

Abstract
β-lapachone (β-lap) is a novel anticancer agent that selectively induces cell death in human cancer cells, by activation of the NQO1 NAD(P)H dehydrogenase and radical oxygen species (ROS) generation. We characterized the gene expression profile of budding yeast cells treated with β-lap using cDNA microarrays. Genes involved in tolerance to oxidative stress were differentially expressed in β-lap treated cells. β-lap treatment generated reactive oxygen species (ROS), which were efficiently blocked by dicoumarol, an inhibitor of NADH dehydrogenases. A yeast mutant in the mitocondrial NADH dehydrogenase Nde2p was found to be resistant to β-lap treatment, despite inducing ROS production in a WT manner. Most interestingly, DNA damage responses triggered by β-lap were abolished in the nde2Δ mutant. Amino acid biosynthesis genes were also induced in β-lap treated cells, suggesting that β-lap exposure somehow triggered the General Control of Nutrients (GCN) pathway. Accordingly, β-lap treatment increased phosphorylation of eIF2α subunit in a manner dependent on the Gcn2p kinase. eIF2α phosphorylation required Gcn1p, Gcn20p and Nde2p. Gcn2p was also required for cell survival upon exposure to β-lap and to elicit checkpoint responses. Remarkably, β-lap treatment increased phosphorylation of eIF2α in breast tumor cells, in a manner dependent on the Nde2p ortholog AIF, and the eIF2 kinase PERK. These findings uncover a new target pathway of β-lap in yeast and human cells and highlight a previously unknown functional connection between Nde2p, Gcn2p and DNA damage responses.
Keywords: antitumoral drug, DNA damage, integrated stress response, reactive oxygen species
Abbreviations
- β-lap
β-lapachone
- ROS
Reactive Oxygen Species
- ISR
Integrated Stress Response
- GO
Gene Ontology
- GCN
General Control of Nutrients
Introduction
Having accumulated mutations that overcome cell-cycle and apoptotic checkpoints, the main obstacle to survival faced by a cancer cell is the restricted supply of nutrients and oxygen. Positron-emission tomography (PET) imaging of various solid tumors, including head and neck cancer, soft tissue sarcoma, breast cancer, and glioblastoma multiforme, has revealed that they all have some degree of hypoxia.1 Although tumors secrete angiogenic factors to promote vasculature growth, this often is not sufficient to provide optimal oxygen and nutrients to the tumor or dispose of wastes.2 Therefore, in addition to hypoxia, cells in a developing tumor can also experience low glucose, pH and amino-acid supplies. Hypoxic tumor cells are known to be more resistant to current treatment modalities3 and numerous studies have shown that the low oxygen availability within solid tumors correlates with a poor clinical outcome.4,5 Therefore, understanding the mechanisms which tumor cells employ to survive adverse conditions is crucial to the development of more effective therapies.
Under nutrient deprivation conditions, eukaryotic cells transiently inhibit protein synthesis to avoid misfolding of proteins which could compromise cell viability. This response forms part of a protective mechanism triggered by several stimuli and is generically known as “integrated stress response” (ISR).6 Translation inhibition is achieved by posttranslational modification of the eukaryotic translation initiation factor 2 (eIF2). Nutrient stress activates protein kinases that phosphorylate eIF2 in Ser51 of its α subunit (eIF2α).6 Phosphorylated eIF2α behaves as a competitive inhibitor of the eIF2B exchanger, thus blocking eIF2 recycling. As the concentration of eIF2 normally exceeds that of eIF2B, very modest increases of eIF2α phosphorylation can completely inhibit protein synthesis initiation and favor selective translation of some mRNAs.6 Some of these mRNAs encode transcription factors that regulate a gene expression program that allows cells to adapt to nutrient stress.7
ISR is a well-conserved pathway throughout evolution. In budding yeast, Gcn2p protein kinase is the responsible of phosphorylating eIF2α at serine 51 under amino-acid deprivation.8 Stimuli other than amino acid have been reported to trigger Gcn2p, such as purine starvation,9 glucose limitation and ethanol,10 high salinity,11 rapamycin12 and volatile anesthetics.13 In mammals, mGcn2p is the kinase in charge of responding to nutrient deprivation, but there are 3 other additional eIF2α kinases responding to several stresses: PERK (misfolded proteins in the ER), PKR (double-stranded RNA) and HRI (heme deprivation) (reviewed in14). Interestingly, ISR activation has been found linked to DNA damage in both yeast 15,16 and mammals.17,18
β-lapachone (β-lap), a natural product from the lapacho tree (Tabebuia avellaneda), induces cell death in a wide spectrum of human cancer cells but not in normal cells.19 Potent anti-tumor activity in xenografted human cancer models has also been reported19,20. Although β-lap inhibits the catalytic activity of topoisomerase I, the concentrations required are above the ones that induce apoptosis19. Its cytotoxicity has also been reported to be dependent on the activity of NADPH:Quinone oxidoreductase enzyme NQO121. However, β-lap treatment triggered apoptosis in cells that are deficient in NQO121. We have previously reported that β-lap activates in yeast a G1/S checkpoint in a Mec1p/Tel1p and XMR complex related fashion, what could be of significance for β-lap anti-tumor activity, given the conserved nature of this checkpoint.22
Here we present a challenge for the reported mechanism of action of β-lap. Inhibition of ROS production induced by β-lap in yeast did not relieve the G1/S checkpoint activation. Moreover, a mutant lacking the dehydrogenase Nde2p showed tolerance to the compound without abolishing ROS production. IRS activation is demonstrated to mediate β-lap action, as mutants for this pathway modulate its toxicity. Furthermore, the dehydrogenase Nde2p is necessary for β-lap-induced full activation of IRS, this way linking 2 pathways previously unrelated. Finally, our study shows that these responses are as significant in mammalian cells as in yeast.
Results
Analysis of β-lap transcriptomal program triggered in yeast
We have previously reported that β-lap was cytotoxic in budding yeast and triggered the DNA damage-dependent Mre11-Tel1p checkpoint in yeast cells.22 This finding was later confirmed in human cells,23 thus validating our experimental approach. To gain further insights into the molecular mechanism of β-lap action, we analyzed the gene expression profile of yeast cells treated with β-lap using cDNA microarrays. We used Significance Analysis of Microarrays to identify differentially expressed genes between β-lap/vehicle treated yeast cells. Any given gene was considered to be differentially regulated when its expression was at least 1.5 fold higher o lower in the β-lap treated cells versus untreated cells. Following these criteria, 212 genes were induced and 52 genes were repressed in β-lap treated cells. We performed a functional classification of induced/repressed genes according to Gene Ontology (GO) terms using the GO Term Finder tool (Table 1). In the set of differentially expressed genes the “oxidative stress response” functional category was clearly represented. Within this category, most of the genes were categorized in the “cellular response to oxidative stress” and “cell redox homeostasis” processes, and mainly functionally grouped as “oxidoreductase activity”. As β-lap cytotoxity in tumor cells was reported to be associated to NQO1-mediated ROS generation, these categories were somehow expected. Surprisingly, another prominent functional category overrepresented was “amino acid biosynthesis” (Table 1), partially overlapping and including other related categories as “amine biosynthesis”, “amino acid metabolism” and many biosynthetic pathways for each amino acid in particular. This noteworthy representation suggested that the drug was altering amino acid homeostasis.
Table 1.
