

Abstract
The T296V mutant of amorpha-4, 11-diene synthase catalyzes the abortive conversion of the natural substrate (E,E)-farnesyl diphosphate mainly into the acyclic product (E)-β-farnesene (88%) instead of the natural bicyclic sesquiterpene amorphadiene (7%). Incubation of the T296V mutant with (3R,6E)-nerolidyl diphosphate resulted in cyclization to amorphadiene. Analysis of additional mutants of amino acid residue 296 and in vitro assays with the intermediate analogue (2Z,6E)-farnesyl diphosphate as well as (3S,6E)-nerolidyl diphosphate demonstrated that the T296V mutant can no longer catalyze the allylic rearrangement of farnesyl diphosphate to the normal intermediate (3R,6E)-nerolidyl diphosphate, while retaining the ability to cyclize (3R,6E)-nerolidyl diphosphate to amorphadiene. The T296A mutant predominantly retained amorphadiene synthase activity, indicating that neither the hydroxyl nor the methyl group of the Thr296 side chain is required for cyclase activity.
TOC image
Introduction
Sesquiterpene synthases convert the universal acyclic precursor (2E,6E)-farnesyl diphosphate ((E,E)-FPP) into more than 300 types of acyclic, monocylic, bicyclic, and tricyclic sesquiterpenes.1–4 Although the cyclization mechanisms have been extensively studied, and the structures of several sesquiterpene synthases of bacterial, fungal, and plant origin have been determined,1–16 knowledge of the precise protein structural factors that control the initiation of the cyclization cascade and the resultant product structure is still limited.
Cyclization of stereospecifically deuterated samples of FPP by recombinant Artemisia annua amorpha-4,11-diene synthase (ADS) expressed in Escherichia coli followed by NMR and GC-MS analysis of the derived labeled samples of amorphadiene has provided strong support for a mechanism involving the initial isomerization of FPP to the transoid conformer of the tertiary allylic isomer (3R,6E)-nerolidyl diphosphate ((3R,6E)-NPP), followed by C-2,3-bond rotation to the corresponding cisoid conformer of NPP which then undergoes ionization and cyclization to generate the intermediate (6R)-bisabolyl cation (Scheme 1).17,18 A 1,3-hydride shift followed by 1,10-ring closure and deprotonation generates the eponymous product (1S,6R,7R,10R)-amorpha-4,11-diene, the precursor of the widely used antimalarial agent artemisinin, accompanied by minor amounts of four bisabolyl cation-derived sesquiterpene side products (<2% total) (Figure S1). Recently, it has been reported that amino acid L374 of amorphadiene synthase is not essential for the cyclization activity, while a single Y402L substitution at the homologous site in the acyclic (E)-β-farnesene synthase from A. annua (BFS) was sufficient to confer significant cyclase activity on this mutant, resulting in the formation of 15 β-bisabolyl cation-derived sesquiterpenes, constituting 75% of the total product mixture.10
Scheme 1.
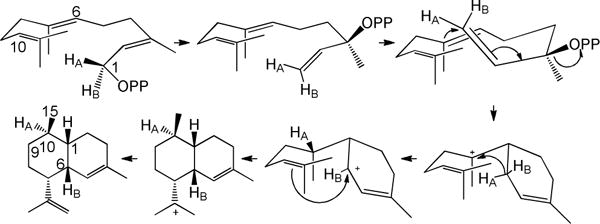
Mechanism for the ADS-catalyzed formation of amorpha-4, 11-diene from FPP.
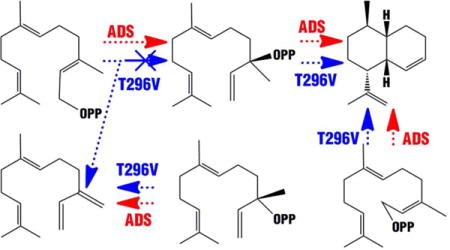
We now report that the single T296V mutation in amorphadiene suppresses 90% of the natural farnesyl diphosphate cyclization activity, predominantly giving rise instead to the acyclic hydrocarbon β-farnesene. Cyclization to form amorphadiene can once again be observed, however, when the T296V ADS mutant is incubated with either synthetic acyclic (3R,6E)-NPP, the presumed intermediate of the wild-type cyclization, or (2Z,6E)-farnesyl diphosphate ((Z,E)-FPP), a geometric analogue whose ionization will give rise to the essential cisoid allylic cation – pyrophosphate ion pair. These results establish that the T296V mutant is defective in the initial allylic isomerization of (E,E)-FPP to (3R,6E)-NPP, but retains the ability to cyclize the natural tertiary allylic diphosphate intermediate.
