

Abstract
Aims
Lipid phosphate phosphatase 3; type 2 phosphatidic acid phosphatase β (LPP3; PPAP2B) is a transmembrane protein dephosphorylating and thereby terminating signalling of lipid substrates including lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P). Human LPP3 possesses a cell adhesion motif that allows interaction with integrins. A polymorphism (rs17114036) in PPAP2B is associated with coronary artery disease, which prompted us to investigate the possible role of LPP3 in human endothelial dysfunction, a condition promoting atherosclerosis.
Methods and results
To study the role of LPP3 in endothelial cells we used human primary aortic endothelial cells (HAECs) in which LPP3 was silenced or overexpressed using either wild type or mutated cDNA constructs. LPP3 silencing in HAECs enhanced secretion of inflammatory cytokines, leucocyte adhesion, cell survival, and migration and impaired angiogenesis, whereas wild-type LPP3 overexpression reversed these effects and induced apoptosis. We also demonstrated that LPP3 expression was negatively correlated with vascular endothelial growth factor expression. Mutations in either the catalytic or the arginine-glycine-aspartate (RGD) domains impaired endothelial cell function and pharmacological inhibition of S1P or LPA restored it. LPA was not secreted in HAECs under silencing or overexpressing LPP3. However, the intra- and extra-cellular levels of S1P tended to be correlated with LPP3 expression, indicating that S1P is probably degraded by LPP3.
Conclusions
We demonstrated that LPP3 is a negative regulator of inflammatory cytokines, leucocyte adhesion, cell survival, and migration in HAECs, suggesting a protective role of LPP3 against endothelial dysfunction in humans. Both the catalytic and the RGD functional domains were involved and S1P, but not LPA, might be the endogenous substrate of LPP3.
Keywords: Endothelial dysfunction, Angiogenesis , Apoptosis , Atherosclerosis
1. Introduction
Endothelial cells have a key role in vascular homeostasis as under quiescent conditions they maintain an anti-inflammatory, anti-thrombotic, and anti-proliferative environment. However, several risk factors activate endothelial cells inducing what is known as endothelial dysfunction. This leads to increased vascular permeability, leucocyte adhesion, and inflammation, all of which underlie atherosclerosis, the major cause of cardiovascular disease.1
LPP3 is an integral membrane protein that degrades various lipid phosphate mediators. LPP3, is encoded by the type 2 phosphatidic acid phosphatase β (PPAP2B) gene which is located in a locus associated with the risk of coronary artery disease (CAD) in recent genome-wide association studies.2 The major allele A (f = 0.91) of the rs17114036 variant located in the fifth intron of PPAP2B was associated with an increased risk of CAD (odds ratio, 1.17; P = 3.81 × 10−19).2 The major allele of rs6588635, a proxy SNP of rs17114036, was associated with lower PPAP2B mRNA levels in human aortic endothelial cells (HAECs)3 a finding later confirmed directly using the rs17114036 probe in microarray expression studies using the same HAECs by co-authors of this study.4 Interestingly, it was shown recently that in macrophages, the CAD risk variant is rs72664324 rather than rs17114036 (both in high LD) which operates to alter the response of macrophages to oxLDL by altering binding of CCAAT/enhancer-binding protein beta (C/EBP) beta to an enhancer site regulating PPAP2B expression.5 Finally, an up-regulation of Ppap2b expression in aortic endothelial cells and in atherosclerotic lesions isolated from Apoe−/− mice was reported.3
The in vitro and in vivo studies performed by Panchatcharam et al.6,7 in mice are consistent with a role of LPP3 in atherosclerosis. First, targeted inactivation of Ppap2b in smooth muscle cells (SMCs) enhanced intimal hyperplasia and promoted SMC dedifferentiation. After vascular injury LPP3 expression limited cellular response to lysophosphatidic acid (LPA) and impaired SMC phenotype modulation.6 Second, targeted Ppap2b inactivation in endothelial cells enhanced inflammatory responses and vascular permeability.7
LPP3 belongs to the phosphatidic acid-phosphatase (PAP) enzymes family. In humans, three PAP genes are known: PPAP2A, PPAP2C, and PPAP2B, which encode LPP1, LPP2, and LPP3, respectively.8,9 LPP3 is an integral membrane protein, with six transmembrane domains that is found in both plasma and intracellular membranes. The third hydrophilic loop contains the catalytic domain that localizes to the extracellular side of the plasma membrane or luminal side of internal membranes. Ppap2b inactivation in mice results in early embryonic lethality due to impaired vasculogenesis, indicating that LPP3 is essential for normal vascular development10,11 (for review12). LPPs dephosphorylate and thereby terminate the signalling of a broad range of lipid substrates including phosphatidic acid, LPA, ceramide-1-phosphate, sphingosine-1-phosphate (S1P), and diacylglycerol pyrophosphate.13,14 LPA and S1P are implicated in several signalling pathways involved in atherosclerosis.15 S1P contributes to vascular development and endothelial barrier functions through the regulation of cellular proliferation, differentiation, migration, and angiogenesis and acts as an intra- and extra-cellular mediator.16–19 LPA is involved in cell migration, proliferation, and differentiation, contributing to neovascularization and to the induction and the release of proteases leading to cell invasiveness.20,21 Human LPP3 (hLPP3) possesses a cell adhesion motif of arginine-glycine-aspartate (RGD) that allows interactions with αvβ3 and α5β1 integrins,22,23 promoting cell adhesion and intracellular signalling.
Our study was focused on human primary endothelial cells, as they constitute the first barrier of the vessel wall and their dysfunction is crucial in atherosclerosis. The rs17114036 in the last intron of PPAP2B constitutes an expression quantitative trait locus (eQTL), associated with a decreased PPAP2B expression in homozygotes bearing the major A allele.3 On the other hand, when using the public eQTL database (GTEx portal), we found that the rs17114036 in the PPAP2B locus was not associated with the expression of PPAP2B in various other tissues, thus pointing out the specific role of LPP3 in human primary endothelial cells.
The absence of functional studies on the role of LPP3 in primary HAECs, with the exception of a paper published during the revision of our manuscript,5 prompted us to determine the downstream mechanisms implicating LPP3 in endothelial dysfunction by combining LPP3 knockdown and overexpression, as well as targeted mutations of the RGD motif. We found that in HAECs, PPAP2B/LPP3 down-regulates the expression and production of the inflammatory cytokines Interleukin (IL)6, IL8 and the expression of the chemokine monocyte chemoattractant protein 1 (CCL2 chemokine (C-C Motif) ligand 2) (MCP1), as well as diminishing cell viability through induction of apoptosis and S1P-mediated migration. LPP3 also prevents leucocyte adhesion to HAEC monolayers. Together, these data strongly support a protective role of LPP3 against endothelial dysfunction and atherosclerosis in humans.
2. Methods
2.1 Cell culture
For the functional studies, HAECs were purchased (PromoCell, Germany; all males of the rs17114036 AA genotype) and used at passages 3–7. To study the correlations according to the genotype of HAECs between LPP3 expression and those of selected genes, we used the database established with HAECs isolated from the aortic explants of 147 heart transplant donors of anonymous origin (108 males as deduced from DNA analysis) through the UCLA transplant program approved by UCLA Institutional Review Board.24
2.2 ELISA assays for cytokines and MCP1
ELISA assays were performed in cell supernatants to detect soluble forms of IL1β, IL6, and IL8 and MCP1 using the respective human DuoSet ELISA kits (R&D systems).
