

Abstract
The ubiquitin-proteasome system (UPS) is one of the major protein degradation pathways, where abnormal UPS function has been observed in cancer and neurological diseases. Many neurodegenerative diseases share a common pathological feature, namely intracellular ubiquitin-positive inclusions formed by aggregate-prone neurotoxic proteins. This suggests that dysfunction of the UPS in neurodegenerative diseases contributes to the accumulation of neurotoxic proteins and to instigate neurodegeneration. Here, we review recent findings describing various aspects of UPS dysregulation in neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease.
Keywords: ubiquitin, deubiquitinating enzyme, proteasome, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease
Abnormal Protein Aggregation in Neurodegenerative Diseases
Many neurodegenerative diseases are associated with aggregate-prone neurotoxic proteins (aggresomes or inclusion bodies) that perturb cellular homeostasis and neuronal function (Popovic et al., 2014). These diseases are also categorically classed as proteinopathies, and comprise aggregation-prone components such as β-amyloid (Aβ) and tau in Alzheimer’s disease (AD), α-synuclein in Parkinson’s disease (PD), and polyglutamine (polyQ) repeat diseases.
Alzheimer’s disease is the most common form of progressive neurodegeneration and represents the main cause of senile dementia affecting about 10% of the population over the age of 65 and about 50% of the population over 85. AD can be divided into two forms: familial AD and sporadic AD. Familial AD accounts for 5%∼10% of AD patients, where inherited autosomal dominant mutations in three genes have been considered as the primary causes of familial AD: these include mutations in the genes encoding β-amyloid precursor protein (APP) and presenilins (PS1 and PS2; Goate et al., 1991; Levy-Lahad et al., 1995; Sherrington et al., 1995). In addition, sporadic AD is associated with polymorphisms in the Apolipoprotein E (APOE) gene, where the epsilon four allele is a strong genetic risk factor for late-onset AD (Strittmatter et al., 1993; Bu, 2009; Fu et al., 2016). Accumulation and aggregation of neurotoxic proteins, such as β-amyloid (Aβ), hyperphosphorylated tau, ubiquitinated proteins, and other unfolded proteins in vulnerable brain regions such as the hippocampus and cortex in AD brain is central to disease pathogenesis (Selkoe, 2001). Two major pathological hallmarks in AD are extracellular amyloid plaques and intraneuronal neurofibrillary tangle (NFT; Zhang et al., 2011). NFTs are comprised of hyperphosphorylated microtubule-associated protein tau (Song et al., 2015; Yamada et al., 2015; Zempel and Mandelkow, 2015). Amyloid plaques are primarily comprised of Aβ which is derived from sequential cleavages of APP by β- and γ-secretases (Zhang et al., 2011; Audrain et al., 2016). Cumulative evidence demonstrates that Aβ and tau are neurotoxic and can trigger a cascade of neurodegenerative processes ending in neuronal death, suggesting that overproduction/accumulation of Aβ and tau in vulnerable brain regions is the primary influence driving AD pathogenesis (Hardy and Selkoe, 2002; Gautam et al., 2015; Golovyashkina et al., 2015; Popugaeva et al., 2015).
Parkinson’s disease is the second most common neurodegenerative disease, clinically characterized by motor abnormalities, including resting tremor, rigidity, hypokinesia, and postural instability (Dauer and Przedborski, 2003; More and Choi, 2015). The loss of dopaminergic neurons in the substantial nigra subregion of midbrain is the major cause of motor deficits (Chen et al., 2015). Major pathological features of PD include intraneuronal accumulation of Lewy bodies and dystrophic neurites (Lewy neurites), both of which comprise aggregated proteins, such as α-synuclein, parkin, and ubiquitinated proteins (Hughes et al., 1992; Braak et al., 2003; Chiasserini et al., 2015; Xiang et al., 2015). Sporadic PD accounts for more than 90% of PD cases (Lang and Lozano, 1998), and only less than 10% of familial PD cases are caused by monogenic mutations (Benitez et al., 2016). Mutations in the gene encoding α-synuclein (Polymeropoulos et al., 1997; Helferich et al., 2015; Majbour et al., 2016; Schonherr et al., 2016) represent autosomal dominant familial PD, and mutations in genes encoding parkin (Kitada et al., 1998; Cha et al., 2015), UCHL1 (Leroy et al., 1998; Maraganore et al., 1999), DJ-1 (Bonifati et al., 2003), PINK1 (Valente et al., 2004), and LRRK2 (Zimprich et al., 2004; Saha et al., 2015) cause autosomal recessive familial PD.
Huntington’s disease (HD) is an autosomal dominant progressive neurodegenerative disorder, clinically characterized by cognitive impairment, chorea and dystonia, bradykinesia, uncoordinated movement, mood disorder and other psychiatric symptoms, and behavioral difficulties (Huntington Study Group, 1996). As the most common of the polyQ disorders, HD is caused by expansion of a CAG repeat coding for polyQ in the N-terminus of the huntingtin protein (MacDonald et al., 1993; Evers et al., 2015; Nekrasov et al., 2016). There is a remarkable repeat length threshold effect, where polyQ chain expansion of 36 repeats triggers HD onset, whereas repeats of less than 35 are non-pathological (Chong et al., 1997). The polyQ expansion in the N-terminus of huntingtin protein confers a gain-of-function phenotype, and is associated with significant neurotoxicity (Tallaksen-Greene et al., 2003).
In addition to huntingtin, expansion of polyQ-encoding CAG repeats in several genes have also been found in other polyQ repeat diseases, such as Ataxin-1 in spinocerebellar ataxia type 1 (SCA1), Ataxin-2 in SCA2, Atantin-3 in SCA3, α1A-voltage-dependent calcium channel (CACNA1A) in SCA6, Ataxin-7 in SCA7, Atrophin-1 in dentatorubral-pallidoluysian atrophy (DRPLA), and androgen receptor in SBMA (Zoghbi and Orr, 2000; Del Cano-Espinel et al., 2015). All polyQ repeat disorders are dominant neurodegenerative diseases and are associated with pathological intracellular inclusion bodies in the affected brain regions (Ross et al., 1997; Zoghbi and Orr, 2000).
Ubiquitin-Proteasome System
Ubiquitin is an evolutionarily conserved 76-amino acid moiety covalently tandemly linked to target protein components for degradation by the ubiquitin-proteasome system (UPS), and is required for degradation of about 80% intracellular proteins in eukaryotes (Pickart, 2001). The presence of ubiquitin in intracellular inclusions has been found in various neurodegenerative diseases, for example, amyloid plaques and NFT in AD, Lewy bodies in PD, and intranuclear inclusions in polyQ disorders (Todi and Paulson, 2011).
Protein ubiquitination occurs through the coordinated activity of several enzymes, including an ubiquitin activating enzyme (E1 ligase), conjugating enzyme (E2 ligase), and E3 ligase. Initially, a single ubiquitin moiety is attached to an active-site Cysteine residue within the E1 ligase through a thioester bond in an ATP dependent manner. The activated ubiquitin is then transferred to an ubiquitin conjugating enzyme-E2 ligase. Finally, the E2 ligase will cooperatively transfer the ubiquitin chain with a specified E3 ligase to a particular substrate, whereby the 26S proteasome will target the polyubiquitinated protein for degradation (Figure 1).
FIGURE 1.
