

Abstract
Antibiotic resistance is a major public health threat, further complicated by unexplained treatment failures caused by bacteria that appear antibiotic susceptible. We describe an Enterobacter cloacae isolate harbouring a minor subpopulation that is highly resistant to the last-line antibiotic colistin. This subpopulation was distinct from persisters, became predominant in colistin, returned to baseline after colistin removal and was dependent on the histidine kinase PhoQ. During murine infection, but in the absence of colistin, innate immune defences led to an increased frequency of the resistant subpopulation, leading to inefficacy of subsequent colistin therapy. An isolate with a lower-frequency colistin-resistant subpopulation similarly caused treatment failure but was misclassified as susceptible by current diagnostics once cultured outside the host. These data demonstrate the ability of low-frequency bacterial subpopulations to contribute to clinically relevant antibiotic resistance, elucidating an enigmatic cause of antibiotic treatment failure and highlighting the critical need for more sensitive diagnostics.
Antibiotic resistance threatens the delivery of safe and effective healthcare1 and is projected to lead to 10 million annual deaths worldwide by 20502. Failure of antibiotic treatment results in increased length of patient stay, healthcare costs and mortality2. Multi-drug resistant Enterobacter spp. have increasingly emerged as a cause of hospital-acquired infections3–5, with the drug colistin being relied on as a last line of treatment6,7. However, colistin-resistant strains have emerged, further limiting treatment options8. Further complicating the treatment of some bacterial infections is the failure of antibiotic therapy in strains that are classified as susceptible; these infections may be non-responsive to treatment in ~10% of cases9. Although relatively little is known about the causes of treatment failures, we show here that they can be mediated by antibiotic-resistant subpopulations in Enterobacter cloacae. Furthermore, such antibiotic-resistant subpopulations can be undetectable by current diagnostic tests.
Results
Phenotypically resistant subpopulation
A strain of E. cloacae was isolated from a renal transplant recipient10 and was observed to harbour a distinct subpopulation with resistance to colistin, visualized as numerous colonies within the zone of inhibition on testing by colistin Etest (we refer to the strain as ‘R/S’, to indicate the presence of both resistant and susceptible subpopulations) (Fig. 1a). This was not observed with either colistin-susceptible or -resistant (Supplementary Fig. 1) clinical strains. A population analysis profile (PAP) of R/S, in which a strain is assayed for survival on agar plates with increasing amounts of antibiotics, revealed a major proportion of the bacteria (>90%) susceptible to 1 μg ml−1 colistin and a highly resistant subpopulation able to withstand at least 500 μg ml−1 colistin (Fig. 1b). This was in contrast to the susceptible strain, which was uniformly killed by 1 μg ml−1 colistin, and the resistant strain, which was uniformly killed by 200 μg ml−1 colistin. The proportion of the R/S colistin-resistant subpopulation was increased to upwards of 80% on exposure to colistin (Fig. 1c). Further analysis revealed that this increase was due to an initial selection against the colistin-susceptible population over the first 2 h of antibiotic exposure, followed by robust replication and expansion of the resistant population in the presence of the drug (Fig. 1d). Importantly, this suggests that the resistant cells are not persisters, which do not significantly expand in number during antibiotic treatment11–13. The increase in the resistant subpopulation was reversible, as subsequent growth after subculture in antibiotic-free media led to a return of these cells to pre-treatment levels (Fig. 1c). This suggests that the resistant subpopulation is not the result of a stable mutation. Furthermore, bacteria from within the zone of inhibition (where antibiotic levels are high) and outside this region (where antibiotic is low or not present) on a colistin Etest plate exhibited identical levels of susceptible and resistant populations after serial culturing in the absence or presence of colistin (Supplementary Fig. 2), suggesting that the bacteria from these two growth conditions are identical. Indeed, deep sequencing of R/S grown with and without colistin (conditions in which the resistant population accounted for the vast majority or minority of the total population, respectively, as summarized in Supplementary Fig. 3) revealed identical genomes. Taken together, these data show that a minor antibiotic-resistant subpopulation is capable of replicating in the presence of antibiotic, becoming predominant and mediating resistance to high levels of drug.
Figure 1. A colistin-resistant subpopulation increases in frequency during in vivo infection.
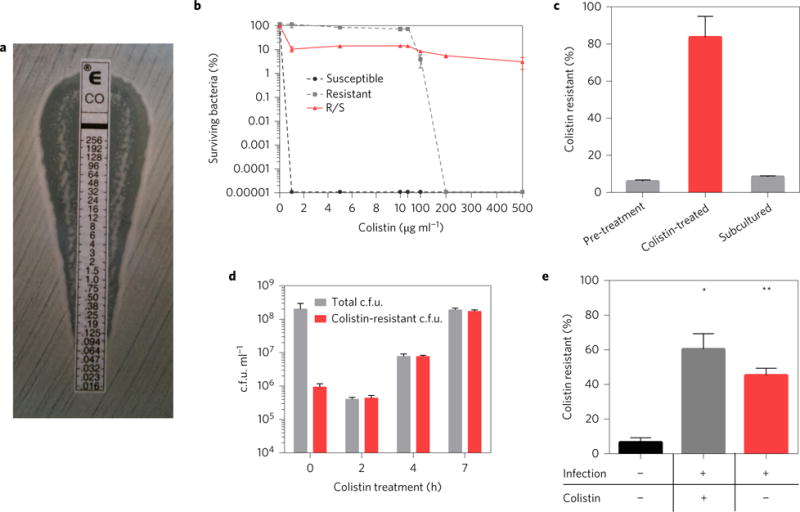
a, Testing of E. cloacae clinical isolate R/S by colistin Etest, with drug concentration indicated in μg ml−1. Colonies within the zone of inhibition indicate a colistin-resistant subpopulation. Data are representative of more than ten Etests. b, Population analysis profile of R/S as well as colistin-susceptible and -resistant E. cloacae clinical isolates (n = 3). c, Percentage of the colistin-resistant subpopulation in R/S in antibiotic-free media, after 24 h treatment with 100 μg ml−1 colistin and after 8 h subculture of the colistin-treated culture in antibiotic-free media. ‘Colistin resistant (%)’ represents the number of c.f.u. in each culture that can grow on media containing 100 μg ml−1 colistin, as a percentage of the total c.f.u. in the culture (n = 3). d, Colistin-resistant c.f.u. and total c.f.u. of R/S during 7 h treatment with 100 μg ml−1 colistin in liquid culture (n = 3). e, Pre-infection inoculum (black bar) was used to infect mice and peritoneal lavage was performed and collected 24 h later and plated to calculate per cent colistin-resistant c.f.u. (n = 5). Mice were treated at 8, 14 and 20 h with colistin (grey bar) or PBS (red bar). Error bars represent standard error of the mean (s.e.m.) (Mann–Whitney test, *P < 0.05, **P < 0.01).
