

Abstract
Pho84, a major facilitator superfamily (MFS) protein, is the main high-affinity Pi transceptor in Saccharomyces cerevisiae. Although transport mechanisms have been suggested for other MFS members, the key residues and molecular events driving transport by Pi:H+ symporters are unclear. The current Pho84 transport model is based on the inward-facing occluded crystal structure of the Pho84 homologue PiPT in the fungus Piriformospora indica. However, this model is limited by the lack of experimental data on the regulatory residues for each stage of the transport cycle. In this study, an open, inward-facing conformation of Pho84 was used to study the release of Pi. A comparison of this conformation with the model for Pi release in PiPT revealed that Tyr179 in Pho84 (Tyr150 in PiPT) is not part of the Pi binding site. This difference may be due to a lack of detailed information on the Pi release step in PiPT. Molecular dynamics simulations of Pho84 in which a residue adjacent to Tyr179, Asp178, is protonated revealed a conformational change in Pho84 from an open, inward-facing state to an occluded state. Tyr179 then became part of the binding site as was observed in the PiPT crystal structure. The importance of Tyr179 in regulating Pi release was supported by site-directed mutagenesis and transport assays. Using trehalase activity measurements, we demonstrated that the release of Pi is a critical step for transceptor signaling. Our results add to previous studies on PiPT, creating a more complete picture of the proton-coupled Pi transport cycle of a transceptor.
Keywords: cell signaling, membrane transport, molecular dynamics, Saccharomyces cerevisiae, yeast metabolism, Pho84, Phosphate transport, Transceptor
Introduction
The major facilitator superfamily (MFS)4 is one of the largest families of secondary active transporters (1). Currently, 64 crystal structures from 10 different families are available, andseveral of these structures were resolved in substrate-free and substrate-bound conformational states, therefore providing significant insights into the molecular mechanisms involved in nutrient transport (2). The key feature of MFS transporters is that transport across the membrane occurs via an alternating-access mechanism, the “rocker-switch” model (3), which involves a variety of distinct conformations including outward-open, substrate-bound occluded, and inward-open states. A limitation of the rocker-switch model, however, is that because it is based solely on rigid body rotation, it does not explain the presence of the occluded state. To account for this limitation, the “clamp-and-switch” model has recently been proposed (2). This model suggests that the conformational transition process is more dynamic than previously assumed and that the switch between conformations is driven by pore-lining helices that bend to form the occluded state and that allow for outward-facing to inward-facing rotations and vice versa.
Inorganic phosphate (Pi) is required for numerous cellular functions, such as synthesis of DNA and membrane lipids, intracellular signaling, and the generation of high-energy phosphate esters in ATP. Because of the central role of Pi, cells have adopted strategies to ensure rapid cellular responses to internal and external fluctuations in Pi levels that may disturb cellular phosphate homeostasis. Moreover, several Pi transporters have been shown to transport Pi by H+-coupled symport (4).
Currently, the only available crystal structure of a Pi:H+ transporter is the eukaryotic Piriformospora indica transporter PiPT, which was resolved in the substrate-bound inward-facing occluded conformation (5). The current model for Pi:H+-transport is based on this crystal structure and describes three major conformational variations (i.e. substrate-docking, binding, and release) (5). A weakness of this model is that it lacks experimental data on the regulatory residues in each stage of the transport cycle.
The Saccharomyces cerevisiae Pho84 high-affinity phosphate transporter is a member of the phosphate:H+ symporter family (2.A.1.9) and is the main transporter during Pi-limiting conditions. Furthermore, Pho84 activates the protein kinase A (PKA) pathway, which senses and signals the uptake of external Pi in phosphate-starved cells. This dual functionality of Pho84 has led to its classification as a transceptor (6). A three-dimensional model of Pho84 in the open inward-facing conformation has been created by homology modeling using the glycerol 3-phosphate transporter GlpT as a template (7). This model has been used to verify the roles of key residues in Pi binding, transport, and signaling (8). A comparison of the Pho84 and PiPT structures revealed a series of conserved residues in the Pi binding site (5). Interestingly, in contrast to Tyr150 in PiPT, Tyr179 in Pho84 is not in the binding site of the open inward-facing conformation. This difference may result from protonation/deprotonation of the nearby Asp178 residue (Asp149 in PiPT), which may affect the active conformation of the transporter. In addition, Asp178 has been shown to participate in H+-transfer (8). Nevertheless, the protonation/deprotonation mechanism of regulating conformational changes in Pi:H+ transporters remains putative.
In this study, we examined the role of Tyr179 on the Pi release step in the open inward-facing conformation of Pho84. To address the finer details of the release mechanism and to build on the transport model previously suggested for PiPT, we performed a series of unrestrained molecular dynamics (MD) and steered molecular dynamics (SMD) simulations using different protonation states of Pi and Asp178.
These simulations revealed that the protonation state of Asp178 alters the conformational state of Pho84. Upon protonation of Asp178, Tyr179 underwent a rotameric change to become part of the binding site. This agrees with the contact found between Pi and Tyr150 in the PiPT occluded inward-facing conformation. Based on SMD simulations, different Pi release routes were suggested. The lowest-energy release route was found with H2PO4− and deprotonated Asp178.
We also confirmed the importance of Tyr179 in regulating Pi transport by a series of site-directed mutagenesis studies and biochemical assays. Finally, we measured trehalase activity to determine whether Tyr179 regulated PKA signaling, and we demonstrated that the release of Pi is critical for signaling. Altogether, our data contributes to a more complete picture of the dual functions of phosphate transceptors.
Results and Discussion
Tyr179 Is Crucial for the Substrate Release Step of Pi Transport
The two-dimensional topology of Pho84 consists of a C-domain and an N-domain, each domain is made up of six transmembrane segments (Table 1), and both the N and C termini are oriented toward the cytoplasm. The three-dimensional in silico model displays a Mayan temple shape (3) (Fig. 1A) with a distinct N- and C-terminal domain organization and a clearly visible transport channel in the center of the protein (Fig. 1B). The transport channel is a structural feature common in MFS proteins.
TABLE 1.