Materials and Methods
Reagents
(E,E)-Farnesyl diphosphate was purchased from Sigma-Aldrich. (3S,6E)-nerolidyl diphosphate and (2E,6Z)-farnesyl diphosphate were purchased from Isoprenoids LC. Geranyl acetone and chlorovinylmagnesium were purchased from Aladdin Industrial Inc.
Protein expression and purification
The full-length open reading frame of amorpha-4,11-diene synthase (GenBank ID AAK15696.1) was amplified and inserted into a BamHI/XhoI-digested pET30a vector. Recombinant plasmid and mutants were transformed into E. coli Rosetta (DE3) and protein production was induced with 0.25 mM β-D-1-thiogalactopyranoside at 18 °C overnight. Protein purification was carried out using traditional Ni-NTA chromatography methods. The full-length open reading frame of α-bisabolol synthase (GenBank ID: AFV40969.1) was optimized for E. coli expression, synthesized and inserted into a BamHI/XhoI-digested pET30a vector. The same expression and purification protocol was used as described for amorpha-4,11-diene synthase.
Site-directed mutagenesis
Site-directed mutagenesis of amorpha-4,11-diene synthase and α-bisabolol synthase were carried out according to the QuikChange Site-Directed Mutagenesis Kit Instruction Manual (Stratagene) using the primers in Table S1. Positive mutants were identified by sequencing purified plasmid DNA.
In vitro enzyme incubations
The assay was performed in 2-ml screw-top glass vials with 500 μL of reaction buffer containing 50 mM phosphate buffer (pH 7.2), 5 mM MgCl2, 25 mM 2-sulfanylethanol, 100 μM prenyl diphosphate substrates and 5 μM enzyme. Reactions were mixed and overlaid with 200 μL of n-hexane. After an overnight incubation at 30 °C, the hydrocarbon products were extracted by vortexing for 30 s, followed by GC-MS analysis.
Malachite green assay for kinetic measurements
Kinetic assays were performed according to the methods previously reported,19 except the volume of the assay in the 96-well flat-bottomed plate was changed to 100 μL in order to acquire enough signal and better reproducibility of results.
Synthesis of racemic nerolidol
Under nitrogen, 3 ml (15.4 mmol) of geranyl acetone (racemic mixture of trans and cis) was added to a 30 ml solution of vinyl magnesium chloride (30.8 mmol) in anhydrous THF, and the mixture was stirred under reflux for 90 min at 60 °C, followed by the addition of 30% sulfuric acid for a further 180 min at 50 °C. The reaction was quenched with saturated ammonium chloride solution (20 mL). THF was removed under vacuum and the nerolidol was extracted with petroleum ether. The collected organic portion was dried with Na2SO4, filtered and concentrated. The crude product was analyzed by TLC (petroleum ether /ethyl acetate: 8/1) and then purified by flash chromatography (petroleum ether/ethyl acetate: 20/1). Purified nerolidol was analyzed by chiral gas chromatography (GC) and GC-MS (Figure S7).
Purification of enantiomerically pure (3R,6E)-nerolidol
A Shimadzu Prominence LC-20AD with switching valve FCV-20AH2 was used to prepare enantiomerically pure (3R,6E)-nerolidol using a Daicel Chiralpak IA column with the mobile phase: petroleum ether/ethanol: 98.5/1.5 and a flow rate of 7 ml/min. The chromatogram of a typical preparation is shown in Figure S8.
Synthesis of (3R,6E)-nerolidol from (2E,6E)-farnesol
Sharpless epoxidation of (2E,6E)-farnesol, as previously described, gave (2S,3R)-epoxyfarnesol which was converted to (3R,6E)-nerolidol according to the previously published procedures (Scheme S1).20,21 Chiral GC-MS analysis of the (3R,6E)-nerolidol product is shown in Figure S9.