To determine whether the increase in the proportion of the resistant subpopulation occurs during antibiotic treatment in vivo, we infected mice with R/S and treated them with colistin or PBS. In colistin-treated mice, we observed a significant increase in the frequency of the resistant subpopulation of bacteria isolated from the peritoneum (Fig. 1e) and liver (Supplementary Fig. 4). Surprisingly, there was also a robust increase in the resistant subpopulation during in vivo infection in the absence of colistin treatment (Fig. 1e and Supplementary Fig. 4). By 48 h, the percentage of the resistant subpopulation increased from <10% to >80% (Supplementary Fig. 5). These results highlight the process of infection as leading to a significant increase in the frequency of an antibiotic-resistant subpopulation of bacteria.
Resistance to innate immune defences
Various host pressures could be responsible for the increase in the colistin-resistant subpopulation during infection. As macrophages are a major component of the early immune response14, we tested their role by depleting these cells with clodronate liposomes15 (Supplementary Fig. 6) and subsequently infecting mice with R/S. In contrast to bacteria recovered from mice treated with control liposomes, which demonstrate a robust increase in the frequency of the resistant subpopulation, those recovered from macrophage-depleted mice showed no such increase (Fig. 2a). Based on these results, we next determined whether macrophages were sufficient to cause the increase in the resistant subpopulation, by infecting them in vitro. During macrophage infection, the colistin-resistant subpopulation increased to 40% within only 2 h (Fig. 2b). This rise was dependent on internalization of the bacteria, because preventing phagocytosis with cytochalasin D abrogated this phenomenon (Fig. 2b). Therefore, macrophages are both required and sufficient for the increased frequency of the resistant subpopulation during infection, underlining a role for a specific innate immune cell type in this process.
Figure 2. Innate immune host defences are required for the increased frequency of the colistin-resistant subpopulation during infection.
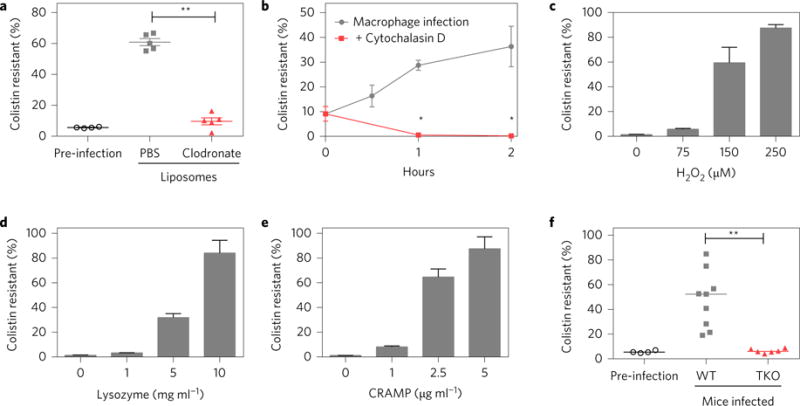
a, Mice pretreated with PBS liposomes (control, grey) or clodronate liposomes (to deplete macrophages, red) were infected with R/S (pre-infection, black). After 8 h, peritoneal lavage fluid was collected and plated to calculate per cent colistin resistance (n = 5). b, Murine bone-marrow-derived macrophages were untreated or pretreated with cytochalasin D, infected with R/S, and per cent colistin resistance was calculated at the indicated time points (n = 6). c–e, R/S was either untreated or treated with the indicated amounts of H2O2 (c), lysozyme (d) or CRAMP (e) for 5 h, and per cent colistin resistance was calculated (n = 3). f, Wild-type (WT, grey) or triple knockout (TKO, red) mice lacking the gp91 subunit of the NADPH oxidase, lysozyme and CRAMP were infected with R/S (pre-infection, black). At 8 h post-infection, peritoneal lavage fluid was collected and plated to calculate per cent colistin resistance (n = 5). Data are compiled from two independent experiments. Error bars represent s.e.m. Mann–Whitney test (**P < 0.01) in a and f; Student’s two-tailed t-test (*P < 0.05) in b.
Macrophages possess many antimicrobials16, and we hypothesized that specific components would be required for the increase in the frequency of the resistant subpopulation, testing reactive oxygen species (formed after treatment with hydrogen peroxide), lysozyme and the murine cationic antimicrobial peptide CRAMP. All of these antimicrobials resulted in a dose-dependent increase in the frequency of the colistin-resistant subpopulation in vitro (Fig. 2c–e), as did LL-37, the human orthologue of CRAMP (Supplementary Fig. 7). These results led us to test whether the antimicrobials were responsible for the increase in the resistant subpopulation during in vivo infection. We infected wild-type (WT) and triple knockout (TKO) mice lacking a functional NADPH oxidase (which leads to the production of reactive oxygen species17), lysozyme and CRAMP. TKO mice were more susceptible to infection by R/S as they harboured over ten times more bacteria than WT mice (Supplementary Fig. 8), demonstrating the importance of these antimicrobials in host defence. Although a robust increase in the frequency of the resistant subpopulation was observed in wild-type mice, this was abrogated in TKO mice (Fig. 2f). The frequency of the resistant subpopulation in mice lacking one of these three antimicrobials was not significantly different from that in WT mice, but it was decreased in double KO mice lacking the NADPH oxidase and CRAMP or lysozyme (Supplementary Fig. 9). These data identify a role for specific host innate immune antimicrobials in the increase of an antibiotic-resistant subpopulation during in vivo infection.
Subpopulation-mediated antibiotic failure
To determine the relevance of the increase in frequency of the resistant subpopulation during in vivo infection, we tested whether the R/S strain was able to resist colistin treatment. We infected mice with either R/S or a colistin-susceptible strain, and treated the mice with PBS (as a control) or high doses of colistin after establishment of infection to simulate the progression of infection and treatment in the clinic. The levels of the susceptible strain in the peritoneum (Fig. 3a) and liver (Fig. 3b) were significantly reduced by colistin treatment. In contrast, the R/S strain was refractory to treatment with colistin as its levels were unchanged between the treated and untreated groups (Fig. 3a,b). In a timecourse experiment, the level of the susceptible strain was reduced by 3 logs at 42 h, whereas the level of the R/S strain was not diminished by colistin treatment, but instead increased by roughly tenfold (Supplementary Fig. 10). These data suggest that the presence of the resistant subpopulation results in inefficacy of colistin to reduce bacterial levels in vivo. Furthermore, these results provide in vivo evidence that the resistant subpopulation does not behave like persisters, which do not significantly expand in number during antibiotic treatment.
Figure 3. R/S is refractory to colistin during infection and leads to colistin treatment failure.