Amino acid sequences of transmembrane (TM) domains, loops (L), and the N and C termini according to Lagerstedt and co-workers (7)
| Amino acid sequence (the position is given in superscript) | |
|---|---|
| Transmembrane domain | |
| TM-I | Asp55-Glu-Gly-Phe-Gly-Trp-Gln-Gln-Val-Lys-Thr-Ile-Ser-Ile-Ala-Gly-Val-Gly-Phe-Leu-Thr-Asp-Ser-Tyr-Asp-Ile-Phe-Ala-Ile-Asn-Leu-Gly-Ile87 |
| TM-II | Gln104-Thr-Leu-Leu-Lys-Val-Ser-Thr-Ser-Val-Gly-Thr-Val-Ile-Gly-Gln-Phe-Gly-Phe-Gly-Thr-Leu-Ala126 |
| TM-III | Gly130-Arg-Lys-Arg-Ile-Tyr-Gly-Met-Glu-Leu-Ile-Ile-Met-Ile-Val-Cys-Thr-Ile-Leu-Gln-Thr-Thr151 |
| TM-IV | Val163-Leu-Thr-Phe-Tyr-Arg-Ile-Val-Met-Gly-Ile-Gly-Ile-Gly-Gly-Asp-Tyr-Pro-Leu-Ser-Ser-Ile-Ile-Thr186 |
| TM-V | Ala201-Val-Phe-Ala-Asn-Gln-Ala-Trp-Gly-Gln-Ile-Ser-Gly-Gly-Ile-Ile-Ala-Leu-Ile-Leu220 |
| TM-VI | Gly254-Leu-Gly-Thr-Val-Leu-Gly-Leu-Ala-Cys-Leu-Tyr-Phe-Arg-Leu-Thr-Ile-Pro-Glu-Ser-Pro-Arg-Tyr-Gln-Leu278 |
| TM-VII | Leu347-Leu-Gly-Thr-Ala-Gly-Ser-Trp-Phe-Thr-Leu-Asp-Val-Ala-Phe-Tyr-Gly-Leu-Ser-Leu-Asn-Ser-Ala-Val-Ile371 |
| TM-VIII | Tyr388-Asp-Thr-Ala-Val-Gly-Asn-Leu-Ile-Leu-Ile-Cys-Ala-Gly-Ser-Leu-Pro-Gly-Tyr-Trp-Val-Ser-Val-Phe-Thr-Val-Asp-Ile-Ile416 |
| TM-IX | Gln422-Leu-Ala-Gly-Phe-Ile-Ile-Leu-Thr-Ala-Leu-Phe-Cys-Val-Ile-Gly-Phe-Ala-Tyr440 |
| TM-X | Gly447-Leu-Leu-Ala-Leu-Tyr-Val-Ile-Cys-Gln-Phe-Phe-Gln-Asn-Phe-Gly-Pro-Asn-Thr-Thr-Thr-Phe-Ile-Val-Pro-Gly-Glu473 |
| TM-XI | Tyr479-Arg-Ser-Thr-Ala-His-Gly-Ile-Ser-Ala-Ala-Ser-Gly-Lys-Val-Gly-Ala-Ile-Ile-Ala-Gln-Thr-Ala-Leu-Gly-Thr504 |
| TM-XII | Met525-Glu-Ile-Phe-Ala-Leu-Phe-Met-Leu-Leu-Gly-Ile-Phe-Thr-Thr-Leu-Leu-Ile-Pro-Glu-Thr-Lys-Arg-Lys-Thr-Leu-Glu551 |
| Loop | |
| L-I | Thr88-Met-Met-Ser-Tyr-Val-Tyr-Trp-His-Gly-Ser-Met-Pro-Gly-Pro-Ser103 |
| L-II | Asp127-Ile-Val129 |
| L-III | Val152-Ala-His-Ser-Pro-Ala-Ile-Asn-Phe-Val-Ala162 |
| L-IV | Ser187-Glu-Phe-Ala-Thr-Thr-Lys-Trp-Arg-Gly-Ala-Ile-Met-Gly200 |
| L-V | Val221-Ala-Ala-Tyr-Lys-Gly-Glu-Leu-Glu-Tyr-Ala-Asn-Ser-Gly-Ala-Glu-Cys-Asp-Ala-Arg-Cys-Gln-Lys-Ala-Cys-Asp-Gln-Met-Trp-Arg-Ile-Leu-Ile253 |
| L-VI | Asp279-Val-Asn-Ala-Lys-Leu-Glu-Leu-Ala-Ala-Ala-Ala-Gln-Glu-Gln-Asp-Gly-Glu-Lys-Lys-Ile-His-Asp-Thr-Ser-Asp-Glu-Asp-Met-Ala-Ile-Asn-Gly-Leu-Glu-Arg-Ala-Ser-Thr-Ala-Val-Glu-Ser-Leu-Asp-Asn-His-Pro-Pro-Lys-Ala-Ser-Phe-Lys-Asp-Phe-Cys-Arg-His-Phe-Gly-Gln-Trp-Lys-Tyr-Gly-Lys-Ile346 |
| L-VII | Leu372-Gln-Thr-Ile-Gly-Tyr-Ala-Gly-Ser-Lys-Asn-Val-Tyr-Lys-Lys-Leu387 |
| L-VIII | Gly417-Arg-Lys-Pro-Ile421 |
| L-IX | His441-Lys-Leu-Gly-Asp-His446 |
| L-X | Cys474-Phe-Pro-Thr-Arg478 |
| L-XI | Leu505-Ile-Asp-His-Asn-Cys-Ala-Arg-Asp-Gly-Lys-Pro-Thr-Asn-Cys-Trp-Leu-Pro-His-Val524 |
| Termini | |
| N-terminal | Met1-Ser-Ser-Val-Asn-Lys-Asp-Thr-Ile-His-Val-Ala-Glu-Arg-Ser-Leu-His-Lys-Glu-His-Leu-Thr-Glu-Gly-Gly-Asn-Met-Ala-Phe-His-Asn-His-Leu-Asn-Asp-Phe-Ala-His-Ile-Glu-Asp-Pro-Leu-Glu-Arg-Arg-Arg-Leu-Ala-Leu-Glu-Ser-Ile-Asp54 |
| C-terminal | Glu552-Ile-Asn-Glu-Leu-Tyr-His-Asp-Glu-Ile-Asp-Pro-Ala-Thr-Leu-Asn-Phe-Arg-Asn-Lys-Asn-Asn-Asp-Ile-Glu-Ser-Ser-Ser-Pro-Ser-Gln-Leu-Gln-His-Glu-Ala587 |
FIGURE 1.

The N-domain (transmembrane helices I-VI) and C-domain (transmembrane helices VII-XII) of Pho84 are shown in blue and orange, respectively. A, frontal view of the Pho84 model. B, periplasmic views of the three-dimensional Pho84 model. All of the loops and non-transmembrane domains are shown in gray.
A multiple sequence alignment (MSA) analysis of Pho84 with other Pi:H+ transporters revealed that Tyr179 is highly conserved (Fig. 2). Furthermore, the corresponding Tyr150 is proposed to be in the Pi binding site of the PiPT crystal structure. In the inward-open conformation of the Pho84 model, Tyr179 orients toward the cytoplasm and points away from the binding cavity. A growth spot test with strains expressing mutant alleles of PHO84 was performed to assess changes in the transport activity of Pho84 (Fig. 3). When compared with the wild-type strain (CEN.PK 113–7D PHO84-6xHis-2xmyc), the strains expressing Pho84 Tyr179-Ala and Tyr179-Gly exhibited drastic reductions in growth in low-Pi (LPi) conditions indicating that Tyr179 is crucial for transport. The growth of the strains expressing Pho84 Tyr179-Ser and Tyr179-Phe was equivalent to the growth of the Pho84 wild-type strain. In addition, in vivo radiolabeled Pi uptake assays showed a drastic reduction in transport activity in the Tyr179-Ala mutant and abolished activity in the Tyr179-Gly mutant (Fig. 4A). The Tyr179-Ser and Tyr179-Phe mutant strains had uptake activities equivalent to the uptake activity in the Pho84 wild-type strain. Immunoblot analysis confirmed that the site-directed mutations yielded full-length recombinant proteins (Fig. 4B).
FIGURE 2.

