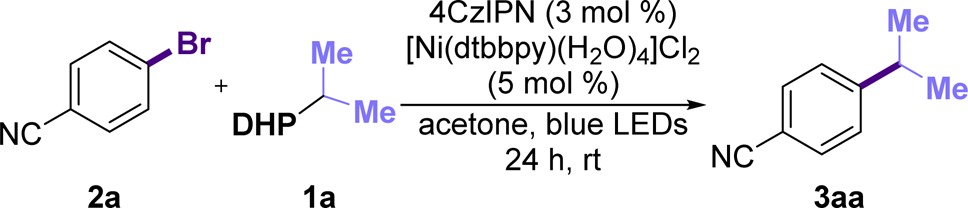
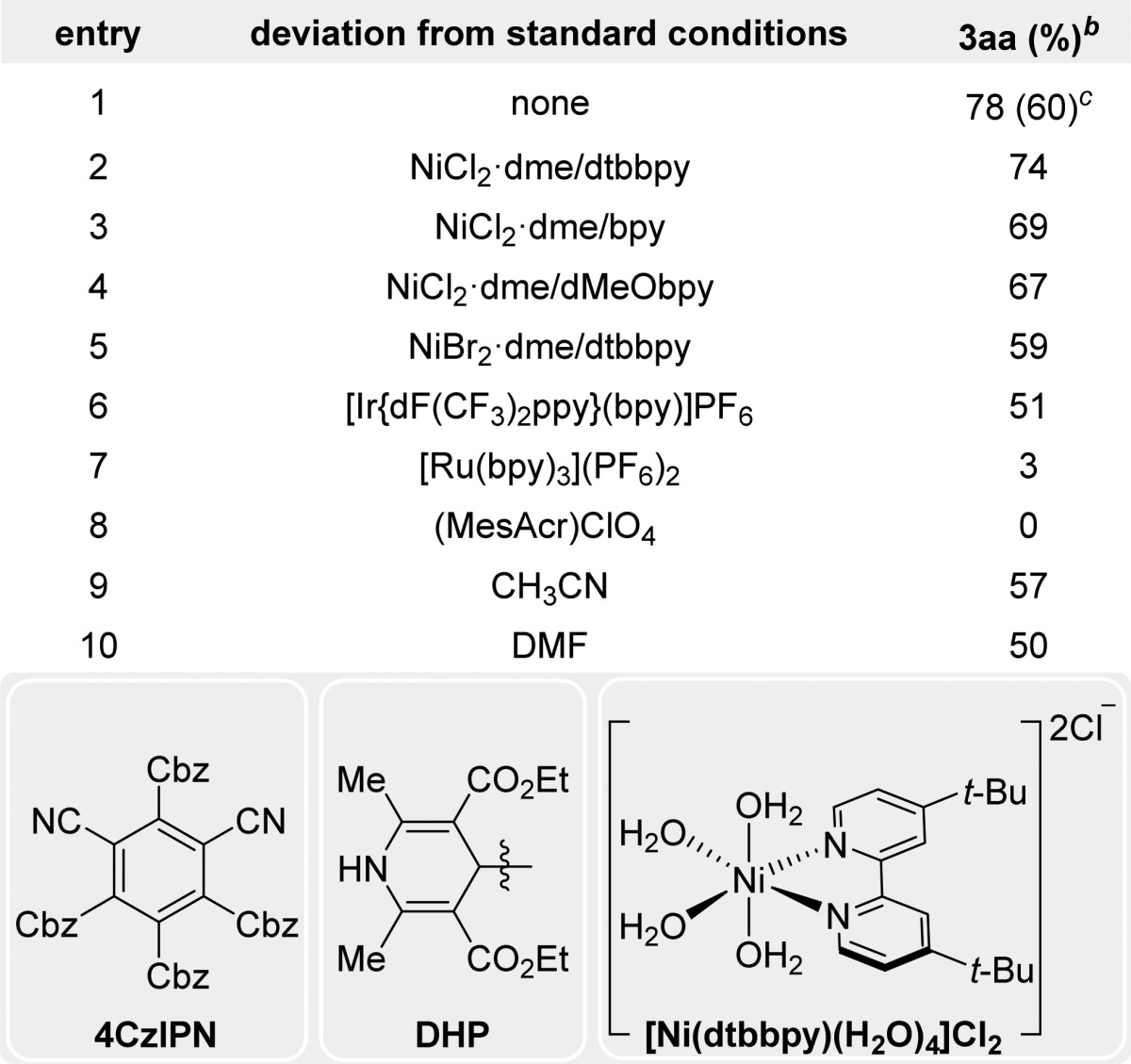
2a (0.1 mmol), 1a (0.12 mmol), 4CzIPN (3 mol %), [Ni(dtbbpy)(H2O)4]Cl2 (5 mol %) in dry, degassed solvent (2.0 mL, 0.05 M) under blue LED irradiation for 24 h. bHPLC yield using 4,4′-di-tert-butylbiphenyl as an internal standard. cIsolated yield.