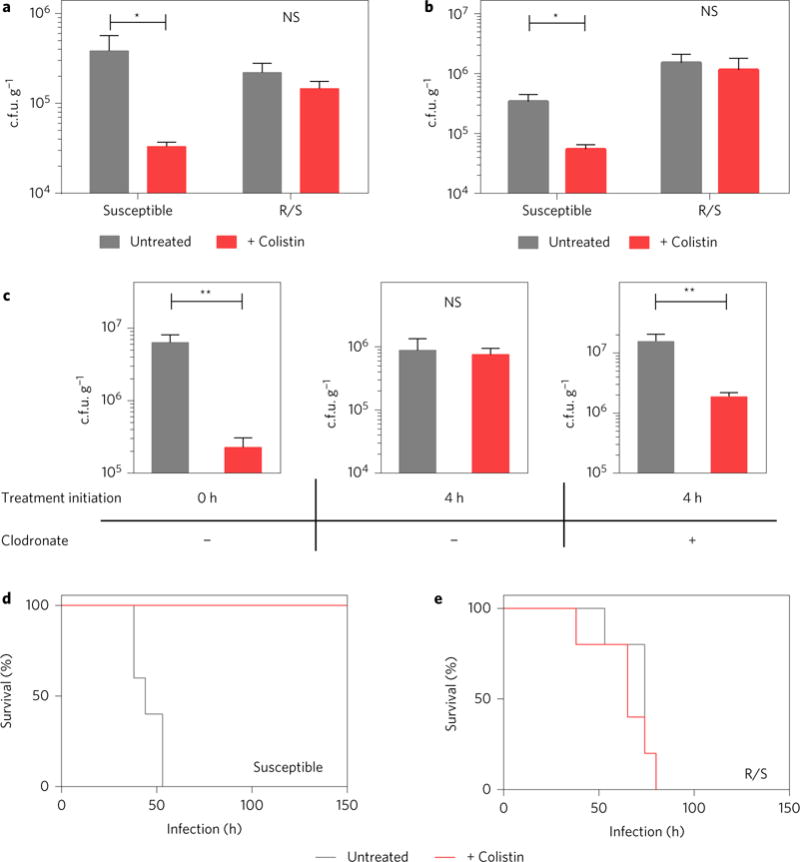
a,b, Mice infected with R/S or the susceptible isolate were treated with colistin at 8, 14 and 20 h. Numbers of c.f.u. were quantified at 24 h in the peritoneal lavage fluid (a) and liver (b) (n = 5). c, Mice pretreated with PBS (left and middle panels) or clodronate (right panel) liposomes were infected with R/S and treated with colistin at 0 h (left panel) or 4 h (middle and right panels). A second dose of colistin was administered 2 h after the first; 2 h later, peritoneal lavage fluid was plated to enumerate c.f.u. (n = 5). d,e, Survival of mice infected with R/S or the colistin-susceptible isolate. Mice were treated with colistin or PBS starting at 12 h post-infection, with additional doses given every 6 h thereafter. Surviving mice were monitored until day 24 (n = 5). Error bars represent s.e.m.; centre values represent the median. Mann–Whitney test: *P < 0.05, **P < 0.01, NS = not significant.
We next tested whether the role of the host immune system in the increase of the resistant subpopulation was directly responsible for the inefficacy of antibiotic therapy. We first found that colistin treatment of R/S-infected mice could cause a significant reduction in bacterial levels if initiated at the time of infection (before the increase in the frequency of the resistant subpopulation), but not if it was delayed until only 4 h after infection (Fig. 3c). However, in macrophage-depleted mice, treatment with colistin at 4 h became effective, leading to a reduction in bacterial levels (Fig. 3c) and indicating that the host-driven increase in the frequency of the resistant subpopulation is responsible for the inefficacy of antibiotic treatment.
To further test the relevance of this phenomenon in vivo, we infected mice with a lethal dose of bacteria and treated with either PBS or colistin after 12 h. Both the susceptible and R/S strains led to lethal infections in the absence of colistin (Fig. 3d,e). In the presence of colistin, only mice infected with the susceptible strain were rescued (Fig. 3d), whereas those infected with R/S still succumbed to infection within 100 h (Fig. 3e). These data demonstrate the impact of an antibiotic-resistant subpopulation in mediating a lethal infection during high-dose antibiotic treatment.
PhoQ-dependent resistant subpopulation
We next set out to determine the molecular mechanism underlying the phenotype of the resistant subpopulation. RNAseq analysis was conducted (Supplementary Fig. 3 and Supplementary Table 1) to determine whether there were transcriptional differences between the susceptible and resistant subpopulations of R/S. In total, this analysis revealed 325 genes upregulated and 360 genes downregulated in the resistant subpopulation as compared to the susceptible subpopulation (Supplementary Table 2). This approach should detect differences between the two subpopulations, but it may also identify expression differences due to colistin treatment. Among the upregulated genes, we noticed a signature (Supplementary Table 3) associated with the two-component histidine kinase PhoQ (refs 18–25), which has previously been implicated in polymyxin resistance, in part through its role in modification of the lipid A portion of lipopolysaccharide26. To validate the RNAseq data, we confirmed that the resistant subpopulation expressed higher levels of the predicted lipid A modification genes arnB and eptA (ref. 27) (Supplementary Fig. 11). These data suggested that R/S displayed a modified lipid A profile, which we confirmed by thin-layer chromatography (Supplementary Fig. 12). Furthermore, modified lipid A species increased in abundance during growth of R/S in the presence of colistin, consistent with their expression by the resistant subpopulation (Supplementary Fig. 12). To test whether the lipid A modifications were dependent on PhoQ, we constructed an R/S deletion mutant lacking phoQ (ΔphoQ). Indeed, lipid A from the ΔphoQ strain lacked the specific lipid A modifications observed in WT R/S that were enhanced in the presence of colistin, which were restored in a phoQ complemented strain (Supplementary Fig. 12). Thus, the R/S resistant subpopulation exhibits PhoQ-dependent lipid A modifications and is transcriptionally distinct from the susceptible subpopulation.
To interrogate the potential contribution of PhoQ to the R/S resistance phenotype, we examined the colistin resistance profile of ΔphoQ. Strikingly, the ΔphoQ strain exhibited a complete absence of the resistant subpopulation by Etest, while the susceptible subpopulation remained unaffected, as the border of the zone of clearing was unaltered from that of WT R/S (Fig. 4a). Complementation with phoQ restored the presence of the resistant subpopulation (Fig. 4a). This was also confirmed by PAP, where ΔphoQ lacked the resistant subpopulation present in R/S and behaved in a similar manner to the susceptible strain (Fig. 4b). Importantly, R/S and ΔphoQ harboured equivalent levels of persisters, clearly indicating that the colistin-resistant subpopulation (which depends on PhoQ) is not made up of persister cells (Supplementary Fig. 13). The phoQ mutant also exhibited no colistin-resistant subpopulation after exposure to host antimicrobials (Supplementary Fig. 14a), during macrophage infection (Supplementary Fig. 14b) or during in vivo infection (Supplementary Fig. 14c). Without the presence of the resistant subpopulation, ΔphoQ was susceptible to colistin treatment in vivo, exhibiting a significantly decreased bacterial load (Fig. 4c). Furthermore, the ability of colistin to rescue mice from an otherwise lethal inoculum was restored during infection with ΔphoQ (Fig. 4d). Thus, the presence of the colistin-resistant subpopulation is dependent on PhoQ, which is required for a lethal drug-resistant infection.