2.3 Human peripheral blood mononuclear cell adhesion assay
Human peripheral blood mononuclear cells (hPBMC) were obtained from the Etablissement Français du Sang, Rungis, France and were isolated from six donors of Buffy coats using a Ficoll density gradient. hPBMC were labelled with calcein-AM, then incubated with HAEC monolayers and the number of fluorescent adherent hPBMC was assessed with an inverted epi-fluorescence microscope NIKON ECLIPSE Ti/Intensilight C-HGFIE Precentered Fiber Illuminator using an fluorescein isothiocyanate (FITC) filter and 4 × 10 magnification. Adherent hPBMC were quantitated in each well by automated counting using ImageJ software (NIH). In controls (siCtrl, Ctrl) 1000–1800 cells/well were counted.
2.4 Cell viability assay
Cell viability was performed using the tetrazolium salt WST-1 assay. Cells were treated with VEGF-165 (vascular endothelial growth factor, VEGF) or LPP3 substrates during the last 24 h prior to addition of WST-1 solution for 3 h, the absorbance was measured at 450 nm.
2.5 Cell proliferation assay
Cell proliferation was assayed using the addition of 5-bromo-2′-deoxyuridine (BrdU) to the plates for 6 h and the absorbance was quantified at 450 nm.
2.6 Cell migration
Transfected HAECs were cultured to reach confluence. Cells were treated with VEGF or LPP3 substrates during the last 24 h. Linear scratches were performed in the cell monolayers using a 1000 µL pipette tip and then the cells were incubated for further 24 h. Images of the gap were obtained at 0 and 16 h with fully motorized inverted microscope (Nikon Eclipse TiS) using bright field at 4 × 10 magnification. The wounded area was analysed using ImageJ software (NIH) by quantification of the surface of wounded area at 16 h as compared with 0 h.
2.7 Angiogenesis tube formation in Matrigel
Angiogenesis assays were performed using 15-well μ-angiogenesis slides (Ibidi, Germany). The slides were coated with Matrigel and HAECs were seeded at 10 000 cells/well and incubated for 24 h. Images of newly structured tubes were captured and tube formation was quantified by counting the number of tubular and branching point structures.
2.8 Genome-wide expression analysis and pre-processing of expression data
Transcriptomic analysis of total RNA was performed using the Illumina HT-12 v4 BeadChip. Briefly, RNA was extracted from HAECs and 250 ng of total RNA was reverse transcribed, amplified, and biotinylated using the Illumina TotalPrep RNA Amplification Kit. Each biotinylated cRNA was hybridized to a single BeadChip and scanned using the Illumina Hiscan array.
2.9 Immunohistochemistry staining
Immunohistochemistry (IHC) was performed using standard protocols. Briefly, IHC was performed on 5 μm sections of Tissue MicroArray blocks. Deparaffinized and rehydrated sections were incubated with primary antibodies, and then with a kit UltraVision LP Detection System (Thermo Fisher Scientific, UK). Slides were counterstained with aqueous hematoxylin and mounted with Immunomount (Shandon, Cergy-Pontoise, France).
2.10 Statistical analysis
All data points were obtained from the mean of the experimental points from two or three replicates. Comparisons between two groups were performed using the Wilcoxon paired test. Comparisons between more than two groups were performed using the Kruskall–Wallis test followed by Dunn post-hoc test (XLStat 2013, Addinsoft, NY, USA). A significance threshold at P ≤ 0.05 was used for all tests.
3. Results
3.1 LPP3 is expressed in human vascular wall
Immunostaining of human atherosclerotic aortas (tissues microarrays) showed that LPP3 is expressed in endothelial cells, intimal macrophages, and in the SMCs of the media (see Supplementary material online, Figure S1). The LPP3 transcript was also present in human atherosclerotic carotid tissue as assessed by RT–qPCR (data not shown).
3.2 Transcriptional analysis of HAECs silenced for LPP3
To investigate the function of hLPP3 we proceeded by knockdown or overexpression strategies in HAECs from different donors. Two LPP3 siRNAs [siLPP3(1) and siLPP3(2)] or hLPP3 over-expression plasmids were transiently transfected into HAECs. mRNA knockdown or overexpression was confirmed by RT–qPCR, and its impact on LPP3 protein level was evaluated by immunoblotting. siLPP3(1), suppressed the expression of LPP3 mRNA by 80–90% (Figure 1A) and the corresponding protein level strongly diminished compared with the control siRNA (Figure 1B). siLPP3(2) was also effective as it suppressed the expression of LPP3 mRNA by about 80% (see Supplementary material online, Figure S2) compared with the control siRNA, finally we preferentially used siLPP3(1) for the functional analysis. When LPP3 was overexpressed in HAECs, a mean two-fold increase in mRNA (Figure 1A) and a substantial increase in protein levels was observed (Figure 1B).
Figure 1.

Modulation of lipid phosphate phosphatase 3 (LPP3) expression in HAECs. HAECs were transfected for 48 h with either siRNA (siCtrl or siLPP3) or with the expression plasmids (Ctrl or hLPP3); siCtrl transfected cells were treated or not with 100 nM VEGF for additional 24 h (A). The level of LPP3 mRNA was determined by RT–qPCR. The results (mean of duplicate measurements) are shown as relative LPP3 mRNA levels as compared with individual respective controls set at 1, each dot within vertical scatter plots represents a single donor (n = 8); the mean +/− standard error of mean (SEM) is depicted. *P < 0.05, ***P < 0.0001. Lysates from HAECs transfected either with siLPP3 or siCtrl (treated or not with VEGF) and either hLPP3 expression plasmids or Ctrl plasmids were analysed by immunoblotting using a rabbit polyclonal anti-LPP3 antibody and re-probed with anti-tubulin antibody to ensure equal loading and transfer. Immunoblot from one representative experiment out of two is shown (B).
We explored the consequences of siRNA-mediated LPP3 silencing in HAECs from five donors by global gene expression profiling using the HumanHT-12 v4 Expression BeadChip which targets more than 47 000 probes. Using an adjusted P-value threshold of 0.05, we found 1941 genes that were differentially expressed between siLPP3 transfected HAECs and the control siRNA (scrambled) (see Supplementary material online, Table S1). Among the genes differentially expressed, 342 had a fold change higher than 1.5. Statistical enrichment of specific biological processes and molecular functions annotated in gene ontology (GO, http://www.geneontology.org/, November 2015, date last accessed) was assessed by gene set enrichment analysis (GSEA) using the Genetrail software. Significant pathways were identified (false discovery rate, FDR-corrected P-values <0.05). Among them, cell cycle, cell adhesion, cell migration, angiogenesis, blood vessel development, leucocyte activation, and differentiation and inflammatory response were found (see Supplementary material online, Table S2). All these signalling pathways are related to either endothelial cell dysfunction or neovessel formation.
3.3 LPP3 down-regulates VEGF expression
VEGF and several proinflammatory cytokines induce LPP3 expression in mice.25 We showed that both LPP3 mRNA and protein levels (n = 2) were up-regulated, in HAECs after 24 h treatment with VEGF165 (Figure 1A and 1B). Next, we evaluated the expression of VEGF mRNA in HAECs silenced for LPP3 or overexpressing it. We observed a two-fold increase in VEGF expression in cells silenced for LPP3 and a small significant decrease of VEGF expression in hLPP3 overexpressing cells (Figure 2). In addition, LPP3 knockdown increased the expression level of the VEGFA receptor (kinase insert domain receptor, KDR) 1.3-fold in our microarray data (adj. P-value = 0.0005) (see Supplementary material online, Table S1; line 562). These observations indicate a possible retrocontrol mechanism between VEGF and LPP3 in HAECs.