A. Functional classification of β-lap differentially induced genes
| Biological process | p-value |
|---|---|
| Amine biosynthesis | 1.6 exp-11 |
| Amino acid biosynthesis | 1.6 exp-11 |
| Amino acid metabolism | 9.3 exp-10 |
| Biosynthesis of amino acids of the glutamine family | 6.1 exp-08 |
| Lysine metabolism | 5.3 exp-07 |
| Lysine biosynthesis | 6.7 exp-07 |
| Arginine biosynthesis | 1.6 exp-06 |
| Arginine metabolism | 1.6 exp-06 |
| Metabolism of amino acids of the glutamine family | 1.9 exp-05 |
| Metabolism of amino acids of the aspartate family | 1.0 exp-02 |
| Metabolism of non protein amino acids | 1.3 exp-02 |
| Catabolism of amino acids | 1.5 exp-02 |
| Response to reactive oxygen species | 1.8 exp-02 |
| Biosynthetic processes of amino acids of the aspartate family | 2.3 exp-02 |
| Amine catabolic processes | 2.9 exp-02 |
| Metabolism of ornithine | 3.6 exp-02 |
| Biosynthesis of glutamate | 3.9 exp-02 |
| B. Functional classification of β-lap differentially repressed genes. | |
| Transport of electrons | 1.2 exp-02 |
β-lap cytotoxicity is not mediated by ROS production in yeast
To validate the findings observed in the microarrays experiment, we measured the ability of β-lap treatment to generate ROS in budding yeast. As expected, β-lap treatment induced ROS in yeast cells (Fig. 1A), which were efficiently inhibited by dicoumarol, a competitive inhibitor of NQO1 NADH dehydrogenase, further extending the parallelism between yeast and tumor cells. Analysis of the differentially expressed genes using the YEASTRACT analysis software, revealed that most of the regulated genes were under the control the Yap1p (38.8% of the analyzed genes) and Skn7p (24.4%) transcription factors, known to mediate transcription in response to oxidative stress. Accordingly, yeast yap1Δ and skn7Δ mutants, but not an unrelated tor1Δ mutant, were hypersensitive to β-lap treatment (Fig. 1B). Surprisingly, preincubation with dicoumarol did not alleviate β-lap cytotoxicity in any strain tested (Fig. 1B), suggesting that β-lap cytoxicity was due to mechanisms other than oxidative stress.
Figure 1.
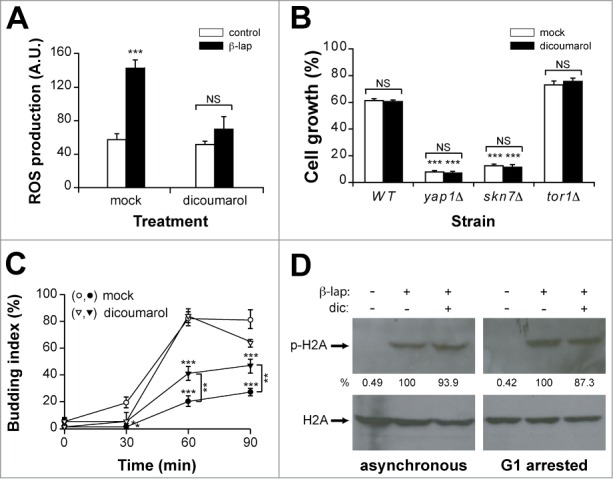
Oxidative stress does not mediate cell responses to β-lap in yeast. (A) ROS production in a WT strain treated with or without β-lap in the presence or absence (mock) of dicoumarol. (B) Growth of the WT, yap1Δ, skn7Δ and tor1Δ strains treated during one hour with β-lap, in the presence or absence of dicoumarol. (C) G1/S progression of a WT strain synchronised in G1 with α-factor and treated (closed symbols) or not (open symbols) with β-lap during 1 hour in the presence (triangles) or absence (circles) of dicoumarol. (D) Immunodetection of phospho-H2A (p-H2A) in asynchronous and G1-arrested cultures of the WT strain treated with β-lap ± dicoumarol. Even loading of the gels was confirmed by immunodetection of H2A protein levels. Quantitation of the phosphorylated band is shown below as a percent of the intensity of the highest signal, arbitrary assigned as 100. β-lap: 10 μg/ml; dicoumarol: 50 μM.
We reported previously that in budding yeast β-lap treatment triggered a G1/S checkpoint and increased histone H2A phosphorylation.22 We explored whether ROS generation was involved in these responses, preincubating cells with dicoumarol before β-lap exposure. Preincubation with dicoumarol partially relieved but did not eliminate the checkpoint responses triggered by β-lap exposure (Fig. 1C). Furthermore, β-lap-induced histone H2A phosphorylation remained unaffected by dicoumarol either in asynchronous or G1 arrested cells (Fig. 1D). Taken together these data indicated that β-lap treatment generated ROS in yeast cells, which were permissive to either β-lap cytotoxicity or the DNA damage response triggered by the drug. These findings clearly challenged the reported mechanism of drug action.
Nde2p mediates β-lap responses in yeast
The fact that dicoumarol, an NADH analog and a competitive inhibitor of NADH dehydrogenases, efficiently blocked ROS generation, suggested the involvement of a NADH oxidoreductase in ROS production. To identify such target we screened β-lap toxicity in a set of strains deleted for the 9 nonessential candidate NADH oxidoreductases in yeast, which could somehow mediate β-lap toxicity. These included Oye2,3p enzymes, the mitochondrial Nde1p, −2p, Ndi1p oxidoreductases, the Cbr5p and Mcr1p cytochrome b oxidoreductases, and the Lpd1 encoding dihydrolipoamide dehydrogenase. Interestingly, the nde2Δ mutant strain was resistant to β-lap cytotoxicity (Fig. 2A). This resistance was not due to a decrease in ROS production, as ROS levels generated by β-lap treatment in the nde2Δ mutant were similar to the WT (Fig. 2B). Surprisingly, β-lap-induced ROS production was significantly lower in the oye2Δ mutant strain. This result indicated that i) the tolerance of the nde2Δ mutant to β-lap was independent of oxidative stress and ii) Oye2p dehydrogenase partially mediated ROS generation in yeast.
Figure 2.