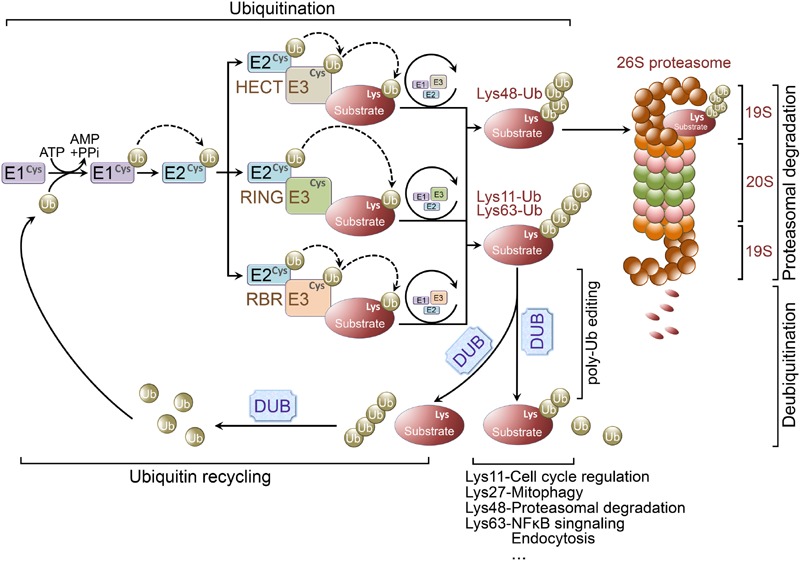
Ubiquitination and ubiquitin-proteasome system. Ubiquitination is a posttranslational modification that the ubiquitin is covalently conjugated to a Lysine residue of the substrate proteins. Ubiquitin is first attached to a Cysteine residue (the active-site) of ubiquitin activating enzyme (E1 ligase) in an ATP-dependent reaction. Subsequently, the activated ubiquitin will be transferred to a Cysteine residue of conjugating enzyme (E2 ligase). Finally, working with a specific E3 ligase, E2 ligase can transfer the (poly)ubiquitin to a Lysine residue of the substrate. There are three major classes of E3 ligases, including RING E3 ligases, HECT E3 ligases, and RBR E3 ligases. RING E3 ligases catalyze the transfer of ubiquitin directly from E2 ligases to the substrate. However, the activated ubiquitin is first transferred from E2 ligases to HECT or RBR E3 ligases and then be transferred to the substrate from the E3 ligases. Lys48-linked polyubiquitin chains usually target proteins for proteasomal degradation, whereas Lys63-linked polyubiquitination is involved in regulation of NFκB signaling. Lys27-linked polyubiquitination is important in regulating mitophagy, and Lys11-linked polyubiquitin chains are implicated in cell cycle regulation. Ubiquitination is reversed by deubiquitinating enzymes (DUBs). DUBs are crucial for determining the fate of ubiquitinated proteins through removing/editing the length/type of polyubiquitin chains. In addition, DUBs can disassemble unanchored ubiquitin chains for ubiquitin recycling.
Ubiquitin E3 ligases determine the specificity of ubiquitination process. E3 ligases are grouped into three classes according to their unique domains, including Really Interesting New Gene (RING) or U-box domain-containing E3 ligases, Homologous to E6AP C-terminus (HECT) domain-containing E3 ligases, and RING-between-RING (RBR) domain-containing E3 ligases (Atkin and Paulson, 2014; Morreale and Walden, 2016). RING E3 ligases catalyze the transfer of ubiquitin directly from E2 ligases to the substrates by binding both of them. Whereas HECT E3 ligases comprise a conserved Cysteine residue as an ubiquitin acceptor, where the activated ubiquitin is initially transferred from the E2 ligases to HECT E3 ligases prior to E3-mediated substrate conjugation (Ravid and Hochstrasser, 2008). Unlike RING E3 ligases, RBR E3 ligases contain two RING domains (RING1 and RING2) separated by an in-between-RING (IBR) domain. Functionally, RBR E3 ligases are similar with HECT E3 ligases, both catalyze ubiquitin first transfer from E2 ligases to a catalytic Cysteine residue on E3 ligases then from the E3 ligases to the substrates (Spratt et al., 2014) (Figure 1).
The fate of an ubiquitin-linked protein is dependent on Lysine chain-specific polyubiquitination (Grabbe et al., 2011; Kulathu and Komander, 2012; Rieser et al., 2013). For example, Lys48-linked polyubiquitin chains usually target proteins for degradation (Husnjak et al., 2008), whereas Lys63-linked polyubiquitination is involved in regulation of NFκB signaling (Laplantine et al., 2009), Lys27-linked polyubiquitin chains are important for mitophagy (Geisler et al., 2010), and Lys11-linked polyubiquitination is implicated in cell cycle regulation (Bremm and Komander, 2011). Therefore, ubiquitination is not only important for protein degradation, but is also implicated in non-proteolytic functions, such as endocytosis, protein-protein interaction, intracellular trafficking, inflammatory signaling, autophagy, and DNA repair (Grabbe et al., 2011; Kulathu and Komander, 2012; Rieser et al., 2013).
The UPS is involved in protein quality control and removal of misfolded and aggregated proteins, and dysfunction of the UPS is implicated in the pathogenesis of neurodegenerative diseases (Popovic et al., 2014).
Deubiquitinating Enzymes
The process of ubiquitination is reversible and the reverse process is called deubiquitination which is accomplished by deubiquitinating enzymes (DUBs; Rubinsztein, 2006; Reyes-Turcu et al., 2009; Popovic et al., 2014). Increasing evidence indicates the importance of DUBs in regulating UPS function, including proofreading ubiquitin-protein conjugates, removing ubiquitin from conjugated proteins as key examples (Amerik and Hochstrasser, 2004) (Figure 1). Pivotal roles for DUBs have been demonstrated in maintaining neuronal functions and regulating the pathogenesis of various neurodegenerative diseases (Rubinsztein, 2006; Todi and Paulson, 2011; Popovic et al., 2014).
More than 100 DUBs have been identified (Popovic et al., 2014), and can be subdivided into five subfamilies. DUBs can be further categorized into two classes, the Cysteine proteases and the zinc-dependent metalloproteases (Todi and Paulson, 2011). Cysteine protease-associated DUBs include the ubiquitin carboxyl-terminal hydrolases (UCHs), ubiquitin specific proteases (USPs), Machado Joseph Diseases (MJD) proteases, and ovarian tumor (OTU) protease. Metalloprotease DUBs comprise the JAB1/MPN/Mov34 metalloenzyme (JAMM) subfamily.
UCHL1, a member of the UCH subfamily, is one of the most abundant proteins in the brain. UCHL1 is known to interact with and stabilize monomeric ubiquitin, and facilitates the E3 ligase activity in its dimeric form (Doran et al., 1983; Wilkinson et al., 1989). An Aplysia UCHL1 ortholog, ubiquitin carboxyl-terminal hydrolase (AP-Uch) has been found to be important in maintaining synaptic function (Hegde et al., 1997). Inhibiting the expression or function of AP-Uch blocks induction of long-term facilitation, which is thought to be the molecular basis for learning and memory (Hegde et al., 1997). Mammalian UCHL1 is required for normal synaptic structure and function in hippocampal neurons (Cartier et al., 2009). Activation of NMDA receptor upregulates UCHL1 activity and the levels of free ubiquitin monomers. Inhibition of UCHL1 disrupts distribution of synaptic proteins, increases dendritic spine size and reduces spine density. However, restoration of ubiquitin in UCHL1-inhibited neurons rescues impaired synaptic structure (Cartier et al., 2009). UCHL1 is also essential for normal structure and function of neuromuscular junctions (NMJs), reduction of synaptic vesicles and accumulation of tubulovesicular structures at the presynaptic nerve terminals have been observed in Uchl1 knockout mice (Chen et al., 2010). UCHL3, a homolog of UCHL1, is involved in spatial and working memory (Wood et al., 2005).
