

Abstract
MicroRNAs have been identified to be involved in center stage of cancer biology. They accommodate cell proliferation and migration by negatively regulate gene expression either by hampering the translation of targeted mRNAs or by promoting their degradation. We characterized and identified the novel miR-9600 and its target in human non–small-cell lung cancer (NSCLC). Our results demonstrated that the miR-9600 were downregulated in NSCLC tissues and cells. It is confirmed that signal transducer and activator of transcription 3 (STAT3), a putative target gene, is directly inhibited by miR-9600. The miR-9600 markedly suppressed the protein expression of STAT3, but with no significant influence in corresponding mRNA levels, and the direct combination of miR-9600 and STAT3 was confirmed by a luciferase reporter assay. miR-9600 inhibited cell growth, hampered expression of cell cycle-related proteins and inhibited cell migration and invasion in human NSCLC cell lines. Further, miR-9600 significantly suppressed tumor growth in nude mice. Similarly, miR-9600 impeded tumorigenesis and metastasis through directly targeting STAT3. Furthermore, we identified that miR-9600 augmented paclitaxel and cisplatin sensitivity by downregulating STAT3 and promoting chemotherapy-induced apoptosis. These data demonstrate that miR-9600 might be a useful and novel therapeutic target for NSCLC.
Keywords: drug resistance, miR-9600, non–small-cell lung cancer, STAT3, tumorigenesis
Introduction
Lung cancer is the main reason of cancer-related death both in men and women all over the world. There are nearly 1.82 million of new cases, and leading nearly 1.59 million people to death in the year 2012 globally, and the 5-year prevalence cases are estimated to be 1.89 million, referring from GLOBOCAN 2012: Estimated Cancer Incidence, Mortality, and Prevalence Worldwide in 2012 (http://globocan.iarc.fr/Pages/fact_sheets_population.aspx). Lung cancer is classified histopathologically as non–small-cell lung cancer (NSCLC) (~85%) and small-cell lung cancer (SCLC) (~15%). The initiation and development of NSCLC is attributed to aberrant expression of proto-oncogenes and tumor-suppressive genes, which leading to tumor cell growth, metastasis, and eventually tumor progression.1,2,3,4,5 Up to date, surgery and platinum-based chemotherapy are the most applications in clinical treatment of NSCLC,6 while they are not very optimistic considering the fact that the 5-year overall survival (OS) rate of them is only 16% at all stages.7
MicroRNAs (miRNAs), a class of small, endogenous, and noncoding RNAs (usually 18–25 nt in length), regulate post-transcriptional gene expression by directly binding to special sites of 3'-untranslated region (UTR) in targeted mRNA, which contributes to suppression of these genes expression.8,9 miRNAs are derived from introns, intergenic regions, or even exons.10 It has been reported that miRNAs are crucially regulatory factors in numerous of cancers.11,12,13,14 miRNAs have been involved in various of biological processes corrected with tumor initiation, development and progression, including tumor cell growth and metastasis, cell identity, senescence, apoptosis, and stem cell maintenance.15,16,17,18,19 Cui and his colleagues reported that miR-224 promoted tumor progression in non–small-cell lung cancer partially antagonist functions of SMAD4 and TNFAIP1.20 And another miRNA, namely miR-31, predicted the presence of lymph node metastases and survival in patients with lung adenocarcinoma.21 In spite of current publications of miRNAs have brought encouraging perception into human cancers, numerous of uncertain details are still required to be further elaborated.
miR-9600, 20 nt in length, is a new miRNA that located in 12q21.1. It is discovered by our previous high-through sequence in three pairs of NSCLC and pair-matched normal lung tissues. Our previous results of high-through sequence demonstrated that, miR-9600 was marked down-regulation in NSCLC lung tissues than that of in their counterparts, suggesting that miR-9600 might be a useful factor in NSCLC.22
Signal transducer and activator of transcription proteins 3 (STAT3) is well demonstrated to be crucial in tumor development and cancer-related inflammation.23 STAT3 is also linked to inflammation-related oncogenesis initiated by genetic alterations and environmental factors,24,25,26 and is constitutively activated in various cancers.27,28 Persistent activation of STAT3 is involved in promoting tumor cell proliferation, survival, tumor invasion, angiogenesis and immunosuppression, inducing and maintaining a procarcinogenic inflammatory microenvironment.29 Growing studies identified novel tumor-promoting functions of STAT3 in mitochondria metabolism,30 drug resistance,31,32 epigenetic regulation,33 and cancer stem cells.34,35 STAT3 has also been revealed to benefit for tumorigenesis via accommodating cell apoptosis through facilitating Bcl-2 and Bcl-xL expression.36 Given the pivotal role in tumor development, STAT3 represents an attractive therapeutic target for solid tumors.
In this study, we identified and characterized the novel miR-9600 in NSCLC. Firstly, we confirmed the downregulation of miR-9600 in lung cancer tissue and NSCLC. Then we examined the roles of miRNA-9600 in NSCLC proliferation, invasion, migration and apoptosis and identified its target gene. Finally we explored the molecular mechanism of miR-9600 in tumorgenesis. Our results uncovered that miR-9600 accommodated certain tumor cell functions, including cell growth, metastasis, and apoptosis, and promotes paclitaxel sensitivity by targeting 3'-UTR of STAT3 in NSCLC.
Results
Identification of miR-9600, the novel miRNA, in NSCLC
In the present study, we intended to ascertain and characterize novel miRNAs expressed in NSCLC. To explore the novel miRNAs profiles, NCBI Basic Local Alignment Search Tool was used to analyze the sequences of miR-9600, and RNAfold program (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) was used to identify its secondary structure formations (Supplementary Figure S1A). The miR-9600, encoded by a gene located on chromosome 12q21 (71498465-71498512), is located in the introns of the LGR5 gene. The sequences of its stem-loop are 5′-ACCAACUUCACAUUGUAUCCUUAACAUGGUUCCAUAGUGUAGUGGUUA-3′, and its mature sequences are 5′-GGUUCCAUAGUGUAGUGGUUA-3′. The miR-9600 is conserved in other mammals, such as Gorilla, Orangutan, Gibbon, Rhesus, Crab-eating macaque, Baboon, Marmoset, Squirrel monkey, and Chimp, as demonstrated by results of Multiz Alignments of 100 Vertebrates in UCSC (Supplementary Figure S1B).
miR-9600 is decreased expressed in NSCLC lung tissues and cell lines, and is a favorable factor for prognosis
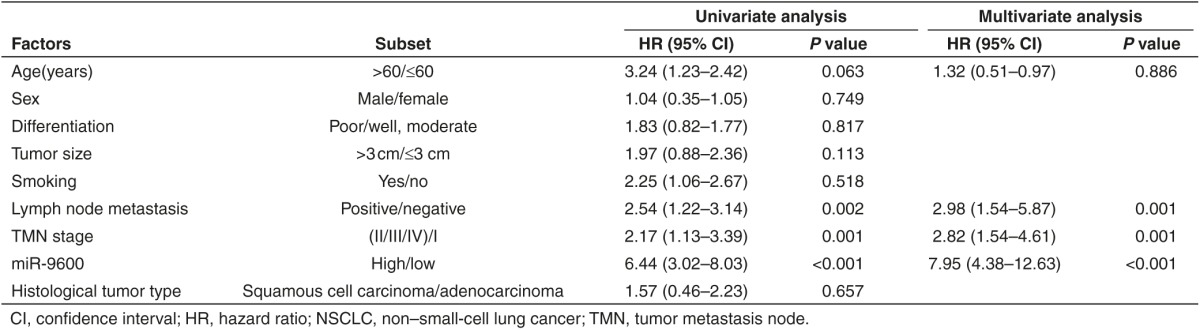
To validate whether miR-9600 is decreased expressed in NSCLC, quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to examine the mature miR-9600 level in human NSCLC lung tissues and their counterparts. We found that miR-9600 levels in 124 NSCLC lung tissues were markedly lower than that of in 124 counterparts (P < 0.05) (Figure 1a). The ideal cutoff level of miR-9600 was 0.32-fold (NSCLC/Normal) with the largest Youden's index (0.813) according to patients' overall survival (Figure 1b). Subsequently, NSCLC patients were classified into a high group (≥0.32-fold, n = 36) and a low group (<0.32-fold, n = 78) on the basis of the cutoff value of miR-9600 expression. Next, we tested miR-9600 levels in NSCLC cell lines, and found that miR-9600 was downexpressed in NSCLC cell lines, including A549, H1299, SK-MES-1, NCI-H520, 95D, and SPC-A-1 cells, in comparison to that of in 16 human bronchial epithelial (16HBE), a normal lung cell lines (Figure 1c). Among the six NSCLC cell lines, miR-9600 decreased the most in A549 and SPC-A-1 cells; thus, we chose A549 and SPC-A-1 cells to perform the following experiments. Moreover, to assess the clinical significance of miR-9600, we evaluated the association between its level and clinic-pathological parameters. Results revealed that miR-9600 levels in NSCLC were remarkably corrected with lymph node metastasis (P = 0.0104), TNM stage (P = 0.0003), smoking history (P = 0.0103), and tumor size (P < 0.0001). Nevertheless, miR-9600 expression level was not associated with other clinical characteristics, including gender (P = 0.5409), differentiation (P = 0.4886), histological tumor type (P = 0.9898), or age (P = 0.1694) in NSCLC (Table 1). Furthermore, multivariate Cox regression analysis revealed that low (<0.32-folds, n = 78) miR-9600 expression, positive lymph node metastasis, and advanced stage are independent predictors of OS in NSCLC patients (Table 2). Kaplan-Meier analysis indicated that low miR-9600 expression was related to a poorer OS (log-rank test, P =0.001, Figure 1d). These data verified that decreased expression of miR-9600 was related to poor prognosis, and downregulated expression of miR-9600 might be crucial in NSCLC initiation, progression, and development.