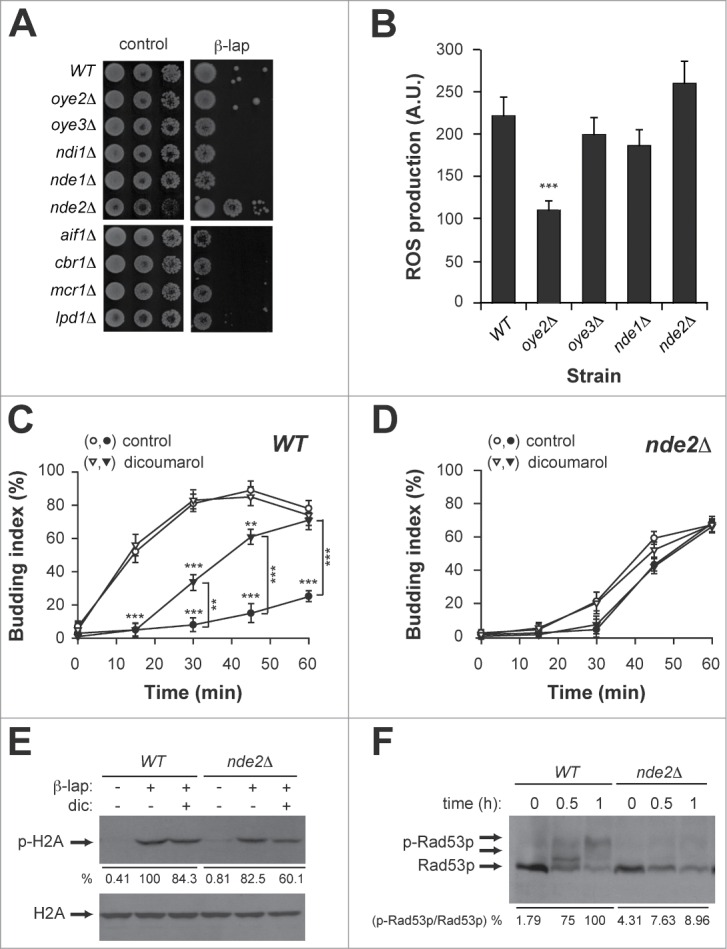
The NADH oxidoreductase Nde2p mediates cell responses to β-lap in yeast. (A) Drop test of NADH oxidorreductase deletion strains and their isogenic WT treated with β-lap (B) ROS production in WT, oye2Δ, oye3Δ, nde1Δ, and nde2Δ strains treated with β-lap. (C and D) G1/S progression of WT (C) and nde2Δ (D) cultures synchronised in G1 with α-factor in the presence (closed symbols) or absence (open symbols) of β-lap and dicoumarol. (E) Immunodetection of phospho-H2A (p-H2A) in WT and nde2Δ strains treated with β-lap ± dicoumarol. Even loading of the gels was confirmed by immunodetection of total H2A protein. Quantitation of the phosphorylated band is shown below as a percent of the intensity of the highest signal, arbitrary assigned as 100. (F) Immunodetection of phospho-Rad53p (p-Rad53p) levels in WT and nde2Δ strain treated with β-lap for the indicated times. Quantitation of the phosphorylated bands is shown below as a percent of the intensity of the highest signal (expressed as relationship between phosphorylated and no-phosphorylated forms), arbitrary assigned as 100. β-lap: 10 μg/ml; dicoumarol: 50 μM.
We next explored the β-lap dependent checkpoint responses in the WT and nde2Δ mutant strains. To do so, G1-synchronised WT and nde2Δ strains were treated with β-lap for 1 hour and then released into S-phase. Remarkably, the nde2Δ mutant already exhibited a G1 delay in the G1/S transition, which was not further exacerbated by β-lap treatment regardless the presence or absence of dicoumarol (Fig. 2C, D). β-lap-induced histone H2A phosphorylation was only partially affected in the nde2Δ mutant (Fig. 2E). However, β-lap treatment failed to trigger rad53p phosphorylation in the nde2Δ mutant (Fig. 2F). Therefore, an intact Nde2p was required for the DNA damage response and cytotoxicity triggered by β-lap.
The GCN pathway mediates β-lap cytotoxicity
As mentioned above, one of the functional categories significantly enriched in the set of differentially induced genes was “amino acid biosynthesis”. In fact, a transcription factor revealed by YEASTRACT tool analysis was Gcn4p (25.5% of the analyzed genes). In yeast, amino acid starvation favors the accumulation of uncharged tRNAs and triggers the GCN pathway, a nutrient-sensing pathway that transiently inhibits translation initiation and, simultaneously, favors selective translation of the GCN4 mRNA. The major transducer of the GCN pathway is the Gcn2p kinase that phosphorylates eIF2 in its α-subunit in response to amino acid limitation. Gcn1p and Gcn20p are adaptor proteins that bind to ribosomes and interact with Gcn2p, mediating its activation by uncharged tRNAs.6-9
During the first steps of translation initiation, the initiator methionyl tRNA is recruited to the small ribosomal subunit in a ternary complex containing activated, GTP-bound eIF2. AUG recognition triggers GTP hydrolysis and dissociation of inactive eIF2-GDP. The binary complex eIF2-GDP dissociates slowly and eIF2 binds more tightly to GDP than GTP. eIF2B is a heteropentameric guanine nucleotide exchange factor that recycles eIF2 from a GDP- to a GTP-bound form that is competent for translation initiation. eIF2B comprises a regulatory subcomplex composed of Gcd2p, Gcd7p, and Gcn3p, and a catalytic subcomplex containing Gcd1p and Gcd6p. The regulatory complex (Gcd2p/Gcd7p/Gcn3p) interacts stably with eIF2 with a binding preference for phosphorylated eIF2 (p-eIF2α) which, in its GDP-bound form, is an inhibitor of eIF2B. Gcd2p, Gcd7p, and Gcn3p together mediate this inhibition of eIF2B activity by p-eIF2α.24,25
In response to amino acid limitation Gcn2p is activated and produces p-eIF2α at levels that block (but not fully) general translation but selectively increase translation of GCN4 mediated by short open reading frames (uORFs) in the leader of GCN4 mRNA.
To explore whether β-lap treatment activated the GCN pathway we measured the phosphorylation status of eIF2α in WT yeast cells by western blot using a commercially available polyclonal antibody that specifically recognizes eIF2α phosphorylated at serine 51 (see methods). We observed that β-lap exposure incremented eIF2α phosphorylation in WT yeast cells (Fig. 3A). As expected, eIF2α phosphorylation was dependent on Gcn2p, as well as Gcn1-20p (Fig. 3A). Taken together, these data indicated that β-lap somehow was able to induce a stress condition, which triggered the activation of the GCN pathway.
Figure 3.
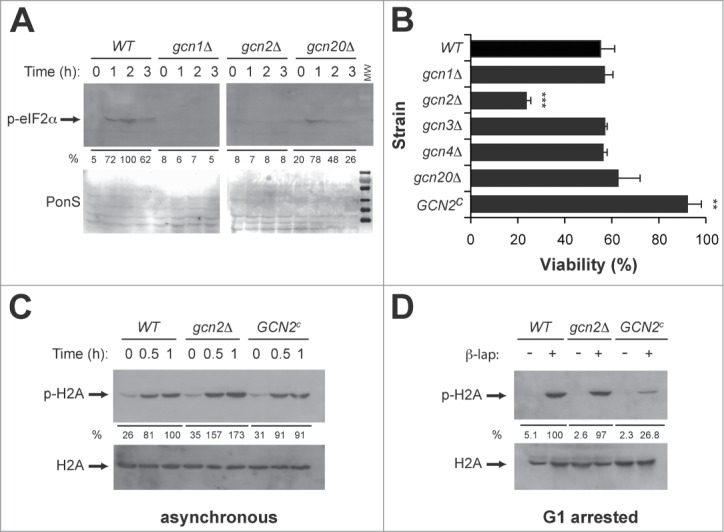
The Gcn2p kinase mediates β-lap toxicity in yeast. (A) Immunodetection of phospho-eIF2α (p-eIF2α) in WT, gcn1Δ, gcn2Δ and gcn20Δ strains treated with β-lap at the indicated times. Even loading of the gels was confirmed by PonceauS staining of membranes after transfer. Quantitation is shown below as a percent of the intensity of the highest signal, arbitrary assigned as 100. (B) Viability of the WT and GCN pathway mutant strains (gcn1Δ, gcn2Δ, gcn3Δ, gcn4Δ, gcn20Δ and GCN2c) treated during one hour with β-lap. (C and D) Immunodetection of phospho-H2A (p-H2A) in asynchronous (C) and G1-arrested (D) cultures of WT, nde2Δ and GCN2c strains treated with β-lap for the indicated times. Even loading of the gels was confirmed by immunodetection of total H2A protein. Quantitation is shown below as a percent of the intensity of the highest signal for WT cells, arbitrary assigned as 100.