19S proteasome-associated DUBs comprise three members: USP14, UCH37, and RPN11, which show differences in ubiquitin chains removals and consequent protein degradation (Lee et al., 2011). It has been reported that both USP14 and UCH37 prevent substrate degradation by removing ubiquitin chains and promoting proteasomal substrate dissociation. In contrast, RPN11 cleaves at the base of the ubiquitin-linked substrates and promotes substrate degradation (Lee et al., 2011). USP14 is essential for synaptic development and normal function of NMJs (Chen et al., 2009), which is mediated through ubiquitination and activation of c-Jun N-terminal kinase signaling (Vaden et al., 2015). Pharmacological inhibition of USP14 enhances proteasomal degradation of several neurotoxic proteins, including tau, TDP14, and Ataxin-3 (Lee et al., 2010). Usp14-deficient mice display motor neuron deficits and developmental NMJ defects (Chen et al., 2009). USP16, a chromosome 21-encoded gene, is involved in chromatin remodeling and cell cycle progression through regulated ubiquitination of histone H2A (Joo et al., 2007). USP16 interacts with HERC2 and modulates the ubiquitination in DNA repair machinery components (Zhang et al., 2014). USP16 is upregulated in Down’s syndrome (DS) cells due to extrachromosomal triplication in trisomy 21, downregulation of USP16 partially restores the impaired proliferation in DS somatic stem cells (Adorno et al., 2013). It has been reported that USP16 acts as a H2A DUB and regulates hematopoiesis and hematopoietic stem cell function (Gu et al., 2016). Moreover, USP16 is required for embryonic stem cell differentiation, where USP16 deficiency leads to embryonic lethality in mouse deletion models (Yang et al., 2014).
The gene encoding USP25 also resides on human chromosome 21 (Valero et al., 1999), USP25 is involved in protein degradation by the 26S proteasome (Valero et al., 1999). USP25 is also implicated in inflammation, and it has been found to facilitate removal of Lys63-linked ubiquitin chains from tumor necrosis factor-receptor associated factor 5/6 (TRAF5/6), Usp25-deficient mice show higher sensitivity to IL-17 dependent inflammation and autoimmunity (Zhong et al., 2012).
Dysregulation of Ubiquitination in Neurodegenerative Diseases
Accumulating evidence indicates that dysfunction of the UPS is a key factor to initiate and aggravate the pathogenesis of neurodegenerative diseases. We will focus on the dysregulation of ubiquitination and proteasomal degradation in neurodegenerative pathology in AD, PD, and HD (Table 1).
Table 1.
Ubiquitin-proteasome system is implicated in neurodegenerative diseases pathogenesis.
| Disease | Protein | Disease Relevance | Pathogenesis | Reference |
|---|---|---|---|---|
| AD | Parkin (E3 ligase) | Decreased in AD | Parkin overexpression reduces Aβ and restores impaired LTP and behavioral abnormalities of APP/PS1 mouse model. | Rosen et al., 2010; Hong et al., 2014 |
| AD | UCHL1 (DUB) | Decreased in AD | UCHL1 overexpression improves contextual memory and restores synaptic functions of APP/PS1 mouse model. | Choi et al., 2004; Gong et al., 2006 |
| AD | HRD1 (E3 ligase) | Decreased in AD | HRD1 reduces Aβ generation through interacting with APP and facilitating APP ubiquitination and proteasomal degradation. | Kaneko et al., 2010 |
| AD | CHIP (E3 ligase) | Upregulated in AD | CHIP ubiquitinates phosphorylated tau. Deletion of CHIP leads to the accumulation of non-aggregated, hyperphosphorylated, as well as caspase-3-cleaved tau species. Overexpression of CHIP and Hsp70/90 decreases the steady-state Aβ levels and promotes Aβ degradation. |
Petrucelli et al., 2004; Shimura et al., 2004; Dickey et al., 2006, 2007; Kumar et al., 2007 |
| PD | Parkin (E3 ligase) | Decreased in PD with loss-of-function mutations | More than 100 different PARK2 (encoding parkin) mutations have been found in PD patients, including missense, truncation, copy number variations, deletions, and insertions. Loss-of-function mutations in parkin impair its E3 ligase activity and lead to the accumulation of α-synuclein and the formation of Lewy bodies. | Shimura et al., 2001; Lesage and Brice, 2009; Lu et al., 2009; Chung et al., 2013 |
| PD | UCHL1 (DUB) | Mutation on I93M and S18Y. | UCHL1I93M mutation promotes its dimerization and inhibits the proteasomal degradation of α-synuclein through the Lys63-linked polyubiquitination of α-synuclein. | Leroy et al., 1998; Liu et al., 2002; Maraganore et al., 2004; Setsuie et al., 2007 |
| PD | CHIP (E3 ligase) | NA | CHIP can enhance parkin’s E3 ligase activity. Moreover, CHIP ubiquitinates LRRK2 and α-synuclein and promotes their degradation. | Imai et al., 2002; Ko et al., 2009; Kalia et al., 2011 |
| PD | TRAF6 (E3 ligase) | Upregulated in PD | TRAF6 promotes Lys6-, Lys27-, and Lys29-linked ubiquitination of DJ-1 and α-synuclein. TRAF6 induces Lys63-linked ubiquitination of PINK1, and stabilizes PINK1 on the depolarized mitochondria. | Zucchelli et al., 2010; Chung et al., 2013; Murata et al., 2013 |
| HD | CHIP (E3 ligase) | NA | CHIP suppresses aggregation and toxicity of polyQ-huntingtin. Knockdown of CHIP in HD transgenic mice aggravates disease pathology. | Miller et al., 2005 |
| SCA3 | Ataxin-3 (E3 ligase) | Gain-of-function mutation with polyQ expansion | Ataxin-3 overexpression protects Drosophila from polyQ-huntingtin-induced neurodegeneration. MJD-linked Ataxin-3 mutations promote ubiquitination and autophagic degradation of parkin. |
Warrick et al., 2005; Durcan et al., 2011 |
Dysregulation of the UPS in Alzheimer’s Disease
The presence of ubiquitin in NFTs and amyloid plaques in AD brain has been described as early as 1987 (Cole and Timiras, 1987; Mori et al., 1987; Perry et al., 1987). Ubiquitin-targeted proteasomal activity declines with age Tg2576 AD mouse model brain, and Aβ treatment markedly attenuates proteasomal activity in cultured neurons (Oh et al., 2005). Aβ induced UPS dysfunction contributes to the accumulation of reticulon 3 (RTN3) in dystrophic neurites, and the presence of dystrophic neurites and clustering of tubular endoplasmic reticulum has been observed in RTN3 transgenic mouse brain (Hu et al., 2007; Sharoar et al., 2016). Interestingly, intracellular Aβ accumulation and impaired proteasome function can be reversed by the ubiquitin E3 ligase parkin (Rosen et al., 2010). Parkin expression is downregulated in AD brains, and reduced parkin expression may contribute to the accumulation of intracellular Aβ. Moreover, parkin expression results in Aβ reduction which can be dampened by proteasome inhibitors (Rosen et al., 2010). In addition, overexpression of parkin in APP/PS1 mouse models restores impaired long-term potentiation (LTP) and rescues behavioral abnormalities by reducing Aβ load and neuroinflammation (Hong et al., 2014).