Synthesis of (3R,6E)-nerolidyl diphosphate
Enantiomerically pure (3R,6E)-nerolidol was used for the synthesis of (3R,6E)-nerolidyl diphosphate. The method of Keller22 was used with modifications. The reaction was stirred overnight at room temperature and the products were purified by flash chromatography (isopropyl alcohol/NH4OH/H2O:9/4/1). The TLC was developed using a mobile phase of iso-propyl alcohol/NH4OH/H2O (6/3/1) and stained with anisaldehyde. TLC and MS analyses are shown in Figure S10.
Analytical equipment and methods
Gas chromatographic (GC) analyses were carried out using an Agilent 7890A gas chromatograph with a FID detector. Commercial geranyl acetone was analyzed on a CycloSil-B column (30 m × 0.32 mm × 0.25 μm) with the method: 50 °C for 2 min, 3 °C/min gradient to 220 °C, 220 °C for 10 min. Chiral analysis of nerolidol used the same column and same procedure. GC-MS analyses of enzyme products from pentane extractions were carried out on an Agilent 7890A gas chromatograph and Agilent 5977A mass spectrometer with an Agilent HP-5MS column (30 m × 0.25 mm × 0.25 μm) with the method: 60 °C for 2 min, 20 °C/min gradient to 280 °C, 280 °C for 2 min. GC-MS analyses of crude nerolidol was carried out on an Agilent 7890A gas chromatograph and Agilent 5975C mass spectrometer with an Agilent HP-5MS column (30 m × 0.25 mm × 0.25 μm) with the method: 45 °C for 2 min, 8 °C/min gradient to 220 °C, 220 °C for 10 min. Chiral GCMS analysis of nerolidol synthesized by the Sharpless epoxidation method was performed on an Agilent 6890A gas chromatograph and Agilent 5973N mass spectrometer using a CP-Chirasil-Dex CB column (25 m × 0.32 mm × 0.25 μm) with the method: 60 °C for 2 min, 3 °C/min gradient to 220 °C, 220 °C for 2 min. GC-MS data was analyzed using the MassFinder 4 software and database. HPLC chiral analyses and preparations were carried out with a Daicel Chiralpak IA column on a Thermo Scientific Dionex UltiMate 3000 and a Shimadzu Prominence LC-20AD with switching valve FCV-20AH2, respectively, with the mobile phase: Petroleum ether/ethanol: 98.5/1.5. The UV detector was set at 210 nm.
Results and discussions
Homology models of both ADS and BFS were generated based on the crystal structure of a γ-humulene-producing mutant of A. annua α-bisabolol synthase (AaBOS, PDB ID 4GAX) (Figure 1).13 By comparison of the homology models for ADS and BFS, we chose to mutate T296 of ADS to the nearly isosteric but less polar V296, corresponding to amino acid residue V324 in BFS. This amino acid residue is located on the wall of the active site cavity just left below D299 of ADS (Figure 1), the first amino acid of the universally conserved aspartate-rich DDxxD motif, which has been implicated in binding of essential divalent Mg2+ ions that are in turn coordinated to the pyrophosphate moiety of the FPP substrate.9,23–25 Incubation of this T296V mutant with (E,E)-FPP resulted in a major change in product distribution which now consisted of 88.5% (E)-β-farnesene, 7.4% amorpha-4,11-diene, and 4% of a mixture of minor products (Figures 2 and S2, and Table 1).
Figure 1.
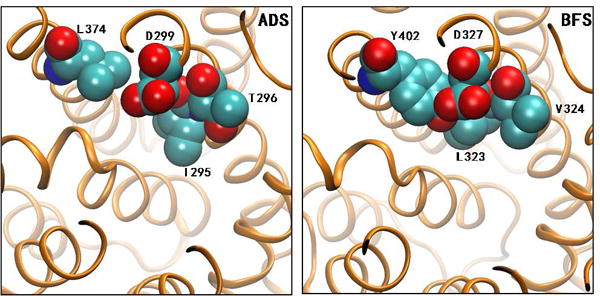
Active Sites of Homologous Structures of ADS and BFS. The spatial positions of I295, T296, L374 and D299 in ADS and the corresponding sites in BFS are illustrated.