We next addressed whether the GCN pathway was mediating β-lap toxicity, by measuring cell viability in the above-mentioned GCN mutants treated with β-lap. Interestingly only the gcn2Δ mutant was sensitive to the drug (Fig. 3B). Accordingly, a yeast strain GCN2c expressing a constitutively active mutant of the Gcn2p kinase was significantly more resistant to β-lap than the WT (Fig. 3B). These data indicated that the Gcn2p kinase specifically and not a fully functional GCN pathway mediated β-lap toxicity. We then analyzed the involvement of Gcn2p in β-lap dependent checkpoint responses. Thus, we measured β-lap-induced histone H2A phosphorylation, in asynchronous (Fig. 3C) and G1 arrested (Fig. 3D) WT, gcn2Δ and GCN2c strains. Remarkably, histone H2A phosphorylation was significantly decreased in the GCN2c gain-of-function mutant, both in asynchronous/G1 arrested cultures. Indeed, in absence of Gcn2p β-lap treatment promotes higher levels H2A phosphorylation in asynchronous culture. Therefore, Gcn2p was required both for the cytotoxicity and for the DNA damage responses triggered by exposure to β-lap.
Given the fundamental nature of the GCN pathway, and the activation of Gcn2p by β-lap in yeast, we analyzed the eIF2α phosphorylation status in human MCF7 breast cancer cells treated with the drug. As shown, β-lap treatment efficiently increased eIF2α phosphorylation in MCF7 cells (Fig. 4B). Interestingly, eIF2α phosphorylation correlated with β-lap cytotoxicity (Fig. 4A). In human cells, phosphorylation of eIF2α is controlled by 4 protein kinases: the double-stranded (ds) RNA-activated protein kinase (PKR), the hemin-regulated inhibitor kinase (HRI), the pancreatic eIF2α or PKR-like endoplasmic reticulum (ER)-related kinase (PEK/PERK), and the general control nonderepressible-2 (GCN2) kinase. Each kinase is activated by distinct stresses that decrease protein synthesis by an appropriate response: PKR by double-stranded RNA [dsRNA], HRI by heme deficiency, PEK/PERK by misfolded proteins in the ER, and GCN2 by amino acid deprivation/UV irradiation. To identify the eIF2α kinase involved in β-lap cytotoxicity, we tested β-lap cytotoxicity in GCN2-/- and PERK-/- knockout mice embryonic fibroblasts (MEFs). Remarkably, PERK-/- (Fig. 4D) but not GCN2-/- (Fig. 4C) MEFs were highly sensitive to β-lap treatment. Therefore, β-lap treatment activated the ISR pathway in human cells, being PERK kinase the major determinant of β-lap cytotoxicity, thus validating our findings in yeast.
Figure 4.
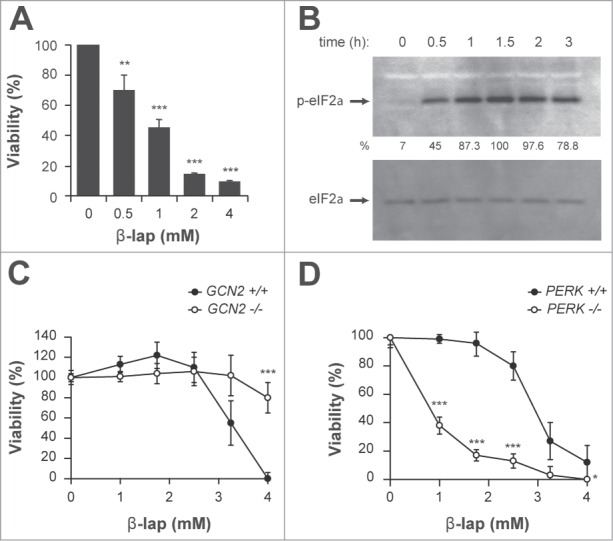
The GCN pathway mediates β-lap toxicity in human and mouse cells. (A) Cell viability of MCF-7 cells treated with increasing doses of β-lap. (B) Immunodetection of phospho-eIF2α (p-eIF2α) in MCF-7 cells treated with 4 μg/ml of β-lap for the indicated times. Even loading of the gels was confirmed by total eIF2α immunodetection. Quantitation is shown below as a percent of the intensity of the highest signal, arbitrary assigned as 100. (C and D) Cell viability of mouse embrionyc fibroblasts (MEFs) of GCN2−/− (C) and PERK −/− (D) knock-out mice and their correspondent wild-type cell line treated with the indicated doses of β-lap.
β-lap requires Nde2p for full Gcn2p activation and eIF2 phosphorylation
As both Nde2p and Gcn2p were required for β-lap toxicity in yeast, we next evaluated whether they were somehow functionally connected. We analyzed the status of eIF2α phosphorylation in β-lap treated WT and nde2Δ cells. We showed that β-lap dependent eIF2α phosphorylation dramatically decreased in nde2Δ cells (Fig. 5A). The closest human orthologues of Nde2p are the AIF and AMID NADH dehydrogenases by sequence comparison. To test whether β-lap induced phosphorylation of eIF2α was dependent on AIF and/or AMID, we downregulated AIF and AMID expression by transfecting MCF7 cells with specific siRNAs for their respective mRNAs. Transfection with either siAIF1 or siAMID siRNAs significantly decreased AIF1 and AMID mRNA levels as measured by quantitative real time PCR (Fig. 5B, C). Remarkably, β-lap dependent eIF2α phoshorylation was markedly reduced in siAIF1 silenced MCF7 cells but not in siAMID cells (Fig. 5D) or cells transfected with a control unrelated siRNA (siCONT, Fig. 5D). Therefore, these data revealed a conserved functional link between Nde2p and Gcn2p, which was conserved throughout evolution.
Figure 5.