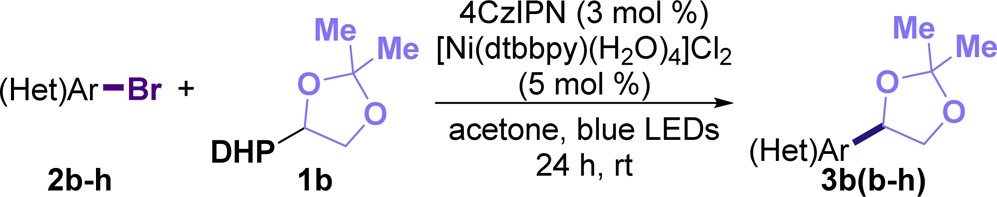
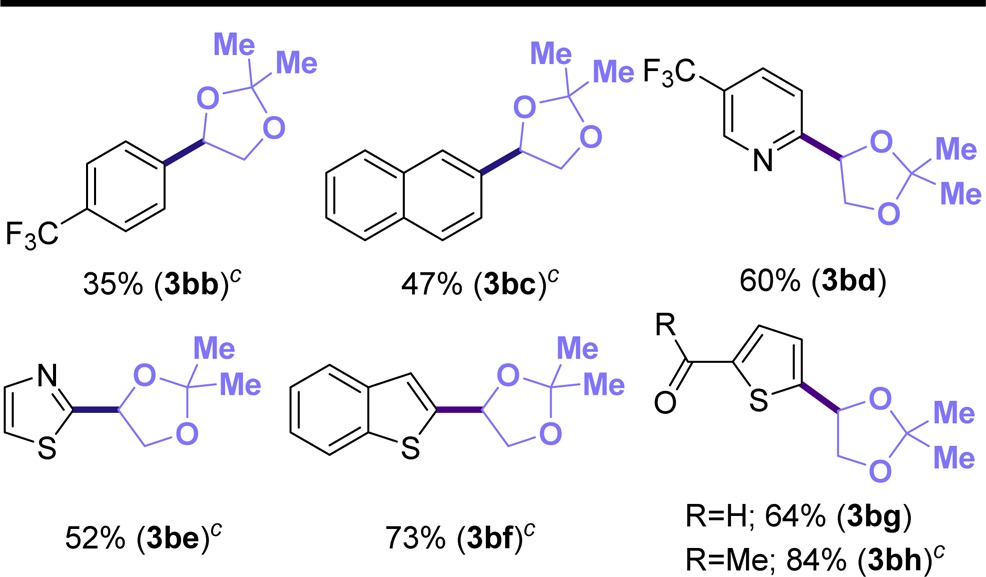
Encouraged by these results, we turned our attention to study the influence of several (hetero)aryl bromides in the reaction. As illustrated in Table 2, different (hetero)aryl bromides were well accommodated with aromatic substrates (3bb and 3bc) and heteroaromatic entities such as pyridine (3bd), thiazole (3be), and thiophene (3bf–3bh). Notably, we observed that 2-bromoheteroarenes were more reactive than other heteroaryl bromides.24 Similarly, bifunctional heteroaryl bromides bearing carbonyl moieties (2g, 2h) proved to be useful coupling partners, and the products of these reactions thus contain reactive groups that can be used for further elaboration.