UCHL1 has been observed in ubiquitin-enriched inclusion bodies in AD brains (Lowe et al., 1990). In addition, decreased soluble UCHL1 protein levels was detected in the brains of postmortem AD patients and APP/PS1 mouse models (Choi et al., 2004; Gong et al., 2006), and soluble UCHL1 is inversely proportional to the number of NFTs in AD brains (Choi et al., 2004). Upregulation of UCHL1 improves contextual learning and restores synaptic functions in APP/PS1 mouse models, where rescue effects are dependent on enzymatic UCHL1 activity (Gong et al., 2006). Moreover, overexpression of UCHL1 reduces the levels of β-secretase BACE1, and consequent BACE1 cleavage products (APP C-terminal fragment C99 and Aβ). Depletion of UCHL1 increases the levels of BACE1, C99, and Aβ in the Uchl1-null gad mice (Zhang et al., 2012).
A direct interaction between APP and ubiquitin ligases also contributes to amyloid pathology. HRD1, an endoplasmic reticulum-associated degradation (ERAD) ubiquitin E3 ligase, is reduced in AD brains (Kaneko et al., 2010). HRD1 interacts with APP through its Proline-rich region, and this interaction reduces Aβ generation through promoting APP ubiquitination and degradation (Kaneko et al., 2010). In addition, APP can bind to ubiquitin E3 ligases Stub1 and CRL4CRBN through the APP cytosolic region (ACR; Del Prete et al., 2016). ACR promotes the ubiquitination of several presynaptic proteins and regulates neurotransmitter release (Del Prete et al., 2016).
Polyubiquitinated tau has also been found in the brains of AD patients (Perry et al., 1987), where ubiquitin conjugation usually occurs at Lys254, Lys311, and Lys353 within the tau microtubule-binding domain (Cripps et al., 2006). In AD brains, accumulation of hyperphosphorylated, ubiquitinated tau protein has been found at both presynaptic and postsynaptic terminals, which is associated with dysfunction of the UPS (Tai et al., 2012). Recently, Myeku et al. reported that tau can induce dysfunction of the 26S proteasome which can be prevented by activating cAMP-PKA signaling. Protective effects derived from cAMP-PKA signaling are mediated through phosphorylation of the 26S proteasomal subunit (Myeku et al., 2016). Han et al. directly delivered purified proteasomes into cells through mesoporous silica nanoparticle-mediated endocytosis, and they found that extraproteasomal transfer reduces tau aggregates and promotes cell survival against tau-induced proteotoxic stress (Han et al., 2014). C-terminus of Hsp70-interacting protein (CHIP), an ubiquitin E3 ligase, is also implicated in AD pathogenesis. It has been found that both CHIP and Hsp70 are upregulated in the brains of AD patients (Sahara et al., 2005). CHIP and its binding protein Hsp70 can induce the ubiquitination and degradation of phosphorylated tau (Petrucelli et al., 2004; Shimura et al., 2004), deletion of CHIP leads to the accumulation of non-aggregated, hyperphosphorylated, as well as caspase-3-cleaved tau (Dickey et al., 2006). Moreover, inhibition of Hsp90 reduces hyperphosphorylated tau through an interaction with CHIP (Dickey et al., 2007). In addition, overexpression of CHIP and Hsp70/90 decreases steady-state Aβ levels and promotes Aβ degradation (Kumar et al., 2007).
Dysregulation of UPS in Parkinson’s Disease
The presence of ubiquitin-immunopositive Lewy bodies in the brains of PD patients has been observed about 30 years ago (Kuzuhara et al., 1988). Impaired proteasomal function in the substantia nigra of PD brains was later observed (McNaught and Jenner, 2001). Importantly, genetic evidence reveals a clear involvement of UPS dysfunction in PD pathogenesis: familial PD-associated genetic variations have been identified on PARK2 (Kitada et al., 1998) and UCHL1 (Leroy et al., 1998; Maraganore et al., 1999), which encode the ubiquitin E3 ligase parkin and deubiquitinating enzyme UCHL1, respectively. Recessive early-onset familial PD can also be caused by mutations in genes encoding DJ-1 (Abou-Sleiman et al., 2003; Bonifati et al., 2003) and PINK1 (Valente et al., 2004), presumably through a loss-of-function mechanism.
An interaction between parkin and its substrate α-synuclein is a crucial regulatory component of dopaminergic neuron degeneration, loss-of-function mutations in PARK2 usually cause early-onset autosomal recessive PD (Kitada et al., 1998), whereas disease-associated mutations in α-synuclein are usually associated with autosomal dominant PD (Polymeropoulos et al., 1997). Mutant parkin fails to interact with α-synuclein, leads to the accumulation of α-synuclein and the formation of Lewy bodies (Shimura et al., 2001). Transgenic mice carrying mutant parkinQ311X develop progressive hypokinetic motor deficits, accumulation of α-synuclein and age-dependent loss of dopaminergic neurons in the substantia nigra and striatum (Lu et al., 2009).
Parkin is an E3 ligase mediating multiple forms of ubiquitination, including monoubiquitination, Lys48- and Lys63-linked polyubiquitination (Moore, 2006). In addition, parkin can form an E3 ligase complex with PINK1 and DJ-1 to promote unfolded protein degradation (Xiong et al., 2009): PD mutations in each component impair E3 ligase activity (Xiong et al., 2009). CHIP can promote parkin-mediated ubiquitination of Pael receptor and inhibit the Pael receptor-induced cell death (Imai et al., 2002). In addition, CHIP ubiquitinates and facilitates proteasomal degradation of LRRK2, where knockdown of CHIP exacerbates neurotoxicity mediated by mutant LRRK2 (Ko et al., 2009). CHIP can ubiquitinate α-synuclein and reduce toxic α-synuclein oligomers (Kalia et al., 2011). TRAF6 is upregulated in the brains of PD patients (Chung et al., 2013), and TRAF6 promotes Lys6-, Lys27-, and Lys29-linked ubiquitination of DJ-1 and α-synuclein. This interaction may cause aggregation of insoluble and polyubiquitinated mutant DJ-1 proteins (Zucchelli et al., 2010). Moreover, TRAF6 ubiquitinates PINK1 at Lys433 residue as a poly-Lys63 conjugated form which stabilizes PINK1 in depolarized mitochondria. Down-regulation of TRAF6 disrupts PINK1 localization, thereby causing mitochondria damage through parkin recruitment (Murata et al., 2013). Mutations in UCHL1 have been reported in rare cases of early-onset familial PD (Leroy et al., 1998; Maraganore et al., 1999). Most notably, the UCHL1I93M PD variant shows reduced deubiquitinating activity (Leroy et al., 1998). Ubiquitin-positive inclusion bodies and the loss of dopaminergic neurons have been observed in the substantia nigra of UCHLI93M transgenic mouse brains (Setsuie et al., 2007). In addition, UCHL1I93M mutation promotes dimerization and Lys63-linked polyubiquitination of α-synuclein. Lys63-linked polyubiquitination may aggravate PD pathology by inhibiting proteasomal degradation of α-synuclein (Liu et al., 2002).
Dysregulation of UPS in Huntington’s Disease
HD patients manifest polyQ repeats of over 40 residues within the N-terminus of huntingtin protein. Ubiquitin-linked mutant huntingtin has been found in neuronal intranuclear inclusions and dystrophic neurites in the cortex and striatum of HD patients (Difiglia et al., 1997). However, it seems that eukaryotic proteasomes fail to cleave and eliminate pathogenic protein species with repeats greater than 25Q residues; this results in accumulation and aggregation of huntingtin protein in the neuronal nucleus (Venkatraman et al., 2004). However, it has been reported that a selective histone deacetylase (HDAC) inhibitor 4b ameliorates cognitive impairment in N171-82Q HD mouse model by preventing formation of nuclear huntingtin aggregates. Mechanistically, HDAC inhibition upregulates expression of multiple genes that are fundamental to protein phosphorylation and ubiquitination, including Ube2K, Ubqln, Ube2e3, Usp28, Sumo2, as well as the genes encoding components of the inhibitor of κB kinase (IKK) complex (Jia et al., 2012). It has been reported that CHIP suppresses polyQ aggregation and toxicity, and knockdown of CHIP in HD transgenic mice aggravated disease pathology (Miller et al., 2005).