Figure 2.

LPP3 down-regulates VEGF expression in HAECs. HAECs were transfected with either siRNA (siCtrl or siLPP3) or with hLPP3 expression plasmids (Ctrl or LPP3) for 48 h. The level of VEGF mRNA was determined by RT–qPCR. The results (mean of duplicate measurements) are shown as relative VEGF mRNA levels as compared with individual respective controls set at 1, each dot within vertical scatter plots represents a single donor (n = 8); the mean +/− SEM is depicted. *P < 0.05, ***P < 0.0001.
3.4 siLPP3 up-regulates the expression of pro-inflammatory cytokines
To determine the effects of LPP3 levels on inflammatory cytokines in HAECs, we assessed the mRNA levels of major proinflammatory mediators: IL6, IL8, IL1β, and MCP1 (or CCL2) following knockdown of LPP3. We found that siLPP3 significantly increased the mRNA levels of these mediators, reaching nearly four-fold for IL6 and IL8 and two-fold for MCP1, as compared with control siRNA (Figure 3A). IL1β mRNA level was up-regulated up to four-fold in LPP3 silenced HAECs (see Supplementary material online, Figure S3A). hLPP3 overexpression, on the other hand, did not dramatically change the IL6, IL8, and MCP1 mRNA expression (see Supplementary material online, Figure S4A), except for IL1β a decrease of 20% was observed (see Supplementary material online, Figure S3B); suggesting that a basal level of LPP3 mRNA is sufficient to maintain a low level of inflammatory molecules. Next, in order to assess whether the up-regulation of cytokine- and MCP1-specific mRNAs in LPP3 silenced cells lead to an increased secretion of these compounds, we measured their presence in culture media using ELISA. As shown in Figures 3B, the level of IL8 was increased in the media of LPP3 silenced HAECs as compared with control siRNA and the level of IL6 (P = 0.055) and MCP1 were only slightly increased without reaching significance. In agreement with no change in mRNA levels observed in HAECs overexpressing hLPP3, no change in the cytokine levels was observed in these cells (see Supplementary material online, Figure S4B). IL1β level was not affected by siLPP3 (see Supplementary material online, Figure S3C) which may suggest that LPP3 silencing is independent of inflammasome activation, or that an additional signal could be required for the IL1β secretion.
Figure 3.

siLPP3 up-regulates the expression of pro-inflammatory mediators in HAECs. HAECs were transfected with siRNA (siCtrl or siLPP3) for 48 h. mRNA relative levels of IL6, IL8, and MCP1 were determined by RT–qPCR (A). The results (mean of duplicates) are shown as relative mRNA levels as compared with individual respective controls set at 1, each dot within vertical scatter plots represents a single donor (n = 8); the mean +/− SEM is depicted. The IL6, IL8, and MCP1 concentrations were determined in supernatants of HAECs cultured for 24 h in serum-free media using ELISA (B). The results (mean of triplicates) are shown, each dot within vertical scatter plots represents a single donor (n = 5); the mean +/− SEM is depicted. *P < 0.05, **P < 0.001, ***P < 0.0001. In panel B we also give the P-value of 0.055 for IL6 to show the trend.
Among the genes affected by the LPP3 silencing, prostaglandin-endoperoxide synthase 2 (cyclooxygenase 2, COX2) (PTGS2) exhibited among the highest fold change of expression (see Supplementary material online, Table S1). PTGS2 (or COX2) in concert with phospholipase A2, group IVA (PLA2G4A) are implicated in atherosclerosis and cardiovascular disease. We tested the impact of LPP3 on PLA2G4A and PTGS2 expression in HAECs, and observed that siRNA-mediated LPP3 silencing increased mRNA levels of both genes (see Supplementary material online, Figure S5A), on the other hand, hLPP3 overexpression decreased PTGS2 level, but not that of PLA2G4A (see Supplementary material online, Figure S5B).
We tested whether or not the expression of the above genes could be affected by the rs17114036 in PPAP2B, in a collection of HAECs from 147 donors (119 homozygotes for the major allele A and 28 heterozygotes AG). We compared the correlations of LPP3 expression with these genes, according to their genotype. Indeed, we found a strong correlation between LPP3 expression and IL6, IL8, PLA2G4A, PTGS2, vascular cell adhesion molecule 1 (VCAM1), and selectin E (SELE) only in HAECs bearing the protective G allele. The AA homozygotes showed no correlation between PPAP2B and these genes. Correlation values of PPAP2B with selected genes are represented in (Figure 4), although in statistical analysis, only the correlation between PPAP2B and IL8 and PTGS2 remained significant. These data support the hypothesis of the protective role of LPP3 in inflammatory response, although such significant correlations do not necessarily imply a causal relationship.
Figure 4.
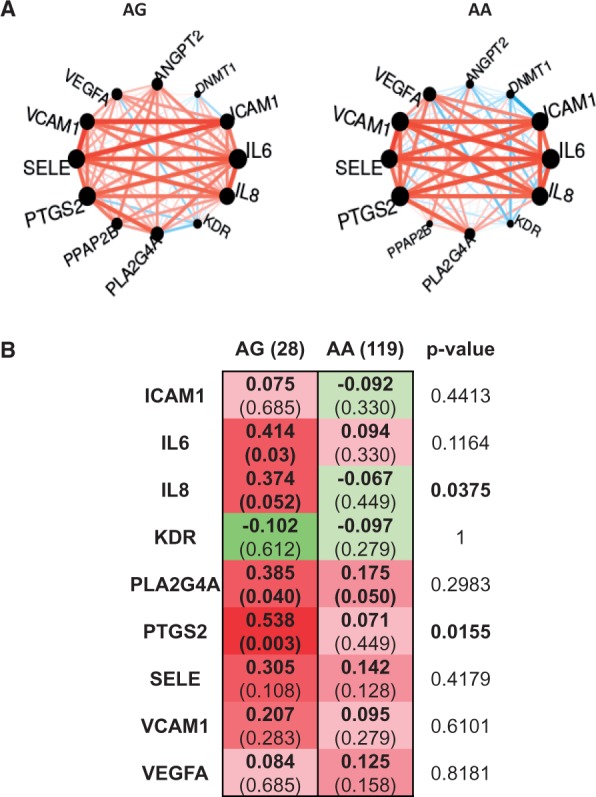
Correlation between PPAP2B/LPP3 expression and other genes for the AA and AG genotypes. (A) Circle plots of the correlations. Edges are coloured in red/blue for positive/negative correlations, respectively, and are weighted according to the correlation value. (B) Statistical comparison of the correlation coefficients between the two genotypes (red, positive and green, negative correlation). Values of the correlations are reported for each genotype, with the P-value into brackets. The P-value related to the comparison tests is given in the last column.
3.5 Role of LPP3 in leucocyte adhesion
As a consequence of vascular inflammation, leucocytes are recruited to endothelial surface and transmigrate into the intimal space of the arteries. Induction of adhesion molecules and chemokine expression in response to inflammatory stimuli plays a critical role in leucocyte adhesion to the endothelium.1 To investigate whether or not LPP3 expression impacts leucocyte adhesion, we first evaluated adhesion molecules and chemokine expression in HAECs. Using RT–qPCR, we measured the expression of three major adhesion molecules, SELE, intercellular adhesion molecule 1 (ICAM1), and VCAM1 and showed that LPP3 silencing as compared with the control siRNA treatment induced a 11-, 3-, and 1.5-fold increase of their respective mRNA levels (Figure 5A), accompanied by the increase in their protein levels (Figure 5C). hLPP3 overexpression significantly decreased ICAM1 mRNA as compared with the control plasmid, whereas VCAM1 and SELE mRNAs were not affected by hLPP3 overexpression (Figure 5B).