Figure 1.

miR-9600 is downregulated in primary human lung cancer and non–small-cell lung cancer (NSCLC) cell lines, and benefits for prognosis. (a) miR-9600 is significantly decreased in primary human lung cancer tissues in comparison to adjacent-normal lung cancer tissues. n = 124 for each group. (b) The cutoff value of miR-9600 was analyzed by ROC analysis according to postoperative survival time. The Kaplan-Meier survival curve and the log-rank test were used to determine associations between miR-9600 expression. (c) The expression level of miR-9600 in six NSCLC cell lines and normal 16HBE cells. Assays were performed in triplicate. (d) Kaplan-Meier survival analysis revealed that down-regulated miR-9600 is associated with poor prognosis in patients with non-small cell lung cancer. *P < 0.001, Means ± SEM was shown. Statistical analysis was conducted using student t-test.
Table 1. Correlation between miR-9600 expression and clinicopathological parameters of NSCLC patients (n = 124).
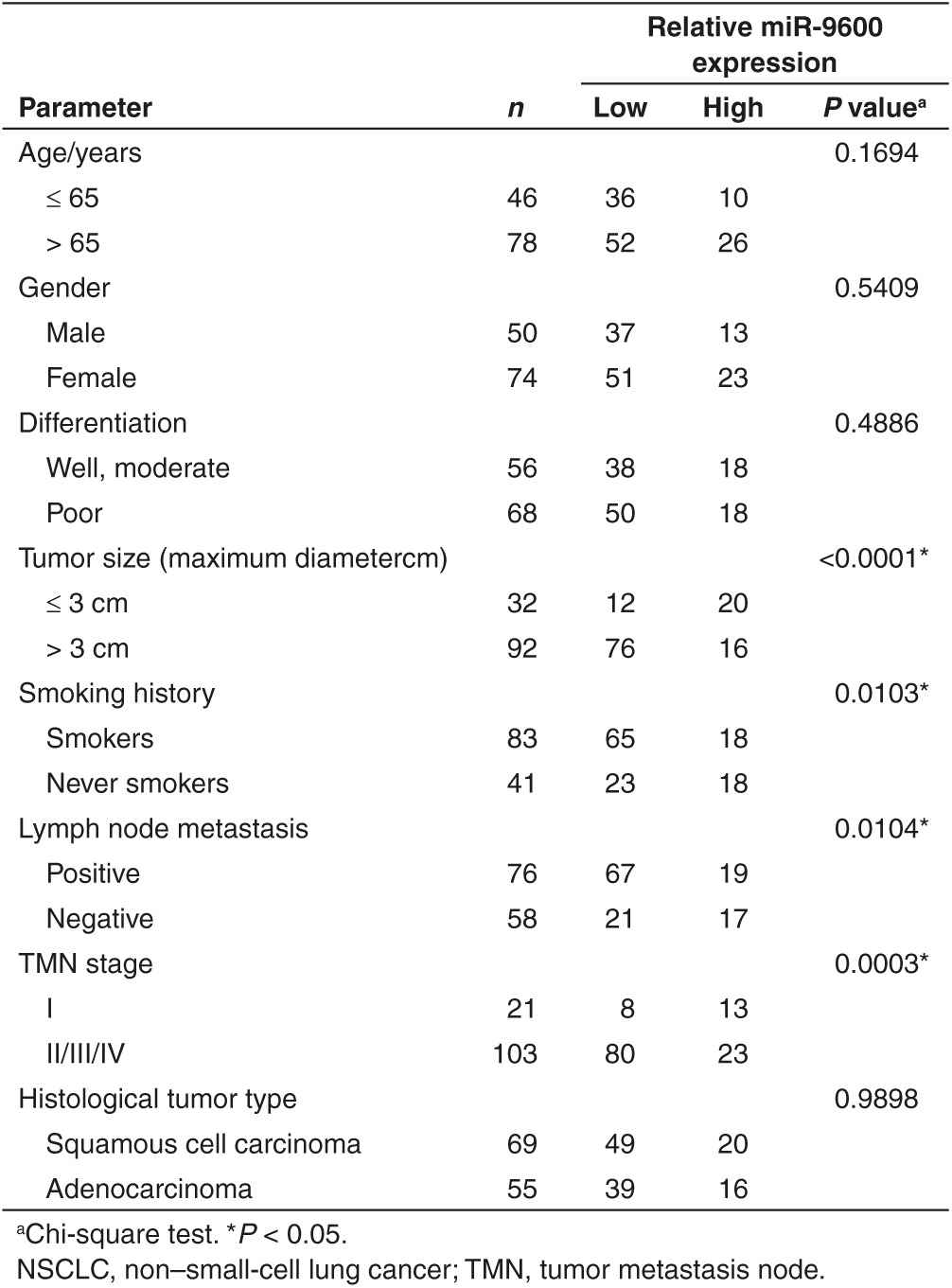
Table 2. Influence of miR-9600 expression and clinical characteristics on overall survival in NSCLC patients.
STAT3 is upregulated in NSCLC lung tissues and its expression is reversely corrected to miR-9600
STAT3, a crucial oncogene with strong oncogenicity, facilitates cell growth, metastasis, and epithelial mesenchymal transition, as accompanies with suppression of cell apoptosis in various types of tumors including NSCLC.24,29,36 Hence, we then explored STAT3 expression in NSCLC lung tissues and their counterparts, and western-blot revealed that STAT3 protein level was increased in NSCLC lung tissues in comparison to their counterparts (3.8-fold of increase in average) (Figure 2a,b), which were verified by examination of STAT3 mRNA expression using qRT-PCR (Figure 2c). On account of the fact that STAT3 is the critical factor on controlling cell cycle, dysregulation of its protein expression might initiate and accelerate development and progression of NSCLC. Additionally, we assessed the relationship between STAT3 mRNA expression and miR-9600 level in 124 NSCLC lung tissues, and results of Pearson correlation analysis (r2 = 0.4124, P < 0.0001) indicated that the expression of STAT3 mRNA and miR-9600 showed a remarkably reverse correlation (Figure 2b).
Figure 2.

Expression of STAT3 is up-regulated in primary human lung cancer and negatively expressed related to miR-9600. (a–c) Western-blot of STAT3 protein and quantitative reverse transcription polymerase chain reaction of STAT3 mRNA in lung cancer tissues and adjacent-normal lung cancers. n = 20 for each group. (d) Scatter plots showing the inverse association between miR-9600 level and STAT3 mRNA expression. *P < 0.001, Means ± SEM was shown. Statistical analysis was conducted using student t-test.
STAT3 expression is positively correlated with the outcome of NSCLC patients
To further explore the critical efficiency of STAT3 in the survival of NSCLC patients, we analyzed the relationship between the STAT3 mRNA expression level and the survival of NSCLC patients from 2437 lung tumors using publicly available datasets (2015 version) (http://kmplot.com/analysis/index.php?p=service&cancer=lung). The Kaplan-Meier analyses demonstrated that higher STAT3 mRNA expression in NSCLC patients is correlated with an improvement of the OS, progression-free survival (FP), as well as post progression survival of patients (Figure 3a–f). These analyses further confirmed the tumor suppressor role of STAT3 in NSCLC.
Figure 3.

Prognostic significance of STAT3 in lung cancer. (a–c) The effect of STAT3 mRNA expression level on the overall survival (OS), progression-free survival (FP), and post progression survival (PPS) in lung cancer patients was analyzed and the Kaplan-Meier plots were generated by Kaplan-Meier Plotter (http://www.kmplot.com). (d–f) The effect of STAT3 mRNA expression level on the overall survival (OS), progression-free survival (FP), and PPS in lung adenocarcinoma patients was analyzed and the Kaplan-Meier plots were generated by Kaplan-Meier Plotter (http://www.kmplot.com).
miR-9600 directly targeting STAT3 by binding at special 3'-UTR sites of its mRNA
Having known decreased miR-9600 expression contributed to the upregulation of predicted target genes in NSCLC lung tissues. We next identified the upregulated genes in the NSCLC using http://www.ebi.ac.uk/gxa/E-MTAB-37. We also identified if the predicted target genes harbored putative binding sites of miR-9600 within 3'-UTR of their mRNA. And STAT3 was sought out as an underlying target gene of miR-9600, since results of a dual luciferase reporter assay in A549 and SPC-A-1 cells demonstrated that the miR-9600 directly bound at the certain sites of 3'-UTR of STAT3 mRNA. To explore the STAT3 certain binding sites (Figure 4a), we established a mutant STAT3 construct, namely, STAT3-MUT, in which the sequences of binding sites at the 3'-UTR of STAT3 mRNA was changed by a site-directed mutagenesis kit. Results demonstrated miR-9600 remarkably decreased the luciferase activity of the STAT3-WT construct, whereas no significant change of luciferase activity was shown in the STAT3-MUT construct (Figure 4b). We also discovered that miR-9600 suppressed the protein expression but had no influence on mRNA expression of STAT3 (Figure 4c,d). Our data revealed that miR-9600 might decrease STAT3 protein expression by directly targeting 3'-UTR of STAT3 mRNA.