Table 2. Scope of (Hetero)Aryl Bromidesa,b.
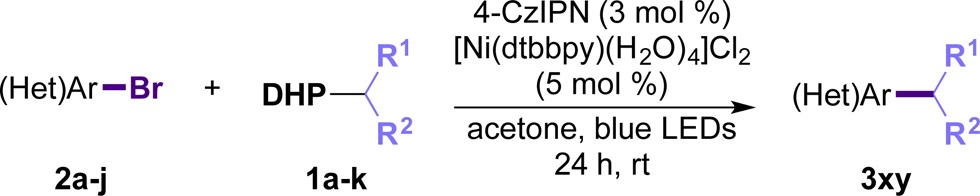
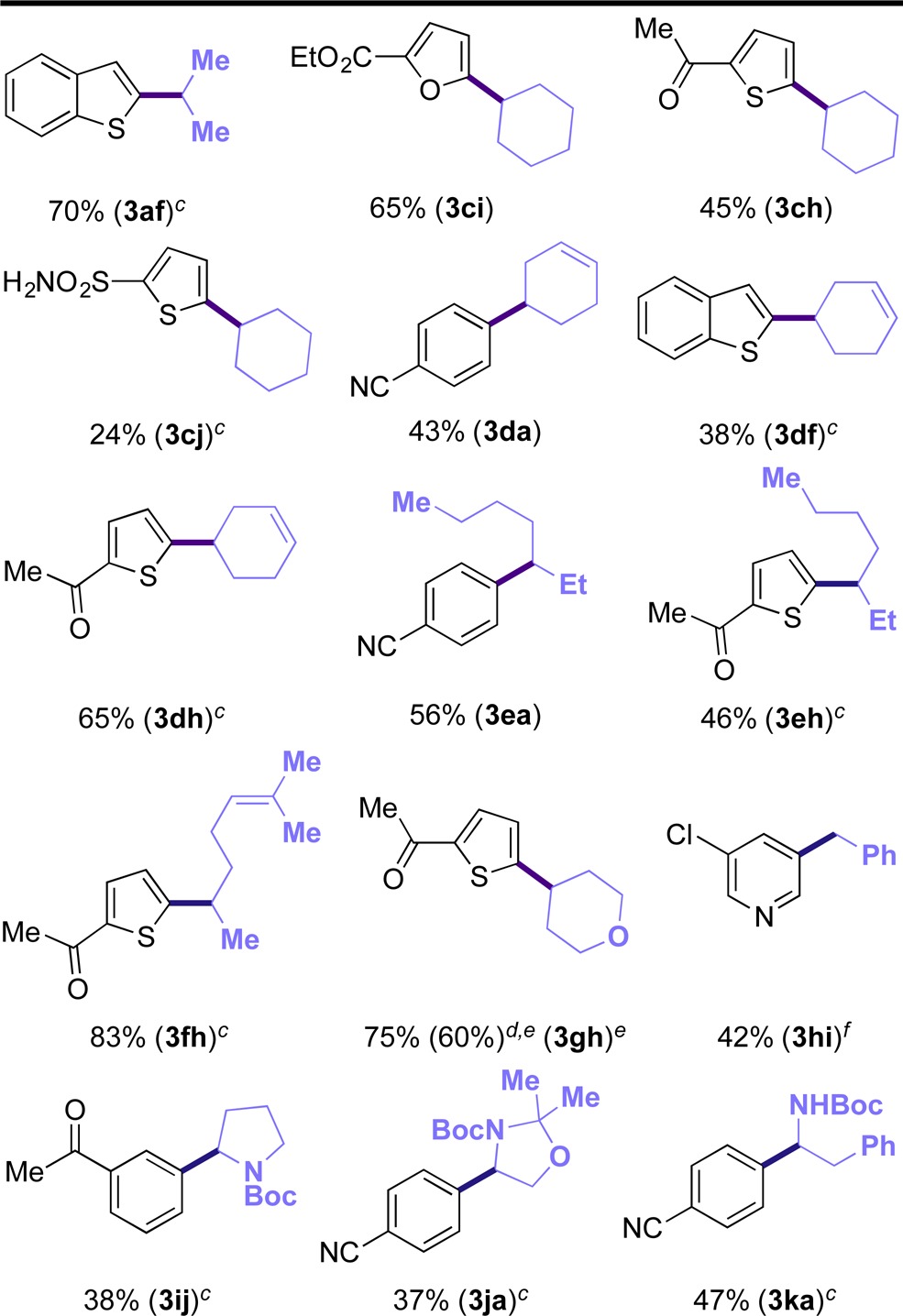
Once the versatility of the protocol was demonstrated against different (hetero)aryl bromides, we decided to focus our attention on both the DHP radical precursors and the aryl bromide partners simultaneously in an effort to showcase the “real-world” utility of this method in cases where both the nucleophilic and electrophilic partner present structural and/or electronic challenges (Table 3). The developed reaction conditions were highly general, and various substitution patterns were well accommodated. A diverse range of alkylated arenes and heteroarenes were isolated in modest to high yields. Unactivated secondary alkyl DHPs could be coupled independently of the cyclic or acyclic nature of the radical. More interestingly, alkyl radicals bearing distal alkenes (3da–3dh) were well tolerated. No evidence of radical cyclization was observed for the melonal-derived DHP (1h). A pyran-derived DHP could likewise be used, and the resulting product was isolated in good yield (3gh). Importantly, even at almost 10-fold higher scale, the reaction went to 60% yield without further optimization. Similar results were achieved for a benzylic DHP when coupled with a challenging pyridine (3hi). In addition to unactivated alkyl radicals, α-heteroalkyl substrates could be employed. Pyrrollidine (1i), as well as protected amino alcohol (1j) and amino acid-derived (1k) DHPs all succeeded in delivering the cross-coupled product, although in modest yields (Table 3).