Figure 5.

LPP3 down-regulates leucocyte recruitment to HAEC monolayers. HAECs were transfected with siRNA (siCtrl or siLPP3) or with hLPP3 expression plasmids (Ctrl or LPP3) for 48 h. mRNA relative levels of adhesion molecules SELE, VCAM-1, and ICAM-1 were determined by RT–qPCR (A, B). The results (mean of duplicates) are shown as relative mRNA levels as compared with individual respective controls set at 1, each dot within vertical scatter plots represents a single donor (n = 7–8); the mean +/− SEM is depicted. Lysates were analysed by immunoblotting with specific antibodies and re-probed with an anti-tubulin antibody (the SELE membrane) to ensure equal loading and transfer (C). Leucocyte adhesion was assessed using a hPBMC adhesion assay as described in Methods. HAECs were transfected with either siRNA (siCtrl or siLPP3) or with hLPP3 expression plasmids (Ctrl or LPP3) for 48 h, and then incubated with calcein-AM labelled hPBMC. Images of fluorescent cells were captured with an epi-fluorescence microscope (D) and the adherent cells were quantified by automated counting using ImageJ and represented as a relative change over the control (E). The results (mean of duplicates) are shown, each dot within vertical scatter plots represents a single leucocyte donor (n = 5–6) tested on three HAECs donors; the mean +/− SEM is depicted. *P < 0.05, **P < 0.001. Bar scale in panel D indicates 500 µm.
In western blots SELE and ICAM1 proteins were decreased whereas VCAM1 protein level remained unchanged compared with LPP3 silenced cells (Figure 5C). We further investigated whether LPP3 expression affects the recruitment of leucocytes, after transfection of HAECs with either siLPP3 or hLPP3 plasmids. HAECs were incubated for 1 h with fluorescent calcein-AM labelled human blood leucocytes, and as shown in Figure 5D and E, leucocyte attachment was markedly enhanced when the cells were transfected with siLPP3 as compared with control siRNA. Conversely, overexpression of hLPP3 significantly decreased leucocyte adhesion to the HAECs monolayer, as compared with control plasmid (Figure 5D and E). These observations are concordant with the increased protein expression of adhesion molecules in siLPP3 silenced cells and with the decreased expression in LPP3 overexpressing cells (Figure 5C).
3.6 LPP3 down-regulates cell survival and proliferation
In order to assess the role of LPP3 in survival and proliferation of HAECs, we performed the mitochondrial activity WST1 assay to determine cell viability and a BrDU incorporation assay to measure cell proliferation rate. LPP3 silencing significantly increased cell viability by 30% (Figure 6A). VEGF treatment only slightly decreased cell survival in control cells (P = 0.072) but did not modify it in siLPP3 silenced cells (Figure 6A) suggesting that LPP3 itself is crucial to control cell viability. Interestingly, neither LPP3 silencing nor VEGF treatment modified HAECs’ proliferation rate as compared with control; however, when siLPP3 HAECs were stimulated with VEGF we observed a small increase of cell proliferation as compared with controls stimulated with VEGF (see Supplementary material online, Figure S6A). Overexpression of hLPP3 decreased both cell survival (−30%; Figure 6B) and proliferation rate measured by BRDU incorporation (−40%; see Supplementary material online, Figure S6B) as compared with the control plasmid.
Figure 6.

LPP3 affects viability of HAECs. HAECs were transfected with either siRNA (siCtrl or siLPP3) (A) or with hLPP3 expression plasmids (Ctrl or LPP3) (B) for 48 h. Cell viability was measured using the WST-1 assay and the results (percentage of respective controls; mean of duplicates) are shown, each dot within vertical scatter plots represents a single donor (n = 7); the mean +/− SEM is depicted. To show the variability of controls each point was calculated as percentage of the mean of controls set as 100%. HAECs transfected with either siLPP3 or siCtrl were treated with inhibitors of either LPA formation (PF8380) or S1P receptor (FTY720) (C). HAECs transfected with Ctrl- or LPP3-containing plasmids were treated with LPA mimic [(2S)-OMPT] or S1P (D). HAECs were transfected with either control plasmids, WT LPP3, or with mutants of the catalytic domain (H249P or H251P) or of the adhesion motif (RGD->RAD), subsequently the WST-1 assays were performed. The results (mean of triplicates) in panels C, D, and E (percentage of respective controls) are shown, each dot within vertical scatter plots represents a single donor (n = 3); the mean +/− SEM is depicted. Caspase-3 and -7 activity was assessed using Apo-ONE® Homogeneous Caspase-3/7 Assay (F) and the relative fluorescence was measured using a microplate reader. The results (mean of duplicates) are shown as percentage of respective controls set at 100%, each dot within vertical scatter plots represents a single donor (n = 8); the mean +/− SEM is depicted. *P < 0.05, **P < 0.001. When more than three groups were compared a global P-value is: panel A, P = 0.006; panel C, P = 0.08; panel D, P = 0.04. In panel A we also give the P-value of 0.072 for si Ctrl vs siCtrl + VEGF and in panel E the P = 0.054 for siLPP3 to show the trend.
To investigate which LPP3 substrate is involved in the survival of HAECs, we performed the WST1 assay on HAECs (n = 3) under silencing or overexpression of hLPP3. siLPP3 transfected cells were treated with 1 µM of either PF8380 (an inhibitor of LPA synthesis) or FTY720 (2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol, hydrochloride, a S1P receptor inhibitor), while hLPP3 overexpressing cells were treated with LPP3 substrates (1 µM) either (2S)-OMPT ((2S)-3-[(hydroxymercaptophosphinyl)oxy]-2-methoxypropyl ester, 9Z-octadecenoic acid, triethyl ammonium salt, LPA mimetic resistant to dephosphorylation) or S1P (S1P receptor agonist). siLPP3 increased WST1 labelling but treatment of the silenced cells with either PF8380 (4-[3-(2,3-dihydro-2-oxo-6-benzoxazolyl)-3-oxopropyl]-(3,5-dichlorophenyl)methyl ester-1-piperazinecarboxylic acid) or FTY720 prevented the siLPP3-induced increase of cell survival (Figure 6C) Conversely, when HAECs were transfected with an hLPP3 plasmid, neither (2S)-OMPT nor S1P compensated the decrease of survival induced by LPP3 overexpression (data not shown).
Next, HAECs were transfected with plasmids carrying PPAP2B cDNA mutations obtained by site-directed mutagenesis in two major functional domains. Two mutations H249P or H251P were introduced in the catalytic domain, and one mutation was introduced into the ligand domain by transforming RGD to arginine-alanine-aspartate (RAD). Both H249P and H251P as the RGD-to-RAD mutation reduced cell survival to some extent as compared with the wild-type (Figure 6D); however, only RGD-to-RAD mutation induced a small decrease of cell proliferation rate as measured by BrDU assay (see Supplementary material online, Figure S6C). These results indicate that both the catalytic and RGD domains are probably involved in the regulation of cell survival.