A section of a multiple amino acid sequence alignment of the S. cerevisiae Pho84 protein (PM0076296) with homologues from P. indica (A8N031), A. thaliana Pht1–1 (Q8VYM2), A. thaliana Pht 1–2 (Q96243), O. sativa (Q8GSD9), H. vulgare (Q8H6E0), M. truncatula (Q8GSG4), G. intraradices (Q96VN6), G. versiforme (Q00908), and P. nameko (Q96X52). The blue line indicates the part of the sequence motif that is shared among proton-coupled phosphate transporters in plants, fungi, bacteria, and mammals (TLCFFRFWLGFGIGGDYPLSATIMSE) (43). This signature sequence contains the phosphate binding sequence GXGXGG. The residue Tyr179 of the Pho84 transporter is indicated by an inverted triangle. Created with CLC workbench 7 (Qiagen, Aarhus, Denmark).
FIGURE 3.

Yeast growth spot test images were taken after 2 days of growth under HPi conditions and after 3 days of growth under LPi conditions. To visualize the reduced growth of the pho84Δ and mutant strains under LPi conditions, images were acquired with a higher illumination setting resulting in a brighter background. All of the strains were dilution-plated as indicated.
FIGURE 4.

A, total transport activity was measured using radioactive phosphate in a short-term uptake assay with a saturating phosphate concentration (110 μm KH2PO4). Cells were grown in LPi conditions. Experiments were performed in triplicate. Data are expressed as percentages of WT activity (14.658 nmol min−1 mg−1 cells, dry weight set as 100%), mean ± S.D. **, p < 0.05, significantly different (Student's t test). B, immunoblot with anti-c-Myc antibody to detect membrane enrichment. CEN.PK 113 7D strain was used as the (+) control, the CEN.PK 5D PHO84Δ was used as the (−) control. The band for the recombinant Pho84–6xHis-2Myc (apparent molecular weight of 66 kDa) is indicated with a square bracket. C, loading controls using complete yeast extract (10 μg) and anti-β-actin. The band for β-actin is indicated with a square bracket.
Furthermore, expression of a functional Pho84 transporter is required to suppress the repressible acid phosphatase (rAPase) activity of PHO5, the major acidic phosphatase (9). A loss-of-function strain, pho84Δ, results in constitutive expression of PHO5 independent of the external Pi conditions (Fig. 5). The wild-type strain was analyzed under either high-Pi (HPi) or low-Pi (LPi) conditions (8) revealing reduced and elevated secreted phosphatase activity, respectively. Strains expressing Pho84 Tyr179-Ala and Tyr179-Gly exhibited pronounced increases in secreted phosphatase activity, which is consistent with the significantly reduced transport activities of these mutants. Strains expressing Pho84 Tyr179-Ser and Tyr179-Phe had secreted phosphatase activities similar to the wild-type level. These results show that full-length expressed, plasma membrane localized, non-functional Pho84 suppresses rAPase activity resulting in less activity relative to the activity in pho84Δ (9).
FIGURE 5.

Acid phosphatase (rAPase) activity assays of strains expressing mutant forms of Pho84 grown under HPi (closed bars) and LPi (open bars) conditions. All measurements were performed in duplicate with three technical repetitions each and the results are shown as mean ± S.D.
Our data for Tyr179 clearly shows that this residue plays a crucial role in Pi transport. By introducing a side chain that is relatively inert, such as a residue that is non-charged, weakly polar, or lacking an aromatic residue, transport activity was largely abolished. In contrast, by introducing side chains with increased polarity (Tyr179-Ser) or hydrophobicity (Tyr179-Phe), transport activity was restored to the wild-type level.
Protonation/Deprotonation of Asp178 and Its Impact on Pi Release
A protonation/deprotonation cycle has been shown to be crucial for the functionality of many transporters. The Escherichia coli lactose permease LacY serves as a paradigm for understanding the importance of protonation/deprotonation in the transport cycle that begins in the outward-open conformation and requires functional protonation to facilitate substrate binding (10). MD simulations of the protonation/deprotonation of Glu325 showed significant changes in the structure leading to a transition from the inward-facing to the occluded conformation (11). MD simulations of the E. coli sugar fucose:H+ symporter FucP also showed that protonation of Glu135 is required to trigger conversion from the outward-open to the inward-open conformation (12). These examples show that protonation/deprotonation events are crucial to the alternating-access dynamics of MFS transporters.
In a previous site-directed mutagenesis study, the Pho84 residue Asp178 was mutated to Glu and Asn, and Asn was found to mimic a protonated aspartate (8). These results led to the hypothesis that protonation of Asp178 plays a role in a late phosphate release step of the transport cycle. To investigate this hypothesis, a series of unrestrained MD simulations were performed on Pho84 models with protonated or deprotonated Asp178. Pi was inserted in the Pho84 binding site at approximately equal distances from Lys492 and Asp178. This allowed us to investigate the roles of these residues on Pi binding and on proton transfer. In addition, simulations were conducted using different protonation states of Pi (H3PO4, H2PO4−, and HPO42−) to resolve the role of charge balance in the binding site and its influence on the energetics of cytosolic release of Pi (see supplemental Table S1 for information on the number of molecules in each system and the denotations that are used throughout this paper). All of the systems were subsequently subjected to 30 ns of equilibration to reach stable energy, temperature, and density values (supplemental Figs. S1–S16). Equilibration of the transmembrane helices (Fig. 1) in the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer were verified by recording root mean square deviation values over time for backbone (N, Cα, and C) atoms. These values reached a plateau after ∼15 ns with values of 3.5 Å or less (supplemental Fig. S17).
Extracted snapshots of the Pho84 binding site after 30 ns of equilibration revealed the position of Pi in the binding site, the location of residues important for Pi binding, and the location of proton transfer (Fig. 6). Analysis of the position of H3PO4 and its variations in distance over time (Fig. 6, A1, and supplemental Fig. S18) revealed stable interactions between H3PO4, Lys492, and Asp178. Models using H2PO4− or HPO42− revealed weakened interactions with Asp178 due to repulsion between mono- and divalent species of Pi and the deprotonated state of Asp178 (Fig. 6, B1 and C1, and supplemental Fig. S18). Moreover, systems with either H2PO4− or HPO42− in the binding site and a protonated state of Asp178 (Fig. 6, B2 and C2, supplemental Figs. S18 and S19) revealed the formation of a hydrogen bond between Asp178 and the adjacent Asp76 residue resulting in less distance between helix I that harbors Asp76 and helix IV that harbors Asp178. Similar interactions were seen in the absence of Pi indicating that the proton is not donated by Pi (supplemental Fig. S19). These data confirm the role of Asp178 in proton-coupling (8). In addition, it has been suggested that the symport of protons through Pho84 occurs at a Pi:H+ stoichiometry of 1:3 (13).
FIGURE 6.

Snapshots from simulations of various protonation states of Pi and Asp178 after 30 ns of unrestrained equilibration. A1, H3PO4; A2, H3PO4 and protonated Asp178; B1, H2PO4−; B2, H2PO4− and protonated Asp178; C1, HPO42−; C2, HPO42− and protonated Asp178. Selected residues are shown. Lys492 and Tyr179 are proposed to be involved in Pi binding (van der Waals representation). Asp178, Asp76, and Asp79 are proposed to be involved in proton shuffling. Arg267 is proposed to form a salt bridge with Asp76, and Arg168 is proposed to form a salt bridge with Asp79.