Figure 4. PhoQ is required for the presence of the colistin-resistant subpopulation.
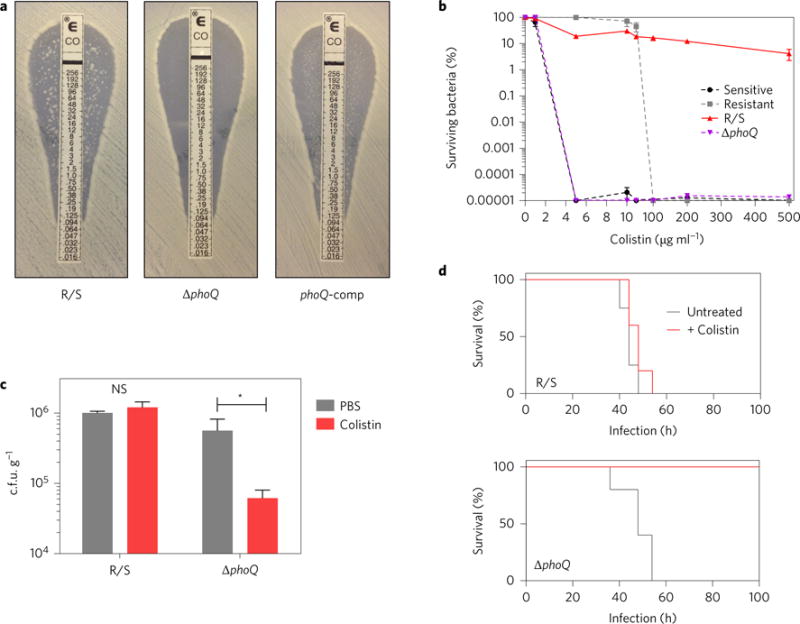
a, Colistin Etest of R/S, ΔphoQ and the complement (phoQ-comp) strains, with drug concentration indicated in μg ml−1. Colonies within the zone of inhibition indicate a colistin-resistant subpopulation. Data are representative of two Etests. b, Population analysis profile of R/S, ΔphoQ and colistin-susceptible and -resistant E. cloacae strains (n = 3). c, Mice infected with R/S or ΔphoQ were treated with colistin at 8, 14 and 20 h. Values of c.f.u. were quantified at 24 h in the peritoneal lavage fluid (n = 5). d, Survival of mice infected with R/S (top) or ΔphoQ (bottom). Mice were treated with colistin or PBS starting at 12 h post-infection, with additional doses given every 6 h thereafter (n = 5). Error bars represent s.e.m.; centre values represent the median. Mann–Whitney test: *P < 0.05, NS = not significant.
Undetected resistant subpopulation
The size of the resistant subpopulation can vary greatly between strains, as exemplified by a distinct E. cloacae clinical isolate (termed R/S-lo) that harbours a colistin-resistant subpopulation comprising between 0.01 and 0.001% of the total population (Fig. 5a), over 1,000-fold less prevalent than that of R/S when grown in media without antibiotic. Similar to R/S, the increase of the R/S-lo resistant subpopulation in the presence of colistin (Supplementary Fig. 15) was due to initial selection against the susceptible subpopulation followed by expansion of the resistant subpopulation (Supplementary Fig. 16). The frequency of the resistant subpopulation was similarly increased by treatment with H2O2, lysozyme, CRAMP and LL-37 (Supplementary Fig. 17), during macrophage infection (Supplementary Fig. 18) and during in vivo infection of mice (Supplementary Fig. 19), and was greatly diminished in macrophage-depleted (Supplementary Fig. 20) and TKO mice (Supplementary Fig. 21). These data revealed that, similar to R/S, the frequency of the resistant subpopulation of R/S-lo is increased by colistin as well as the activity of specific host innate immune components. During in vivo infection, although the levels of a susceptible strain were significantly reduced by colistin treatment, the levels of R/S-lo were unaffected (Supplementary Fig. 22). These data directly correlated with a failure of colistin therapy to rescue R/S-lo infected mice from a lethal infection (Fig. 5c), whereas mice infected with a susceptible strain were completely rescued (Fig. 5b). Importantly, unlike R/S, R/S-lo was clinically classified as being susceptible to colistin, as the resistant subpopulation (present at a frequency of only 1 in 10,000 c.f.u.) was not detected by Etest (Fig. 5d). Therefore, this seemingly colistin-susceptible strain, harbouring an undetected resistant subpopulation, is capable of causing an antibiotic-resistant and lethal infection in vivo.
Figure 5. Clinical isolate harbouring an undetected colistin-resistant subpopulation causes a lethal, antibiotic-resistant infection.
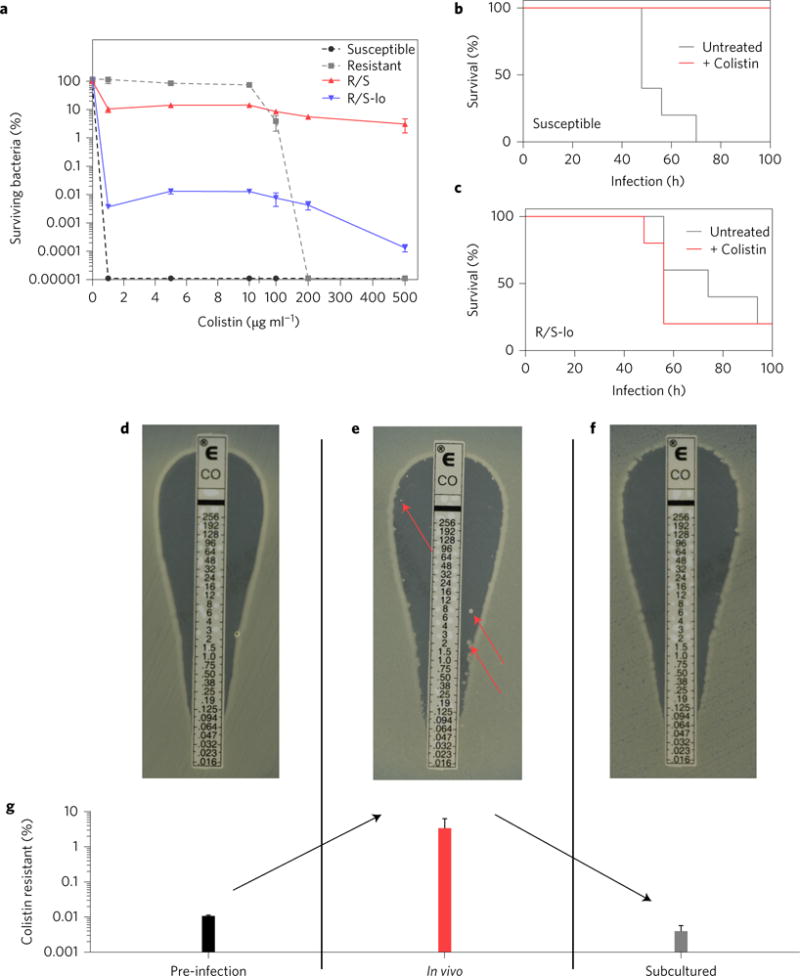
a, PAP of E. cloacae clinical isolate R/S-lo compared to R/S and the colistin-susceptible and -resistant isolates (n = 3). b,c, Infection of mice with the colistin-susceptible isolate (b) or R/S-lo with or without colistin treatment (c) every 6 h and initiated 12 h post-infection (n = 5). Surviving mice were monitored until day 24. d–f, Colistin Etest results, with drug concentration indicated in μg ml−1, of R/S-lo from pre-infection inoculum (d), peritoneal lavage sample from a mouse infected for 8 h (e) and the peritoneal lavage sample subcultured overnight in drug-free media (f) (n = 5). Colonies in the zone of inhibition (e, red arrows) indicate resistant bacteria. Images are representative of five individual samples. g, Samples from d–f were plated to determine per cent colistin resistance (n = 5). Error bars represent s.e.m.