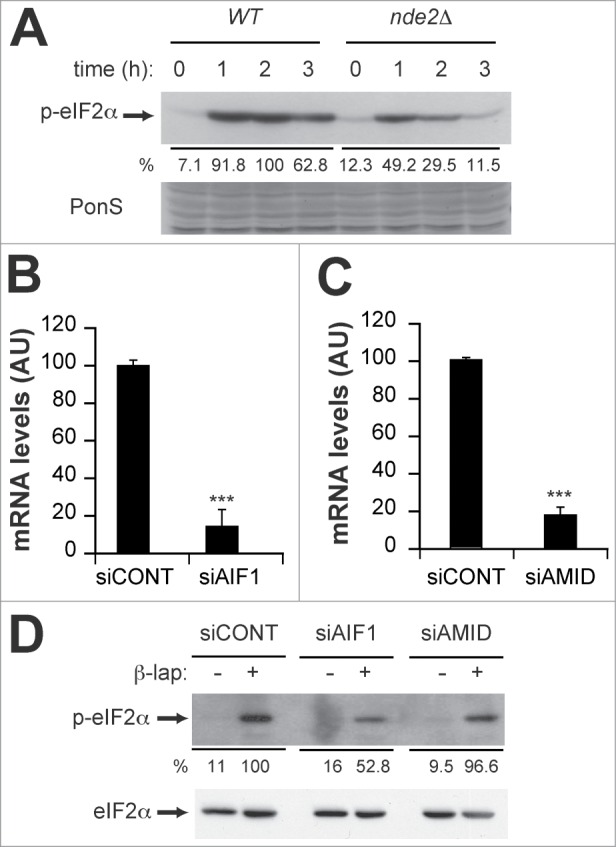
β-lap requires Nde2p/Aif1p for full GCN pathway activation. (A) Immunodetection of phospho-eIF2α (p-eIF2α) in WT and nde2Δ strains treated with 4 μg/ml β-lap for the indicated times. Even loading of the gels was confirmed by PonceauS staining of the membranes after transfer. Quantitation is shown below as a percent of the intensity of the highest signal, arbitrary assigned as 100. (B and C) Real-time PCR quantitation of AIF1 (B) and AMID (C) mRNA levels in MCF-7 cells transfected with control (siCONT), AIF1 (siAIF1) and AMID (siAMID) siRNAs, respectively. (D) Immunodetection of phospho-eIF2α (p-eIF2α) in MCF-7 cells transfected with control (siCONT), AIF1 (siAIF1) and AMID (siAMID) siRNAs, treated or not with 4 μg/ml β-lap. Even loading of the gels was confirmed by immunodetection of total eIF2α protein. Quantitation is shown below as a percent of the intensity of the highest signal, arbitrary assigned as 100.
Discussion
β-lapachone is a potential anticancer agent that induces cell death in human cancer cells with a wide spectrum of activity selectively inducing apoptosis in transformed cells but not in proliferating normal cells, an unusual property that is not shared by conventional chemotherapeutic agents. Initially it was reported that β-lap cytotoxicity did not involve DNA damage, but recently we and others groups have demonstrated that β-lap-induced cell death is initiated by the induction of DNA damage and checkpoint activation.22,26 To further characterize the molecular mechanism of β-lap-induced cell death, we analyzed the profile of gene expression of WT yeast cells treated with the drug. As expected, categories as ROS response and electron transport were present in this analysis (Table 1). Nevertheless, this strong response involving ROS production, although important, is not the primary cause of β-lap toxicity in budding yeast as indicated by the fact that dicoumarol does not affect viability, and it does not abolish the G1/S transition delay or histone H2A phosphorylation triggered by β-lap (Fig. 1).
It was reported that anticancer effects of β-lap are due to the NQO1-dependent oxidoreduction of the drug, resulting in a futile cycling wherein β-lap is reduced to an unstable hydroquinone that spontaneously reverts to its parent structure using 2 oxygen molecules. This cycle leads to ROS generation, DNA damage, γ-H2AX foci formation, PARP-1 hyperactivation, and subsequent loss of ATP and NAD+.27 A screening for yeast NADH oxidoreductases revealed that ROS generation is dependent on Oye2p (Fig. 2B), although deficiency in this activity does not modulate β-lap toxicity. In a similar manner it was described in Trypanosoma cruzi that TcOYE (Oye2p homolog) specifically catalyzed one-electron reduction of menadione and β-lap to semiquinone-free radicals with concomitant generation of superoxide radical anions.28
The dehydrogenase activity screening also revealed that a deficiency in Nde2p leads to resistance to β-lap treatment without affecting ROS production (Fig. 2A, B). Furthermore, this deficiency abrogated the G1/S delay and Rad53p phosphorylation normally triggered by β-lap, partially decreasing the levels of histone H2A phosphorylation observed in WT cells (Fig. 2C–F). This fact could mean that in the absence of Nde2p, β-lap treatment is still able to promote DNA damage, but this damage is either easily repaired or it has not enough magnitude to trigger a complete checkpoint response. At this regard it was proposed by Rouse and Jackson 29 that DNA damage could be either quickly repaired or it persists depending on the nature of the damage and/or the genomic context. If the lesion is not repaired quickly enough, Mec1p-Lcd1p complex is recruited and Mec1p phosphorylates targets in the vicinity of the lesion, such as H2A (local response). Then, if full repair occurs the global DNA damage response is averted. If not, a global response is triggered that includes Rad53p phosphorylation and cell-cycle arrest. Thus, the relevance of Nde2p in modulating the grade of lesion could be a reflection of its role in bioactivating the drug, working at this regard in a similar way than NQO1.
Alternatively Nde2p could play a role in the proper coupling between DNA damage detection and later events involved in slowering cycle. Supporting this idea, a Δnde2 mutant has higher repair efficiency in experiments with linearized plasmids (Fig. S1) and increased basal levels of histone H2A phosphorylation (Fig. 2E). This could count for the hypersensitivity of this strain to genotoxic agents such as phleomycin, UV or hidroxyurea (Fig. S2). In a similar way, it was proposed that NQO1 plays a role in p53 stabilization in oxidative stress conditions, mediating p53-dependent responses after DNA damage.30
Microarray analysis also indicated that a relevant category of genes induced by β-lap included amino acid biosynthesis (Table 1). This category was previously observed to be equally represented in genes differentially expressed by a mutant GCN2c with the GCN pathway constitutively active,15 suggesting a potential activation of this pathway by the drug. Exploring this possibility we observed phosphorylation of the initiation factor eIF2α in response to β-lap treatment (Fig. 3A). This phosphorylation was totally dependent on Gcn2p, and on the activators Gcn1-20p (Fig. 3A). Nevertheless, β-lap toxicity does not seem to be mediated by a functional GCN pathway, but by Gcn2p itself (Fig. 3B). Indeed, it was demonstrated that a GCN2c gain of function mutant was resistant to treatment showing a significant decrease in the level of phosphorylation of histone H2A both in asynchronous and G1 arrested cells (Fig. 3B–D). All these data suggest that β-lap activation of Gcn2p mediates toxicity in conditions where no amino acid deprivation occurs. In a similar way, it was reported before that the genotoxic agent MMS also activates Gcn2p in a Gcn1/20p dependent manner.15 It is tempting to speculate that the activation of Gcn2p by the interaction of Gcn1/20p with an anomalous structure in DNA could explain the activation of this pathway. Supporting this idea, we have observed that Gcn2p activation by β-lap is fully dependent on the Xrs2-Mre11-Rad50 checkpoint complex in a Mec1p- Tel1p independent way (Fig. S3). Once active, Gcn2p could modulate effectors that strengthen checkpoint/repair responses.
In a similar approach, we observed that β-lap also triggered phosphorylation of the initiation factor eIF2α in MCF7 breast cancer at doses that compromise cell viability (Fig. 4A, B). Our studies in mammalian cells indicated that is PERK and not GCN2 the kinase responsible of β-lap toxicity (Fig. 4C, D). At this respect, it was recently described in MDA-MB-231 cells that death induced by β-lap treatment could be mainly due to induction of ER stress.31 Although with our data we cannot discard a difference in the nature of stress induced by the drug between yeasts and mammalian cells, as it was reported that ER stress inhibitors attenuates mitochondria-mediated cell death caused by β-lap,31 it is important to remark that recent evidences suggest a strong connexion between DNA damage and ER stress.32-34 Indeed, the integrated stress and DNA damage responses are known to share components. For example, cell cycle arrest after UV irradiation is mediated in part by phosphorylation of eIF2α by GCN2 or PERK, with a mechanism of kinase activation not fully understood.35,36 It was also short time ago described that in mammalian cells, CHK1 was activated by agents that induce ER stress, which resulted in a G2 cell cycle delay.32
Finally, we explored the potential connection between Nde2p and Gcn2p, as they both were involved in modulating β-lap toxicity, demonstrating that Nde2p is required for a proper activation of Gcn2p (Fig. 5A). Actually, in mammalian cells this effect was mediated by the Nde2p ortholog AIF but not AMID (Fig. 5D). Accordingly, it was recently described in mammalian cells that β-lap treatment caused AIF translocation to nucleus.31 Moreover, siRNAs targeting AIF attenuated cell death caused by β-lap treatment, highlighting the similarity between yeast Nde2p and AIF.