The largest shift in the distance between Asp178 and Asp76 upon protonation of Asp178 was observed for H2PO4− rather than for HPO42− suggesting that H2PO4− has the largest impact on the conformational flexibility of Pho84. Simulations with H3PO4 in the binding site and protonated Asp178 resulted in a helical rotation that affected helices I and IV such that Asp76, rather than Asp178, bound to Pi (Fig. 6, A2, and supplemental Fig. S18). These data demonstrate that the charge distribution in the Pho84 binding site regulates the active conformational state of Pho84.
In addition to the observed roles of Asp178 in regulating Pi binding and proton-coupling, the change in the conformational state of helix IV in Pho84 that occurred upon protonation of Asp178 led to Tyr179 becoming more accessible for Pi binding (Fig. 6, B2 and C2, and supplemental Fig. S20). X-ray crystallographic analysis of PiPT in the substrate-bound inward-facing occluded conformation shows that the corresponding tyrosine (Tyr150) is involved in Pi binding (5). As shown in a comparison of Fig. 6, panels A1/A2 versus panels B1/B2/C1/C2, the shift in the position of Tyr179 as correlated to the protonation state of Asp178 was not as prominent for models that incorporated H3PO4.
We investigated the stability of salt bridges involving Asp76 and Asp79 and their link to Pho84 conformational changes (Fig. 6). We found stable contacts between Asp76-Arg267 and Asp79-Arg168 in all of the simulations with the exception of the model incorporating H3PO4 and protonated Asp178 in which Asp178 and Asp76 exchanged positions (Fig. 6, A2). Moreover, a Pho84 simulation without Pi in the binding site showed a disruption of the Asp79-Arg168 salt bridge after 15 ns of equilibration. Because protonation of Asp178 led to a stable Asp79-Arg168 salt bridge, we suggest that the presence of Pi in the binding site may increase the conformational stability of Pho84 (data not shown).
Characterizing Alternative Release Routes Using SMD Simulations
A series of SMD simulations were performed to investigate alternative Pi release routes in Pho84. For each model investigated, five separate SMD simulations were performed to sample the configurational space provided by the flexible Lys492 residue and to identify possible Pho84 exit routes using various protonation states of both Asp178 and Pi. Although, according to the Jarzynski equality method that requires a large number of samplings to be performed (14–16), five simulations for each system is not enough to estimate the free energies of the possible release pathways, we used the simulations to qualitatively, rather than quantitatively, describe the alternative release routes and their corresponding forces and energies.
Calculation of force profiles (supplemental Figs. S21–S22), work (supplemental Figs. S23–S24), as well as hydrogen bond contacts between residues and Pi (supplemental Tables S2–S5 and Figs. S25–S28) and water molecules and Pi (supplemental Fig. S29 –S30) along the series of routes populated allowed for a deeper understanding of the molecular basis of the regulation of Pi release in Pho84. The steered MD simulations suggested that the lowest-energy release route involved H2PO4− and deprotonated Asp178 (supplemental Figs. S23–S34). SMD (Fig. 7) and unrestrained simulations demonstrated that protonation of Asp178 resulted in a rotation of helix IV, which increased the work required to release Pi via this route. The increased work required could be a result of the increase in hydrogen bond contacts and the number of water molecules encountered by H2PO4− when Asp178 is protonated. A 30-ns unrestrained simulation showed that rotation of Tyr179 into the binding site made it more accessible for Pi binding. This increased binding between Pi and Tyr179 upon protonation of Asp178 indicated that Pho84 regulates its conformational state preceding Pi release. The lowest-energy route for the release of H2PO4− from Pho84 involves helices IV and XI and a minimum number of contacts with residues in L-VI. Upon protonation of Asp178, however, the transport channel of Pho84 narrowed as a result of increased contacts with Gln119 in helix II and increased contacts with L-VI residues. In this protonated Pho84, H2PO4− must be released via a higher energy route (Fig. 7). A detailed analysis of the number of hydrogen bond contacts formed between H2PO4− and water along the release route (Fig. 7) demonstrated a reduction in hydrogen bonding between Pi and water. This reduction resulted from increased hydrogen bonding to Tyr179 in the binding site upon protonation of Asp178; these data support the observation that the release route narrowed. In addition, H3PO4 was found to contact Gln119 in helix II and to make contacts with L-VI residues suggesting that this alternate high-energy release route is used for H3PO4 as well as for H2PO4−. SMD simulations of the cytosolic release of HPO42− showed that this protonation state of Pi binds more strongly to Pho84 than any of the other investigated protonation states, thus charge balance in the transport channel appears to regulate Pi release routes (supplemental Figs. S22, S28, S30, and S34 and Table S5).
FIGURE 7.
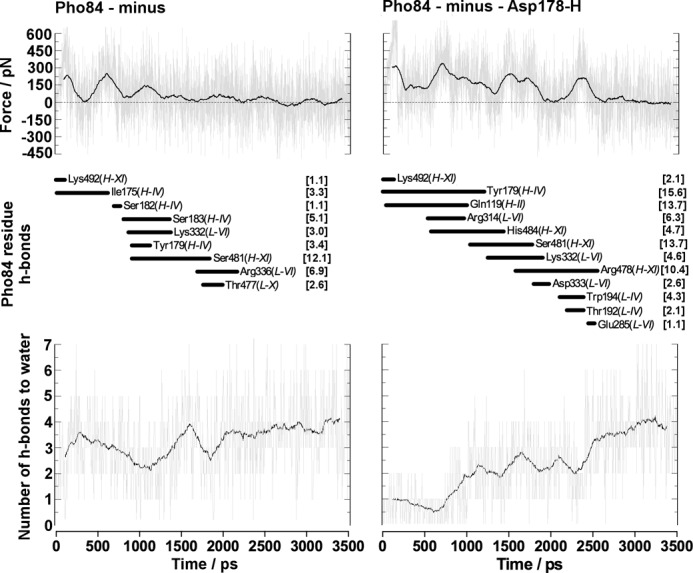
Representative data from steered dynamics simulations for the release of Pi from the Pho84 binding site after initially restraining the Lys492-Pi distance for 0.1 ns. The calculated force profiles and the Pho84 residues (and their transmembrane helix) that form hydrogen bonds with Pi along the release pathway or the average number of hydrogen bonds formed with water along the route for models involving deprotonated and protonated Asp178. Numbers in brackets represent hydrogen bonding between the residue and H2PO4− as a percentage of the total simulation time.
The Proposed Mechanism for Pi Release in Pi:H+ Transporters Using Pho84 as a Model
Results from unrestrained and steered MD simulations showed that the protonation states of Pi and Asp178 and the resulting orientations of Tyr179 impact regulation of the Pi release pathway. Protonation of Asp178 is a crucial step in triggering the conformational change of Pho84 from an open inward-facing conformation to a more occluded conformation. However, this switch in conformation appears to be independent of the protonation state of Pi. As a result of Pho84 adopting the occluded conformation, Tyr179 becomes part of the binding site as seen in the PiPT crystal structure.