Figure 2.
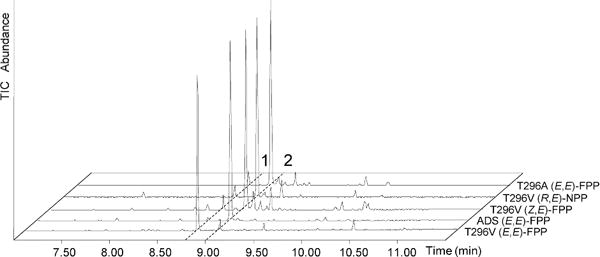
GC-MS (TIC) Analysis of Products of Incubation of ADS and its T296 Mutants with FPP and NPP Substrates. Peak 1: (E)-β-farnesene; Peak 2: amorpha-4,11-diene.
Table 1.
Amorpha-4, 11-diene Synthase Mutants: Kinetic Parameters and Product Ratios for (E, E)-FPP
| protein | Steady-state kinetic
parameters |
Relative Proportion of
sesquiterpenes (TIC %) |
||||
|---|---|---|---|---|---|---|
|
kcat (10−3s−1) |
Km (μM) |
kcat/Km (103s−1M−1) |
(E)-β-farnesene | amorpha-4,11-diene | Others | |
| ADSwt | 16.3 ± 0.6 | 15.3 ± 2.0 | 1.06 | 1.74 | 92.67 | 5.59 |
| T296A | 5.9 ± 0.4 | 10.8 ± 2.4 | 0.55 | 5.15 | 78.18 | 16.67 |
| T296S | 7.5 ± 1.0 | 33.0 ± 10.1 | 0.23 | 1.57 | 90.32 | 8.11 |
| T296V | 1.5 ± 0.2 | 28.2 ± 9.2 | 0.05 | 88.54 | 7.38 | 4.08 |
| T296I | 1.3 ± 0.1 | 19.8 ± 5.7 | 0.07 | 100 | 0 | 0 |
| T296L | – | – | – | 54.05 | 35.24 | 10.74 |
It has been reported that substitution of hydroxyl-bearing amino acid residues for aliphatic amino acids can short-circuit complex cyclizations catalyzed by diterpene synthases, suggesting that the side-chain hydroxyl group may stabilize specific carbocation intermediates.26 By analogy, we first speculated that the β-hydroxyl group of T296 might be necessary for cyclization of farnesyl diphosphate to amorpha-4, 11-diene due to an H-bonding interaction with the pyrophosphate moiety of the substrate or ion-dipole stabilization of one or more intermediate carbocations. Both of these possibilities were ruled out, however, by the finding that the corresponding T296A mutant of ADS converted (E,E)-FPP to a mixture containing 78% amorphadiene and only 5% (E)-β-farnesene (Figures 2 and S3, Table 1).
To further investigate the influence of the side chain of T296 on enzyme activity, we generated the corresponding T296S, T296I and T296L mutants of ADS and determined both the product ratios and steady-state kinetic parameters for each (Table 1, Figures S3 and S4). The main product of the T296S mutant was amorpha-4,11-diene (90%), essentially the same as for wild type ADS. It is therefore clear that neither the methyl nor the hydroxyl of T296 is essential for control of the cyclization. For the T296I mutant, with the larger Ile side chain, the sole product was the acyclic (E)-β-farnesene. When T296 was mutated to Leu, the relative proportion of amorpha-4,11-diene (35%) was greater than that produced by the T296V mutant. These results strongly indicate that both the steric volume and the stereochemistry of the side-chain of amino acid residue 296 are critical for control of the natural cyclization reaction. (E)-β-farnesene can in principle be generated by deprotonation of the 3-methyl group of either the transoid or cisoid conformers of the farnesyl/nerolidyl allylic cation intermediate that normally undergoes cyclization to the bisabolyl cation and thence to amorpha-4,11-diene (Schemes 1 and 2)17,18 There have been a small number of previous reports in which specific terpene synthase mutants generate the acyclic product β-farnesene in place of the wild-type cyclase product. For example, the Y92A mutant aristolochene synthase, which can no longer generate aristolochene, instead converts farnesyl diphosphate to (E)-β-farnesene, thought to be due to aberrant binding of (E,E)-FPP in an extended, non-cyclizable conformation.27
Scheme 2. Incubation of ADS and Mutants with Allylic Diphosphate Substrates.