Figure 4.

STAT3 proto-oncogene is a target of miR-9600 at specific 3'-UTR sites. (a) The 3'-UTR of STAT3 harbors one miR-9600 cognate site. (b) Relative luciferase activity of reporter plasmids carrying wild-type or mutant STAT3 3'-UTR in A549 and SPC-A-1 cells cotransfected with negative control (NC) or miR-9600 mimic. (c) mRNA expression of STAT3 in A549 and SPC-A-1 cells after transfected with NC, miR-9600, si-STAT3, and ASO-9600. (d) Protein expression of STAT3 in A549 and SPC-A-1 cells after transfected with NC, miR-9600, si-STAT3, and ASO-9600. Assays were performed in triplicate. *P < 0.001, Means ± SEM was shown. Statistical analysis was conducted using student t-test.
miR-9600 represses the proliferation in vitro
To validate if miR-9600 represses cell proliferation in A549 and SPC-A-1 cells, trypan blue staining as well as CCK8 assay were performed (Figure 5a–c). miR-9600 remarkably inhibited cell proliferation at 48 hours treatment, and a same pattern was occurred in si-STAT3-treated groups. The knockdown efficiency of si-STAT3 is shown in Supplementary Figure S2. These results were confirmed by colony formation assays (Figure 5d,e), which indicated that miR-9600 retarded cell growth by suppression of STAT3 expression. Further, flow cytometry was conducted to analyze cell cycle. And results indicated both miR-9600 and si-STAT3 treatment retarded the G1/S transition, namely, they promoted the number of cells in the G1 phase while reduced the number of cells in the S and G2/M phases, in comparison to NC group (Figure 5f). Additionally, we examined downstream genes expression of the STAT3 signaling pathway which was relative to cell cycle (Figure 5g–m). Results indicated suppression of STAT3 by miR-9600 remarkably inhibited protein expression of cyclin E, cyclin D1, phosphorylated-Rb (p-Rb), and CDK2. In addition, overexpression of STAT3 in miR-9600-over-expressed A549 and SPC-A-1 cells reversed the inhibitory role of miR-9600 on NSCLC cell growth, as demonstrated by trypan blue staining, CCK8, as well as colony formation assays (Supplementary Figure S3A–D). These results indicate miR-9600 may repress cell growth by suppression of STAT3 expression in A549 and SPC-A-1 cells.
Figure 5.

The miR-9600 affects cell proliferation and cell cycle. A549 and SPC-A-1 cells were analyzed after transfection. (a) The proportion of viable cells was assessed using the trypan blue dye exclusion assay. (b,c) CCK8 assays of A549 and SPC-A-1 cells after transfected with NC, miR-9600, si-STAT3, and ASO-9600. (d) Shown are representative photomicrographs of colony formation assay after transfected with NC, miR-9600, si-STAT3, and ASO-9600 for 14 days. (e) Statistical analysis of colony formation assay. Assays were performed in triplicate. (f) Cell-cycle analysis was performed 48 hours following the treatment A549 and SPC-A-1 cells with NC, miR-9600, si-STAT3, and ASO-9600. The DNA content was quantified by flow cytometric analysis. (g–m) Expression of RB, p-RB, CDK2, cyclin D1, and cyclin E protein in transfected A549 and SPC-A-1 cells. Assays were performed in triplicate. *P < 0.001, Means ± SEM was shown. Statistical analysis was conducted using student t-test.
miR-9600 inhibits cell metastasis of NSCL C in vitro
Next, we explored the efficiency of miR-9600 on migration and invasion in A549 and SPC-A-1 cells. Results showed that both miR-9600 and si-STAT3 repressed cell motility, migration and invasion in comparison to NC or ASO-miR-9600 groups (Figure 6a–c). In detail, treatment with miR-9600 and si-STAT3 nearly suppressed 45–60% of the cells' migratory activity in A549 and SPC-A-1 cells, and inhibited 50–65% of the cells' invasion activity in A549 and SPC-A-1 cells.
Figure 6.

Ectopic expression of miR-9600 in A549 and SPC-A-1 cells reduces cell migration and invasion motility. (a) A549 and SPC-A-1 cells were loaded onto the top well of a transwell inserts for cell migration or invasion assay. After 24 hours, cells that migrated to the bottom chamber containing serum-supplemented medium were stained with 0.1% crystal violet, visualized under a phase-contrast microscope, and photographed. Bar = 50 μm. (b,c) Total number of cells in five fields was counted manually. (d–h) Expression of MMP-2, MMP-7, and MMP-9 protein in A549 and SPC-A-1 cells after transfection. Assays were performed in triplicate. *P < 0.001, Means ± SEM was shown. Statistical analysis was conducted using student t-test.
MMP2, MMP-7, and MMP-9 are downstream genes participated in the STAT3 signaling pathway, and all of them are critical for tumor metastasis. Hence, we then examined these proteins expression in NSCLC cells following transfecting with miR-9600, si-STAT3, ASO-miR-9600, and NC. Results indicated both miR-9600 and si-STAT3 treatment repressed the protein expression of MMP-2, MMP-7, and MMP-9 in A549 and SPC-A-1 cells (Figure 6d–h). Nevertheless, loss of miR-9600 markedly increased the protein expression of MMP2, MMP-7, and MMP-9 in A549 and SPC-A-1 cells (Figure 6d–h).
In addition, overexpression of STAT3 in miR-9600-over-expressed A549 and SPC-A-1 cells reversed the inhibitory role of miR-9600 on NSCLC cell migration and invasion, as demonstrated by transwell migration/invasion assays (Supplementary Figure S3E–F).
These results, taken together, clearly indicated that miR-9600 expression significantly retarded cell migration and invasion motility in NSCLC.
miR-9600 facilitates cell apoptosis in NSCLC
We also investigated the efficiency of miR-9600 on cell apoptosis in vitro. And miR-9600 led to a ~2.8-folds and ~1.9-folds of increase in apoptotic cell death of A549 and SPC-A-1 cells, as analyzed by flow cytometric analysis (Figure 7a), separately. However, ASO-9600 treatment significant repressed the percentage of apoptotic cells (Figure 7a). Moreover, si-STAT3 treatment resulted in a ~3.3-folds and ~3.7-folds of increase in apoptotic cell death of A549 and SPC-A-1 cells (Figure 7a), separately. In addition, we also tested the caspase-3/7 activity after transfection, and results demonstrated that miR-9600 markedly augmented the caspase-3/7 activity in A549 and SPC-A-1 cell lysate, by nearly 4.4- and 3.6-folds increase (caspase-3 activity), 3.7- and 4.5-folds increase (caspase-7 activity), in comparison to that of in each NC group (Figure 7b,c), respectively. Nevertheless, loss of miR-9600 by treated with ASO-9600 significantly inhibited the caspase-3/7 activity in A549 and SPC-A-1 cell lysate, compared with that of in each NC group (Figure 7b,c), respectively. Moreover, si-STAT3 treatment markedly augmented the caspase-3/7 activity in A549 and SPC-A-1 cell lysate, by nearly 5.5- and 4.9-folds of increase (caspase-3 activity), 4.7- and 5.8-folds of increase (caspase-7 activity), than that of in each NC group (Figure 7b,c), respectively. Additionally, miR-9600 and si-STAT3 treatment also inhibited the protein expression levels of downstream of STAT3 signaling pathway, anti-apoptotic protein Bcl2, Mcl-1, and Bcl-xL (Figure 7d–l), and increased the protein expression of total caspase-3/7, and cleaved-caspase-3/7 (Figure 7d–l) in A549 and SPC-A-1 cells. And ASO-9600 treatment markedly increased protein expression of Bcl2, Mcl-1, and Bcl-xL, and decreased the protein expression of total caspase-3/7, and cleaved-caspase-3/7 (Figure 7d–l) in A549 and SPC-A-1 cells. In addition, overexpression of STAT3 in miR-9600-over-expressed A549 and SPC-A-1 cells reversed the favorable role of miR-9600 on NSCLC cell apoptosis, as demonstrated by flow cytometric analysis, as well as Caspase-3/7 activity assays (Supplementary Figure S3G–I). These data revealed that miR-9600 actually facilitated cell apoptosis in A549 and SPC-A-1 cells.
Figure 7.