Taken together all our data indicate that β-lap toxicity is mediated by a ROS-independent alternative pathway at least partially conserved in yeast and human cells. This toxicity involves the action of the dehydrogenase Nde2p and the activity of eIF2 kinase Gnc2p in yeast, being the corresponding mammalian homologues AIF and PERK. The insights on the mechanism of action of this drug uncover a novel putative link between Nde2p, Gcn2p and DNA damage response mechanisms, highlighting new potential therapeutic uses for the drug associated to these described pathways such as diabetes, aging and neurodegeneration.
Materials and Methods
Yeast growth assays
Yeast strains are described in Table S1. Standard methods for yeast culture and manipulations were used.37 YPD medium contained 2% glucose, 2% peptone and 1% yeast extract. β-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyan-5,6-dione; Sigma-Aldrich) was dissolved in DMSO and diluted in YPD at a final dose of 10 μg/ml, unless indicated. Dicoumarol (3,3′-Methylene-bis[4-hydroxycoumarin]; Sigma-Aldrich) was dissolved in mild basic water and diluted in YPD at the corresponding dose.
For analysis of cell growth by drop test, cells growing logarithmically in liquid YPD medium were 10-fold serially diluted, and volumes of around 3 μl were dropped with a stainless steel replicator (Sigma-Aldrich) on solid plates containing 2% Bacto-Agar (Pronadisa) and YPD medium with the corresponding treatments as indicated. Growth was recorded after 2–5 days in all cases.
For viability assays, exponentially growing cultures in liquid YPD were collected by centrifugation, and an equal number of cells were plated onto YPD plates containing the corresponding treatments. Colonies were quantitated after 2–5 days in all cases. Data are represented as mean ± SEM of at least 2 independent experiments, each one done in duplicate.
Microarray analysis
Yeast cells were grown to mid-log phase and treated for an hour with DMSO or 4 μg/ml β-lap. Total RNA was purified by standard procedures and labeled by incorporation through reverse transcription of 5-(3-aminoallyl)-2-deoxi-UTP (aa-dUTP) to single-stranded cDNA. Cy3 or Cy5 flurophores (Amersham) were coupled to aa-cDNA.
Microarrays were kindly supplied by Dr. J. Ariño. Hybridization was carried out manually using Telechem hybridization chambers, according to manufacturer instructions. Briefly, 30 minutes prehybridization was performed with 0,1% SDS and 0,1 mg/ml BSA. Then, hybridization was done over-night at 50°C with 0,1% SDS and 0,1 mg/ml salmon sperm DNA. Arrays were washed at 42°C and dried by centrifugation at 600 g. Microarray images were registered with a scanner (Axon Instruments) at 10 μm resolution. Then, the images were analyzed and normalized with GenePix software (Axon Instruments). Points with twice more intensity at least in a channel than average base were selected. Acuity (Axon Instruments) and/or SAM 38 were used to normalize and analyze data. One gene was considered differentially regulated when its expression was at least 1,5 higher or lower than control (FDR < 6%).
Collected data were deposited at NCBI Gene Expression Omnibus (GEO, accession number GSE15412)
The groups of genes with changes in expression regarding functional biological processes were evaluated through Gene Ontology and FatiGo plus 39 databases. For detection of genes under control of transcription factors YEASTRACT 40 database was used (http://www.yeastract.com).
G1/S checkpoint analysis
The yeast cells used in this study have all BY4741 genetic background (MATa). Cultures of these cells were arrested in G1 phase of cell cycle with 4 μg/ml mating pheromone α-factor (Sigma-Aldrich) and incubated or not with the corresponding treatments for an hour. For quantification of re-entrance in cell-cycle, cells were washed twice with saline buffer and resuspended in culture medium without α-factor. At time points indicated, 1 ml samples were harvested and fixed with 75 μL formaldehyde. The percentage of budded cells (G1 assay) was scored by microscopic observations at different time points after release. Figures shown are representative of at least 3 independent experiments.
Cell lines culture
Cells were incubated at 37°C with 5% CO2. MCF7 cell line and GCN2−/−, PERK−/− and their wild type counterparts MEFs were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Lonza) with 4,5 g/l glucose and supplemented with 10% Fetal Bovine Serum (FBS), 1 mM glutamine, penicillin (10 μg/ml), and streptomycin (100 μg/ml). GCN2−/−, PERK−/− and their wild type counterparts MEFs were kindly provided by Dr. RC Wek and were described elsewhere.41
ROS production measurements
Intracellular concentration of Reactive Oxygen Species (ROS) was measured using the dihydrorhodamine-123 method.42 Briefly, yeast cells in exponential growth phase were collected by centrifugation and washed twice with water to eliminate all traces of culture medium. The cells were then resuspended en 1 ml distilled water plus 5 μg of the fluorescent probe dihydrorhodamine 123 (Sigma-Aldrich) from a 2,5 mg/ml ethanol stock solution. Correspondent treatment was added at the same time of dihydrorhodamine 123. ROS production was monitored by analysis of fluorescent spectra between 500 and 550 nm in a Perkin Elmer LS50B fluorimeter.
Immunoblotting
Yeast strains were grown to mid-log phase and then submitted to treatment. After treatment, equal number of cells was collected and proteins were extracted by the Trichloroacetic acid (TCA) method as described previously.22 Protein extracts were then centrifuged and resuspended in alkaline Laemmli buffer. Samples were boiled for 5 minutes and soluble extracts were recovered after centrifugation. 20 μg of total cellular protein was subjected to SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose (Hybond™, Amersham Biosciences) filters. Uniform gel loading was confirmed by Ponceau S staining of membranes after transfer.
For MCF7 experiments, cells seeded on p100 plates were scraped with 750 μl/plate of 50 mM HEPES pH 7.5, 400 mM KCl, 1% NP40, 10% glycerol, 200 nM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinine, 5 μg/ml leupeptine, 5 μg/ml pepstatine and phosphatase inhibitors cocktails 1 and 2 (Sigma-Aldrich). 25 μg of soluble protein extract was subjected to SDS-PAGE and transferred to PVDF (Immobilon-P; Millipore) filters. Uniform gel loading was confirmed by Ponceau S staining of membranes after transfer.
Phosphorylated H2A was detected with an antiphospho-H2A antibody (Ser129) from Abcam. An antiphospho-Rad53p antibody43 was used for detection of phosphorylation of kinase Rad53p. Phosphorylated eIF2α was detected with an antiphospho-eIF2α antibody (Ser51) from New England Biolabs. Immunocomplexes were visualized by enhanced chemiluminiscence detection (Amersham) using a HRP-conjugated anti-rabbit IgG (BioRad) or anti-mouse IgG (Abcam).