Table 3. Scope of (Hetero)Aryl Bromides and Dihydropyridinesa,b.
To demonstrate further the applicability of the developed method, the protocol was tested for the synthesis of two saccharide derivatives. Previous literature reports for the synthesis of C-aryl glycosides via cross-coupling relied on the employment of alkenyl stannane25 or alkenyl boronate26 derivatives, presumably because the alkyllithium intermediates required for the synthesis of the requisite alkyl glycosidic stannanes and alkyl boronates would suffer β-elimination of the adjacent alkoxy group. Thus, current approaches require a cumbersome synthesis of the dihydropyranylmetallics, followed by coupling and hydrogenation. By contrast, for the present transformation, 1,4-dihydropyridines from saccharides are readily available,14a and the particularly mild reaction conditions allow the formation of saccharide-containing product (3lf) in good yield and excellent diastereoselectivity. Access to 3fk demonstrates that a fully deprotected carbohydrate core can be coupled, albeit in modest yield. (See Figure 1.)
Figure 1.
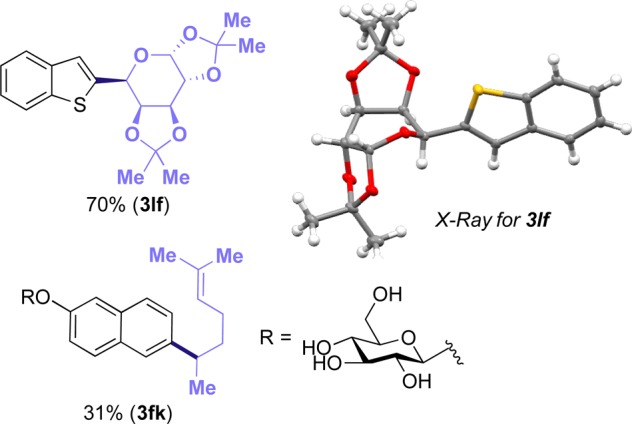
Synthesis of aryl-containing saccharides, as in Table 1 (entry 1), 0.50 mmol scale. Isolated yield, average of at least two independent runs. Using [Ni(dtbbpy)(H2O)4]Cl2 (10 mol %) and 1- (1.5 equiv). For 3lf and 3fk, diastereomeric ratio > 20:1.