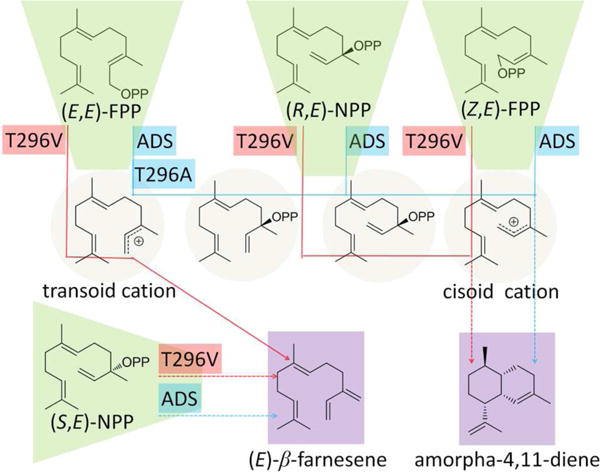
Broken lines denote multiple steps.
The Km values of the mutants, which ranged from 11–33 μM, were all within a factor of 2.2 of the Km of wild-type amorphadiene synthase (15 μM), indicating that none of the mutations had a significant effect on the overall structure of the protein, in spite of the variations in the size and hydrophobicity of the amino acid 296 side chains (Table 1). (The Km of the T296L mutant could not be determined due to the extremely low catalytic activity.) The four most active mutants showed modest decreases in turnover number, with kcat values that ranged from 3–12-fold lower than wild-type. These results confirm that the hydroxyl group of T296 is not important for overall catalytic activity.
In order to elucidate the mechanistic basis for the lesion in amorphadiene formation in the T296V mutant and to identify the immediate precursor of (E)-β-farnesene, we carried out incubations of wild-type ADS and the T296V mutant with (3R,6E)-NPP, (3S,6E)-NPP and (Z,E)-FPP. Although either (3R,6E)- or (3S,6E)-NPP could in principle serve as a precursor of the cisoid conformer of the allylic farnesyl/nerolidyl cation, we have previously predicted, based on stereochemical analysis of the results of cyclization of chirally deuterated samples of FPP that the true intermediate is (3R,6E)-NPP (Scheme 1).17 It was also expected that (Z,E)-FPP might serve as an unnatural, surrogate precursor of the cisoid allylic cation.
Both wild-type ADS and the T296V mutant converted (3S,6E)-NPP exclusively into (6E)-β-farnesene (Scheme 2, Figures S1 and S2), indicating that (3S,6E)-NPP is not an intermediate in the ADS-catalyzed cyclization of (2E,6E)-FPP to amorpha-4,11-diene. By contrast, incubation of (3R,6E)-NPP with either wild-type ADS or the T296V mutant gave predominantly amorphadiene in essentially the same relative proportions (89% and 83% of total product mixture), with nearly identical yields of β-farnesene (4% and 5%) and a small increase in the proportion of minor sesquiterpene side products (7% to 12%) (Scheme 2, Figures 2 and S2, Table 2). When (3R,6E)-NPP was incubated with the corresponding T296I and T296L mutants of ADS, amorphadiene was generated as the major product, although its proportion of total sesquiterpenes decreased slightly (52% and 63%) compared to both wild-type and the T296V mutant (Table 2). These results confirm that 1) (3R,6E)-NPP is the natural intermediate of the ionization-isomerization-ionization-cyclization of farnesyl diphosphate to amorphadiene via the bisabolyl cation, as previously suggested,17 and 2) the T296V mutant is defective in the isomerization of (2E,6E)-farnesyl diphosphate to (3R,6E)-nerolidyl diphosphate.
Table 2.