We conclude that LPP3 silencing substantially increases the viability of HAECs and supplementation with the specific drugs that impede LPA or S1P signalling results in an inhibition of cell viability. In contrast, overexpression of LPP3 significantly decreased cell survival and this effect could not be compensated by adding (2S)-OMPT (LPA mimetic) or S1P, suggesting that the RGD motif of hLPP3 may play a role in the regulation of cell viability in HAECs.
In order to examine whether or not LPP3 level impacts apoptosis we performed several assays. In the first assay, annexin V/7-AAD binding was performed in hLPP3 overexpressing cells using two different plasmid concentrations and showed that there was an increased fraction of both annexin V+/7-AAD− expressing HAECs showing early apoptosis (data not shown) and annexin V+/7-AAD+ expressing HAECs showing late apoptosis (see Supplementary material online, Figure S7A and B). However, LPP3 knockdown only slightly decreased annexin V/7-AAD labelling. In the second assay, LPP3 knockdown induced a marginal (P = 0.054) decrease of caspase-3/7 activity, but when hLPP3 was overexpressed the caspase-3/7 activity increased approximately two-fold as compared with the control plasmid (Figure 6E).
Finally, to further understand the mechanism underlying the LPP3 involvement in cell viability, we assessed whether or not LPP3 levels affect ERK1/2 phosphorylation. LPP3 knockdown increased ERK1/2 phosphorylation as compared with control siRNA, whereas, hLPP3 overexpression did not affect ERK1/2 phosphorylation (see Supplementary material online, Figure S7C). Therefore, LPP3 regulates cell viability through the induction of apoptosis via the activation of caspase-3 and -7 and ERK1/2 phosphorylation.
3.7 LPP3 down-regulates cell migration
Migration of endothelial cells is a key initiating event in the formation of new blood vessels. To investigate whether LPP3 is involved in HAECs migration, we performed wound-healing assays. To compare the differences in migratory behaviour, the surface of the wounded area was determined after 16 h of various treatments. Cells were treated or not with VEGF which is an activator of endothelial cell migration. When HAECs were silenced for LPP3 the migration distance in the dish increased 1.6-fold as compared with the siRNA control (Figure 7A). HAECs silenced for LPP3 and treated by VEGF showed slightly increased migration as compared siCtrl (see Supplementary material online, Figure S8). hLPP3 overexpression decreased the wounded area up to 50% as compared with the control plasmid (Figure 7A). To determine if the catalytic or the ligand domain of LPP3 is involved in cell migration, we used the same approach as described above to study cell survival. When LPP3 was silenced, treatment of HAECs with PF8380 did not modify the migration of HAECs, indicating that inhibition of LPA is not sufficient to restore the LPP3 silencing effect (Figure 7B). When siLPP3 cells were treated with FTY720 the wounded area was reduced by 30%, as compared with siLPP3, pointing to an important role for S1P in cell migration in this model (Figure 7B). When (2S)-OMPT was added to hLPP3 over-expressing cells, no change in the migration was observed, as compared with the control plasmid (Figure 7C). This observation is in agreement with our results obtained with PF8380, suggesting that LPA production is not required for LPP3-regulated endothelial cell migration. In contrast, addition of S1P to HAECs overexpressing LPP3 tended to increase (P = 0.075) migration 3.6-fold as compared with the control plasmid (Figure 7C). Together, these results suggest that LPP3 may down-regulate HAEC migration through S1P degradation.
Figure 7.

LPP3 down-regulates HAECs migration. HAECs were transfected with either siRNA (siCtrl or siLPP3) or with hLPP3 expression plasmids (Ctrl or LPP3) for 48 h. Cell monolayers were wounded with 1000 µL pipette tips (right panel) and incubated for 16 h to assess cell migration. Wounded areas were imaged at 0 and 16 h. Results expressed in percentage of the respective controls (mean of duplicates) are shown, each dot within vertical scatter plots represents a single donor (n = 7); the mean +/− SEM is depicted (A). Bar scale in micrographs in panel A indicates 500 µm. siLPP3 and siCtrl transfected HAECs were treated with inhibitors of either LPA formation (PF8380) or S1P receptor (FTY720) (B). LPP3-containing plasmid or Ctrl plasmid transfected cells were treated with LPA mimics [(2S)-OMPT] or S1P (C). The results (mean of duplicates) in panel B and C are shown, each dot within vertical scatter plots represents a single donor (n = 3); the mean +/− SEM is depicted. To show the variability of controls each point was calculated as percentage of the mean of individual controls set as 100%. *P< 0.05, ***P< 0.0001. In panel C we give the P-value of 0.075 to show the trend. When more than three groups were compared a global P-value is: panel B, P = 0.02; panel C, P = 0.03
3.8 LPP3 regulates angiogenesis
To investigate whether or not LPP3 participates in the formation of tubes, a hallmark of angiogenesis, we performed a Matrigel tube formation assay in HAECs under- or over-expressing hLPP3. At early times (3–6 h), LPP3 silenced cells started to organize into tube structures more effectively than the siRNA control cells and this process was delayed when hLPP3 was overexpressed (not shown), consistent with our results obtained both on cell viability (Figure 6) and migration (Figure 7).
After 24 h LPP3 silencing strongly decreased tube formation (−63%) as compared with the control siRNA (Figure 8A), suggesting that the angiogenic structures formed at 6 h (data not shown) became disorganized. On the contrary, overexpression of hLPP3 increased the formation of tubes by 50% as compared with the control plasmid (Figure 8B). Branching point structure number was regulated in the same proportion as the number of tubes (data not shown). In order to determine which LPP3 substrate is involved in the regulation of angiogenesis, we treated the LPP3 silenced cells with the PF8380 and showed that inhibiting LPA supply did not affect the number of tube structures as compared with the control (dimethylsulfoxyde, DMSO), whereas FTY720 treatment nearly restored the number of tubes in LPP3 siRNA transfected cells toward the level of control siRNA (Figure 8C). Treatment of hLPP3 overexpressing cells with either S1P or (2S)-OMPT decreased LPP3-induced angiogenesis (Figure 8D). Next, we investigated which of the LPP3 functions is responsible for this regulation using either catalytic or RAD LPP3 mutants. We observed that both catalytic mutants (H249P or H251P) as well as RAD mutant decreased the number of tubes as compared with the plasmid carrying the wild-type LPP3 (Figure 8E). Our results suggest that both RGD ligand and the enzymatic domains of LPP3 are involved in LPP3-regulated angiogenesis. Among LPP3 substrates, S1P is likely the one implicated in LPP3-mediated regulation of angiogenesis.
Figure 8.

LPP3 is a positive regulator of angiogenesis. HAECs were transfected with either siRNA (siCtrl or siLPP3) (A) or with hLPP3 expression plasmids (Ctrl or LPP3) (B) for 48 h. Cells were harvested and seeded on Matrigel and incubated for an additional 48 h. Microphotographs of cultures were taken at 24 h and angiogenesis was quantified by counting the tube-like and branching point structures, using an automated ImageJ program. Bar scale in panel A and B indicates 500 µm. siLPP3 and siCtrl transfected HAECs were treated with inhibitors of either LPA formation (PF8380) or S1P receptor (FTY720) (C). HAECs transfected with either hLPP3 expression plasmids or Ctrl plasmids were treated with LPA mimics [(2S)-OMPT] or S1P (D). HAECs were transfected with control plasmid, wild-type LPP3 or mutants of the catalytic domain (H249P or H251P) or the adhesion motif RGD->RAD (E). The results expressed as percentage of respective controls (mean of duplicates) are shown, each dot within vertical scatter plots represents a single donor (n = 4–6); the mean +/− SEM is depicted. To show the variability of controls each point was calculated as percentage of the mean of individual controls set as 100%. *P < 0.05, ***P < 0.0001. When more than three groups were compared a global P-value is: panel C, P = 0.0002; panel D, P = 0.06, and panel E, P = 0.0004.