The developed method does present a few limitations,27 because of the intrinsic stability of the radical intermediate formed. Consequently, primary alkyl or cyclopropyl DHPs did not succeed in delivering the cross-coupling product, because oxidation of the DHP delivers only the 4-alkylated pyridine byproduct.15 Nonetheless, this is arguably a minor limitation, considering complementary cross-coupling protocols available for primary or cyclopropyl motifs.2a
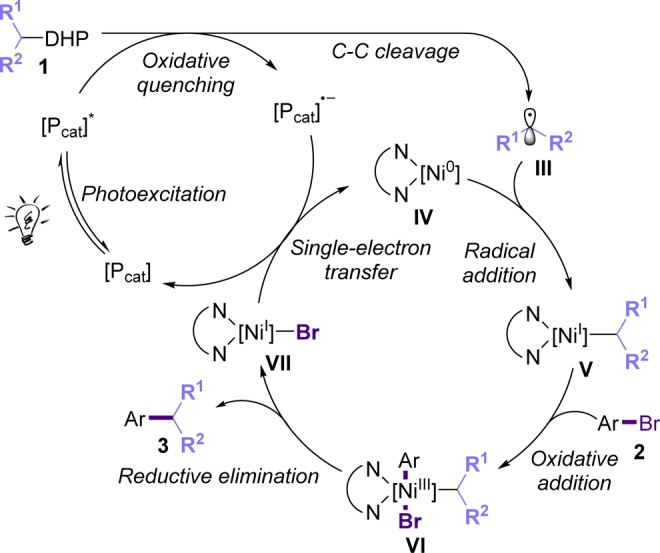
Based on our previous studies with alkyltrifluoroborates28 as well as previous studies detailing the photochemical oxidative cleavage of DHPs,15,16 we propose the mechanistic scenario depicted in Scheme 2. First, photoexcitation of the organic photocatalyst to its excited state generates a species that is a sufficient SET oxidant.21 At this point, the photocatalyst is oxidatively quenched by the DHP derivative (1), thus forming a radical cation (not pictured), which undergoes homolysis, delivering the carbon-centered alkyl radical III. This radical reacts with active Ni(0) catalyst (IV), forming the alkyl nickel(I) intermediate V, which undergoes further oxidative addition with the aryl bromide (2), forming the Ni(III) complex VI.29,30 Subsequently, reductive elimination delivers the cross-coupled product (3), along with Ni(I) complex VII, which can be reduced to Ni(0) by the reduced photocatalyst, thus regenerating both active catalysts. We believe that the homolysis of the DHP unit occurs via formation of a radical cation, followed by a deprotonation step, forming an aminyl radical that will then undergo homolysis.15,16 Notably, we observed that 1c-N-Me (the N-methylated analogue of 1c) failed to deliver the cross-coupling product under reaction conditions, thus supporting the deprotonation prior to the C–C cleavage event hypothesis.15a
Scheme 2. Nickel/Photoredox Dual Catalysis: Mechanistic Rational.
In summary, the use of DHPs as radical precursors has allowed the successful and general introduction of an interesting feedstock into the dual Ni/photoredox cross-coupling toolbox. Because DHPs are derived from their corresponding, commercially available aldehydes, previously unrepresented radicals can be accessed, thereby expanding the chemical space of Csp2–Csp3 cross-couplings. Importantly, the reaction is characterized by its sustainability, as the only metal introduced in the entire process is the base metal cross-coupling catalyst. The transformation furthermore proceeds under extremely mild reaction conditions using visible light, allowing the inclusion of diverse and unexplored coupling partners, such as carbohydrate cores.31
Acknowledgments
We sincerely thank P. Carroll (University of Pennsylvania) for X-ray crystallographic data, C. W. Ross III (University of Pennsylvania) for HRMS analysis, and J. Gu (University of Pennsylvania) for NMR advice. Similarly, Johnson Matthey is acknowledged for a gift of metal sources.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b02786.
Characterization data for the X-ray crystal structures of [Ni(dtbbpy)(H2O)4]Cl2 (CIF)
Characterization data for compound 3lf (CIF)
Experimental details for the synthesis of 1,4-dihydropyridines, cyclic voltammetry measurements, and procedures for all synthetic studies. Copies are provided of the 1H- and 13C-NMR spectra of all materials prepared (PDF)
We thank the National Institutes of Health (No. NIGMS 1 R01 GM113878) for support.