Spinocerebellar ataxia type 3 (SCA3) is another polyQ repeat disease, which is caused by a polyQ-encoding CAG repeat expansion in the gene encoding Ataxin-3 (Kawaguchi et al., 1994). SCA3 is characterized by progressive ataxia with degeneration in the cerebellum, brain stem, substantia nigra, and globus pallidus interna. Clinical symptoms of SCA3 include impaired eye movement, speech and swallowing defects (Todi and Paulson, 2011). Ataxin-3 is a DUB from MJD subfamily of proteases which contain an ubiquitin interacting motif flanking the polyQ domain. Ataxin-3 preferentially cleaves Lys63 linkages in polyubiquitinated chains containing both Lys48 and Lys63 linkages (Winborn et al., 2008). Overexpression of normal human Ataxin-3 can protect Drosophila from polyQ-huntingtin-induced neurodegeneration (Warrick et al., 2005). Unlike wild-type Ataxin-3, MJD-linked mutant Ataxin-3 can promote ubiquitination and autophagic degradation of parkin probably through regulating the levels of Lys27- and Lys29 ubiquitin-linked parkin (Durcan et al., 2011).
Conclusion
Although UPS-mediated protein degradation has been well-studied in the last few decades, regulation of ubiquitination and proteasomal degradation in normal brain function and pathogenesis of neurodegenerative diseases remains largely unknown. The regulation of polyubiquitination processes may vary at different physiological and/or pathologic conditions. This review surveyed dysregulation of UPS in several major neurodegenerative diseases, more research is required to understand how the UPS is disrupted in neurodegenerative pathogenesis. By enriching our understanding of protein degradation, we can begin to formulate strategies to treat neurodegenerative diseases remediating aspects of UPS dysregulation.
Author Contributions
QZ, LZ, and XW wrote the manuscript, TH, YZ, HL, and HX edited the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by National Natural Science Foundation of China (81571176 to XW, 91332114 and U1405222 to HX), the Thousand Young Talents Program of China, the Fundamental Research Funds for the Chinese Central Universities (20720150061 to XW), US National Institutes of Health grants (R01 AG038710, R01 AG021173, R01 NS046673, R01 AG030197, and R01 AG044420 to HX) and grants from the Alzheimer’s Association (to HX) and the Global Down Syndrome Foundation (DSADIIP-13-283543, to HX), BrightFocus Foundation (to HX), and Cure Alzheimer’s Fund (to HX).
References
- Abou-Sleiman P. M., Healy D. G., Quinn N., Lees A. J., Wood N. W. (2003). The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann. Neurol. 54 283–286. 10.1002/ana.10675 [DOI] [PubMed] [Google Scholar]
- Adorno M., Sikandar S., Mitra S. S., Kuo A., Nicolis Di Robilant B., Haro-Acosta V., et al. (2013). Usp16 contributes to somatic stem-cell defects in Down’s syndrome. Nature 501 380–384. 10.1038/nature12530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerik A. Y., Hochstrasser M. (2004). Mechanism and function of deubiquitinating enzymes. Biochem. Biophys. Acta 1695 189–207. 10.1016/j.bbamcr.2004.10.003 [DOI] [PubMed] [Google Scholar]
- Atkin G., Paulson H. (2014). Ubiquitin pathways in neurodegenerative disease. Front. Mol. Neurosci. 7:63 10.3389/fnmol.2014.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audrain M., Fol R., Dutar P., Potier B., Billard J. M., Flament J., et al. (2016). Alzheimer’s disease-like APP processing in wild-type mice identifies synaptic defects as initial steps of disease progression. Mol. Neurodegener. 11:5 10.1186/s13024-016-0070-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez B. A., Davis A. A., Jin S. C., Ibanez L., Ortega-Cubero S., Pastor P., et al. (2016). Resequencing analysis of five Mendelian genes and the top genes from genome-wide association studies in Parkinson’s Disease. Mol. Neurodegener. 11:29 10.1186/s13024-016-0097-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V., Rizzu P., Van Baren M. J., Schaap O., Breedveld G. J., Krieger E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299 256–259. 10.1126/science.1077209 [DOI] [PubMed] [Google Scholar]
- Braak H., Del Tredici K., Rub U., De Vos R. A. I., Steur E., Braak E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24 197–211. 10.1016/s0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Bremm A., Komander D. (2011). Emerging roles for Lys11-linked polyubiquitin in cellular regulation. Trends Biochem. Sci. 36 355–363. 10.1016/j.tibs.2011.04.004 [DOI] [PubMed] [Google Scholar]
- Bu G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10 333–344. 10.1038/nrn2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier A. E., Djakovic S. N., Salehi A., Wilson S. M., Masliah E., Patrick G. N. (2009). Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci. 29 7857–7868. 10.1523/jneurosci.1817-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha S. H., Choi Y. R., Heo C. H., Kang S. J., Joe E. H., Jou I., et al. (2015). Loss of parkin promotes lipid rafts-dependent endocytosis through accumulating caveolin-1: implications for Parkinson’s disease. Mol. Neurodegener. 10:63 10.1186/s13024-015-0060-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Sugiura Y., Myers K. G., Liu Y., Lin W. (2010). Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc. Natl. Acad. Sci. U.S.A. 107 1636–1641. 10.1073/pnas.0911516107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. C., Qin L. N., Li X. M., Walters B. J., Wilson J. A., Mei L., et al. (2009). The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 29 10909–10919. 10.1523/jneurosci.2635-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zhang D. Q., Liao Z., Wang B., Gong S., Wang C., et al. (2015). Anti-oxidant polydatin (piceid) protects against substantia nigral motor degeneration in multiple rodent models of Parkinson’s disease. Mol. Neurodegener. 10:4 10.1186/1750-1326-10-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasserini D., Paciotti S., Eusebi P., Persichetti E., Tasegian A., Kurzawa-Akanbi M., et al. (2015). Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 10:15 10.1186/s13024-015-0010-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Levey A. I., Weintraub S. T., Rees H. D., Gearing M., Chin L. S., et al. (2004). Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 279 13256–13264. 10.1074/jbc.M314124200 [DOI] [PubMed] [Google Scholar]
- Chong S. S., Almqvist E., Telenius H., Latray L., Nichol K., Bourdelat-Parks B., et al. (1997). Contribution of DNA sequence and CAG size to mutation frequencies of intermediate alleles for Huntington disease: evidence from single sperm analyses. Hum. Mol. Genet. 6 301–309. 10.1093/hmg/6.2.301 [DOI] [PubMed] [Google Scholar]
- Chung J. Y., Park H. R., Lee S. J., Lee S. H., Kim J. S., Jung Y. S., et al. (2013). Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab. Invest. 93 663–676. 10.1038/labinvest.2013.60 [DOI] [PubMed] [Google Scholar]
- Cole G. M., Timiras P. S. (1987). Ubiquitin-protein conjugates in Alzheimer’s lesions. Neurosci. Lett. 79 207–212. 10.1016/0304-3940(87)90698-7 [DOI] [PubMed] [Google Scholar]
- Cripps D., Thomas S. N., Jeng Y., Yang F., Davies P., Yang A. J. (2006). Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 281 10825–10838. 10.1074/jbc.M512786200 [DOI] [PubMed] [Google Scholar]