3.9 LPP3 degrades both intra- and extra-cellular S1P
We showed that S1P, but not LPA, was the major substrate of LPP3 involved in cell migration and angiogenesis. To determine whether S1P is degraded by LPP3 under our experimental conditions, and to assess if LPP3 degrades preferentially extra- or intra-cellular pools of S1P, we measured its levels using reverse phase HPLC, in both culture media and cellular extracts from LPP3-silenced or overexpressing HAECs. The S1P content in intact HAECs was low, ranging from 3.1 to 4.2 pmol/106 cells (intracellular) and 1.1 nM (extracellular). However, in LPP3 silenced HAECs (n = 3) both intra- (see Supplementary material online, Figure S9A) and extra-cellular (see Supplementary material online, Figure S9B) S1P levels increased, whereas in hLPP3 overexpressing HAECs the intracellular levels of S1P decreased (see Supplementary material online, Figure S9A) and became undetectable in supernatants (see Supplementary material online, Figure S9B). As the number of donors was too low to apply the statistical analysis, these results must be confirmed in the future on a larger number of donors. In parallel, we measured the level of LPA in the same HAEC supernatants by mass spectrometry, using two different protocols and appropriate internal standards, and in neither of the experimental conditions (+/− VEGF), could we detect LPA (data not shown). Thus in our experiments, LPP3 might control both intra- and extra-cellular pools of S1P in HAECs.
4. Discussion
The novelty of our study is the use of a new integrated approach, combining transcriptomics and functional cellular studies to demonstrate that LPP3 constitutes an important component of HAEC physiology. Indeed, silencing LPP3 in HAECs readily enhanced inflammation, cell survival and migration, and impaired angiogenesis, whereas, hLPP3 overexpression had the opposite effects. In various human cells LPP3 expression is induced by growth factors and inflammatory cytokines, including VEGF.6 In this study, we demonstrated for the first time that modulation of LPP3 expression by either knockdown or overexpression negatively correlated with VEGF expression suggesting a negative feedback control loop between VEGF and LPP3 in HAECs. This is in agreement with former studies showing that VEGF increases LPP3 protein expression in HUVECs.25 Additionally, LPA, one of the LPP3 substrates, was shown to induce VEGF expression in ovarian cancer cells; however LPP3 expression was not studied in this context.26
4.1. Anti-inflammatory role of LPP3 in HAECs
We showed here that LPP3 silencing in HAECs, promoted their pro-inflammatory response by increasing IL1β mRNA and MCP1 expression and both expression and secretion of IL6 and IL8. Basal LPP3 is sufficient to maintain a low level of inflammatory cytokines because hLPP3 overexpression did not modify cytokine expression. In addition, we showed that LPP3 silencing significantly increased the expression of adhesion molecules, which correlated with an increased human blood leucocytes adhesion to HAEC monolayers. In the future work we plan to use the specific blocking antibodies directed against adhesion molecules to ascertain that LPP3 knockdown is indeed involved in leucocyte adhesion via these molecules. Interestingly, it was shown previously in HUVECs that a high level of S1P induces the expression of SELE and VCAM1 via NFκB.27 Moreover, in epithelial cells, LPP3 down-regulates the inflammatory response by degrading S1P and LPA and thus inhibiting mitogen activated protein kinase and NFκB activation,28–30 and the production of IL6,31 IL832, and IL1β.30 Of note, several genes involved in the I-κB kinase/NF-κB pathway were up-regulated in LPP3-silenced HAECs (see Supplementary material online, Table S2), which is in accordance with a LPP3 protective role in inflammation, although in some settings in vivo NFκB readily contributed to the resolution of inflammation.33 Recently, it was reported that targeted inactivation of Ppap2b in mice increases inflammation and vascular permeability induced by LPS mainly due to the accumulation of LPA.7 Altogether, these results confirm that LPP3 has a key role in inflammation and endothelial adhesiveness for leucocytes.
4.2. Differential co-expression of PPAP2B with proinflammatory genes stratified by PPAP2B genotype in HAECs
In our study, a comparative analysis, in a collection of HAECs from 147 donors (119 AA homozygotes and 28 AG heterozygotes)3 of the two genotypes of rs17114036 showed a strong correlation of the expression level of PPAP2B with the inflammatory genes (IL6, IL8, PLA2G4A, and PTGS2) when the protective minor allele G was present. However, in AA homozygotes, there was no correlation. During endothelial dysfunction, there is an increase of VEGF expression concomitant with the increase of these pro-inflammatory mediators and adhesion molecules. Therefore an increased LPP3 expression could be a defensive mechanism tending to reduce the expression of proinflammatory genes. The lack of such correlation in the AA homozygotes might therefore be explained by the fact that the expression of LPP3 is too low in these cells to counteract the inflammatory response.
4.3. LPP3 is involved in HAECs’ survival
In the present study, LPP3 overexpression strongly decreased HAECs’ survival by inducing apoptosis and decreased their proliferation rate. Several studies have shown that degradation of LPA and S1P by LPP334 alters both activation of ERK1/2 and caspases 3/7,35 indeed, in the present work we showed that in HAECs overexpressing hLPP3, the caspases 3/7 were activated and accompanied by an increased annexin V binding to apoptotic cells, thus pointing to the LPP3 pro apoptotic potential. In parallel, LPP3 knockdown increased ERK 1/2 phosphorylation favouring cell survival. This is consistent with the previous study showing that S1P inhibits the activation of caspases 3/7-dependent apoptotic pathway downstream of ceramide,36 a pathway called the ‘sphingolipid rheostat’ that governs the cell fate leading either to survival or to apoptosis.37
Here, we demonstrated that the loss of either catalytic or adhesion function of LPP3 decreased to the similar extent the number of viable cells. Furthermore, both LPA supply and S1P receptor inhibitors blocked cell survival induced by LPP3 silencing, consistent with the previous studies showing that LPP3 inhibits survival via degradation of its lipid substrates. However, when hLPP3 was overexpressed, neither LPA mimic nor S1P was able to reverse the inhibition of cell survival, indicating that the RGD domain was involved. Although, the precise mechanism remains to be defined, we suggest that LPP3 may regulate cell survival via both catalytic and RGD domains.
4.4. S1P plays a major role in HAECs’ migration and angiogenesis
We showed that LPP3 down-regulates cell migration as measured by wound healing assay. Since the S1P receptor inhibitor blocked siLPP3-induced migration and conversely, the addition of the S1P nearly reversed the inhibition of migration mediated by LPP3 overexpression, we conclude that the anti-migratory effect of LPP3 in HAECs depends solely on the degradation of S1P. Therefore the role of S1P in HAECs is consistent with the former studies which demonstrated that S1P stimulates migration through its receptor S1PR1 (previously endothelial differentiation gene 1) in human umbilical endothelial and SMCs.38,39 Indeed, S1PR1 was up-regulated under LPP3 knockdown conditions in our transcriptomic data (see Supplementary material online, Table S1; line 313). In contrast, Panchatcharam et al.6 showed that LPP3 is involved in SMC migration in mice through degradation of LPA; this discrepancy could be due to different cells and different species.