Amorpha-4, 11-diene Synthase and Mutants: Product Ratios from (Z,E)-FPP and (R,E)-NPP
| protein | Relative Proportion with
(R,E)-NPP (TIC %)
|
Relative Proportion with
(Z,E)-FPP (TIC %)
|
||||
|---|---|---|---|---|---|---|
| (E)-β-farnesene | amorpha-4,11-diene | Others | (E)-β-farnesene | amorpha-4,11-diene | Others | |
| ADSwt | 3.99 | 89.12 | 6.89 | 0.26 | 89.53 | 10.21 |
| T296V | 5.05 | 82.54 | 12.41 | 5.30 | 66.10 | 28.60 |
| T296I | 16.79 | 51.87 | 31.34 | 25.63 | 53.53 | 20.84 |
| T296L | 5.05 | 63.31 | 31.64 | 0.78 | 76.90 | 22.32 |
Incubation of (2Z,6E)-FPP with both wild-type and T296V mutant of ADS produced amorpha-4,11-diene as the major product, although there was some decrease in the proportion (90% to 66%) of the bicyclic product and an increase in the amounts of other sesquiterpene side products (10% – 29%) (Figures 2 and S1, Table 2, Scheme 2). The ability of the T296V mutant to cyclize (Z,E)-FPP to amorpha-4,11-diene indicates that this mutant is capable of generating the cisoid farnesyl/nerolidyl allylic carbocation from (Z,E)-FPP and cyclizing it to amorpha-4,11-diene via the bisabolyl cation. The formation of increased amounts of anomalous products, however, reflects the fact that ADS and its mutants must bind the unnatural geometric isomer (2Z,6E)-FPP in a manner that differs from that of the native substrate (2E,6E)-FPP. These findings reinforce the conclusion that the T296V mutation blocks allylic isomerization of (2E,6E)-FPP to the requisite, conformationally flexible (3R,6E)-NPP intermediate. Instead of recapturing the paired pyrophosphate ion, the T296V mutant deprotonates the intermediate allylic cation to give (E)-β-farnesene. Two previous reports have implicated amino acids that may be crucial for the allylic isomerization of farnesyl diphosphate, but these conclusions were supported only by incubations with the unnatural substrate isomer (Z,E)-FPP.28,29
We also carried an analogous set of experiments with A. annua α-bisabolol synthase (AaBOS). Recombinant AaBOS, which has 82% amino acid sequence identity to ADS, cyclizes (2E,6E)-FPP to α-bisabolol (~95%), accompanied by minor amounts of a variety of monocyclic bisabolyl cation derivatives and a trace of β-farnesene (Figure 3).13 We found that incubation of the T296V mutant of AaBOS with (2E,6E)-FPP yielded a mixture of (E)-β-farnesene (~61%) and α-bisabolol (~36%), accompanied by 3% of other minor sesquiterpenes, indicating that the replacement of the active site Thr residue by Val resulted in an increase in the formation of the acyclic (E)-β-farnesene product formed by deprotonation of the allylic cation – pyrophosphate ion pair at the expense of isomerization–cyclization to the bisabolyl cation, analogous to the effect of the same mutation on amorphadiene synthase. Importantly incubation of both wild-type and T296V mutant of AaBOS with (3R,6E)-NPP resulted in efficient formation of the native cyclization product α-bisabolol (Figure 3). By contrast, both the wild-type and T296V mutant of AaBOS converted the enantiomeric (3S,6E)-NPP almost exclusively to the acyclic deprotonation product (E)-β-farnesene.
Figure 3.
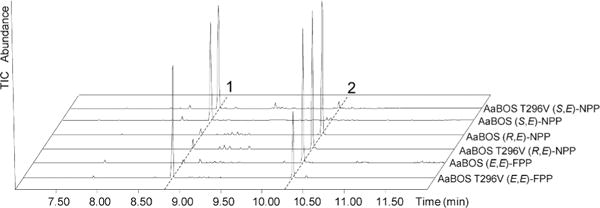
GC-MS (TIC) of the Incubation of AaBOS and its T296V mutant with (E,E)-FPP and (3R,6E)-NPP. Peak 1: (E)-β-farnesene; Peak 2: α-bisabolol (See Figure S10 for MS spectra).