Ectopic expression of miR-9600 promotes apoptosis in A549 and SPC-A-1 cells. (a) Shown are statistical analysis of flow cytometric analysis. (b,c). Quantitative representation of caspase-3 and caspase-7 activity in A549 and SPC-A-1 cells transfected with NC, miR-9600, si-STAT3, and ASO-9600 for 48 hours. (d–l) Western-blot of Bcl2, Mcl-1, Bcl-xL, caspase-3, cleaved-caspase-3, caspase-7, and cleaved-caspase-7 protein in A549 and SPC-A-1 cells after transfection. Assays were performed in triplicate. *P < 0.001, Means ± SEM was shown. Statistical analysis was conducted using student t-test.
miR-9600 retards tumor growth in vivo
To validate the tumor-suppressive efficiency of miR-9600 in vivo, we constructed a BALB/c nude mouse xenograft model by A549 cells. Results demonstrated the tumor volume and weight of tumors in nude mice treated with miR-9600 were markedly suppressed (52% of decrease in tumor weight) relative to that of treated with NC (Figure 8a,b). And the tumor volume and weight of tumors in nude mice treated with si-STAT3 were also significantly reduced (55% of decrease in tumor weight) relative to that of treated with NC (Figure 8a,b). However, the tumor volume and weight of tumors in mice treated with ASO-9600 were significantly increased (~1.4-folds of increase in tumor weight) relative to that of treated with NC (Figure 8a,b). These data indicated that miR-9600 markedly inhibited the tumorigenicity of A549 cells in the nude mouse xenograft model. In addition, western-blot revealed that the decreased protein expression of STAT3 (48% of decrease) and p-STAT3 (42% of decrease) in tumors derived from miR-9600-treated nude mice in comparison to that of in control tumors (Figure 8d–f). In accordant with results of miR-9600 treatment, si-STAT3 treatment inhibited protein expression of STAT3 and p-STAT3 in tumors developed from si-STAT3-treated nude mice relative to that of control tumors (Figure 8d–f). While ASO-9600 increased the protein expression of STAT3 and p-STAT3 in tumors developed from ASO-9600-treated nude mice relative to that of control tumors (Figure 8d–f). Moreover, immunehistochemical staining of isolated tumor tissues revealed that tumors derived from miR-9600-treated A549 cells showed reduced positivity (46% of decrease) for Ki67 (brown stains) compared with those in NC group (Figure 8g,h). And si-STAT3-transfected A549 cells exhibited reduced positivity (64% of decrease) for Ki67 compared with those formed from control cells (Figure 8g,h). However, ASO-9600-transfected A549 cells exhibited increased positivity (~1.9-folds of increase) for Ki67 compared with those formed from control cells (Figure 8g,h). Collectively, miR-9600 reduces the growth of established NSCLC xenografts.
Figure 8.

Ectopic expression of miR-9600 suppresses tumor growth in vivo. (a) Tumor volume in nude mice. (b) Tumor weight in nude mice. Each group contained six mice (n = 6); the data are presented as the mean ± SEM; *P < 0.001, compared with the NC group. (c) The expression of miR-9600 in nude mice. (d–f) The expression of STAT3 and p–STAT3 protein in nude mice. (g,h) Immunohistochemistry showed miR-9600 decreased the proliferation index Ki67. Assays were performed in triplicate. *P < 0.001, Means ± SEM are shown. Statistical analysis was conducted using student t-test.
The miR-9600 activity is dicer dependent
To validate whether the mature sequence of miR-9600 is generated through dicer-dependent processing, we transfected A549 and SPC-A-1 cells with either NC or si-Dicer. As expected, si-Dicer treated A549 and SPC-A-1 cells showed a lower Dicer expression than that of in NC (Figure 9a). And we examined the let-7a and miR-9600 expression levels were assessed using qRT-PCR. Results indicated the mature sequence of miR-9600 is generated through dicer-dependent processing (Figure 9b).
Figure 9.

The miR-9600 activity is dicer dependent. (a) The mRNA levels of Dicer were examined via 1% agarose gel electrophoresis, and 18S rRNA was used as an internal control. (b) A549 and SPC-A-1 cells were transfected with either NC or si-Dicer. The let-7a and miR-9600 expression levels were assessed using quantitative reverse transcription polymerase chain reaction. The expression data were normalized to RNU6. Assays were performed in triplicate. *P < 0.001, Means ± standard error of the mean are shown. Statistical analysis was conducted using student t-test.
miR-9600 alters paclitaxel and cisplatin sensitivities in vitro
To identify the molecular mechanisms of miR-9600 in paclitaxel sensitivity of NSCLC cells, we first analyzed the relevance between miR-9600 expression and the IC50 of paclitaxel. The IC50 values developed from paclitaxel-treated A549, SPC-A-1, A549/PR and SPC-A-1/PR were 11 ± 0.78, 42 ± 3.64, 15 ± 1.15, and 45 ± 2.5 nmol/l, respectively (Supplementary Figures S4A,B and S5B). miR-9600 expression was reversely correlated to the IC50 values of paclitaxel in all four above cell lines (P = 0.0178, r2 = 0.9647) (Supplementary Figure S5C). To investigate the relation between miR-9600 expression and paclitaxel-resistance, miR-9600 was induced into A549/PR and SPC-A-1/PR cells, and then treated them with related increasing concentrations of paclitaxel. And results indicated miR-9600 augmented the paclitaxel sensitivity in A549/PR and SPC-A-1/PR cells (Figure 10a). Additionally, inhibition of miR-9600 with ASO-9600 aggrandized the paclitaxel-resistance in A549 and SPC-A-1 cells (Figure 10b). Cisplatin is another important agent for treating NSCLC, and our results also revealed that miR-9600 correlated with cisplatin, and paclitaxel-resistant cells acquired cisplatin resistance (Supplementary Figure S4A,B). In addition, similar functions were obtained that miR-9600 augmented cisplatin resistance as paclitaxel (Figure 10a,b). Our data reveal that miR-9600 expression is favorable with the sensitivity of NSCLC cells to cisplatin and paclitaxel.
miR-9600 augments paclitaxel sensitivity of NSCLC via altering apoptosis mediated by decreased STAT3 expression
Lee et al. reported that STAT3 suppressed cell apoptosis in lung cancer.37 In the present study, we explored the protein expression of STAT3, p-STAT3, Bcl-2, Bcl-xL, and Mcl-1 by western-blot analysis in paclitaxel-sensitive and -resistant NSCLC cell lines. Results demonstrated that the protein expression of STAT3 and p-STAT3 were upregulated in paclitaxel-resistant cells (A549/PR and SPC-A-1/PR) in comparison to paclitaxel-sensitive cells (A549 and SPC-A-1). Same tendencies were obtained in the protein expression of downstream effectors of STAT3, including Bcl-2, Mcl-1, and Bcl-xL (Supplementary Figure S6).
Since miR-9600 could directly target 3'-UTR of STAT3 mRNA and then enhance paclitaxel and cisplatin sensitivity, we suggested that miR-9600 enhanced paclitaxel and cisplatin sensitivity in NSCLC by mediating cell apoptosis by down-regulation of STAT3. To validate this hypothesis, we used flow cytometry analysis to explore the roles of miR-9600 and chemotherapeutic agents on A549/PR cell apoptosis. Results indicated that miR-9600, paclitaxel or cisplatin treatment, individually, enhanced A549/PR cell apoptosis to 7.3, 7.5, and 9.7%, separately, in comparison to that of in NC group (3.6%) (Figure 11a,b). Moreover, A549/PR cells with overexpressed miR-9600 and handled with paclitaxel or cisplatin treatment showed a markedly higher ratio of apoptotic cells (46.9 and 54.3%, separately) (Figure 11a,b). Analogous outcomes were observed in SPC-A-1/PR cells with overexpressed miR-9600 or handled with paclitaxel or cisplatin treatment (Figure 11a,b). Additionally, paclitaxel or cisplatin treatment, or overexpression of miR-9600 repressed the protein expression of p-STAT3, Bcl-2, Mcl-1, and Bcl-xL (Figure 11e,f), butSTAT3 protein expression was only inhibited by miR-9600 overexpression. The coinstantaneous treatment with overexpression of miR-9600 plus paclitaxel or cisplatin contributed to the main suppression in protein expression of p-STAT3, Bcl-2, Bcl-xL, and Mcl-1 (Figure 11e,f). Recovering the expression of STAT3 reversed the roles of miR-9600 overexpression and chemotherapeutic reagents treatment-mediated apoptosis in the NSCLC cells (Figure 11a–f). Our results demonstrated that miR-9600 inhibited NSCLC cell paclitaxel and cisplatin sensitivity by mediating cellular apoptosis pathway through downregulation of STAT3.
Figure 11.

miR-9600 suppresses paclitaxel resistance in A549/PR or SPC-A-1/PR cells by downregulating STAT3 resulting in altered expression of apoptosis-related genes. (a) Representative flow cytometry analysis of annexin V (1:1,000) and propidium iodide (1:1,000) staining in A549/PR or SPC-A-1/PR cells transfected with miR-9600 or miR-9600 plus STAT3 and treated with or without paclitaxel (20 nmol/l) or cisplatin (10 μmol/l) for 24 hours. The proportion of apoptotic cells is shown as the means ± standard error of the mean from three independent experiments (*P < 0.05, **P < 0.01). (b) Representative western blots using the indicated antibodies in A549/PR or SPC-A-1/PR cells transfected and treated as in a.