LPP3 is a key element in cell–cell interactions due to its RGD motif which interacts with integrins,40 and it allows initiation of angiogenesis either by basic fibroblast growth factor (bFGF) or VEGF.25 Our work demonstrated that LPP3 tightly regulates the number of tubes and branching point structures, consistent with its pro-angiogenic role. We showed that both the catalytic and RGD domains were necessary to sustain angiogenesis. When LPP3 was silenced, inhibiting S1P receptor, but not LPA formation, tended to restore the angiogenesis. These results suggest that LPP3 regulates angiogenesis possibly by controlling the availability of S1P.
We showed that although the LPP3 silencing in HAECs increased the expression of the pro-angiogenic factor VEGF, their angiogenic response was strongly decreased, possibly because of the excessive accumulation of S1P and the up-regulation of S1PR1 expression, as shown in our transcriptomic data (see Supplementary material online, Table S1). However, the loss of S1pr1 gene in mice is lethal in utero due to a defect in vessel maturation,41 indicating that S1P is indispensable for vessel stabilization termination of angiogenesis.42 Indeed, there is a gradual expression of S1PR1 from the mature sprouting region to the growing vascular front during blood vessel formation.43 In this process S1P activates S1PR1 which in turn inhibits VEGF signalling and suppresses sprouting, promotes junction formation, and recruitment of mural cells to prevents excessive sprouting.43 Our results on angiogenesis in HAECs support these studies, as LPP3 silencing prevented angiogenesis, because the S1P level was likely too high to initiate sprouting. In contrast, when hLPP3 was overexpressed, sprouting was favoured.
4.5. Summary and limitations of our study
To summarize (see Supplementary material online, Figure S10), our transcriptomic and functional studies using primary HAECs from 3 to 13 different donors show that LPP3 down-regulates both inflammation and leucocyte adhesion by reducing expression of cytokines and adhesion molecules in these cells. LPP3 also down-regulates cell growth by degrading both S1P and LPA. LPP3 inhibits cell migration by controlling the levels of S1P and the expression of its receptor S1PR1. We finally showed that LPP3 may degrade intra- and extra-cellular S1P in HAECs, the latter result due to the limited number of samples (n = 3) should be confirmed in the future on a larger number of HAECs donors. In contrast to S1P, we were unable to detect LPA in the supernatants of HAECs; however we cannot exclude an intracellular role for LPA. Indeed, in HAECs silenced for LPP3 addition of an inhibitor of LPA decreased cell survival to the similar extent as the antagonist of S1P receptors.
The main limitation of our study was the access to a large collection of cultivable HAECs with different genotypes in respect to the coronary artery disease PPAP2B leading SNP rs171140363 permitting the functional studies which could reveal the difference between those genotypes. In the future, such an approach will shed a light on the transcriptional control of LPP3 and open the way to novel pharmacological interventions to combat endothelial dysfunction.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Acknowledgements
We are grateful to Prof. F. Koskas (Vascular Surgery Department, Groupe Hospitalier Pitié-Salpétrière, Paris) for the human vessel arteriosclerotic samples, A. Lessot and Dr F. Charlotte (Pathology Department Groupe Hospitalier Pitié-Salpêtrière, Paris) and Prof. I. Brochériou (Hôpital Tenon, Paris) for tissue microarrays preparation, immunohistochemistry staining, and interpretation. We thank: (1) the Post-genomics Platform P3S Paris, France for the microarrays, QPCR data acquisition, and western blot scanning; (2) the Flow Cytometry Core CyPS, Paris, France for the apoptotic cell analysis, and (3) Prof. O. Quehenberger from the Lipid Maps Lipidomics Core/University of California San Diego, La Jolla, CA, USA for LPA analysis. Dr V. Baecker, ImageJ User and Developer Conference 2012. Luxembourg: Centre de Recherche Public Henri Tudor; 2012; ISBN: 2-919941-18-6.
Conflict of interest: none declared.
Funding
This work was supported by the Institut National de la Santé et de la Recherche Médicale and the Transatlantic Networks of Excellence, Fondation Leducq (12CVD02) (France); National Institutes of Health (USA) grants (HL28481 and K99HL121172). E.N is Director of Research in Centre National de la Recherche Scientifique.
References
- 1.Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev 2013;93:1317–1542. [DOI] [PubMed] [Google Scholar]
- 2.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AFR, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Cardiogenics, Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJ, Khaw KT, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Mühleisen TW, Muhlestein JB, Münzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nöthen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schäfer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups K, Stoll M, Tang WH, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJ, Wells GA, Wichmann HE, Wild PS, Willenborg C, Witteman JC, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, März W, Hengstenberg C, Blankenberg S, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, O'Donnell CJ, McPherson R, Erdmann J, CARDIoGRAM Consortium, Samani NJ. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011;43:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erbilgin A, Civelek M, Romanoski CE, Pan C, Hagopian R, Berliner JA, Lusis AJ. Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res 2013;54:1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu C, Huang R-T, Kuo C-H, Kumar S, Kim CW, Lin Y-C, Chen Y-J, Birukova A, Birukov KG, Dulin NO, Civelek M, Lusis AJ, Loyer X, Tedgui A, Dai G, Jo H, Fang Y. Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ Res 2015;117:e41–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reschen ME, Gaulton KJ, Lin D, Soilleux EJ, Morris AJ, Smyth SS, O’Callaghan CA. Lipid-induced epigenomic changes in human macrophages identify a coronary artery disease-associated variant that regulates PPAP2B expression through altered C/EBP-beta binding. PLoS Genet 2015;11:e1005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panchatcharam M, Miriyala S, Salous A, Wheeler J, Dong A, Mueller P, Sunkara M, Escalante-Alcalde D, Morris AJ, Smyth SS. Lipid phosphate phosphatase 3 negatively regulates smooth muscle cell phenotypic modulation to limit intimal hyperplasia. Arterioscler Thromb Vasc Biol 2013;33:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panchatcharam M, Salous AK, Brandon J, Miriyala S, Wheeler J, Patil P, Sunkara M, Morris AJ, Escalante-Alcalde D, Smyth SS. Mice with targeted inactivation of ppap2b in endothelial and hematopoietic cells display enhanced vascular inflammation and permeability. Arterioscler Thromb Vasc Biol 2014;34:837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kai M, Wada I, Imai S i, Sakane F, Kanoh H. Cloning and characterization of two human isozymes of Mg2+-independent phosphatidic acid phosphatase. J Biol Chem 1997;272:24572–24578. [DOI] [PubMed] [Google Scholar]
- 9.Roberts R, Sciorra VA, Morris AJ. Human type 2 phosphatidic acid phosphohydrolases. Substrate specificity of the type 2a, 2b, and 2c enzymes and cell surface activity of the 2a isoform. J Biol Chem 1998;273:22059–22067. [DOI] [PubMed] [Google Scholar]
- 10.Escalante-Alcalde D, Morales SL, Stewart CL. Generation of a reporter-null allele of Ppap2b/Lpp3and its expression during embryogenesis. Int J Dev Biol 2009;53:139–147. [DOI] [PubMed] [Google Scholar]