The figures show an experiment representative of at least 2 independent ones with essentially identical results.
Quantitations of Western Blot band intensities were perfomed using the ImageJ program. When possible, intensities were normalized with the respective control. In all the cases, values were expressed as percentages of the strongest intensity.
MTT viability assays
We followed the procedure published in44 with slight modifications. Briefly, the day before treatment medium from logarithmic phase growing cells was removed and substituted for 0,5% FBS DMEM. Cells were then treated with β-lap during 3 hours, after which medium was removed and substituted with fresh medium without drug. The day after treatment, medium was removed and cells were incubated with 1 mg/ml MTT (Sigma-Aldrich) in culture medium for 2–3 hours. Dye was then extracted from the intact, viable cells with a solution of 0.1N HCl and 10% Triton (v/v) in isopropanol. The absorbance of solubilised dye at 570 nm was then determined using an ELx800 microplate reader (Biotek Instruments). Quantitation of the extracted MTT by spectrophotometry was normalized to mock-treated cultures.
siRNA transfection
Small interfering RNA (siRNA) technology was used to knock down AIF1 and AMID expression. A control ON-TARGETplus Non-Targeting siRNA or specific ON-TARGETplus SMARTpool-Human AIF or AMID siRNAs (Thermo Scientific Dharmacon) were transfected into MCF7 cells with Lipofectamine 2000 (Invitrogen) (100 nM final siRNA concentration). After 48 h in culture with siRNA, cells were harvested and protein extracts were analyzed by immunoblotting as described above.
The efficiency of RNA interference was confirmed by quantitative real-time PCR. Total RNA was extracted using TRI-reagent (Sigma-Aldrich) and treated with RNAse-free DNase (Invitrogen). Gene expression was analyzed by real-time RT-PCR using Brilliant SYBR-Green QPCR Master-Mix (Stratagene). The human-specific primers used were: AMID-forward; AAGATCAACAGCTCCGCCTA, AMID-reverse; CGCTGCTTCACAGAGTTGAC, AIF1-forward; TCTTCCCCGAGAAAGGAAAT, AIF1-reverse; AACTCAACATTGGGCTCCAG. The constitutively expressed L19 ribosomal gene was used as control to normalize mRNA expression (L19-forward; 5′-GCGGAAGGGT- ACAGCCAAT-3′, L19-reverse; GCAGCCGGCGCAAA).
Statistics
Depending on the type of experiment, data were analyzed with either the Student's t-test or the Mann-Whitney test. Values are given as means ± SEM. In all of the cases, differences were considered statistically significant at P ≤ 0.05. P values of ≤0.05, 0.01 and 0.01 were indicated by *, **, and *** in all figures, respectively. Unless otherwise stated, all data represent the media of independent experiments, each performed in triplicate.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the Ministerio de Ciencia e Innovación (SAF2010-21013).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Rajendran JG, Mankoff DA, O'Sullivan F, Peterson LM, Schwartz DL, Conrad EU, Spence AM, Muzi M, Farwell DG, Krohn KA. Hypoxia and glucose metabolism in malignant tumors: evaluation by [18F]fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin Cancer Res 2004; 10:2245-52; PMID:15073099 [DOI] [PubMed] [Google Scholar]
- 2. Sivridis E, Giatromanolaki A, Koukourakis MI. The vascular network of tumours–what is it not for? J Pathol 2003; 201:173-80; PMID:14517833; http://dx.doi.org/ 10.1002/path.1355 [DOI] [PubMed] [Google Scholar]
- 3. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004; 4:437-47; PMID:15170446; http://dx.doi.org/ 10.1038/nrc1367 [DOI] [PubMed] [Google Scholar]
- 4. Evans SM, Judy KD, Dunphy I, Jenkins WT, Hwang WT, Nelson PT, Lustig RA, Jenkins K, Magarelli DP, Hahn SM, et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin Cancer Res 2004; 10:8177-84; PMID:15623592; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-1081 [DOI] [PubMed] [Google Scholar]
- 5. Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, Becker A, Adam M, Molls M, Dunst J, et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol 2005; 77:18-24; PMID:16098619; http://dx.doi.org/ 10.1016/j.radonc.2005.06.038 [DOI] [PubMed] [Google Scholar]
- 6. Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell 2007; 28:721-9; PMID:18082597; http://dx.doi.org/ 10.1016/j.molcel.2007.11.018 [DOI] [PubMed] [Google Scholar]
- 7. Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 2005; 59:407-50; PMID:16153175 [DOI] [PubMed] [Google Scholar]
- 8. Hinnebusch AG. Translational regulation of yeast GCN4. A window on factors that control initiator-trna binding to the ribosome. J Biol Chem 1997; 272:21661-4; PMID:9268289; http://dx.doi.org/ 10.1074/jbc.272.35.21661 [DOI] [PubMed] [Google Scholar]
- 9. Rolfes RJ, Hinnebusch AG. Translation of the yeast transcriptional activator GCN4 is stimulated by purine limitation: implications for activation of the protein kinase GCN2. Mol Cell Biol 1993; 13:5099-111; PMID:8336737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang R, Wek SA, Wek RC. Glucose limitation induces GCN4 translation by activation of Gcn2 protein kinase. Mol Cell Biol 2000; 20:2706-17; PMID:10733573; http://dx.doi.org/ 10.1128/MCB.20.8.2706-2717.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goossens A, Dever TE, Pascual-Ahuir A, Serrano R. The protein kinase Gcn2p mediates sodium toxicity in yeast. J Biol Chem 2001; 276:30753-60; PMID:11408481; http://dx.doi.org/ 10.1074/jbc.M102960200 [DOI] [PubMed] [Google Scholar]
- 12. Valenzuela L, Aranda C, Gonzalez A. TOR modulates GCN4-dependent expression of genes turned on by nitrogen limitation. J Bacteriol 2001; 183:2331-4; PMID:11244074; http://dx.doi.org/ 10.1128/JB.183.7.2331-2334.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palmer LK, Shoemaker JL, Baptiste BA, Wolfe D, Keil RL. Inhibition of translation initiation by volatile anesthetics involves nutrient-sensitive GCN-independent and -dependent processes in yeast. Mol Biol Cell 2005; 16:3727-39; PMID:15930127; http://dx.doi.org/ 10.1091/mbc.E05-02-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 2006; 34:7-11; PMID:16246168 [DOI] [PubMed] [Google Scholar]
- 15. Menacho-Marquez M, Perez-Valle J, Arino J, Gadea J, Murguia JR. Gcn2p regulates a G1/S cell cycle checkpoint in response to DNA damage. Cell Cycle 2007; 6:2302-5; PMID:17890903; http://dx.doi.org/ 10.4161/cc.6.18.4668 [DOI] [PubMed] [Google Scholar]
- 16. Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol 2001; 21:4347-68; PMID:11390663; http://dx.doi.org/ 10.1128/MCB.21.13.4347-4368.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol 2002; 12:1279-86; PMID:12176355; http://dx.doi.org/ 10.1016/S0960-9822(02)01037-0 [DOI] [PubMed] [Google Scholar]