Based on our observations, we propose a sequential mechanism for the release of Pi (Fig. 8).
FIGURE 8.
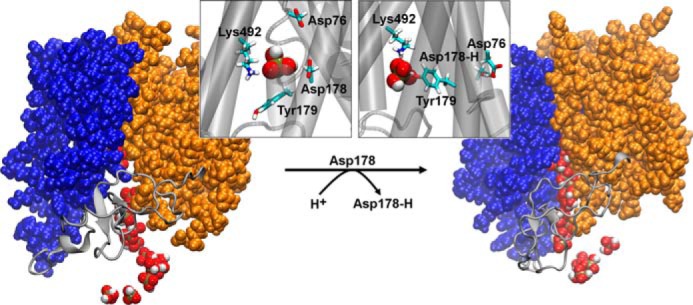
A proposed mechanism for the release step in Pi:H+ transporters using Pho84 as a model system. Snapshots representing the inward-facing open (image on the left) and occluded (image on the right) conformational states of Pho84 after simulating protonated (Asp178-H) or deprotonated states (Asp178) of Asp178. Detailed views of the binding site (helices are represented as gray cylinders) in both conformational states of Pho84 clearly show H2PO4− in the binding site and residue Tyr179 either oriented away from the binding site (Asp178) or toward the binding site (Asp178-H). The structures shown were obtained after 3.5 ns of steered MD simulations and multiple superimposed structures of Pi (van der Waals representation, extracted every 0.175 ns). Moreover, the N (transmembrane helices I-VI)- and C (transmembrane helices VII-XII)-domains of Pho84 are shown as blue and orange van der Waals representations, respectively. The connecting loop between these domains, L-VI, is shown as a gray schematic representation.
(i) The open inward-facing conformation of Pho84 containing deprotonated Asp178 is the active form that releases Pi. We found that H2PO4− is the most favorable protonation state of Pi to be released due to the distribution of the charges of the residues along the release pathway. In this active conformation of Pho84, Tyr179 points away from the binding site and may regulate the possible H2PO4− release routes and/or may act as a gatekeeper, which blocks more hydrophobic phosphate substrates.
(ii) Protonation of Asp178 results in a conformational change from an open inward-facing conformation to an occluded conformation in which Tyr179 is reoriented into the binding site and Pho84 is locked into a closed and inactive conformation, which is unable to release H2PO4−.
Transceptor Function Is Coupled to the Transport Cycle
In addition to its well known transport function, the Pho84 transceptor has the unique property of PKA signaling (17, 18). Previous point mutation studies have shown that these functions can be uncoupled resulting in a non-transporting protein that is still able to activate PKA upon phosphate detection (8). These studies focused on mutating residues that bind Pi or that could bind phosphate analogues that trigger PKA activation. In this study, we addressed whether limited release of Pi from the Pho84 binding site influenced the ability of Pho84 to activate PKA (Fig. 9). Because transport activity data clearly showed that Tyr179 is crucial for Pho84 functionality, trehalase activity assays were performed with relevant mutants.
FIGURE 9.

Trehalase activation after addition of 1 mm phosphate to phosphate-starved cells. pho84Δ (closed circles), wild-type (closed squares), Tyr179-Ala (closed triangles), Tyr179-Gly (inverted triangles), Tyr179-Ser (closed diamond shape), and Tyr179-Phe (open circles) are shown.
The ability of the Tyr179-Ala mutant strain to activate PKA was slightly reduced, whereas the ability of the Tyr179-Gly mutant strain to activate PKA was abolished. The Tyr179-Phe and Tyr179-Ser mutant strains were able to activate PKA equivalent to the wild-type strain. Modest to strong reductions in transport activity in Asp178 and Asp358 mutants, respectively, were shown to not affect signaling. Abolishing transport by introducing an Asp358-Glu mutation resulted in a 50% reduction in signaling capacity (8). These observations show that, although transport and signaling are tightly intertwined, there are subtle yet profound differences in the residues that regulate the dual functions of Pho84 (Table 2).
TABLE 2.
Residues involved in substrate transport and sensing/signaling
The importance of residues in Pho84 for transport and signaling/sensing are as follows: +++, crucial; ++, contributing; +, little contribution. This is based on data obtained by site-directed-mutagenesis and relevant biochemical assays performed in this study and in previous work (8).
| Amino acid | Transport | Sensing/signaling |
|---|---|---|
| Asp178 | + | + |
| Asp358 | +++ | + |
| Lys492 | ++ | + |
| Tyr179 | +++ | +++ |
In this study, we showed that Tyr179, which is associated with the substrate release step during transport, has a stronger influence on signaling than Asp358 (Table 2), which has been shown to be crucial for substrate binding (8). We hypothesize that this difference originates from the different phosphate binding and release mechanisms operating in transport and in signaling.
Experimental Procedures
Materials and Strains
[32P]Orthophosphate (carrier-free) was obtained from PerkinElmer Life Sciences. Anti-Myc-HRP antibodies were obtained from Life Technologies Invitrogen (The Netherlands). Anti-actin-HRP antibodies were obtained from Abcam (UK). All other reagent grade materials were obtained from commercial sources. Haploid, prototrophic S. cerevisiae CEN.PK 113–7D (MATa MAL2–8c SUC2) was kindly provided by P. Kötter (Frankfurt, Germany).
Sequence Conservation Analysis
Functionally important amino acid residues in the Pho84 primary amino acid sequence were identified by performing a conservation analysis based on MSA using multiple sequence comparison by log-expectation (MUSCLE) alignment software (19). Pho84 homologues were obtained from a BLAST search of the UniProt database. Sequences representative of fungi and plants were selected. The Swiss-Prot protein sequences were retrieved from the NCBI Protein server (www.ncbi.nlm.nih.gov/protein/). The selected species and their accession numbers are S. cerevisiae Pho84 (P25297), P. indica (A8N031), Arabidopsis thaliana Pht1-1 (Q8VYM2), A. thaliana Pht 1–2 (Q96243), Oryza sativa (Q8GSD9), Hordeum vulgare (Q8H6E0), Medicago truncatula (Q8GSG4), Glomus intraradices (Q96VN6), Glomus versiforme (Q00908), and Pholiota nameko (Q96X52) (Fig. 2). The MSA figure was created using CLC Workbench (CLC Bio-Qiagen, Aarhus, Denmark). The Tyr179 residue was mapped onto the three-dimensional in silico Pho84 model (available at PMDB website, ID PM0076296). All of the figures were created using either PyMOL (Molecular Graphics System, version 1.3, Schrödinger, LLC.) or Visual Molecular Dynamics (VMD, version 1.9.1, University of Illinois at Urbana-Champaign, IL) (20).
Strain Construction
Mutants were created by oligonucleotide-directed site-specific mutagenesis using a plasmid containing the PHO84WT gene as described previously (8). The synthetic oligonucleotides used are listed in Table 3. The mutagenesis was performed using the Stratagene (USA) QuikChangeTM II mutagenesis kit according to the manufacturer's protocol. All of the mutant constructs were confirmed by DNA sequencing the entire PHO84WT gene. Using pU6H2MYC/PHO84WT and pU6H2MYC/PHO84MUT as templates for PCR, cassettes containing the last 1.3 kb of PHO84WT or PHO84MUT, c-myc, a His6 epitope, and a selection (Kanr) marker were transformed into CEN.PK 113–7D and were incorporated into the genome by homologous recombination. Positive transformants were selected on YPD-G418 (200 μg ml−1) plates. Resistant colonies were re-streaked onto fresh YPD-G418 (200 μg ml−1) plates and were verified by PCR, sequencing, and immunoblot analysis.