- Dauer W., Przedborski S. (2003). Parkinson’s disease: mechanisms and models. Neuron 39 889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Del Cano-Espinel M., Acebes J. R., Sanchez D., Ganfornina M. D. (2015). Lazarillo-related Lipocalins confer long-term protection against type I Spinocerebellar Ataxia degeneration contributing to optimize selective autophagy. Mol. Neurodegener. 10:11 10.1186/s13024-015-0009-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete D., Rice R. C., Rajadhyaksha A. M., D’adamio L. (2016). Amyloid Precursor Protein (APP) May act as a substrate and a recognition unit for CRL4CRBN and Stub1 E3 ligases facilitating ubiquitination of proteins involved in presynaptic functions and neurodegeneration. J. Biol. Chem. 291 17209–17227. 10.1074/jbc.M116.733626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey C. A., Kamal A., Lundgren K., Klosak N., Bailey R. M., Dunmore J., et al. (2007). The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 117 648–658. 10.1172/jci29715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey C. A., Yue M., Lin W. L., Dickson D. W., Dunmore J. H., Lee W. C., et al. (2006). Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci. 26 6985–6996. 10.1523/jneurosci.0746-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277 1990–1993. 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- Doran J. F., Jackson P., Kynoch P. A., Thompson R. J. (1983). Isolation of PGP 9.5, a new human neurone-specific protein detected by high-resolution two-dimensional electrophoresis. J. Neurochem. 40 1542–1547. 10.1111/j.1471-4159.1983.tb08124.x [DOI] [PubMed] [Google Scholar]
- Durcan T. M., Kontogiannea M., Thorarinsdottir T., Fallon L., Williams A. J., Djarmati A., et al. (2011). The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 20 141–154. 10.1093/hmg/ddq452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers M. M., Schut M. H., Pepers B. A., Atalar M., Van Belzen M. J., Faull R. L., et al. (2015). Making (anti-) sense out of huntingtin levels in Huntington disease. Mol. Neurodegener. 10:21 10.1186/s13024-015-0018-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Zhao J., Atagi Y., Nielsen H. M., Liu C. C., Zheng H. H., et al. (2016). Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol. Neurodegener. 11:37 10.1186/s13024-016-0099-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam V., D’avanzo C., Berezovska O., Tanzi R. E., Kovacs D. M. (2015). Synaptotagmins interact with APP and promote A beta generation. Mol. Neurodegener. 10:31 10.1186/s13024-015-0028-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S., Holmstrom K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12 119–131. 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- Goate A., Chartier-Harlin M. C., Mullan M., Brown J., Crawford F., Fidani L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349 704–706. 10.1038/349704a0 [DOI] [PubMed] [Google Scholar]
- Golovyashkina N., Penazzi L., Ballatore C., Smith A. B., Bakota L., Brandt R. (2015). Region-specific dendritic simplification induced by A beta, mediated by tau via dysregulation of microtubule dynamics: a mechanistic distinct event from other neurodegenerative processes. Mol. Neurodegener. 10:60 10.1186/s13024-015-0049-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B., Cao Z., Zheng P., Vitolo O. V., Liu S., Staniszewski A., et al. (2006). Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126 775–788. 10.1016/j.cell.2006.06.046 [DOI] [PubMed] [Google Scholar]
- Grabbe C., Husnjak K., Dikic I. (2011). The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 12 295–307. 10.1038/nrm3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y., Jones A. E., Yang W., Liu S. R., Dai Q., Liu Y. D., et al. (2016). The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc. Natl. Acad. Sci. U.S.A. 113 E51–E60. 10.1073/pnas.1517041113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D. H., Na H. K., Choi W. H., Lee J. H., Kim Y. K., Won C., et al. (2014). Direct cellular delivery of human proteasomes to delay tau aggregation. Nat. Commun. 5:5633 10.1038/ncomms6633 [DOI] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Hegde A. N., Inokuchi K., Pei W., Casadio A., Ghirardi M., Chain D. G., et al. (1997). Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89 115–126. 10.1016/S0092-8674(00)80188-9 [DOI] [PubMed] [Google Scholar]
- Helferich A. M., Ruf W. P., Grozdanov V., Freischmidt A., Feiler M. S., Zondler L., et al. (2015). α-synuclein interacts with SOD1 and promotes its oligomerization. Mol. Neurodegener. 10:66 10.1186/s13024-015-0062-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong X., Liu J., Zhu G., Zhuang Y., Suo H., Wang P., et al. (2014). Parkin overexpression ameliorates hippocampal long-term potentiation and beta-amyloid load in an Alzheimer’s disease mouse model. Hum. Mol. Genet. 23 1056–1072. 10.1093/hmg/ddt501 [DOI] [PubMed] [Google Scholar]
- Hu X., Shi Q., Zhou X., He W., Yi H., Yin X., et al. (2007). Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J. 26 2755–2767. 10.1038/sj.emboj.7601707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A. J., Daniel S. E., Kilford L., Lees A. J. (1992). Accuracy Of Clinical-Diagnosis Of Idiopathic Parkinsons-Disease - A Clinicopathological Study Of 100 Cases. J. Neurol. Neurosur. Psychiatry 55 181–184. 10.1136/jnnp.55.3.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington Study Group (1996). Unified huntington’s disease rating scale: reliability and consistency. Mov. Disord. 11 136–142. 10.1002/mds.870110204 [DOI] [PubMed] [Google Scholar]
- Husnjak K., Elsasser S., Zhang N., Chen X., Randles L., Shi Y., et al. (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453 481–488. 10.1038/nature06926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., Soda M., Hatakeyama S., Akagi T., Hashikawa T., Nakayama K., et al. (2002). CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. Cell. 10 55–67. 10.1016/s1097-2765(02)00583-x [DOI] [PubMed] [Google Scholar]
- Jia H., Kast R. J., Steffan J. S., Thomas E. A. (2012). Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: implications for the ubiquitin-proteasomal and autophagy systems. Hum. Mol. Genet. 21 5280–5293. 10.1093/hmg/dds379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo H. Y., Zhai L., Yang C., Nie S., Erdjument-Bromage H., Tempst P., et al. (2007). Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature 449 1068–1072. 10.1038/nature06256 [DOI] [PubMed] [Google Scholar]
- Kalia L. V., Kalia S. K., Chau H., Lozano A. M., Hyman B. T., Mclean P. J. (2011). Ubiquitinylation of alpha-synuclein by Carboxyl Terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS ONE 6:e14695 10.1371/journal.pone.0014695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M., Koike H., Saito R., Kitamura Y., Okuma Y., Nomura Y. (2010). Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci. 30 3924–3932. 10.1523/jneurosci.2422-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., et al. (1994). CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 8 221–228. 10.1038/ng1194-221 [DOI] [PubMed] [Google Scholar]
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 605–608. 10.1038/33416 [DOI] [PubMed] [Google Scholar]
- Ko H. S., Bailey R., Smith W. W., Liu Z. H., Shin J. H., Lee Y. I., et al. (2009). CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc. Natl. Acad. Sci. U.S.A. 106 2897–2902. 10.1073/pnas.0810123106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathu Y., Komander D. (2012). Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 13 508–523. 10.1038/nrm3394 [DOI] [PubMed] [Google Scholar]
- Kumar P., Ambasta R. K., Veereshwarayya V., Rosen K. M., Kosik K. S., Band H., et al. (2007). CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence A beta metabolism. Hum. Mol. Genet. 16 848–864. 10.1093/hmg/ddm030 [DOI] [PubMed] [Google Scholar]