- 11.Escalante-Alcalde D, Hernandez L, Le Stunff H, Maeda R, Lee H-S, Jr-Gang-Cheng, Sciorra VA, Daar I, Spiegel S, Morris AJ, Stewart CL. The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Dev Camb Engl 2003;130:4623–4637. [DOI] [PubMed] [Google Scholar]
- 12.Ren H, Panchatcharam M, Mueller P, Escalante-Alcalde D, Morris AJ, Smyth SS. Lipid phosphate phosphatase (LPP3) and vascular development. Biochim Biophys Acta BBA—Mol Cell Biol Lipids 2013;1831:126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brindley DN, English D, Pilquil C, Buri K, Ling ZC. Lipid phosphate phosphatases regulate signal transduction through glycerolipids and sphingolipids. Biochim Biophys Acta 2002;1582:33–44. [DOI] [PubMed] [Google Scholar]
- 14.Sciorra VA, Morris AJ. Roles for lipid phosphate phosphatases in regulation of cellular signaling. Biochim Biophys Acta 2002;1582:45–51. [DOI] [PubMed] [Google Scholar]
- 15.Brindley DN, Pilquil C. Lipid phosphate phosphatases and signaling. J Lipid Res 2009;50(Suppl):S225–S230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn EH, Schroeder JJ. Induction of apoptosis by sphingosine, sphinganine, and C(2)-ceramide in human colon cancer cells, but not by C(2)-dihydroceramide. Anticancer Res 2010;30:2881–2884. [PubMed] [Google Scholar]
- 17.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest 2009;119:1871–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y-L, Lin H-S, Chen S-U, Lee H. Tyrosine sulphation of sphingosine 1-phosphate 1 (S1P1) is required for S1P-mediated cell migration in primary cultures of human umbilical vein endothelial cells. J Biochem (Tokyo) 2009;146:815–820. [DOI] [PubMed] [Google Scholar]
- 19.Moriue T, Igarashi J, Yoneda K, Nakai K, Kosaka H, Kubota Y. Sphingosine 1-phosphate attenuates H2O2-induced apoptosis in endothelial cells. Biochem Biophys Res Commun 2008;368:852–857. [DOI] [PubMed] [Google Scholar]
- 20.Brindley DN, Lin F-T, Tigyi GJ. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim Biophys Acta 2013;1831:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samadi N, Bekele R, Capatos D, Venkatraman G, Sariahmetoglu M, Brindley DN. Regulation of lysophosphatidate signaling by autotaxin and lipid phosphate phosphatases with respect to tumor progression, angiogenesis, metastasis and chemo-resistance. Biochimie 2011;93:61–70. [DOI] [PubMed] [Google Scholar]
- 22.Jia Y-J, Kai M, Wada I, Sakane F, Kanoh H. Differential localization of lipid phosphate phosphatases 1 and 3 to cell surface subdomains in polarized MDCK cells. FEBS Lett 2003;552:240–246. [DOI] [PubMed] [Google Scholar]
- 23.Kai M, Wada I, Imai S, Sakane F, Kanoh H. Identification and cDNA cloning of 35-kDa phosphatidic acid phosphatase (type 2) bound to plasma membranes. Polymerase chain reaction amplification of mouse H2O2-inducible hic53 clone yielded the cDNA encoding phosphatidic acid phosphatase. J Biol Chem 1996;271:18931–18938. [DOI] [PubMed] [Google Scholar]
- 24.Romanoski CE, Lee S, Kim MJ, Ingram-Drake L, Plaisier CL, Yordanova R, Tilford C, Guan B, He A, Gargalovic PS, Kirchgessner TG, Berliner JA, Lusis AJ. Systems genetics analysis of gene-by-environment interactions in human cells. Am J Hum Genet 2010;86:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wary KK, Humtsoe JO. Anti-lipid phosphate phosphohydrolase-3 (LPP3) antibody inhibits bFGF- and VEGF-induced capillary morphogenesis of endothelial cells. Cell Commun Signal CCS 2005;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song Y, Wu J, Oyesanya RA, Lee Z, Mukherjee A, Fang X. Sp-1 and c-Myc mediate lysophosphatidic acid-induced expression of vascular endothelial growth factor in ovarian cancer cells via a hypoxia-inducible factor-1-independent mechanism. Clin Cancer Res Off J Am Assoc Cancer Res 2009;15:492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia P, Gamble JR, Rye KA, Wang L, Hii CS, Cockerill P, Khew-Goodall Y, Bert AG, Barter PJ, Vadas MA. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci USA 1998;95:14196–14201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saatian B, Zhao Y, He D, Georas SN, Watkins T, Spannhake EW, Natarajan V. Transcriptional regulation of lysophosphatidic acid-induced interleukin-8 expression and secretion by p38 MAPK and JNK in human bronchial epithelial cells. Biochem J 2006;393:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JGN, Watkins T, He D, Saatian B, Natarajan V. Protein kinase Cdelta mediates lysophosphatidic acid-induced NF-kappaB activation and interleukin-8 secretion in human bronchial epithelial cells. J Biol Chem 2004;279:41085–41094. [DOI] [PubMed] [Google Scholar]
- 30.Liu Q, Rehman H, Shi Y, Krishnasamy Y, Lemasters JJ, Smith CD, Zhong Z. Inhibition of sphingosine kinase-2 suppresses inflammation and attenuates graft injury after liver transplantation in rats. PloS One 2012;7:e41834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang W-C, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013;23:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Cummings R, Usatyuk P, Morris A, Irani K, Natarajan V. Involvement of phospholipases D1 and D2 in sphingosine 1-phosphate-induced ERK (extracellular-signal-regulated kinase) activation and interleukin-8 secretion in human bronchial epithelial cells. Biochem J 2002;367:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med 2001;7:1291–1297. [DOI] [PubMed] [Google Scholar]
- 34.Alderton F, Darroch P, Sambi B, McKie A, Ahmed IS, Pyne N, Pyne S. G-protein-coupled receptor stimulation of the p42/p44 mitogen-activated protein kinase pathway is attenuated by lipid phosphate phosphatases 1, 1a, and 2 in human embryonic kidney 293 cells. J Biol Chem 2001;276:13452–13460. [DOI] [PubMed] [Google Scholar]
- 35.Long J, Darroch P, Wan KF, Kong KC, Ktistakis N, Pyne NJ, Pyne S. Regulation of cell survival by lipid phosphate phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1-phosphate pools. Biochem J 2005;391:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuvillier O, Rosenthal DS, Smulson ME, Spiegel S. Sphingosine 1-phosphate inhibits activation of caspases that cleave poly(ADP-ribose) polymerase and lamins during Fas- and ceramide-mediated apoptosis in Jurkat T lymphocytes. J Biol Chem 1998;273:2910–2916. [DOI] [PubMed] [Google Scholar]
- 37.Van Brocklyn JR, Williams JB. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: oxidative stress and the seesaw of cell survival and death. Comp Biochem Physiol B Biochem Mol Biol 2012;163:26–36. [DOI] [PubMed] [Google Scholar]
- 38.Wang F, Van Brocklyn JR, Hobson JP, Movafagh S, Zukowska-Grojec Z, Milstien S, Spiegel S. Sphingosine 1-phosphate stimulates cell migration through a G(i)-coupled cell surface receptor. Potential involvement in angiogenesis. J Biol Chem 1999;274:35343–35350. [DOI] [PubMed] [Google Scholar]
- 39.Kluk MJ, Hla T. Role of the sphingosine 1-phosphate receptor EDG-1 in vascular smooth muscle cell proliferation and migration. Circ Res 2001;89:496–502. [DOI] [PubMed] [Google Scholar]
- 40.Humtsoe JO, Feng S, Thakker GD, Yang J, Hong J, Wary KK. Regulation of cell-cell interactions by phosphatidic acid phosphatase 2b/VCIP. EMBO J 2003;22:1539–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 2000;106:951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011;473:298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung B, Obinata H, Galvani S, Mendelson K, Ding B, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, Hla T. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell 2012;23:600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.