- 18. Powley IR, Kondrashov A, Young LA, Dobbyn HC, Hill K, Cannell IG, Stoneley M, Kong YW, Cotes JA, Smith GC, et al. Translational reprogramming following UVB irradiation is mediated by DNA-PKcs and allows selective recruitment to the polysomes of mRNAs encoding DNA repair enzymes. Genes Dev 2009; 23:1207-20; PMID:19451221; http://dx.doi.org/ 10.1101/gad.516509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pardee AB, Li YZ, Li CJ. Cancer therapy with beta-lapachone. Curr Cancer Drug Targets 2002; 2:227-42; PMID:12188909; http://dx.doi.org/ 10.2174/1568009023333854 [DOI] [PubMed] [Google Scholar]
- 20. Li CJ, Li YZ, Pinto AV, Pardee AB. Potent inhibition of tumor survival in vivo by beta-lapachone plus taxol: combining drugs imposes different artificial checkpoints. Proc Natl Acad Sci U S A 1999; 96:13369-74; PMID:10557327; http://dx.doi.org/ 10.1073/pnas.96.23.13369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem 2000; 275:5416-24; PMID:10681517; http://dx.doi.org/ 10.1074/jbc.275.8.5416 [DOI] [PubMed] [Google Scholar]
- 22. Menacho-Marquez M, Murguia JR. Beta-lapachone activates a Mre11p-Tel1p G1/S checkpoint in budding yeast. Cell Cycle 2006; 5:2509-16; PMID:17106258; http://dx.doi.org/ 10.4161/cc.5.21.3394 [DOI] [PubMed] [Google Scholar]
- 23. Sun X, Li Y, Li W, Zhang B, Wang AJ, Sun J, Mikule K, Jiang Z, Li CJ. Selective induction of necrotic cell death in cancer cells by beta-lapachone through activation of DNA damage response pathway. Cell Cycle 2006; 5:2029-35; PMID:16969131; http://dx.doi.org/ 10.4161/cc.5.17.3312 [DOI] [PubMed] [Google Scholar]
- 24. Dev K, Qiu H, Dong J, Zhang F, Barthlme D, Hinnebusch AG. The beta/Gcd7 subunit of eukaryotic translation initiation factor 2B (eIF2B), a guanine nucleotide exchange factor, is crucial for binding eIF2 in vivo. Mol Cell Biol 2010; 30(21):5218-33; PMID:20805354; http://dx.doi.org/ 10.1128/MCB.00265-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang W, Hinnebusch AG. Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol Cell Biol 1996; 16(11):6603-16; PMID:8887689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bentle MS, Reinicke KE, Dong Y, Bey EA, Boothman DA. Nonhomologous end joining is essential for cellular resistance to the novel antitumor agent, beta-lapachone. Cancer Res 2007; 67:6936-45; PMID:17638905; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0935 [DOI] [PubMed] [Google Scholar]
- 27. Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, Minna JD, Bornmann WG, Gao J, Boothman DA. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci U S A 2007; 104:11832-7; PMID:17609380; http://dx.doi.org/ 10.1073/pnas.0702176104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kubata BK, Kabututu Z, Nozaki T, Munday CJ, Fukuzumi S, Ohkubo K, Lazarus M, Maruyama T, Martin SK, Duszenko M, et al. A key role for old yellow enzyme in the metabolism of drugs by Trypanosoma cruzi. J Eep Med 2002; 196:1241-51; PMID:12417633; http://dx.doi.org/ 10.1084/jem.20020885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297:547-51; PMID:12142523; http://dx.doi.org/ 10.1126/science.1074740 [DOI] [PubMed] [Google Scholar]
- 30. Asher G, Lotem J, Sachs L, Kahana C, Shaul Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc Natl Acad Sci U S A 2002; 99:13125-30; PMID:12232053; http://dx.doi.org/ 10.1073/pnas.202480499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee H, Park MT, Choi BH, Oh ET, Song MJ, Lee J, Kim C, Lim BU, Park HJ. Endoplasmic reticulum stress-induced JNK activation is a critical event leading to mitochondria-mediated cell death caused by β-lapachone treatment. PLoS One. 2011; 6(6):e21533; PMID:21738692; http://dx.doi.org/ 10.1371/journal.pone.0021533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malzer E, Daly ML, Moloney A, Sendall TJ, Thomas SE, Ryder E, Ryoo HD, Crowther DC, Lomas DA, Marciniak SJ. Impaired tissue growth is mediated by checkpoint kinase 1 (CHK1) in the integrated stress response. J Cell Sci. 2010; 123(Pt 17):2892-900; PMID:20682638; http://dx.doi.org/ 10.1242/jcs.070078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuzefovych LV, LeDoux SP, Wilson GL, Rachek LI. Mitochondrial DNA damage via augmented oxidative stress regulates endoplasmic reticulum stress and autophagy: crosstalk, links and signaling. PLoS One. 2013; 8(12):e83349; PMID:24349491; http://dx.doi.org/ 10.1371/journal.pone.0083349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, Marciniak SJ, Goodall JC, Green AR, Wouters BG, Wienholds E, Dick JE. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 2014; 510(7504):268-72; PMID:24776803; http://dx.doi.org/ 10.1038/nature13228 [DOI] [PubMed] [Google Scholar]
- 35. Jiang HY, Wek RC. GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem J 2005; 385, 371-380; PMID:15355306; http://dx.doi.org/ 10.1042/BJ20041348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu S, Hu Y, Wang JL, Chatterjee M, Shi Y, Kaufman RJ. Ultraviolet light inhibits translation through activation of the unfolded protein response kinase PERK in the lumen of the endoplasmic reticulum. J Biol Chem 2002; 277:18077-18083; PMID:11877419; http://dx.doi.org/ 10.1074/jbc.M110164200 [DOI] [PubMed] [Google Scholar]
- 37. Prinz W. Guide to yeast genetics and molecular cell biology. In: Guthrie CF, GR, ed. Guide to Yeast Genetics and Molecular Cell Biology, Part B, Volume 350 (Methods in Enzymology). New York: Academic Press, 2003. [Google Scholar]
- 38. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 2001; 98:5116-21; PMID:11309499; http://dx.doi.org/ 10.1073/pnas.091062498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Al-Shahrour F, Minguez P, Tarraga J, Montaner D, Alloza E, Vaquerizas JM, Conde L, Blaschke C, Vera J, Dopazo J. BABELOMICS: a systems biology perspective in the functional annotation of genome-scale experiments. Nucleic Acids Res 2006; 34:W472-6; PMID:16845052; http://dx.doi.org/ 10.1093/nar/gkl172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teixeira MC, Monteiro P, Jain P, Tenreiro S, Fernandes AR, Mira NP, Alenquer M, Freitas AT, Oliveira AL, Sa-Correia I. The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res 2006; 34:D446-51; PMID:16381908; http://dx.doi.org/ 10.1093/nar/gkj013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 2005; 280:14189-202; PMID:15684420; http://dx.doi.org/ 10.1074/jbc.M413660200 [DOI] [PubMed] [Google Scholar]
- 42. Madeo F, Frohlich E, Ligr M, Grey M, Sigrist SJ, Wolf DH, Frohlich KU. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol 1999; 145:757-67; PMID:10330404; http://dx.doi.org/ 10.1083/jcb.145.4.757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de la Torre-Ruiz MA, Green CM, Lowndes NF. RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J 1998; 17:2687-98; PMID:9564050; http://dx.doi.org/ 10.1093/emboj/17.9.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55-63; PMID:6606682; http://dx.doi.org/ 10.1016/0022-1759(83)90303-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.