The authors declare no competing financial interest.
Supplementary Material
References
- a Corbet J.-P.; Mignani G. Chem. Rev. 2006, 106, 2651–2710. 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]; b Brown D. G.; Boström J. J. Med. Chem. 2016, 59, 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]; c Johansson Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]; d Chemler S. R.; Trauner D.; Danishefsky S. J. Angew. Chem., Int. Ed. 2001, 40, 4544–4568. . [DOI] [PubMed] [Google Scholar]
- a Jana R.; Pathak T. P.; Sigman M. S. Chem. Rev. 2011, 111, 1417–1492. 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lennox A. J. J.; Lloyd-Jones G. C. Angew. Chem., Int. Ed. 2013, 52, 7362–7370. 10.1002/anie.201301737. [DOI] [PubMed] [Google Scholar]
- a Tellis J. T.; Kelly C. B.; Primer D. N.; Jouffroy M.; Patel N. R.; Molander G. A. Acc. Chem. Res. 2016, 49, 1429–1439. 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shaw M. H.; Twilton J.; MacMillan D. W. C. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tellis J. C.; Amani J.; Molander G. A. Org. Lett. 2016, 18, 2994–2997. 10.1021/acs.orglett.6b01357. [DOI] [PubMed] [Google Scholar]; b Karimi-Nami R.; Tellis J. C.; Molander G. A. Org. Lett. 2016, 18, 2572–2575. 10.1021/acs.orglett.6b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Amani J.; Sodagar E.; Molander G. A. Org. Lett. 2016, 18, 732–735. 10.1021/acs.orglett.5b03705. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ryu D.; Primer D. N.; Tellis J. C.; Molander G. A. Chem.—Eur. J. 2016, 22, 120–123. 10.1002/chem.201504079. [DOI] [PubMed] [Google Scholar]; e El Khatib M.; Serafim R. A. M.; Molander G. A. Angew. Chem., Int. Ed. 2016, 55, 254–258. 10.1002/anie.201506147. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Yamashita Y.; Tellis J. C.; Molander G. A. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12026–12029. 10.1073/pnas.1509715112. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294–3297. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195–2198. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Corce V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414–11418. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]; b Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475–478. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Patel N. R.; Kelly C. K.; Jouffroy M.; Molander G. A. Org. Lett. 2016, 18, 764–767. 10.1021/acs.orglett.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jouffroy M.; Kelly C. K.; Molander G. A. Org. Lett. 2016, 18, 876–879. 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Leveque C.; Chenneberg L.; Corce V.; Goddard J.-P.; Ollivier C.; Fensterbank L. Org. Chem. Front. 2016, 3, 462–465. 10.1039/C6QO00014B. [DOI] [Google Scholar]; f Patel N. R.; Molander G. A. J. Org. Chem. 2016, 81, 7271–7275. 10.1021/acs.joc.6b00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; For anhydrides as radical precursors, see:; b Le C.; MacMillan D. W. C. J. Am. Chem. Soc. 2015, 137, 11938–11941. 10.1021/jacs.5b08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang P.; Le C.; MacMillan D. W. C. J. Am. Chem. Soc. 2016, 138, 8084–8087. 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Duan Z.; Li W.; Lei A. Org. Lett. 2016, 18, 4012–4015. 10.1021/acs.orglett.6b01837. [DOI] [PubMed] [Google Scholar]