It is worrisome that R/S-lo was not identified as colistin-resistant, and we wondered whether the resistant population could be detected by diagnostic testing when it is more frequent during host infection. We directly plated peritoneal lavage samples from infected mice in the absence of subculture and were able to detect the R/S-lo resistant subpopulation by Etest, as indicated by colonies within the zone of inhibition (Fig. 5e,g). In contrast, when these samples were processed by the clinical microbiology laboratory (as would occur with a sample from a human patient and including a critical subculture step), Etest could no longer detect the diminished resistant subpopulation (Fig. 5f,g). Strikingly, these data reveal how and when detection of the resistant subpopulation can be missed during routine diagnostic testing and how this can translate into an unexplained failure of antibiotic therapy.
Discussion
The findings presented here highlight the role of a minor colistin-resistant bacterial subpopulation in mediating antibiotic treatment failure in vivo. This resistant subpopulation is genetically identical to the susceptible subpopulation, but exhibits differences in gene expression and lipid A modification. Furthermore, the presence of this resistant subpopulation is dependent on the histidine kinase PhoQ. The data also highlight an unexpected role for specific host innate immune components (lysozyme, CRAMP and H2O2) in the increase of the antibiotic-resistant subpopulation during infection. The increase in the frequency of the resistant subpopulation induced by host immune pressure in vivo was shown to be critical for eventual failure of colistin therapy.
Like bacterial persistence, the phenotypic resistance phenomenon we describe involves a resistant subpopulation, but there are important differences. Persistence involves a small subpopulation of bacteria that are tolerant to a drug due to a state of low metabolic activity, with no or limited replication28. Wakamoto et al. showed that, in some cases, persisters can replicate, although it is at a very low rate and is insufficient to cause an overall increase in the numbers of the population11. In contrast, we describe a resistant subpopulation that rapidly replicates both in vitro and in vivo in the presence of antibiotic and leads to a very significant overall increase in bacterial population level (Fig. 1d and Supplementary Fig. 10). Furthermore, we directly show that the PhoQ-dependent colistin-resistant subpopulation is distinct from persisters, which are also present but independent of PhoQ (Supplementary Fig. 13). Several papers have recently demonstrated the importance of persisters as a reservoir of infection during antibiotic treatment in vivo12, which can continue to replicate after treatment has been stopped, leading to relapse13. In contrast, we demonstrate that the colistin-resistant subpopulation described here facilitates bacterial growth and subsequent host lethality, even in the presence of antibiotic. Persistence has also been linked to immune pressure, as bacterial populations within macrophages can have increased numbers of persisters29. We observe a similar link; both in vitro and in vivo, specific host antimicrobials lead to an increased frequency of the resistant subpopulation. Taken together, both persisters and the resistant subpopulation described here highlight the ability of a minority of a bacterial population to exert a striking effect on the outcome of infection and antibiotic treatment. Persisters are kept at bay by antibiotic treatment and form a reservoir that can cause relapse, but the colistin-resistant subpopulation described here has the ability to cause acute infection and lethality during the course of antibiotic treatment.
We propose to refer to the resistance phenomenon described here as ‘clonal heteroresistance’. The phenomenon of heteroresistance, in which a resistant subpopulation exhibits an increased level of antibiotic resistance relative to the larger susceptible subpopulation, was described as far back as 1947 (ref. 30). However, its relevance to infection and resistance has remained unclear, and even its definition has been debated. We use the term clonal heteroresistance to distinguish the phenomenon we describe from the blanket term heteroresistance, which is often used to refer to mixed populations of genetically distinct bacteria31–34. We show that clonal heteroresistance, in addition to mediating lethal infection in the presence of antibiotic, can also go undetected and cause unexplained treatment failure during in vivo infection (Supplementary Fig. 23). Current widely used methods of antibiotic susceptibility testing rely on in vitro culture and analysis. Our data show that these methods can greatly alter results and present an inaccurate picture of the level of in vivo resistance. Our findings highlight both a need and opportunity for improved diagnostics to detect antibiotic-resistant subpopulations and ultimately prevent such treatment failures.
Methods
Bacterial strains
E. cloacae strain R/S was isolated from a blood sample from a renal transplant recipient at Emory University Hospital, Atlanta, GA. E. cloacae R/S-lo, the colistin-susceptible strain Mu819 and the colistin-resistant strain Mu117 were isolated from urine samples from patients at other Atlanta hospitals.
Bacterial culture
All bacterial strains were streaked on Mueller–Hinton (MH) agar plates and grown in MH medium at 37 °C in a shaking incubator from a single colony before each experiment. Numbers of c.f.u. were determined by plating dilutions on MH agar plates incubated at 37 °C and then counting bacterial colonies at the lowest distinguishable dilution.
Bacterial genetics
To generate strain ΔphoQ, 600–700 bp upstream and downstream fragments of the genomic region surrounding phoQ were PCR-amplified with primers 81 and 118, and 82 and 119, respectively (Supplementary Table 4), and fused with the hygromycin resistance cassette HmR amplified from vector pMQ310 with primers 79 and 80 (ref. 35) using splicing by overlap extension (SOE) PCR (ref. 36). The suicide vector pEXR6K was generated by replacing the pMB1 ori from PCR linearized plasmid pEX100T (ref. 37) using primers 110 and 111 with the R6K ori amplified from plasmid pMQ310 with primers 108 and 109 using the Gibson Assembly Cloning Kit (Invitrogen). The HmR construct was inserted into SmaI (New England Biolabs) digested pEXR6K by Gibson assembly and the resulting plasmid was transformed to strain R/S by electroporation.