TABLE 3.
Synthetic oligonucleotides used in site-directed mutagenesis of PHO84WT
| Amino acid substitution | Mutagenized oligonucleotide (5′ → 3′) |
|---|---|
| Tyr179-Ala | ggtattggtatcggtggtgacgccccactatcttctattattac |
| Tyr179-Gly | ggtattggtatcggtggtgacggcccactatcttctattattac |
| Tyr179-Ser | gtattggtatcggtggtgactccccactatcttcta |
| Tyr179-Phe | tagaagatagtgggaagtcaccaccgataccaatac |
Growth Conditions
Cells expressing Pho84WT-myc or Pho84MUT-myc were precultivated aerobically for 12 h in YPD medium (transformed cells were kept in the presence of 200 μg ml−1 G418) at 30 °C under continuous agitation, washed twice, and inoculated in synthetic complete (SC) high phosphate (HPi, 10 mm KH2PO4) or low phosphate (LPi, 200 μm KH2PO4) media supplemented with 2% glucose. Cells in LPi conditions were grown aerobically at 30 °C with shaking at 200 rpm for 6 h. Samples were withdrawn at the indicated time points for further phosphate assay and Western blotting analyses.
Yeast Growth Spot Tests
Strains were cultured overnight in YPD at 30 °C under continuous agitation. Cells were collected, washed twice with sterile dH2O, and resuspended in sterile dH2O at an A600 of 0.3. A ×10 dilution series was spotted onto HPi and LPi SC medium agar plates. The plates were incubated at 30 °C, and growth was recorded every 24 h for a total of 3 days. To visualize the reduction in growth of the pho84Δ and mutant strains under LPi conditions, images were acquired with a higher illumination setting resulting in a brighter background. The figures were cropped and no further image processing was applied.
Phosphate Transport Measurements
Phosphate uptake in intact S. cerevisiae cells expressing Pho84WT-myc or Pho84MUT-myc was measured as previously described (8). Briefly, cells were grown in LPi medium and assayed by addition of 2 μl of [32P]orthophosphate (carrier-free, 0.18 Ci μmol−1; 1 mCi = 37 MBq) (PerkinElmer, USA) and phosphate (10 mm KH2PO4) to a final concentration of 220 μm. To determine total transport activity, a final phosphate concentration of 110 μm was used. Cells were resuspended to 1 mg ml−1 (wet weight) in buffer containing 25 mm Tris succinate, pH 4.5, and 3% glucose. Aliquots of 30 μl were incubated for 10 min at room temperature. After 10 min, 3 ml of ice-cold 25 mm Tris succinate, pH 4.5, buffer was added to stop the reaction. The cells were rapidly filtered (Whatman GF/F, USA) and the radioactivity retained on the filters was measured by liquid scintillation spectrometry.
Immunoblot Analysis of Pho84 Expression
Immunoblot analysis was performed as previously described (8). Briefly, membrane fractions were collected and 10 μg of protein samples were separated by SDS-PAGE using a 10% Laemmli system (21). Anti-myc HRP-conjugated antibody (anti-myc-HRP, 1:5000, Novex®) was used to detect expression of the Pho84WT/MUT-myc constructs. The (−) control was cut from the blot to avoid visualization of irrelevant samples, but detection was performed simultaneously with the mutant sample blot. As a loading control, 10 μg of total cell extract was loaded onto a separate gel according to the previously described protocol, and anti-β-actin HRP-conjugated antibody (1:5000) (Abcam, UK) was used for detection. After 1 min of incubation with chemiluminescent substrate (GE Healthcare, UK), the membrane-enriched sample blot was exposed to X-ray film for 1.5 min. The control blot was visualized using the Bio-Rad ChemiDocTM MP imaging system with an exposure time of 30 s. The molecular masses of the separated proteins were determined by their mobility relative to the pre-stained protein markers (Fermentas, Germany). Figures were cropped and no further image processing was applied.
Acid Phosphatase Assays
rAPase activity was assayed in liquid using an adapted protocol for the colorimetric Abcam® Acid Phosphatase Assay Kit. Briefly, whole cells were used as the source of the enzyme and p-nitrophenyl phosphate was used as the substrate. Yeast strains were grown overnight in 5 ml of YPD at 30 °C, centrifuged, and washed twice with LPi SC medium. Washed cells were inoculated into 15 ml of LPi or HPi phosphate SC medium to an A600 of 0.4. Cells were grown at 30 °C under continuous agitation for 3 h after which 80 μl of the cell suspension was harvested, washed once with acetate buffer (60 mm, pH 4.5), and resuspended in 80 μl of acetate buffer containing 1 mm p-nitrophenyl phosphate (final concentration). The reaction was incubated at 25 °C for 1 h and was then stopped with the addition of 20 μl of saturated Na2CO3. The cells were removed from the reaction by centrifugation before measuring the A405. The relative rAPase activity was determined by the formula A405/A600 × t, in which t is the time of incubation (min).
Trehalase Activity Measurements
Cells were cultured at 30 °C to exponential phase (A600 = 1.0–1.5) in YP medium with 2% (w/v) glucose. Mid-exponential phase cells were harvested and transferred to phosphate starvation medium (5.7 g liter−1 YNB without phosphate, with ammonium sulfate) with 4% (w/v) glucose and appropriate auxotrophic supplements. Cells were starved of phosphate for 3 days at 30 °C under continuous shaking, and starvation medium was refreshed daily. The phosphate-starved glucose-repressed cells were rapidly cooled on ice and harvested by centrifugation (5000 × g for 5 min at 4 °C). The pellet was washed twice with ice-cold 25 mm MES buffer, pH 6.0, resuspended in phosphate starvation medium with 4% (w/v) glucose, and incubated at 30 °C with shaking. After 30 min of incubation, 1 mm KH2PO4 was added to the culture. 75 mg ml−1 cell samples were taken at the indicated time points. Cells were rapidly cooled by the addition of ice-cold dH2O, centrifuged (5000 × g for 5 min at 4 °C), and re-suspended in 0.5 ml of ice-cold 25 mm MES buffer, pH 7.0. Crude cell extracts were prepared as described previously (22) and dialyzed (BRL microdialysis system) against 25 mm MES buffer, pH 7.0, with 50 μm CaCl2 at 4 °C. Trehalase activity in the dialyzed cell extracts was determined using a coupled enzymatic reaction of glucose oxidase and peroxidase with glucose as described previously (22). The specific activity was expressed as nanomole of glucose liberated per min per mg of protein. The total amount of protein in the samples was determined using the standard Lowry method described previously (23).