- Kuzuhara S., Mori H., Izumiyama N., Yoshimura M., Ihara Y. (1988). Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 75 345–353. 10.1007/BF00687787 [DOI] [PubMed] [Google Scholar]
- Lang A. E., Lozano A. M. (1998). Parkinson’s disease. First of two parts. New Engl. J. Med. 339 1044–1053. 10.1056/nejm199810083391506 [DOI] [PubMed] [Google Scholar]
- Laplantine E., Fontan E., Chiaravalli J., Lopez T., Lakisic G., Veron M., et al. (2009). NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 28 2885–2895. 10.1038/emboj.2009.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H., Lee M. J., Park S., Oh D. C., Elsasser S., Chen P. C., et al. (2010). Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467 179–184. 10.1038/nature09299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J., Lee B. H., Hanna J., King R. W., Finley D. (2011). Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol. Cell. Proteomics 10 R110003871 10.1074/mcp.R110.003871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy E., Boyer R., Auburger G., Leube B., Ulm G., Mezey E., et al. (1998). The ubiquitin pathway in Parkinson’s disease. Nature 395 451–452. 10.1038/26652 [DOI] [PubMed] [Google Scholar]
- Lesage S., Brice A. (2009). Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 18 R48–R59. 10.1093/hmg/ddp012 [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E., Wasco W., Poorkaj P., Romano D. M., Oshima J., Pettingell W. H., et al. (1995). Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269 973–977. 10.1126/science.7638621 [DOI] [PubMed] [Google Scholar]
- Liu Y., Fallon L., Lashuel H. A., Liu Z., Lansbury P. T., Jr., et al. (2002). The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 111 209–218. 10.1016/S0092-8674(02)01012-7 [DOI] [PubMed] [Google Scholar]
- Lowe J., Mcdermott H., Landon M., Mayer R. J., Wilkinson K. D. (1990). Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol. 161 153–160. 10.1002/path.1711610210 [DOI] [PubMed] [Google Scholar]
- Lu X. H., Fleming S. M., Meurers B., Ackerson L. C., Mortazavi F., Lo V., et al. (2009). Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J. Neurosci. 29 1962–1976. 10.1523/jneurosci.5351-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M. E., Ambrose C. M., Duyao M. P., Myers R. H., Lin C., Srinidhi L. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes, Huntington’s Disease Collaborative Research Group. Cell 72 971–983. [DOI] [PubMed] [Google Scholar]
- Majbour N. K., Vaikath N. N., Van Dijk K. D., Ardah M. T., Varghese S., Vesterager L. B., et al. (2016). Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol. Neurodegener. 11:7 10.1186/s13024-016-0072-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore D. M., Farrer M. J., Hardy J. A., Lincoln S. J., Mcdonnell S. K., Rocca W. A. (1999). Case-control study of the ubiquitin carboxy-terminal hydrolase L1 gene in Parkinson’s disease. Neurology 53 1858–1860. 10.1212/WNL.53.8.1858 [DOI] [PubMed] [Google Scholar]
- Maraganore D. M., Lesnick T. G., Elbaz A., Chartier-Harlin M. C., Gasser T., Kruger R., et al. (2004). UCHL1 is a Parkinson’s disease susceptibility gene. Ann. Neurol. 55 512–521. 10.1002/ana.20017 [DOI] [PubMed] [Google Scholar]
- McNaught K. S., Jenner P. (2001). Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 297 191–194. 10.1016/S0304-3940(00)01701-8 [DOI] [PubMed] [Google Scholar]
- Miller V. M., Nelson R. F., Gouvion C. M., Williams A., Rodriguez-Lebron E., Harper S. Q., et al. (2005). CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J. Neurosci. 25 9152–9161. 10.1523/jneurosci.3001-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. J. (2006). Parkin: a multifaceted ubiquitin ligase. Biochem. Soc. Trans. 34 749–753. 10.1042/bst0340749 [DOI] [PubMed] [Google Scholar]
- More S. V., Choi D. K. (2015). Promising cannabinoid-based therapies for Parkinson’s disease: motor symptoms to neuroprotection. Mol. Neurodegener. 10:17 10.1186/s13024-015-0012-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H., Kondo J., Ihara Y. (1987). Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 235 1641–1644. 10.1126/science.3029875 [DOI] [PubMed] [Google Scholar]
- Morreale F. E., Walden H. (2016). Types of Ubiquitin Ligases. Cell 165:e241 10.1016/j.cell.2016.03.003 [DOI] [PubMed] [Google Scholar]
- Murata H., Sakaguchi M., Kataoka K., Huh N. (2013). SARM1 and TRAF6 bind to and stabilize PINK1 on depolarized mitochondria. Mol. Biol. Cell 24 2772–2784. 10.1091/mbc.E13-01-0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myeku N., Clelland C. L., Emrani S., Kukushkin N. V., Yu W. H., Goldberg A. L., et al. (2016). Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22 46–53. 10.1038/nm.4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov E. D., Vigont V. A., Klyushnikov S. A., Lebedeva O. S., Vassina E. M., Bogomazova A. N., et al. (2016). Manifestation of huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 11:27 10.1186/s13024-016-0092-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S., Hong H. S., Hwang E., Sim H. J., Lee W., Shin S. J., et al. (2005). Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 126 1292–1299. 10.1016/j.mad.2005.07.006 [DOI] [PubMed] [Google Scholar]
- Perry G., Friedman R., Shaw G., Chau V. (1987). Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl. Acad. Sci. U.S.A. 84 3033–3036. 10.1073/pnas.84.9.3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L., Dickson D., Kehoe K., Taylor J., Snyder H., Grover A., et al. (2004). CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 13 703–714. 10.1093/hmg/ddh083 [DOI] [PubMed] [Google Scholar]
- Pickart C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70 503–533. 10.1146/annurev.biochem.70.1.503 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276 2045–2047. 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- Popovic D., Vucic D., Dikic I. (2014). Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20 1242–1253. 10.1038/nm.3739 [DOI] [PubMed] [Google Scholar]
- Popugaeva E., Pchitskaya E., Speshilova A., Alexandrov S., Zhang H., Vlasova O., et al. (2015). STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity. Mol. Neurodegener. 10:37 10.1186/s13024-015-0034-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T., Hochstrasser M. (2008). Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9 679–690. 10.1038/nrm2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu F. E., Ventii K. H., Wilkinson K. D. (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78 363–397. 10.1146/annurev.biochem.78.082307.091526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieser E., Cordier S. M., Walczak H. (2013). Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem. Sci. 38 94–102. 10.1016/j.tibs.2012.11.007 [DOI] [PubMed] [Google Scholar]
- Rosen K. M., Moussa C. E., Lee H. K., Kumar P., Kitada T., Qin G., et al. (2010). Parkin reverses intracellular beta-amyloid accumulation and its negative effects on proteasome function. J. Neurosci. Res. 88 167–178. 10.1002/jnr.22178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C. A., Margolis R. L., Rosenblatt A., Ranen N. G., Becher M. W., Aylward E. (1997). Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA). Medicine (Baltimore) 76 305–338. 10.1097/00005792-199709000-00001 [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443 780–786. 10.1038/nature05291 [DOI] [PubMed] [Google Scholar]
- Saha S., Ash P. E., Gowda V., Liu L., Shirihai O., Wolozin B. (2015). Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol. Neurodegener. 10:26 10.1186/s13024-015-0022-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N., Murayama M., Mizoroki T., Urushitani M., Imai Y., Takahashi R., et al. (2005). In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 94 1254–1263. 10.1111/j.1471-4159.2005.03272.x [DOI] [PubMed] [Google Scholar]