Transformants were selected on MH agar containing 150 μg ml−1 hygromycin (Sigma) then passaged to Luria-Bertani (LB) agar containing 20% sucrose and no NaCl to counterselect for vector loss. Chromosomal replacement of phoQ with the hygromycin marker was confirmed by Sanger sequencing. To generate strain phoQ-comp, the promoter region of the phoPQ operon was amplified with primers 142 and 143 and fused by SOE PCR to gene phoQ amplified with primers 144 and 145. The resulting construct was inserted to plasmid pBAV-1K-T5-GFP (ref. 38) PCR linearized with primers 146 and 147 to create the complementation vector. The vector was transformed to strain ΔphoQ by electroporation and selected on MH agar containing 90 μg ml−1 kanamycin (Sigma).
Antibiotic susceptibility testing
The colistin susceptibility of all strains was determined using the Etest method. Briefly, the inoculum was prepared from colonies grown on a 5% sheep blood agar plate (Remel, Lenexa, KS) for 18 h. Several colonies were suspended in 0.9% sterile saline (Remel) and adjusted to a concentration equivalent to a 0.5 McFarland turbidity standard. The suspension was used to streak a 100-mm-diameter MH agar plate and the Etest strip (bioMérieux) was placed on it. The plate was incubated at 35 °C for 20 h and the minimum inhibitory concentration (MIC) was read where inhibition of growth intersected the Etest strip. Small colonies that grew within the zone of inhibition were included in the MIC determination. Etest analyses of samples from mouse infections were plated directly from peritoneal lavage samples without subculturing. PAPs were performed by growing bacteria to mid-log phase and then plating on MH agar containing various concentrations of colistin. Percentage colistin resistance was calculated as the number of bacteria that grew on 100 μg ml−1 colistin divided by the number of bacteria that grew on MH alone.
Mice
WT C57BL/6J mice were purchased from Jackson Laboratories and used at age 8–10 weeks; all experiments used age- and sex-matched mice. TKO mice deficient in the gp91 component of the NADPH oxidase, lysozyme and CRAMP, as well as double knockout mice lacking two of the indicated antimicrobials, were derived by crossing cybb−/− (gp91, Jackson Laboratories), lysM−/− (lysozyme; generously provided by D. Portnoy, UC Berkeley) and cnlp−/− (CRAMP; Jackson Laboratories) mice. TKO mice were investigated for health defects by histology and bacterial culture of various organs, with no overt health differences observed in uninfected TKO mice when compared to WT. Mice were housed under specific-pathogen-free conditions in filter-top cages at Yerkes National Primate Center, Emory University, and provided food and water ad libitum. All experimental procedures were approved by the Emory University Institutional Animal Care and Use Committee (IACUC). Sample size, as reported in the figure legends, was determined by allowing for significance by the Mann–Whitney test (n ≥ 4) while minimizing the number of animals used, and five mice were thus used per group for the majority of experiments. No randomization or blinding was done in the animal studies.
Mouse infections
Approximately 5 × 107 c.f.u. were administered per mouse for infections to quantify the bacterial load, and approximately 2 × 108 c.f.u. were administered for survival experiments. Bacterial inocula were suspended in PBS and 100 μl was inoculated intraperitoneally (i.p.) to each mouse. Colistin methanesulfonate was injected i.p. in 100 μl PBS at a dosage of 10 mg per kg per dose. Mice were monitored by weight and were euthanized if found to be below 80% starting weight, as mandated by the IACUC protocol. Mice were euthanized, and the liver, spleen and peritoneal lavage samples were collected into sterile PBS. Solid organ samples were homogenized using a tissue-tearor (BioSpec), and all samples were plated for c.f.u. and per cent colistin resistance.
Macrophage depletion
Macrophages were depleted from mice using clodronate liposomes (clodronateliposomes.com). Mice were injected with 200 μl liposomes i.p. 3 days before infection and then injected again with 100 μl liposomes i.v. 1 day before infection. Mice were infected i.p. for 8 h before peritoneal lavage fluid was collected and plated for c.f.u. Part of this sample was also used for flow cytometry to confirm macrophage depletion.
Macrophage infection
Macrophages were derived from the bone marrow of mice. Briefly, femurs from mice were removed and whole bone marrow was flushed out. The bone marrow cells were grown in media containing DMEM, 10% fetal bovine serum (FBS) and macrophage colony-stimulating factor (M-CSF), which induces the differentiation and growth of macrophages. After confluent layers of macrophages were derived, cells were plated into 24-well plates at 3 × 105 cells per well. Bacteria were added to the wells at 3 × 106 c.f.u. per well for a multiplicity of infection (MOI) of 10:1. Plates were centrifuged to synchronize the infection. After 30 min, the macrophages were washed and 100 μg ml−1 of gentamicin was added to the media to remove and prevent the growth of extracellular bacteria. At 1, 2 and 4 h post-infection, macrophages were incubated with 1% saponin in PBS for 2 min to lyse open cells and remove bacteria. Samples were then plated for c.f.u., and per cent colistin resistance calculated. To prevent internalization of bacteria, some wells were pretreated with 1 μg ml−1 cytochalasin D for 30 min before the addition of bacteria.
Flow cytometry
Peritoneal lavage fluid was stained with F4/80-PE/Cy7 (BM8) (Biolegend) and CD11b-APC/A700 (M1I70) (eBioscience) antibodies for 35 min. Red blood cells (RBCs) were lysed with RBC lysis buffer (Becton Dickinson) for 5 min. Cells were fixed with 1% paraformaldehyde and analysed on an LSRII flow cytometer (BD). Macrophages were defined as F4/80+CD11b+ cells.
DNA and RNA isolation
An overnight liquid culture of R/S grown at 37 °C in MH broth was back-diluted in triplicate to either fresh MH broth or MH broth containing 100 μg ml−1 colistin to enrich for susceptible or colistin-resistant bacteria, respectively. Cultures were grown to exponential phase at 37 °C and collected for DNA and RNA isolation. Numbers of c.f.u. were calculated as above. DNA was isolated using the DNEasy Blood and Tissue Kit (Qiagen) following the Gram negative bacteria protocol with RNase treatment. RNA was isolated using a modified phase extraction method39 with initial incubation in TriReagent (Zymo) followed by phase separation with chloroform. RNA was precipitated from the aqueous phase with isopropanol and 1.2 M NaCl at 4 °C, and further purified with the Directzol RNA Kit (Zymo) following the recommended DNase treatment step.