Computational Section
An inward-facing open conformation of Pho84 from S. cerevisiae was obtained by structurally fitting (7) it to the helix containing the transmembrane region (TM-I–XII, Table 1) in the crystal structure of the glycerol 3-phosphate/phosphate antiporter GlpT (24) from E. coli. This conformation was used as the starting conformation for the series of MD simulations performed in this work. After performing primary structural alignments between Pho84 (587 amino acids) and GlpT, the N- and C-terminal extensions of Pho84 could not be modeled onto the GlpT structure that contains only 452 amino acids. Hence, we decided not to include the regions of Pho84 (amino acids 1–54 and 552–587) that we could not structurally model in these studies. It must, however, be noted that these regions of Pho84 may be important for biological function and will be the target of future studies.
To investigate the role of proton transfer on translocation of Pi by Pho84, simulations were performed after protonating Asp178, which was previously suggested (8) to participate in the intrinsic proton transfer system of Pho84. A model of the truncated Pho84 embedded in a phospholipid bilayer was constructed after inserting Pho84 into a fully solvated (0.15 m TIP3P/water solution of potassium chloride) 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer using the CHARMM-gui (25).
Systems were also set up where a single inorganic phosphate ligand (H3PO4, H2PO4−, and HPO42−) was inserted into the center of Pho84 between two amino acids believed to be a part of the binding pocket (Lys492) and the proton transfer system (Asp178) using PACKMOL (26). This positioning was based on a recent X-ray crystal structure in which PiPT was co-crystallized with Pi (5). During the system design, titratable amino acids were assigned charges based upon neutral pH conditions. All of the MD simulations were performed using Amber software (version 10, USCF, San Francisco, CA) (27, 28). The Amber14SB force field, which is an evolved continuation of the Amber99SB (29) force field with improved protein backbone parameters, was used with compatible force fields, such as the newly developed lipid force field Lipid14 (30) and GAFF, a force field that has been developed for small organic ligands (31). The parameters used for the simulations of explicitly solvated potassium and chloride ions were those developed by Joung and Cheatham (32). The starting geometries for H3PO4, H2PO4−, and HPO42− were initially built in Avogadro (33), were pre-minimized using MMFF94 (34), and were further optimized in Gaussian09 (35) using the default convergence criteria implemented in the software at the HF/6–31G* level. GAFF parameters were used for simulations that incorporated H3PO4, whereas recently updated parameters for bioorganic phosphates were used for simulations that incorporated H2PO4− and HPO42− (36). The atomic partial charges for the Pi ligands were estimated using the restrained electrostatic potential procedure (37) and the Antechamber module.
Unrestrained Molecular Dynamics Simulations
Each system was initially energetically minimized by removing the high-energy van der Waals contacts with a 500.0 kcal mol−1 Å−2 position restraint on the lipids, Pho84, and the Pi ligand. Subsequently, the restraint for the lipids was removed. In a final energy minimization step, the whole system was allowed to relax without restraints. In this study, a total number of 10,000 steps were undertaken in each round and were divided into 5,000 steps of steepest descent and 5,000 steps of conjugate gradient. In a second step, the temperature of the system was initially raised from 0 to 100 K for 5 ps under conditions of NVT (constant number of particles, volume, and temperature) imposing a 10.0 kcal mol−1 Å−2 restraint on the lipids, Pho84, and the Pi ligand before running an additional 100 ps of simulation under conditions of NPT (constant number of particles, pressure, and temperature) in which the temperature was further raised from 100 to 310 K. The NPT simulation step was conducted using an anisotropic pressure scaling and a pressure relaxation constant (τp) of 2.0 ps while still applying a 10.0 kcal mol−1 Å−2 restraint on the lipid molecules, Pho84, and the Pi ligand. Subsequently, the restraint on the lipids was released and an additional 30 ns of simulation data using a τp of 1.0 ps was collected. During all of the simulations, the temperature was held constant using Langevin dynamics with a collision frequency set to 1.0 ps−1. All of the bonds to hydrogen were constrained using the SHAKE algorithm, which allowed a time step set to 0.002 ps. Periodic boundary conditions were applied in all of the directions using a 10-Å non-bonded interaction cutoff. Long-range electrostatics were treated using the particle mesh Ewald summation method, and long-range van der Waals interactions were corrected using a continuum model correction of both energy and pressure. Data were collected for the membrane system in the absence and presence of the Pi ligand, as well as for systems studying the effect of Asp178 protonation/deprotonation. Data were generally saved every 10 ps.
Steered Molecular Dynamics Simulations
To study the cytosolic release of Pi from the inward-facing open conformation of Pho84, constant velocity steered MD (14) was used to obtain potential release pathways and their respective work profiles. Initially, equilibration at NPT (1 bar, 310 K) and a restraint of 500.0 kcal mol−1 Å−2 was imposed on the distance between the phosphorus atom of Pi and the ϵ-amino nitrogen of Lys492, which set the distance 0.2 Å shorter than the equilibrated distance found after 30 ns of unrestrained simulation. To investigate how the residual motion of the restrained Lys492-Pi pair and the overall dynamics of Pho84 affect the various cytosolic release pathways, five individual simulations were conducted for each system after initially restraining the Lys492-Pi distance for 0.1, 0.2, 0.5, 1.0, or 5.0 ns as described previously. Numerous constant velocity steered MD simulations were then performed using a moving harmonic potential with a spring constant of 5.68 kcal mol−1 Å−2 (790 pN Å−1) and increasing the distance between Lys492-Pi at a constant pulling velocity of 10 Å ns−1. The total extended distance studied in each simulation was 35 Å in which the atomic coordinates and forces were saved every 1 ps. Although, in principle, a steered MD simulation can be considered analogous to a single-molecule atomic force microscopy experiment, the pulling velocity commonly employed is typically several orders of magnitude faster, thus it is difficult to make direct comparisons between calculated ligand pulling work profiles using these techniques (38). Typically, constant velocity steered MD simulations have utilized different pulling velocities to optimize the conditions under which the ligand experiences amino acid contacts on the way out of the receptor site and to ensure an energetically stable trajectory. The constant velocity chosen in this study (0.01 Å ns−1) is among the slowest found in the literature and was selected based on the results of unbinding studies on similar systems (39–42).
Author Contributions
D. S. and J. V. d. V. performed the site-directed mutagenesis, phosphate uptake assays, acid phosphatase activity studies, and the spot tests. G. V. Z. performed the trehalase activity measurements. B. C. G. K. performed all of the theoretical calculations and analyses. B. C. G. K., D. S., and B. P. conceived the idea and the experimental design of the project. B. C. G. K. and D. S. led the writing of this paper. All of the authors have taken part in the preparation of this manuscript, have reviewed the results, and have approved the final version of this manuscript.
Supplementary Material
Acknowledgments
Simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc-Alarik/Aurora (Dnr: 2014/1–404/2015-1–444, Biophysics of Cellular Transport).
This work was supported by the Linnæus University and the Crafoord Foundation. The authors declare that they have no conflicts of interest regarding the contents of this article.

This article contains supplemental Tables S1–S5 and Figs. S1–S35.
- MFS
- major facilitator superfamily
- MD
- molecular dynamics
- SMD
- steered molecular dynamics
- MSA
- multiple sequence alignment
- rAPase
- repressible acid phosphatase
- HPi
- high-Pi
- LPi
- low-Pi.