- Schonherr C., Bien J., Isbert S., Wichert R., Prox J., Altmeppen H., et al. (2016). Generation of aggregation prone N-terminally truncated amyloid beta peptides by meprin beta depends on the sequence specificity at the cleavage site. Mol. Neurodegener. 11:19 10.1186/s13024-016-0084-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (2001). Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81 741–766. [DOI] [PubMed] [Google Scholar]
- Setsuie R., Wang Y. L., Mochizuki H., Osaka H., Hayakawa H., Ichihara N., et al. (2007). Dopaminergic neuronal loss in transgenic mice expressing the Parkinson’s disease-associated UCH-L1 I93M mutant. Neurochem. Int. 50 119–129. 10.1016/j.neuint.2006.07.015 [DOI] [PubMed] [Google Scholar]
- Sharoar M. G., Shi Q., Ge Y., He W., Hu X., Perry G., et al. (2016). Dysfunctional tubular endoplasmic reticulum constitutes a pathological feature of Alzheimer’s disease. Mol. Psychiatry 21 1263–1271. 10.1038/mp.2015.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R., Rogaev E. I., Liang Y., Rogaeva E. A., Levesque G., Ikeda M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375 754–760. 10.1038/375754a0 [DOI] [PubMed] [Google Scholar]
- Shimura H., Schlossmacher M. G., Hattori N., Frosch M. P., Trockenbacher A., Schneider R., et al. (2001). Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293 263–269. 10.1126/science.1060627 [DOI] [PubMed] [Google Scholar]
- Shimura H., Schwartz D., Gygi S. P., Kosik K. S. (2004). CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 279 4869–4876. 10.1074/jbc.M305838200 [DOI] [PubMed] [Google Scholar]
- Song L., Lu S. X., Ouyang X., Melchor J., Lee J., Terracina G., et al. (2015). Analysis of tau post-translational modifications in rTg4510 mice, a model of tau pathology. Mol. Neurodegener. 10:14 10.1186/s13024-015-0011-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt D. E., Walden H., Shaw G. S. (2014). RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem. J. 458 421–437. 10.1042/bj20140006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter W. J., Saunders A. M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G. S., et al. (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90 1977–1981. 10.1073/pnas.90.17.8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai H. C., Serrano-Pozo A., Hashimoto T., Frosch M. P., Spires-Jones T. L., Hyman B. T. (2012). The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 181 1426–1435. 10.1016/j.ajpath.2012.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallaksen-Greene S. J., Ordway J. M., Crouse A. B., Jackson W. S., Detloff P. J., Albin R. L. (2003). Hprt(CAG)146 mice: age of onset of behavioral abnormalities, time course of neuronal intranuclear inclusion accumulation, neurotransmitter marker alterations, mitochondrial function markers, and susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Comp. Neurol. 465 205–219. 10.1002/cne.10855 [DOI] [PubMed] [Google Scholar]
- Todi S. V., Paulson H. L. (2011). Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci. 34 370–382. 10.1016/j.tins.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaden J. H., Bhattacharyya B. J., Chen P. C., Watson J. A., Marshall A. G., Phillips S. E., et al. (2015). Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Mol. Neurodegener. 10:3 10.1186/1750-1326-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M. K., Harvey K., Gispert S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304 1158–1160. 10.1126/science.1096284 [DOI] [PubMed] [Google Scholar]
- Valero R., Marfany G., Gonzalez-Angulo O., Gonzalez-Gonzalez G., Puelles L., Gonzalez-Duarte R. (1999). USP25, a novel gene encoding a deubiquitinating enzyme, is located in the gene-poor region 21q11.2. Genomics 62 395–405. 10.1006/geno.1999.6025 [DOI] [PubMed] [Google Scholar]
- Venkatraman P., Wetzel R., Tanaka M., Nukina N., Goldberg A. L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell. 14 95–104. 10.1016/S1097-2765(04)00151-0 [DOI] [PubMed] [Google Scholar]
- Warrick J. M., Morabito L. M., Bilen J., Gordesky-Gold B., Faust L. Z., Paulson H. L., et al. (2005). Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol. Cell. 18 37–48. 10.1016/j.molcel.2005.02.030 [DOI] [PubMed] [Google Scholar]
- Wilkinson K. D., Lee K. M., Deshpande S., Duerksen-Hughes P., Boss J. M., Pohl J. (1989). The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246 670–673. 10.1126/science.2530630 [DOI] [PubMed] [Google Scholar]
- Winborn B. J., Travis S. M., Todi S. V., Scaglione K. M., Xu P., Williams A. J., et al. (2008). The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 283 26436–26443. 10.1074/jbc.M803692200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. A., Kaplan M. P., Brensinger C. M., Guo W., Abel T. (2005). Ubiquitin C-terminal hydrolase L3 (Uchl3) is involved in working memory. Hippocampus 15 610–621. 10.1002/hipo.20082 [DOI] [PubMed] [Google Scholar]
- Xiang W., Menges S., Schlachetzki J. C., Meixner H., Hoffmann A. C., Schlotzer-Schrehardt U., et al. (2015). Posttranslational modification and mutation of histidine 50 trigger alpha synuclein aggregation and toxicity. Mol. Neurodegener. 10:8 10.1186/s13024-015-0004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H., Wang D., Chen L., Choo Y. S., Ma H., Tang C., et al. (2009). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest. 119 650–660. 10.1172/jci37617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Patel T. K., Hochgrafe K., Mahan T. E., Jiang H., Stewart F. R., et al. (2015). Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol. Neurodegener. 10:55 10.1186/s13024-015-0052-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Lee Y. H., Jones A. E., Woolnough J. L., Zhou D. W., Dai Q., et al. (2014). The histone H2A deubiquitinase Usp16 regulates embryonic stem cell gene expression and lineage commitment. Nat. Commun. 5:3818 10.1038/ncomms4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H., Mandelkow E. M. (2015). Tau missorting and spastin-induced microtubule disruption in neurodegeneration: Alzheimer Disease and Hereditary Spastic Paraplegia. Mol. Neurodegener. 10:68 10.1186/s13024-015-0064-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Deng Y., Luo Y., Zhang S., Zou H., Cai F., et al. (2012). Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J. Neurochem. 120 1129–1138. 10.1111/j.1471-4159.2011.07644.x [DOI] [PubMed] [Google Scholar]
- Zhang Y. W., Thompson R., Zhang H., Xu H. (2011). APP processing in Alzheimer’s disease. Mol. Brain 4:3 10.1186/1756-6606-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Yang H., Wang H. (2014). The histone H2A deubiquitinase USP16 interacts with HERC2 and fine-tunes cellular response to DNA damage. J. Biol. Chem. 289 32883–32894. 10.1074/jbc.M114.599605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B., Liu X., Wang X., Chang S. H., Liu X., Wang A., et al. (2012). Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat. Immunol. 13 1110–1117. 10.1038/ni.2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant Parkinsonism with pleomorphic pathology. Neuron 44 601–607. 10.1016/j.neuron.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Zoghbi H. Y., Orr H. T. (2000). Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 23 217–247. 10.1146/annurev.neuro.23.1.217 [DOI] [PubMed] [Google Scholar]
- Zucchelli S., Codrich M., Marcuzzi F., Pinto M., Vilotti S., Biagioli M., et al. (2010). TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 19 3759–3770. 10.1093/hmg/ddq290 [DOI] [PubMed] [Google Scholar]