Discussion
In the present study, we identified and characterized a novel miRNA, miR-9600, in NSCLC. The primary miR-9600 is ~48 nt in length and has a secondary folding structure (Figure 1a). The miR-9600, located on chromosome 12, is transcribed and derived from an intron region of LGR5 gene, and then develops as a 20 nt-mature sequence, and we find that this process is dicer-dependent (Figure 9). In addition, we found that the mature sequences of miR-9600 are highly conserved in several mammals (Supplementary Figure S1B).
Several researches have suggested that miRNAs may be crucial in cancer-associated processes.5,19,38,39,40,41 miRNAs may act as tumor suppressor and are usually downregulated in cancer tissue. For example, the miR-206 is downregulated in NSCLC and suppresses the proliferation of A549 and SK-MES-1 cells.19 The expression of miR-193b, miR-139-5p, and miR-134 is decreased expressed in NSCLC lung tissues in comparison to their counterparts and controls cell proliferation, colony formation, migration, invasion, and apoptosis in NSCLC.5,38,42 Our previous study revealed that miR-187-3p is downregulated in NSCLC and suppresses the proliferation of A549 cells.41 Here, we also found that miR-9600 was downregulated in NSCLC lung tissues by high-through sequence in our previous study,22 and we also confirmed it by qRT-PCR (Figure 1a). We also identified that the decreased expression of miR-9600 in various types of NSCLC cell lines (Figure 1b), suggesting that miR-9600 was indeed downexpressed in NSCLC cells and the downexpression of miR-9600 might be crucial for cellular biological mechanisms related to NSCLC initiation, development, and progression.
Trough directly targeting the 3'-UTR of targeted mRNA for contributing to their degradation or suppressing targeted-protein expression, miRNAs play a crucial role on controlling post-transcription of their targeted gene.43 On account of the fact that miR-9600 was decreased expression in NSCLC lung tissues and cell lines, we explored the upregulated genes in NSCLC to predict the potential target genes of miR-9600 by the EMBL databases. Since numerous of studies have verified that genes involved in cell growth and metastasis might have greatly potential relevancy with cancer therapy,42 we next selected the putative target genes that are participated in cell growth and metastasis. Since novel miRNAs cannot be indexed with several miRNA-target gene searching programs, including TargetScan, microRNA.org and so on. Hence, we selected and verified perfect pair-matched bases between the 5'-end of the novel miR-9600 and the 3'-UTR of mRNA of predicted genes using Rnahybrid tools to seek out the detail target sites.44 Eventually, we sought out the STAT3 gene as an underlying targeted gene.
We confirmed STAT3 expression was upregulated in NSCLC by a web program (http://www.ebi.ac.uk/gxa/E-MTAB-37), and it has been reported to be activated in NSCLC.45,46,47 Our results of western-blot also revealed STAT3 was higher expressed in NSCLC lung tissues compared with their counterparts (Figure 2a). Further, the mRNA expression of STAT3 was negatively correlated with miR-9600 expression in NSCLC tissues (Figure 2d). We also confirmed that STAT3 expression was positively correlated with the outcome of NSCLC patients, using Kaplan-Meier analyses (Figure 3a–f). These analyses further confirmed the tumor suppressor role of STAT3 in NSCLC. In addition, we established two vectors containing the STAT3-WT or STAT3-MUT 3'-UTR, which contained the sequences from 1395 to 1417 nt, and identified that the STAT3 gene was controlled by miR-9600 by a luciferase reporter assay (Figure 4b). Though STAT3 mRNA expression was not influenced by miR-9600, the STAT3 and p-STAT3 protein expression were found to be inhibited by overexpression of miR-9600 (Figure 4c,d). These results revealed that miR-9600 reversely regulated STAT3 expression by directly binding to 3'-UTR of STAT3 mRNA.
STAT3 expression is related to cell growth, survival, cell cycle, and metastasis in various types of cancers,43,46,47 and decreased expression of p-STAT3 due to STAT3 knockdown significantly influences cell growth.47 Here, we discovered that cell proliferation in the A549 and SPC-A-1 cells was markedly repressed by the overexpression of miR-9600 (Figure 5a–e). It has been reported that STAT3 signaling pathway is participated in lung cancer cell proliferation and survival,47 and STAT3 is also known to involve in cell apoptosis of cervical cancer and glioma.48,49 Our data also demonstrated that miR-9600 overexpression resulted in decreased expression of STAT3 and then led to cell apoptosis or survival in NSCLC (Figure 7). In the present study, we first focused on cell growth and apoptosis and evaluated the functions of miR-9600 on the downstream genes of the STAT3 signaling pathway. Our results demonstrated the protein levels of p-RB, cyclin D1, cyclin E, and CDK2 were suppressed by the overexpression of miR-9600 and downregulation of STAT3 (Figure 5g–m). This is consistent with the report that the suppression of p-STAT3 and STAT3 facilitates the downregulation of cyclin D1.50 Downregulation of cell cycle related regulatory proteins, including Rb, CDK2, cyclin D1, and cyclin E, could mediate cell cycle arrest.51 In the research, miR-9600-transfected A549 and SPC-A-1 cells increased cell numbers in the G1 phase, but suppressed cell numbers in S and G2/M phase, according to results of flow cytometry analysis.
Previous studies revealed STAT3 could accommodate several cancer processes, such as cell motility and migration.48,49,50 Our data also showed that miR-9600 modulates cell motility and metastasis in A549 and SPC-A-1 cells by targeting the STAT3 gene (Figure 6). In addition, miR-9600 expression repressed tumorigenesis in vivo, evidenced by the fact that both mice injected with miR-9600 and si-STAT3 exhibited a decreased tumorigenesis in comparison to the NC- and ASO-9600-injected mice.
STAT3 is crucial for tumor initiation, development, and progression by regulating gene expression, including the downstream genes of STAT3 signaling pathway during tumorigenesis,46,47,48,49 STAT3 activation and expression are frequently contributed to the development of tumorigenesis in various types of cancers.43,47,50 For instance, miR-125a suppresses tumorigenesis in cervical carcinoma through targeting STAT3.48 Additionally, miR-519a effectively reduces cancer cell growth and metastasis by indirectly inhibition of the STAT3 pathway during tumorigenesis.49 In our present study, we also identified the novel miR-9600 can also repress tumor cell metastatic activity (Figure 6). Our results indicate that miR-9600 directly inhibits cell growth and metastasis and facilitates cell apoptosis both in vivo and in vitro.
Additionally, our results also demonstrated that overexpressing miR-9600 enhanced the resistant sensitivity of NSCLC cells to paclitaxel and cisplatin, and knockdown of miR-9600 in paclitaxel-sensitive NSCLC cells contributed to paclitaxel resistance. Moreover, we found that miR-9600 enhanced sensitivity of paclitaxel-resistant NSCLC cells acquired cisplatin resistance. Hence, a reverse correlation between the expression of miR-9600 and chemoresistance was verified in NSCLC.
Taking all into account, we identified and characterized a novel miRNA, miR-9600, which was downregulated in NSCLC tissue samples and cell and lines, as a crucial regulatory factor in NSCLC. Additionally, we identified STAT3 as a direct target of miR-9600, and found that reduction of STAT3 expression by miR-9600 treatment markedly contributed to the suppression of cell growth and metastasis both in vitro and in vivo. Additionally, miR-9600 reduced paclitaxel and cisplatin resistance by inhibition of STAT3, which facilitated cell apoptosis in NSCLC. Enhancing miR-9600 or suppressing STAT3 expression might be crucial in combination paclitaxel with cisplatin for treatment of chemoresistant in NSCLC. Our results suggest miR-9600 has a crucial role in the initiation, development and progression of NSCLC, and it might supply a novel therapeutic target for NSCLC therapy.
Materials and methods
Tissue collection. Fresh and formalin-fixed, paraffin-embedded, NSCLC tumor tissue samples were obtained from patients who were diagnosed with primary NSCLC. Elective surgery was carried out on these patients at People's Hospital of Wuhan University (Wuhan, China). In total, 20 cases of fresh NSCLC tissues were freshly frozen in liquid nitrogen and stored at −80 °C until further use. 124 cases of archived, formalin-fixed, paraffin-embedded NSCLC tissue samples were collected and used in clinicopathological and prognostic investigation of miR-9600. A comprehensive set of clinicopathological data were recorded, including age, gender, size of primary tumor, tumor differentiation, T stage, lymph node metastasis, and distant metastasis. The stage of disease was determined according to the tumor size, lymph node, and metastasis (pTNM) classification system. Complete follow-up, ranging from 1–56 months, was available for the cohort of 116 patients, and the median survival was 32 months. The use of tissues for this study has been approved by the ethics committee of People's Hospital of Wuhan University. Before using these clinical materials for research purposes, all the patients signed the informed consent. None of these patients received any preoperative chemotherapy or radiotherapy.