DNA and RNA sequencing
Sample integrities were verified with the Agilent 2100 Bioanalyzer. DNA libraries were prepared using the NexteraXT DNA kit (Illumina). For RNA libraries, samples were first depleted of ribosomal RNAs using the Ribo-Zero rRNA Removal Kit (Illumina) and libraries prepared using the EpiCentre ScriptSeq Complete (Bacteria) Low Input kit (Illumina). Next-generation short sequence reads were generated with the Illumina HiSeq 1000 platform at the Yerkes National Primate Research Center Nonhuman Primate Genomics Core (http://www.yerkes.emory.edu/nhp_genomics_core/). Long sequence reads were generated with the PacBio II platform using P5-C3 chemistry at the Duke University Sequencing and Genomic Technologies Shared Resource.
De novo genome assembly and sequence analysis
A hybrid de novo assembly was performed using both Illumina and PacBio data using Celera Assembler version 8.2 (ref. 40). The sequence data resolved into two contigs, one representing the chromosome and the other representing the plasmid. The quality of the assembly was confirmed by analysis using the ALE tool41. The assembly was automatically annotated using the NCBI prokaryotic annotation pipeline. Illumina whole shotgun sequences of the samples enriched for colistin resistance (COL) and colistin susceptibility (MH) were aligned against the assembled genome using bwa-0.7.12 (ref. 42) and visualized with the samtools-1.2 mpileup function43. Single nucleotide polymorphisms between the assembled genome and short sequence reads were manually analysed to determine sequence conservation between COL and MH samples.
RNAseq analysis
Single-end Illumina libraries from reverse-transcribed RNA were mapped against the Enterobacter de novo assembled reference using Bowtie2 (ref. 44). Differential gene expression between the three colistin-treated strains and controls was quantified by the cufflinks/cuffdiff tools in CufflinksVersion 2.2.1 (refs 45,46). Sequences of differentially expressed genes with significant q-values were analysed with Blast2Go software version 3.1.3 to identify the Escherichia coli gene orthologue and putative function47.
Quantitative reverse transcriptase PCR (qRT-PCR)
RNA was collected as described above. One-step qRT-PCR was performed using the Power SYBR Green RNA-to-Ct kit (Applied Biosystems) with primers (Supplementary Table 4) on a StepOnePlus Real-time PCR System (Applied Biosystems) according to the manufacturer’s instructions. rpoD was used as the internal control gene48. Relative expression was calculated as 2−(ΔCt) (ref. 49).
Isolation and analysis of 32P lipid A species
E. cloacae strains were grown overnight in MH broth, diluted 1:400 in fresh MH broth containing appropriate selective antibiotics. For induction of the resistant phenotype, 100 μg ml−1 colistin (Sigma) was used. E. coli W3110 and WD101 strains were grown in LB broth overnight followed by a 1:100 dilution in fresh LB medium. After dilutions, cells were immediately labelled with 2.5 μCi ml−1 of inorganic 32P-phosphate (Perkin Elmer) and collected at absorbance at 600 nm (A600) = 0.5 (E. cloacae) or A600 = 1.0 (E. coli). Lipid A extraction, separation and visualization was performed as described previously50. Briefly, lipid A extraction was carried out by mild acidic hydrolysis and spotted onto a silica thin-layer chromatography plate (10,000 c.p.m. per lane). Labelled lipid A species were separated using a solvent mixture of chloroform, pyridine, 88% formic acid and water (50:50:16:5). The thin-layer chromatography plate was exposed to a phosphoimager screen and visualized by phosphoimaging analysis (Bio-Rad PMI). The analysed images were cropped to aid in data analysis (for the full unaltered images see Supplementary Fig. 24).
Statistics
Statistical analyses were performed using Prism 5 (Graphpad Software). The significance of the mouse experiments was determined with the Mann–Whitney test, as not all data were normally distributed, and all in vitro experiments were analysed using the two-tailed student’s t-test (for data with a normal distribution). All experiments were repeated at least two to three times (and up to ten times). All replicates shown are biological replicates.
Supplementary Material
Acknowledgments
The authors thank S. Satola, M. Farley and the Georgia Emerging Infections Program for providing Enterobacter cloacae strains Mu117, Mu819 and R/S-lo, P. Rather for providing plasmid pMQ310, the Yerkes Nonhuman Primate Genomics Core for help with DNA sequencing and analysis, and C.-Y. Chin and D. Bonenberger for breeding and genotyping knockout mice. The authors also thank R. Ahmed, A. Grakoui and W. Shafer for comments and revisions of the manuscript. D.S.W. is supported by a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease award, VA Merit Award I01 BX002788 and National Institutes of Health (NIH) grant AI098800. E.K.C. is supported by the National Institute of Allergy and Infectious Diseases of the NIH under award no. T32AI106699. M.S.T. is supported by the NIH (grants nos RO1AI064184, RO1AI76322 and R21AI11987) and the Army Research Office (grant no. 61789-MA-MUR). T.D.R. and K.V. were supported by 1U01CI000906-05 ‘Emerging Infections Program (EIP) PPACA: Enhancing Epidemiology and Laboratory Capacity’ funding from the Emory Public Health Bioinformatics Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, the Department of Veterans Affairs or the Centers for Disease Control and Prevention.
Footnotes
Accession codes. DNA and RNA sequencing data were deposited at NCBI under Bioproject no. PRJNA263343 as BioSamples SAMN03099688, SAMN04538424 and SAMN04538425.
Author contributions
Experiments were conducted by V.I.B., E.K.C. and B.A.N. The manuscript was prepared by V.I.B., E.K.C. and D.S.W. Clinical microbiological assays were conducted by E.M.B., who also provided the R/S strain. Lipid A analysis was performed by C.M.H. and M.S.T. Sequence analysis was performed by E.K.C., G.K.T., K.V., T.D.R. and S.E.B. J.P. synthesized and purified host antimicrobials. The study was planned and directed by D.S.W.
Additional information
Supplementary information is available online.
Competing interests
The authors declare no competing financial interests.
References
- 1.United States Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. 2013 http://www.cdc.gov/drugresistance/pdf/ar-threats-2013-508.pdf.
- 2.Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014 http://go.nature.com/GLHVCr.