Conclusions
In summary, the nearly isosteric change associated with the single T296V mutation in both amorphadiene synthase and the closely related α-bisabolol synthase largely suppresses cyclization to the bisabolyl cation by blocking the formation of the intermediate tertiary allylic isomer, (3R,6E)-NPP. The native cyclization activity is retained by both mutants, however, and can be observed by incubation with the intermediate (3R,6E)-NPP to give the native products amorpha-4,11-diene (Scheme 2) and α-bisabolol, respectively.
Supplementary Material
Acknowledgments
We thank Dr. Jianrong Wei (Hebei University) for the help with GC analysis.
Funding Sources
This work was supported by the National Natural Science Foundation of China (Grant Nos. 30900111, 31300056, and 31570305) to Z.L., and U. S. National Institutes of Health Grant GM030301 to D.E.C., as well as Hebei University postdoctoral grants and Bioengineering key discipline of Hebei Province.
ABBREVIATIONS
- AaBOS
A. annua α-bisabolol synthase
- ADS
amorpha-4,11-diene synthase
- BFS
(E)-β-farnesene synthase
- FPP
farnesyl diphosphate
- NPP
nerolidyl diphosphate
- THF
tetrahydrofuran
- TLC
thin layer chromatography
Footnotes
Supporting Information
Figures S1–S11, Scheme S1, and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.Cane DE. The stereochemistry of allylic pyrophosphate metabolism. Tetrahedron. 1980;36:1109–1159. [Google Scholar]
- 2.Cane DE. In: Biosynthesis of Isoprenoid Compounds. Porter JW, Spurgeon SL, editors. Vol. 1. J. W. Wiley & Sons; New York: 1981. pp. 283–374. [Google Scholar]
- 3.Cane DE. Enzymatic formation of sesquiterpenes. Chem Rev. 1990;90:1809–1103. [Google Scholar]
- 4.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 5.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 6.Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 7.Li R, Chou WK, Himmelberger JA, Litwin KM, Harris GG, Cane DE, Christianson DW. Reprogramming the chemodiversity of terpenoid cyclization by remolding the active site contour of epi-isozizaene synthase. Biochemistry. 2014;53:1155–1168. doi: 10.1021/bi401643u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rynkiewicz MJ, Cane DE, Christianson DW. Structure of trichodiene synthase from Fusarium sporotrichioides provides mechanistic inferences on the terpene cyclization. Proc Natl Acad Sci U S A. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cane DE, Pawlak JL, Horak RM. Studies of the cryptic allylic pyrophosphate isomerase activity of trichodiene synthase using the anomalous substrate 6,7-dihydrofarnesyl pyrophosphate. Biochemistry. 1990;29:5476–5490. doi: 10.1021/bi00475a011. [DOI] [PubMed] [Google Scholar]
- 10.Salmon M, Laurendon C, Vardakou M, Cheema J, Defernez M, Green S, Faraldos JA, O’Maille PE. Emergence of terpene cyclization in Artemisia annua. Nat Commun. 2015;6 doi: 10.1038/ncomms7143. art No. 6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Maille PE, Malone A, Dellas N, Andes Hess B, Jr, Smentek L, Sheehan I, Greenhagen BT, Chappell J, Manning G, Noel JP. Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nat Chem Biol. 2008;4:617–623. doi: 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noel JP, Dellas N, Faraldos JA, Zhao M, Hess BA, Jr, Smentek L, Coates RM, O’Maille PE. Structural elucidation of cisoid and transoid cyclization pathways of a sesquiterpene synthase using 2-fluorofarnesyl diphosphates. ACS Chem Biol. 2010;5:377–392. doi: 10.1021/cb900295g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li JX, Fang X, Zhao Q, Ruan JX, Yang CQ, Wang LJ, Miller DJ, Faraldos JA, Allemann RK, Chen XY, Zhang P. Rational engineering of plasticity residues of sesquiterpene synthases from Artemisia annua: product specificity and catalytic efficiency. Biochem J. 2013;451:417–426. doi: 10.1042/BJ20130041. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez V, Touchet S, Grundy DJ, Faraldos JA, Allemann RK. Evolutionary and mechanistic insights from the reconstruction of alpha-humulene synthases from a modern (+)-germacrene A synthase. J Am Chem Soc. 2014;136:14505–14512. doi: 10.1021/ja5066366. [DOI] [PubMed] [Google Scholar]