Cell culture and transfection. The human NSCLC cell lines, namely, A549, SPC-A-1, H1299, SK-MES-1, NCI-H520, 95D and 16HBE cells were grown in RPMI 1640 (Gibco) medium containing 10% heat-inactivated (56 °C, 30 minutes) fetal calf serum, 2 mmol/l glutamine, penicillin (100 U/ml), and streptomycin (100 U/ml), which was maintained in an incubator at 37 °C with 5% CO2 in a humidified atmosphere. Paclitaxel-resistant A549/PR and SPC-A-1/PR cells were developed from A549 and SPC-A-1 cell lines by treatment with gradually increasing concentrations of paclitaxel in cell culture medium. Briefly, cells were seeded in six-well plates and reached about 80% confluency in fresh medium before treating with paclitaxel. The dose of paclitaxel range from 0.1 to 20 nmol/l and it was increased by a dose gradient that was 25–50% of the previous dose. The next dose was given until the cells were stable in proliferation without significant death. miR-9600 mimic and mimic negative control, miR-9600 inhibitor and inhibitor negative control were purchased from GenePharma (Shanghai, China). Complete medium without antibiotics was used to culture the cells at least twenty-four hours prior to transfection. The cells were washed with 1× phosphate buffer saline (PBS) (pH7.4) and then transiently transfected with 50 nmol/l NC or miR-9600, 100 nmol/l ASO-9600 or si-STAT3 (sequences for siRNAs are shown in Supplementary Table S2), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Western blot analysis. Forty-eight hours after transfection, total protein was extracted from the A549 and SPC-A-1 cells using RIPA cell lysis reagent containing proteinase and phosphatase inhibitors (Sangon Biotech, Shanghai, China) at 4 °C for 30 minutes. Cell lysates were centrifuged at 12,000× g for 20 minutes at 4 °C, and the protein concentrations of the supernatant were determined using the BCA protein assay reagent kit (Thermo). The supernatants containing total protein were then mixed with a corresponding volume of 5× SDS loading buffer and heated at 100 °C for 10 minutes. Then, the supernatant lysates were run on 10% SDS-polyacrylamide gels (50 μg/lane), and proteins were transferred to poly (vinylidene fluoride) (PVDF) membranes (Hertfordshire, UK) by semidry electroblotting (1.5 mA/cm2). PVDF membranes were then incubated in blocking buffer (Tris-buffered saline (TBS) supplemented with 0.05% (vol/vol) Tween 20; TBST) containing 5% (wt/vol) skimmed milk powder for 120 minutes at room temperature followed by three 10-minute washes in TBST. The PVDF membranes were then incubated with anti-STAT3 (1:1,000 dilutions, Santa Cruz Biotechnology, Santa Cruz, CA), anti-p-STAT3 (1:1,000 dilutions, Santa Cruz), anti-RB (1:1,000 dilutions, Santa Cruz), anti-p-RB (1:1,000 dilutions, Santa Cruz), anti-cyclin D1 (1:1,000 dilutions, Santa Cruz), anti-CDK2 (1:1,000 dilutions, Santa Cruz), anti-cyclin E (1:1,000 dilutions, Santa Cruz), anti-MMP-2 (1:1,000 dilutions, Santa Cruz), anti-MMP-7 (1:1,000 dilutions, Santa Cruz), anti-MMP-9 (1:1,000 dilutions, Santa Cruz), anti-Bcl-2 (1:1,000 dilutions, Santa Cruz), anti-Bcl-xL (1:1,000 dilutions, Santa Cruz), anti-Mcl-1 (1:1,000 dilutions, Santa Cruz), anti-caspase-3 (1:1,000 dilutions, Santa Cruz), anti-cleaved-caspase-3 (1:1,000 dilutions, Santa Cruz), anti-caspase-7 (1:1,000 dilutions, Santa Cruz), anti-cleaved-caspase-7 (1:1,000 dilutions, Santa Cruz), and anti-Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:5,000 dilutions, Santa Cruz) as internal normalizers in TBST containing 5% (wt/vol) skimmed milk powder (antibody buffer) overnight at 4 °C on a three-dimensional rocking table. Then the membranes were washed three times for 10 minutes in TBST and then incubated with goat anti-rabbit IgG conjugated to horseradish peroxidase (1:12,000 dilutions) in antibody buffer for 120 minutes. Finally, membranes were washed three times for 10 minutes in TBST and exposed to ECL Advance reagent (GE Healthcare Biosciences, Buckinghamshire, UK) for 2 minutes as described in the manufacturer's protocol. Then membranes were exposed to Hyperfilm-ECL (GE Healthcare Bio-Sciences) for 2–5 minutes and visualized using a Fluor S Multimager and Quantity One 4.1 (Bio-Rad Laboratories, Hercules, CA). The molecular weights of the bands were calculated by a comparison with prestained molecular weight markers (molecular weight range: 6,500–250,000) that were run in parallel with the samples. Semiquantitative analysis of specific immunolabeled bands was performed using a Fluor S image analyzer and Quantity One 4.1.
RNA isolation and qRT-PCR. Total RNA from the cultured cells was extracted using Trizol reagent (Invitrogen) according to the manufacturer's instructions. miRNA levels were measured by qRT-PCR. For the qRT-PCR detection of mature miR-9600 expression, we purchased the Bulge-Loop miRNA qRT-PCR Primer Set and the miRNA qRT-PCR Control Primer Set (both from RiboBio). RNA (2 µg) was converted into cDNA using the PrimeScript RT reagent kit with gDNA Eraser (Takara, Dalian, China) according to the manufacturer's instructions. qRT-PCR was performed using SYBR Premix Ex Taq II (Takara) in the ABI PRISM 7300 real-time PCR system (Applied Biosystems, Foster City, CA). GADPH and U6 were used as endogenous controls. In addition, melting curves were used to evaluate nonspecific amplification. The relative expression level was calculated using the 2-ΔΔCt method. The primer sequences used in this study are as listed in Supplementary Table S1. The formula and its derivations were obtained from the ABI Prism 7300 sequence detection system user guide. Statistical analysis was performed on the fold change.
Colony formation assay. Cells were transfected with NC, miR-9600, si-STAT3, or the ASO-miR-9600, as described above. Twenty-four hours later, transfected cells were trypsinized, counted, and replated at a density of 500 cells/6 cm dish. Ten days later, colonies resulting from the surviving cells were fixed with 3.7% methanol, stained with 0.1% crystal violet and counted. Colonies containing at least 50 cells were scored. Each assay was performed in triplicates.
Luciferase reporter assays. The 3'-untranslated region (UTR) of human STAT3 was amplified from human genomic DNA and individually inserted into the pmiR-RB-REPORT (Ribobio, Guangzhou, China) using the XhoI and NotI sites. Similarly, the fragment of STAT3 3'-UTR mutant (which the first eleven nucleotides complementary to the miR-9600 seed-region were mutated by site-directed mutagenesis (Stratagene) served as a mutant control) was inserted into the pmiR-RB-REPORT control vector at the same sites. For reporter assays, A549 and SPC-A-1 cells were cotransfected with wild-type (mutant) reporter plasmid and miR-9600 mimics (miR mimic NC) using Lipofectamine 2000 (Invitrogen). Firefly and Renilla luciferase activities were measured in cell lysates using the Dual-Luciferase Reporter Assay system. Luciferase activity was measured 48 hours post-transfection using dual-glo luciferase reporter system according to the manufacturer's instructions (Promega, Madison, WI). Firefly luciferase units were normalized against Renilla luciferase units to control for transfection efficiency.
Transwell migration/invasion assay. A549 and SPC-A-1 cells were grown in RPMI 1640 containing 10% fetal bovine serum to ~60% confluence and transfected with NC, miR-9600, si-STAT3, or the ASO-miR-9600. After twenty-four hours, the cells were harvested by trypsinization and washed once with Hanks' balanced salt solution (Invitrogen). To measure cell migration, 8-mm pore size culture inserts (Transwell; Costar, High Wycombe, UK) were placed into the wells of 24-well culture plates, separating the upper and the lower chambers. In the lower chamber, 500 μl of RPMI 1640 containing 10% FBS was added. Then, serum-free medium containing 5 × 104 cells were added to the upper chamber for migration assays, whereas 1 × 105 cells were used for matrigel invasion assays. After 24 hours of incubation at 37 °C with 5% CO2, the number of cells that had migrated through the pores was quantified by counting 10 independent visual fields under the microscope (Olympus) using a ×20 magnifications, and cell morphology was observed by staining with 0.1% crystal violet. Filters were washed thoroughly with 1× PBS and dissolved in 500 μl of 33% acetic acid, and absorbance was measured at 570 nm. Absorbance of cells incubated in the serum-free medium in the bottom chamber was used as negative control. Each experiment was performed at least three times.
Trypan blue staining. Cell viability was assessed using the trypan blue (Lonza, Basel, Switzerland) exclusion method. The A549 and SPC-A-1 cells were seeded in 24-well culture plates at a density of 3 × 105 cells per well, and the cells were then transfected with NC, miR-9600, si-STAT3, or the ASO-miR-9600. Each cell suspension was mixed with an equal volume of 0.4% trypan blue solution, and the living cells were quantified using a hemocytometer. The cells were also counted using a microscope. The data are representative of three independent experiments performed on different days.
CCK8 assay. Cell growth was measured using the cell proliferation reagent WST-8 (Roche Biochemicals, Mannheim, Germany). After plating cells in 96-well microtiter plates (Corning Costar, Corning, NY) at 1.0 × 103/well, 10 μl of CCK8 was added to each well at the time of harvest, according to the manufacturer's instructions. One hour after adding CCK8, cellular viability was determined by measuring the absorbance of the converted dye at 450 nm.