- 3.Mezzatesta ML, Gona F, Stefani S. Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiol. 2012;7:887–902. doi: 10.2217/fmb.12.61. [DOI] [PubMed] [Google Scholar]
- 4.Davin-Regli A, Pagès JM. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front Microbiol. 2015;6:392. doi: 10.3389/fmicb.2015.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders WE, Sanders CC. Enterobacter spp.: pathogens poised to flourish at the turn of the century. Clin Microbiol Rev. 1997;10:220–241. doi: 10.1128/cmr.10.2.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlet J, Mainardi JL. Antibacterial agents: back to the future? Can we live with only colistin, co-trimoxazole and fosfomycin? Clin Microbiol Infect. 2012;18:1–3. doi: 10.1111/j.1469-0691.2011.03702.x. [DOI] [PubMed] [Google Scholar]
- 7.Nation RL, Li J. Colistin in the 21st century. Curr Opin Infect Dis. 2009;22:535–543. doi: 10.1097/QCO.0b013e328332e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Livermore DM, et al. What remains against carbapenem-resistant Enterobacteriaceae? Evaluation of chloramphenicol, ciprofloxacin, colistin, fosfomycin, minocycline, nitrofurantoin, temocillin and tigecycline. Int J Antimicrob Agents. 2011;37:415–419. doi: 10.1016/j.ijantimicag.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 9.Kuper KM, Boles DM, Mohr JF, Wanger A. Antimicrobial susceptibility testing: a primer for clinicians. Pharmacotherapy. 2009;29:1326–1343. doi: 10.1592/phco.29.11.1326. [DOI] [PubMed] [Google Scholar]
- 10.Napier BA, Band V, Burd EM, Weiss DS. Colistin heteroresistance in enterobacter cloacae is associated with cross-resistance to the host antimicrobial lysozyme. Antimicrob Agents Chemother. 2014;58:5594–5597. doi: 10.1128/AAC.02432-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakamoto Y, et al. Dynamic persistence of antibiotic-stressed mycobacteria. Science. 2013;339:91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 12.Claudi B, et al. Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell. 2014;158:722–733. doi: 10.1016/j.cell.2014.06.045. [DOI] [PubMed] [Google Scholar]
- 13.Kaiser P, et al. Cecum lymph node dendritic cells harbor slow-growing bacteria phenotypically tolerant to antibiotic treatment. PLoS Biol. 2014;12:e1001793. doi: 10.1371/journal.pbio.1001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 15.Van Rooijen N. The liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods. 1989;124:1–6. doi: 10.1016/0022-1759(89)90178-6. [DOI] [PubMed] [Google Scholar]
- 16.Nathan CF. Mechanisms of macrophage antimicrobial activity. Trans R Soc Trop Med Hyg. 1983;77:620–630. doi: 10.1016/0035-9203(83)90190-6. [DOI] [PubMed] [Google Scholar]
- 17.Iles KE, Forman HJ. Macrophage signaling and respiratory burst. Immunol Res. 2002;26:95–105. doi: 10.1385/IR:26:1-3:095. [DOI] [PubMed] [Google Scholar]
- 18.Minagawa S, et al. Identification and molecular characterization of the Mg2+ stimulon of Escherichia coli. J Bacteriol. 2003;185:3696–3702. doi: 10.1128/JB.185.13.3696-3702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alpuche Aranda CM, Swanson JA, Loomis WP, Miller SI. Salmonella typhimurium activates virulence gene transcription within acidified macrophage phagosomes. Proc Natl Acad Sci USA. 1992;89:10079–10083. doi: 10.1073/pnas.89.21.10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwir I, et al. Dissecting the PhoP regulatory network of Escherichia coli and Salmonella enterica. Proc Natl Acad Sci USA. 2005;102:2862–2867. doi: 10.1073/pnas.0408238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merighi M, Ellermeier CD, Slauch JM, Gunn JS. Resolvase— in vivo expression technology analysis of the Salmonella enterica serovar Typhimurium PhoP and PmrA regulons in BALB/c mice. J Bacteriol. 2005;187:7407–7416. doi: 10.1128/JB.187.21.7407-7416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin QY, et al. Serratia marcescens arn, a PhoP-regulated locus necessary for polymyxin B resistance. Antimicrob Agents Chemother. 2014;58:5181–5190. doi: 10.1128/AAC.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monsieurs P, et al. Comparison of the PhoPQ regulon in Escherichia coli and Salmonella typhimurium. J Mol Evol. 2005;60:462–474. doi: 10.1007/s00239-004-0212-7. [DOI] [PubMed] [Google Scholar]
- 24.Oshima T, et al. Transcriptome analysis of all two-component regulatory system mutants of Escherichia coli K-12. Mol Microbiol. 2002;46:281–291. doi: 10.1046/j.1365-2958.2002.03170.x. [DOI] [PubMed] [Google Scholar]
- 25.Choi E, Groisman EA, Shin D. Activated by different signals, the PhoP/PhoQ two-component system differentially regulates metal uptake. J Bacteriol. 2009;191:7174–7181. doi: 10.1128/JB.00958-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Band VI, Weiss DS. Mechanisms of antimicrobial peptide resistance in Gram-negative bacteria. Antibiotics (Basel) 2015;4:18–41. doi: 10.3390/antibiotics4010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in Gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keren I, Minami S, Rubin E, Lewis K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio. 2011;2:e00100–e00111. doi: 10.1128/mBio.00100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helaine S, et al. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science. 2014;343:204–208. doi: 10.1126/science.1244705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander HE, Leidy G. Mode of action of streptomycin on type B Hemophilus influenzae: II. Nature of resistant variants. J Exp Med. 1947;85:607–621. doi: 10.1084/jem.85.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinder H. Hetero-resistance: an under-recognised confounder in diagnosis and therapy? J Med Microbiol. 2001;50:1018–1020. doi: 10.1099/0022-1317-50-12-1018. [DOI] [PubMed] [Google Scholar]
- 32.Zheng C, et al. Mixed infections and rifampin heteroresistance among mycobacterium tuberculosis clinical isolates. J Clin Microbiol. 2015;53:2138–2147. doi: 10.1128/JCM.03507-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El-Halfawy OM, Valvano MA. Antimicrobial heteroresistance: an emerging field in need of clarity. Clin Microbiol Rev. 2015;28:191–207. doi: 10.1128/CMR.00058-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kao CY, et al. Heteroresistance of Helicobacter pylori from the same patient prior to antibiotic treatment. Infect Genet Evol. 2014;23:196–202. doi: 10.1016/j.meegid.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Kalivoda EJ, et al. New vector tools with a hygromycin resistance marker for use with opportunistic pathogens. Mol Biotechnol. 2011;48:7–14. doi: 10.1007/s12033-010-9342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 37.Schweizer HP, Hoang TT. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene. 1995;158:15–22. doi: 10.1016/0378-1119(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 38.Bryksin AV, Matsumura I. Rational design of a plasmid origin that replicates efficiently in both gram-positive and gram-negative bacteria. PLoS ONE. 2010;5:e13244. doi: 10.1371/journal.pone.0013244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Applied Biosystems. TRI Reagent Solution RNA/DNA/Protein Isolation Reagent Manual. Ambion: 2010. [Google Scholar]
- 40.Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark SC, Egan R, Frazier PI, Wang Z. ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics. 2013;29:435–443. doi: 10.1093/bioinformatics/bts723. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Durbin R. Fast and accurate long-read alignment with Burrows– Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 46.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Götz S, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landman D, Salamera J, Quale J. Irreproducible and uninterpretable polymyxin B MICs for Enterobacter cloacae and Enterobacter aerogenes. J Clin Microbiol. 2013;51:4106–4111. doi: 10.1128/JCM.02129-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 50.Herrera CM, Hankins JV, Trent MS. Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid A promoting resistance to antimicrobial peptides. Mol Microbiol. 2010;76:1444–1460. doi: 10.1111/j.1365-2958.2010.07150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.