- 15.Faraldos JA, Antonczak AK, Gonzalez V, Fullerton R, Tippmann EM, Allemann RK. Probing eudesmane cation-pi interactions in catalysis by aristolochene synthase with non-canonical amino acids. J Am Chem Soc. 2011;133:13906–13909. doi: 10.1021/ja205927u. [DOI] [PubMed] [Google Scholar]
- 16.Yoshikuni Y, Ferrin TE, Keasling JD. Designed divergent evolution of enzyme function. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 17.Picaud S, Mercke P, He X, Sterner O, Brodelius M, Cane DE, Brodelius PE. Amorpha-4,11-diene synthase: mechanism and stereochemistry of the enzymatic cyclization of farnesyl diphosphate. Arch Biochem Biophys. 2006;448:150–155. doi: 10.1016/j.abb.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 18.Kim SH, Heo K, Chang YJ, Park SH, Rhee SK, Kim SU. Cyclization mechanism of amorpha-4,11-diene synthase, a key enzyme in artemisinin biosynthesis. J Nat Prod. 2006;69:758–762. doi: 10.1021/np050356u. [DOI] [PubMed] [Google Scholar]
- 19.Vardakou M, Salmon M, Faraldos JA, O’Maille PE. Comparative analysis and validation of the malachite green assay for the high throughput biochemical characterization of terpene synthases. MethodsX. 2014;1:187–196. doi: 10.1016/j.mex.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kigoshi H, Ojika M, Shizuri Y, Niwa H, Yamada K. Isolation of (10R11R)-(+)-squalene-10,11-epoxide from the red alga Laurencia okamurai and its enantioselective synthesis. Tetrahedron. 1986;42:3789–3792. [Google Scholar]
- 21.Le Thanh C, Chauhan KR. Simple and short synthesis of trans-(R)-nerolidol, a pheromone component of fruit spotting bug. Nat Prod Commun. 2014;9:297–298. [PubMed] [Google Scholar]
- 22.Keller RK, Thompson RJ. Rapid synthesis of isoprenoid diphosphates and their isolation in one step using either thin layer or flash chromatography. Chromatography. 1993;645:161–167. doi: 10.1016/0021-9673(93)80630-q. [DOI] [PubMed] [Google Scholar]
- 23.Lin X, Cane DE. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor. Mechanism and stereochemistry of the enzymatic formation of epi-isozizaene. J Am Chem Soc. 2009;131:6332–6333. doi: 10.1021/ja901313v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cane DE, Xue Q, Fitzsimons BC. Trichodiene synthase. Probing the role of the highly conserved aspartate-rich region by site-directed mutagenesis. Biochemistry. 1996;35:12369–12376. doi: 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- 25.Rynkiewicz MJ, Cane DE, Christianson DW. X-ray crystal structures of D100E trichodiene synthase and its pyrophosphate complex reveal the basis for terpene product diversity. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 26.Zhou K, Peters RJ. Electrostatic effects on (di)terpene synthase product outcome. Chem Commun (Camb) 2011;47:4074–4080. doi: 10.1039/c0cc02960b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deligeorgopoulou A, Allemann RE. Evidence for differential folding of farnesyl pyrophosphate in the active site of aristolochene synthase: a single-point mutation converts aristolochene synthase into an (E)-beta-farnesene synthase. Biochemistry. 2003;42:7741–7747. doi: 10.1021/bi034410m. [DOI] [PubMed] [Google Scholar]
- 28.Kollner TG, O’Maille PE, Gatto N, Boland W, Gershenzon J, Degenhardt Two pockets in the active site of maize sesquiterpene synthase TPS4 carry out sequential parts of the reaction scheme resulting in multiple products. J Arch Biochem Biophys. 2006;448:83–92. doi: 10.1016/j.abb.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 29.Kollner TG, Gershenzon J, Degenhardt J. Molecular and biochemical evolution of maize terpene synthase 10, an enzyme of indirect defense. Phytochemistry. 2009;70:1139–1145. doi: 10.1016/j.phytochem.2009.06.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.