- a Shaw M. H.; Shurtleff V. W.; Terrett J. A.; Cuthbertson J. D.; MacMillan D. W. C. Science 2016, 352, 1304–1308. 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the use of tertiary alcohol feedstock as radical precursors, see:Nawrat C. C.; Jamison C. R.; Slutskyy Y.; MacMillan D. W. C.; Overman L. E. J. Am. Chem. Soc. 2015, 137, 11270–11273. 10.1021/jacs.5b07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh E. N. G.; Waugh M. W. ACS Catal. 2013, 3, 2515–2521. and references therein 10.1021/cs400637t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Berman J. D.; Stanley J. H.; Sherman W. V.; Cohen S. G. J. Am. Chem. Soc. 1963, 85, 4010–4013. 10.1021/ja00907a022. [DOI] [Google Scholar]; b Winstein S.; Seubold F. H. Jr. J. Am. Chem. Soc. 1947, 69, 2916–2917. 10.1021/ja01203a513. [DOI] [Google Scholar]; c Harris E. F. P.; Waters W. A. Nature 1952, 170, 212–213. 10.1038/170212a0. [DOI] [Google Scholar]; d Dénès F.; Pichowicz M.; Povie G.; Renaud P. Chem. Rev. 2014, 114, 2587–2693. 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]; For examples not involving thiyl radical mediation, see:; e Esposti S.; Dondi D.; Fagnoni M.; Albini A. Angew. Chem., Int. Ed. 2007, 46, 2531–2534. 10.1002/anie.200604820. [DOI] [PubMed] [Google Scholar]; f Tsujimoto S.; Iwahama T.; Sakaguchi S.; Ishii Y. Chem. Commun. 2001, 2352–2353. 10.1039/b107548a. [DOI] [PubMed] [Google Scholar]
- a Watanabe E.; Kaiho A.; Kusama H.; Iwasawa N. J. Am. Chem. Soc. 2013, 135, 11744–11747. 10.1021/ja406028c. [DOI] [PubMed] [Google Scholar]; b Ellington B. R.; Paul B.; Das D.; Vitek A. K.; Zimmerman P. M.; Marsh N. G. ACS Catal. 2016, 6, 3293–3300. 10.1021/acscatal.6b00592. [DOI] [Google Scholar]; c Shokri A.; Que L. Jr. J. Am. Chem. Soc. 2015, 137, 7686–7691. 10.1021/jacs.5b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Cho J.; Sarangi R.; Nam W. Acc. Chem. Res. 2012, 45, 1321–1330. 10.1021/ar3000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatgilialoglu C.; Crich D.; Komatsu M.; Ryu I. Chem. Rev. 1999, 99, 1991–2070. 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]
- For representative examples, see:; a Tewari N.; Dwivedi N.; Tripathi R. Tetrahedron Lett. 2004, 45, 9011–9014. 10.1016/j.tetlet.2004.10.057. [DOI] [Google Scholar]; b Adibi H.; Samimi H. A.; Beygzadeh M. Catal. Commun. 2007, 8, 2119–2124. 10.1016/j.catcom.2007.04.022. [DOI] [Google Scholar]; c Heydari A.; Khaksar S.; Tajbakhsh M.; Bijanzadeh H. R. J. Fluorine Chem. 2009, 130, 609–614. 10.1016/j.jfluchem.2009.03.014. [DOI] [Google Scholar]
- a Zhang D.; Wu L.-Z.; Zhou L.; Han X.; Yang Q.-Z.; Zhang L.-P.; Tung C.-H. J. Am. Chem. Soc. 2004, 126, 3440–3441. 10.1021/ja037631o. [DOI] [PubMed] [Google Scholar]; b Wei X.; Wang L.; Jia W.; Du S.; Wu L.; Liu Q. Chin. J. Chem. 2014, 32, 1245–1250. 10.1002/cjoc.201400521. [DOI] [Google Scholar]; c Fukuzumi S.; Suenobu T.; Patz M.; Hirasaka T.; Itoh S.; Fujitsuka M.; Ito O. J. Am. Chem. Soc. 1998, 120, 8060–8068. 10.1021/ja9813459. [DOI] [Google Scholar]; d Wang D.; Liu Q.; Chen B.; Zhang L.; Tung C.; Wu L. Chin. Sci. Bull. 2010, 55, 2855–2858. 10.1007/s11434-010-3143-2. [DOI] [Google Scholar]
- Nakajima K.; Nojima S.; Sakata K.; Nishibayashi Y. ChemCatChem 2016, 8, 1028–1032. 10.1002/cctc.201600037. [DOI] [Google Scholar]
- Chen W.; Liu Z.; Tian J.; Li J.; Ma J.; Cheng X.; Li G. J. Am. Chem. Soc. 2016, 138, 12312–12315. 10.1021/jacs.6b06379. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for further details.