References
- 1.Pao S. S., Paulsen I. T., and Saier M. H. (1998) Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 62, 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quistgaard E. M., Löw C., Guettou F., and Nordlund P. (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 [DOI] [PubMed] [Google Scholar]
- 3.Law C. J., Maloney P. C., and Wang D.-N. (2008) Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 62, 289–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samyn D. R., and Persson B. L. (2016) Inorganic phosphate and sulfate transport in S. cerevisiae. in Yeast Membrane Transport (Ramos J., Sychrová H., and Kschischo M. eds) pp. 253–269, Springer International Publishing, Switzerland, 10.1007/978-3-319-25304-6_10 [DOI] [PubMed] [Google Scholar]
- 5.Pedersen B. P., Kumar H., Waight A. B., Risenmay A. J., Roe-Zurz Z., Chau B. H., Schlessinger A., Bonomi M., Harries W., Sali A., Johri A. K., and Stroud R. M. (2013) Crystal structure of a eukaryotic phosphate transporter. Nature 496, 533–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schothorst J., Kankipati H. N., Conrad M., Samyn D. R., Van Zeebroeck G., Popova Y., Rubio-Texeira M., Persson B. L., and Thevelein J. M. (2013) Yeast nutrient transceptors provide novel insight in the functionality of membrane transporters. Curr. Genet. 59, 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagerstedt J. O., Voss J. C., Wieslander A., and Persson B. L. (2004) Structural modeling of dual-affinity purified Pho84 phosphate transporter. FEBS Lett. 578, 262–268 [DOI] [PubMed] [Google Scholar]
- 8.Samyn D. R., Ruiz-Pávon L., Andersson M. R., Popova Y., Thevelein J. M., and Persson B. L. (2012) Mutational analysis of putative phosphate- and proton-binding sites in the Saccharomyces cerevisiae Pho84 phosphate:H+ transceptor and its effect on signalling to the PKA and PHO pathways. Biochem. J. 445, 413–422 [DOI] [PubMed] [Google Scholar]
- 9.Wykoff D. D., and O'Shea E. K. (2001) Phosphate transport and sensing in Saccharomyces cerevisiae. Genetics 159, 1491–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stroud R. M. (2007) Transmembrane transporters: an open and closed case. Proc. Natl. Acad. Sci. U.S.A. 104, 1445–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersson M., Bondar A.-N., Freites J. A., Tobias D. J., Kaback H. R., and White S. H. (2012) Proton-coupled dynamics in lactose permease. Structure 20, 1893–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y., Ke M., and Gong H. (2015) Protonation of Glu(135) facilitates the outward-to-inward structural transition of fucose transporter. Biophys. J. 109, 542–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borst-Pauwels G. W. (1993) Mutual interaction of ion uptake and membrane potential. Biochim. Biophys. Acta 1145, 15–24 [DOI] [PubMed] [Google Scholar]
- 14.Hummer G., and Szabo A. (2010) Free energy profiles from single-molecule pulling experiments. Proc. Natl. Acad. Sci U.S.A. 107, 21441–21446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarzynski C. (1997) Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 78, 2690–2693 [Google Scholar]
- 16.Park S., Khalili-Araghi F., Tajkhorshid E., and Schulten K. (2003) Free energy calculation from steered molecular dynamics simulations using Jarzynski's equality. J. Chem. Phys. 119, 3559–3566 [Google Scholar]
- 17.Holsbeeks I., Lagatie O., Van Nuland A., Van de Velde S., and Thevelein J. M. (2004) The eukaryotic plasma membrane as a nutrient-sensing device. Trends Biochem. Sci. 29, 556–564 [DOI] [PubMed] [Google Scholar]
- 18.Popova Y., Thayumanavan P., Lonati E., Agrochão M., and Thevelein J. M. (2010) Transport and signaling through the phosphate-binding site of the yeast Pho84 phosphate transceptor. Proc. Natl. Acad. Sci. U.S.A. 107, 2890–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson J. D., Higgins D. G., and Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 21.Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 22.Pernambuco M. B., Winderickx J., Crauwels M., Griffioen G., Mager W. H., and Thevelein J. M. (1996) Glucose-triggered signalling in Saccharomyces cerevisiae: different requirements for sugar phosphorylation between cells grown on glucose and those grown on non-fermentable carbon sources. Microbiology 142, 1775–1782 [DOI] [PubMed] [Google Scholar]
- 23.Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 24.Huang Y., Lemieux M. J., Song J., Auer M., and Wang D.-N. (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–620 [DOI] [PubMed] [Google Scholar]
- 25.Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 [DOI] [PubMed] [Google Scholar]
- 26.Martínez L., Andrade R., Birgin E. G., and Martínez J. M. (2009) PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 [DOI] [PubMed] [Google Scholar]
- 27.Case D. A., Cheatham T. E. 3rd., Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Case D. A., Darden T. A., Cheatham T. E. 3rd, Simmerling C., Wang J., Duke R. E., Luo R., Crowley M., Walker R. C., Zhang W., Merz K. M., Wang S., Hayik S., Roitberg A., Seabra G., et al. (2008) AMBER 10, University of California, San Francisco, CA [Google Scholar]
- 29.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickson C. J., Madej B. D., Skjevik A. A., Betz R. M., Teigen K., Gould I. R., and Walker R. C. (2014) Lipid14: the Amber lipid force field. J. Chem. Theory Comput. 10, 865–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 32.Joung I. S., and Cheatham T. E. 3rd (2008) Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112, 9020–9041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanwell M. D., Curtis D. E., Lonie D. C., Vandermeersch T., Zurek E., and Hutchison G. R. (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halgren T. A. (1996) Merck molecular force field: I. basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519 [Google Scholar]
- 35.Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., et al. (2009) Gaussian09, Gaussian Inc., Wallingford, CT [Google Scholar]
- 36.Steinbrecher T., Latzer J., and Case D. A. (2012) Revised AMBER parameters for bioorganic phosphates. J. Chem. Theory Comput. 8, 4405–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J., Cieplak P., and Kollman P. A. (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074 [Google Scholar]
- 38.Gullingsrud J. R., Braun R., and Schulten K. (1999) Reconstructing potentials of mean force through time series analysis of steered molecular dynamics simulations. J. Comput. Phys. 151, 190–211 [Google Scholar]
- 39.Kosztin D., Izrailev S., and Schulten K. (1999) Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophys. J. 76, 188–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wriggers W., and Schulten K. (1999) Investigating a back door mechanism of actin phosphate release by steered molecular dynamics. Proteins 35, 262–273 [PubMed] [Google Scholar]
- 41.Zhang D., Gullingsrud J., and McCammon J. A. (2006) Potentials of mean force for acetylcholine unbinding from the α7 nicotinic acetylcholine receptor ligand-binding domain. J. Am. Chem. Soc. 128, 3019–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y., Shen J., Luo X., Silman I., Sussman J. L., Chen K., and Jiang H. (2003) How does huperzine A enter and leave the binding gorge of acetylcholinesterase? steered molecular dynamics simulations. J. Am. Chem. Soc. 125, 11340–11349 [DOI] [PubMed] [Google Scholar]
- 43.Harrison M. J., Dewbre G. R., and Liu J. (2002) A phosphate transporter from Medicago truncatula involved in the acquisition of phosphate released by arbuscular mycorrhizal fungi. Plant Cell 14, 2413–2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.