To analyze the effects of miR-9600 in combination with paclitaxel or cisplatin, cells transfected with either miR-9600 or ASO-9600 were treated at concentrations of 0, 2.5, 5, 10, 20, 40, and 80 nm with paclitaxel or at concentrations of 0, 2, 4, 8, 16, 32, 64, 128 μm with cisplatin for 24 hours. The IC50 value was calculated as the concentration of paclitaxel that reduced cell viability by 50%.
Tumor formation in BALB/c nude mice. BALB/c athymic nude mice (male, 4–6 weeks old and 16–20 g) were purchased from Hubei Research Center of Laboratory Animal (Wuhan, China). All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of Wuhan University. To establish lung cancer xenograft model, 5 × 105 A549 cells were suspended in 100 μl phosphate-buffered saline and inoculated subcutaneously into the flanks of nude mice. After 8 days, the transplanted nude mice were randomly divided into four groups (n = 6 each). NC, miR-9600, si-STAT3, or the ASO-miR-9600 was directly injected into the implanted tumor at the dose of 10 nmol (in 20 μl phosphate-buffered saline) per mouse every 4 days for seven times. The tumor size was monitored by measuring the length (L) and width (W) with calipers every 4 day, and the volumes were calculated using the formula: (L × W2)/2. Mice were killed by cervical dislocation in day 28, and the tumors were excised and snap-frozen for protein and RNA extraction.
Immunohistochemistry. Immunohistochemistry of the tumor tissues was performed as described previously.52,53,54 3-μm tumor sections were incubated with commercial rabbit polyclonal antibodies against Ki67 (Affinity) at 1/100 dilution overnight at 4 °C. Then, the sections were conjugated with horseradish peroxidase antibody (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) at room temperature for 2 hours, then covered by diaminobenzidine (DAB) (Vector Laboratories, Burlingame, CA), and slides were mounted with Vectashield mounting medium (Vector Laboratories). Subsequently, all fields were observed under light microscopy (Olympus 600 auto-biochemical analyzer, Tokyo, Japan). Control experiments without primary antibody demonstrated that the signals observed were specific.
Flow cytometry
Apoptosis analysis. A549 and SPC-A-1 cells transfected with NC, miR-9600, si-STAT3, or the ASO-miR-9600 were trypsinized and resuspended in 1× binding buffer at 1 × 106 cells/ml. 100 μl of this cell suspension was incubated with 5 μl of FITC-Annexin V and 5 μl propridium iodide (PI) for 15 minutes in the dark. The reaction was terminated with the addition of 400 μl 1× binding buffer and analyzed with FACSCalibur using the CellQuest software (Becton Dickinson). FITC-Annexin V-positive and PI-negative cells were considered as apoptotic and the experiments were carried out in triplicates.
Cell-cycle analysis. Transfected cells were harvested 48 hours after transfection. The cells were fixed in 70% ethanol, washed once with PBS, and then labeled with propidium iodide (Sigma-Aldrich) in the presence of RNase A (Sigma-Aldrich) for 30 minutes in the dark (50 g/ml). Samples were run on a FACSalibur flow cytometer (Becton-Dickinson, FL, NJ), and the percentages of cells within each phase of the cell cycle were analyzed using Cell Quest software.
Caspase-3/7 activity assay. The activity of caspase-3/7 was determined using the caspase-3/7 activity kit (Beyotime Institute of Biotechnology, Haimen, China).To evaluate the activity of caspase-3/7, cell lysates were prepared after their respective treatment with various designated treatments. Assays were performed on 96-well microtitre plates by incubating 10 μl protein of cell lysate per sample in 80 μl reaction buffer (1% NP-40, 20 mmol/l Tris-HCl (pH 7.5), 137 mmol/l Nad and 10% glycerol) containing 10 μl caspase-3 substrate (Ac-DEVD-pNA) (2 mmol/l). Lysates were incubated at 37 °C for 4 hours. Samples were measured with an ELISA reader at an absorbance of 405 nm. The detail analysis procedure was described in the manufacturer's protocol.
Conservation analysis. Multiz Alignments of 100 Vertebrates in UCSC were used for conservation analysis of miR-9600. And conservation results were confirmed using the NCBI BLAST too.
Statistical analysis. All experiments were repeated three times independently. The results are presented as the means ± standard error mean. Two independent sample t-test or one-way analysis of variance was performed using SPSS 19.0 software in order to detect significant differences in measured variables among groups. A value of P < 0.05 was considered to indicate a statistically significant difference.
SUPPLEMENTARY MATERIAL Figure S1. Basic information regarding the novel miR-9600. Figure S2. si-STAT3-1 exerts the most inhibitory efficacy to mRNA expression of STAT3. A549 and SPC. Figure S3. The miR-9600 inhibitedcell growth, migration and invasion, and induced cell apoptosis in NSCLC cells. Figure S4. Characterization of paclitaxel-resistantNSCLC cells. Figure S5. Relationship between miR-9600 expression and paclitaxelresistance in NSCLC cells. Figure S6. Expression of STAT3 and downstream effectors in paclitaxel sensitive and resistant NSCLC cell lines. Table S1. Primer sequences for quantitative reverse transcription(RT)-PCR. Table S2. Sequences for si-STAT3.
Author contributions
Participated in research design: C-C.S., S-J.L., and D-J.L. Conducted experiments: C-C.S., S-J.L., F-Z., Y-D.Z., Z-Y.Z., Y-Y.X., L-W., and D-J.L. Contributed new reagents or analytic tools: C-C.S., S-J.L., and D-J.L. Performed data analysis: C-C.S., S-J.L., and D-J.L. Wrote or contributed to the writing of the manuscript: C-C.S., S-J.L., and D-J.L.
Figure 10.
Altered miR-9600 expression modulates sensitivity to paclitaxel in vitro. (a) Overexpression of miR-9600 in paclitaxel-resistant cervical cells (A549/PR and SPC-A-1/PR) were treated with increasing concentrations of paclitaxel as indicated. After 24 hours, cell viability assays were performed using a CCK-8 kit. The group not treated with paclitaxel was presented as 100% viable cells and was used as an internal control for comparison. Histograms on the right show the level of miR-9600 overexpression. (b) A cell viability assay on paclitaxel-sensitive cells (A549 and SPC-A-1) following knockdown of miR-9600 expression. The cells were treated as in a. Histograms on the right show the level of miR-9600 knockdown. All values shown are the means ± standard error of the mean of triplicate samples. Experiments were repeated three times (*P < 0.05, **P < 0.01).
Acknowledgments
We thank Doctor Huang and Doctor Xie from People's Hospital of Wuhan University, for providing lung cancer tissues and normal lung tissues. The authors declare that they have no competing interests. This work was supported by National Natural Science Foundation of China (No. 81271943) to D-J.L., The plan for Scientific and Technological Innovation Team of High-tech Industries of Wuhan Municipal Science and Technology Bureau (No. 2015070504020219) to D-J.L. and the Fundamental Research Funds for the Central Universities (No. 2015305020202) to C-C.S.