- The value is in agreement with the previously reported: Eox = +0.654 V (Fc+/Fc) for 1a. See:Cheng J.-P.; Lu Y.; Zhu X.-Q.; Sun Y.; Bi F.; He J. J. Org. Chem. 2000, 65, 3853–3857. 10.1021/jo991145v. [DOI] [PubMed] [Google Scholar]
- Interestingly, 1a-CN (Eox = +1.34 V (SCE)) did not succeed in delivering the product. After oxidative fragmentation, the carbon-centered radical appears to react preferentially with the pyridine byproduct, delivering the 4-alkylated pyridine, which was observed in quantitative yield. See the Supporting Information for further details.
- Luo J.; Zhang J. ACS Catal. 2016, 6, 873–877. 10.1021/acscatal.5b02204. [DOI] [Google Scholar]
- [Ni(dtbbpy)(H2O)4]Cl2 was prepared using 1.0 equiv of NiCl2(H2O)6 and 1.0 equiv of dtbbpy (see the Supporting Information for detailed procedure and X-ray crystal structure). [Ni(dtbbpy)(H2O)4]Cl2 is a bench-stable complex that simplifies the reaction setup, making it more user-friendly.
- Ohkubo K.; Mizushima K.; Iwata R.; Souma K.; Suzuki S.; Fukuzumi S. Chem. Commun. 2010, 46, 601–603. 10.1039/B920606J. [DOI] [PubMed] [Google Scholar]
- a Legault C. Y.; Garcia Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2007, 129, 12664–12665. 10.1021/ja075785o. [DOI] [PubMed] [Google Scholar]; b Garcia Y.; Schoenebeck F.; Legault C. Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 6632–6639. 10.1021/ja9004927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Friesen R. W.; Sturino C. F. J. Org. Chem. 1990, 55, 2572–2574. 10.1021/jo00296a005. [DOI] [Google Scholar]; b Dubois E.; Beau J.-M. Tetrahedron Lett. 1990, 31, 5165–5168. 10.1016/S0040-4039(00)97832-8. [DOI] [Google Scholar]; c Friesen R. W.; Sturino C. F.; Daljeet A. K.; Kolaczewska A. J. Org. Chem. 1991, 56, 1944–1947. 10.1021/jo00005a053. [DOI] [Google Scholar]
- a Sakamaki S.; Kawanishi E.; Nomura S.; Ishikawa T. Tetrahedron 2012, 68, 5744–5753. 10.1016/j.tet.2012.05.035. [DOI] [Google Scholar]; b Parkan K.; Pohl R.; Kotora M. Chem.—Eur. J. 2014, 20, 4414–4419. 10.1002/chem.201304304. [DOI] [PubMed] [Google Scholar]
- Electron-rich aryl bromides could not be coupled because of a demanding oxidative addition step under the developed reaction conditions, resulting in recovered starting material. On the other hand, in those cases with moderate yields, the formation of aryl chloride or protodebrominated starting material was observed, thus indicating that the more demanding steps in those cases was not the oxidative addition.
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896–4899. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of Ni(I)-promoted oxidative addition into C–X bonds see:; a Anderson T. J.; Jones G. D.; Vicic D. A. J. Am. Chem. Soc. 2004, 126, 8100–8101. 10.1021/ja0483903. [DOI] [PubMed] [Google Scholar]; b Jones G. D.; McFarland C.; Anderson T. J.; Vicic D. A. Chem. Commun. 2005, 4211–4213. 10.1039/b504996b. [DOI] [PubMed] [Google Scholar]; c Anton M.; Clos N.; Muller G. J. Organomet. Chem. 1984, 267, 213–219. 10.1016/0022-328X(84)80177-1. [DOI] [Google Scholar]
- At this point, we cannot rule out a mechanistic scenario consisting of an oxidative addition of Ni(0) complex IV with the aryl bromide 2 followed by radical addition, delivering Ni(III) complex VI.
- While this work was under revision, Nishibayashi et al. reported the cross-coupling of aryl iodides with DHPs under Ni/photoredox dual catalysisNakajima K.; Nojima S.; Nishibayashi Y.. Angew. Chem., Int. Ed. 2016, in press, DOI: 10.1002/anie.201606513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.