Supplementary Material
References
- Chen, K, He, H, Xie, Y, Zhao, L, Zhao, S, Wan, X et al. (2015). miR-125a-3p and miR-483-5p promote adipogenesis via suppressing the RhoA/ROCK1/ERK1/2 pathway in multiple symmetric lipomatosis. Sci Rep 5: 11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, S, Xu, Z, Lou, F, Zhang, L, Ke, F, Bai, J et al. (2015). NF-κB-induced microRNA-31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in psoriasis. Nat Commun 6: 7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z , Song, Y,Liu, L,Hou, N, An, X, Zhan, D et al. (2015). miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. [DOI] [PMC free article] [PubMed]
- Tang, X , Hou, Y, Yang, G,Wang, X, Tang, S, Du, YE et al. (2015). Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ 23: 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C, Sang, M, Li, S, Sun, X, Yang, C, Xi, Y et al. (2015). Hsa-miR-139-5p inhibits proliferation and causes apoptosis associated with down-regulation of c-Met. Oncotarget 6: 39756–39792. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Schiller, JH, Harrington, D, Belani, CP, Langer, C, Sandler, A, Krook, J et al.; Eastern Cooperative Oncology Group. (2002). Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346: 92–98. [DOI] [PubMed] [Google Scholar]
- Laskin, JJ and Sandler, AB (2005). State of the art in therapy for non-small cell lung cancer. Cancer Invest 23: 427–442. [DOI] [PubMed] [Google Scholar]
- Grosswendt, S, Filipchyk, A, Manzano, M, Klironomos, F, Schilling, M, Herzog, M et al. (2014). Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell 54: 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgardt, BF, Ahmed, K, Spranger, M, Latreille, M, Denzler, R, Kondratiuk, N et al. (2015). The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat Med 21: 619–627. [DOI] [PubMed] [Google Scholar]
- Yoo, JK, Jung, HY, Lee, JM, Yi, H, Oh, SH, Ko, HY et al. (2014). The novel miR-9500 regulates the proliferation and migration of human lung cancer cells by targeting Akt1. Cell Death Differ 21: 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y, Kuscu, C, Banach, A, Zhang, Q, Pulkoski-Gross, A, Kim, D et al. (2015). miR-181a-5p inhibits cancer cell migration and angiogenesis via downregulation of matrix metalloproteinase-14. Cancer Res 75: 2674–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, M, Zhang, T, Liu, C, Badeaux, MA, Liu, B, Liu, R et al. (2014). miRNA-128 suppresses prostate cancer by inhibiting BMI-1 to inhibit tumor-initiating cells. Cancer Res 74: 4183–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C., Li, S, Yang, C, Xi, Y, Wang, L, Zhang, Fet al. (2016). MicroRNA-187-3p mitigates non-small cell lung cancer (NSCLC) development through down-regulation of BCL6. Biochem Biophys Res Commun 471: 82–88. [DOI] [PubMed] [Google Scholar]
- Sun, C, Huang, C, Li, S, Yang, C, Xi, Y, Wang, L et al. (2016). Hsa-miR-326 targets CCND1 and inhibits non-small cell lung cancer development. Oncotarget 7: 8341–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, Q, Xue, G, Li, X, Wu, Z, Li, X, Yan, H et al. (2015). Methylation-induced loss of miR-484 in microsatellite-unstable colorectal cancer promotes both viability and IL-8 production via CD137L. J Pathol 236: 165–174. [DOI] [PubMed] [Google Scholar]
- Díaz-Martín, J, Díaz-López, A, Moreno-Bueno, G, Castilla, MÁ, Rosa-Rosa, JM, Cano, A et al. (2014). A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. J Pathol 232: 319–329. [DOI] [PubMed] [Google Scholar]
- Hsu, CY, Hsieh, TH, Tsai, CF, Tsai, HP, Chen, HS, Chang, Y et al. (2014). miRNA-199a-5p regulates VEGFA in endometrial mesenchymal stem cells and contributes to the pathogenesis of endometriosis. J Pathol 232: 330–343. [DOI] [PubMed] [Google Scholar]
- Sun, CC, Li, SJ, Zhang, F, Pan, JY, Wang, L, Yang, CLet al. (2016). Hsa-miR-329 exerts tumor suppressor function through down-regulation of MET in non-small cell lung cancer. Oncotarget 7: 21510–21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C, Liu, Z, Li, S, Yang, C, Xue, R, Xi, Y et al. (2015). Down-regulation of c-Met and Bcl2 by microRNA-206, activates apoptosis, and inhibits tumor cell proliferation, migration and colony formation. Oncotarget 6: 25533–25574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, R, Meng, W, Sun, HL, Kim, T, Ye, Z, Fassan, M et al. (2015). MicroRNA-224 promotes tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci USA 112: E4288–E4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, W, Ye, Z, Cui, R, Perry, J, Dedousi-Huebner, V, Huebner, A et al. (2013). MicroRNA-31 predicts the presence of lymph node metastases and survival in patients with lung adenocarcinoma. Clin Cancer Res 19: 5423–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C, Sun, C, Liang, X, Xie, S, Huang, J and Li, D (2016). Integrative analysis of microRNA and mRNA expression profiles in non-small-cell lung cancer. Cancer Gene Ther 23: 90–97. [DOI] [PubMed] [Google Scholar]
- Yu, H, Pardoll, D and Jove, R (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9: 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst, M, Najdovska, M, Grail, D, Lundgren-May, T, Buchert, M, Tye, H et al. (2008). STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest 118: 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, SP, Mark, KG, Leslie, K, Pao, W, Motoi, N, Gerald, WL et al. (2007). Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 117: 3846–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortylewski, M, Xin, H, Kujawski, M, Lee, H, Liu, Y, Harris, T et al. (2009). Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15: 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzo, R, Abate, F, Lasorsa, E, Tabbo', F, Gaudiano, M, Chiesa, N et al.; European T-Cell Lymphoma Study Group, T-Cell Project: Prospective Collection of Data in Patients with Peripheral T-Cell Lymphoma and the AIRC 5xMille Consortium “Genetics-Driven Targeted Management of Lymphoid Malignancies”. (2015). Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 27: 516–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, QW, Cheng, CK, Gustafson, WC, Charron, E, Zipper, P, Wong, RA et al. (2013). EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 24: 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H, Lee, H, Herrmann, A, Buettner, R and Jove, R (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14: 736–746. [DOI] [PubMed] [Google Scholar]
- Gough, DJ, Corlett, A, Schlessinger, K, Wegrzyn, J, Larner, AC and Levy, DE (2009). Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324: 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, B, Meng, J, Peyton, M, Girard, L, Bornmann, WG, Ji, L et al. (2011). STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res 71: 3658–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, HJ, Zhuang, G, Cao, Y, Du, P, Kim, HJ and Settleman, J (2014). Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26: 207–221. [DOI] [PubMed] [Google Scholar]
- Ernst, M, Thiem, S, Nguyen, PM, Eissmann, M and Putoczki, TL (2014). Epithelial gp130/Stat3 functions: an intestinal signaling node in health and disease. Semin Immunol 26: 29–37. [DOI] [PubMed] [Google Scholar]
- Guryanova, OA, Wu, Q, Cheng, L, Lathia, JD, Huang, Z, Yang, J et al. (2011). Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 19: 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, E, Kim, M, Woo, DH, Shin, Y, Shin, J, Chang, N et al. (2013). Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23: 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, JH, Kim, C, Sethi, G and Ahn, KS (2015). Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 6: 6386–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, JH, Kim, C, Sethi, G and Ahn, KS (2015). Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 6: 6386–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C, Li, S and Li, D (2016). Hsa-miR-134 suppresses non-small cell lung cancer (NSCLC) development through down-regulation of CCND1. Oncotarget 7: 35960–35978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C, Huang, C, Li, S,Yang, C, Xi, Y, Wang, Let al. (2016). Hsa-miR-326 targets CCND1 and inhibits non-small cell lung cancer development. Oncotarget 7: 8341–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, CC, Li, SJ, Zhang, F, Pan, JY, Wang, L, Yang, CLet al. (2016). Hsa-miR-329 exerts tumor suppressor function through down-regulation of MET in non-small cell lung cancer. Oncotarget 7:21510–21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, C, Li, S, Yang, C, Xi, Y, Wang, L, Zhang, F et al. (2016). MicroRNA-187-3p mitigates non-small cell lung cancer (NSCLC) development through down-regulation of BCL6. Biochem Biophys Res Commun 471: 82–88. [DOI] [PubMed] [Google Scholar]
- Hu, H, Li, S, Liu, J and Ni, B (2012). MicroRNA-193b modulates proliferation, migration, and invasion of non-small cell lung cancer cells. Acta Biochim Biophys Sin (Shanghai) 44: 424–430. [DOI] [PubMed] [Google Scholar]
- Garzon, R, Marcucci, G and Croce, CM (2010). Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov 9: 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky, N (2006). microRNA target predictions in animals. Nat Genet 38 Suppl: S8–13. [DOI] [PubMed] [Google Scholar]
- Grivennikov, S and Karin, M (2008). Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell 13: 7–9. [DOI] [PubMed] [Google Scholar]
- Hong, D, Kurzrock, R, Kim, Y, Woessner, R, Younes, A, Nemunaitis, J et al. (2015). AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 7: 314ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico, A (2015). Lung cancer: Driver-mutation-dependent stratification: learning from STAT3. Nat Rev Clin Oncol 12: 251. [DOI] [PubMed] [Google Scholar]
- Fan, Z, Cui, H, Yu, H, Ji, Q, Kang, L, Han, B et al. (2016). MiR-125a promotes paclitaxel sensitivity in cervical cancer through altering STAT3 expression. Oncogenesis 5: e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, L, Ya-Wei, L, Hai, W,Qiang, Z, Jun-Jie, L, Huang, Aet al. (2016). MiR-519a functions as a tumor suppressor in glioma by targeting the oncogenic STAT3 pathway. J Neurooncol 128: 35–45. [DOI] [PubMed] [Google Scholar]
- Shields, BJ, Hauser, C, Bukczynska, PE, Court, NW and Tiganis, T (2008). DNA replication stalling attenuates tyrosine kinase signaling to suppress S phase progression. Cancer Cell 14: 166–179. [DOI] [PubMed] [Google Scholar]
- Jeong, JB, Hong, SC, Jeong, HJ and Koo, JS (2011). Arctigenin induces cell cycle arrest by blocking the phosphorylation of Rb via the modulation of cell cycle regulatory proteins in human gastric cancer cells. Int Immunopharmacol 11: 1573–1577. [DOI] [PubMed] [Google Scholar]
- Sun, C, Yang, C, Xue, R, Li, S, Zhang, T, Pan, L et al. (2015). Sulforaphane alleviates muscular dystrophy in mdx mice by activation of Nrf2. J Appl Physiol (1985) 118: 224–237. [DOI] [PubMed] [Google Scholar]
- Sun, CC, Li, SJ, Yang, CL, Xue, RL, Xi, YY, Wang, L et al. (2015). Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 (Nrf2)-mediated Inhibition of NF-κB Signaling Pathway. J Biol Chem 290: 17784–17795. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sun, C, Li, S and Li, D (2016). Sulforaphane mitigates muscle fibrosis in mdx mice via Nrf2-mediated inhibition of TGF-beta/Smad signaling. J Appl Physiol (1985) 120:377–390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.