

Abstract
The precise mechanism underlying the conversion of normal prion protein (PrPC) into abnormal prion protein (PrPSc) remains unclear. Protein misfolding cyclic amplification (PMCA), an in vitro technique used for amplifying PrPSc, results in PrPSc replication that preserves the strain-specific characteristics of the input PrPSc; thus, PMCA mimics the process of in vivo PrPSc replication. Previous work has demonstrated that in PMCA, nucleic acids are critical for PrPSc amplification, but little information has been reported on glycosaminoglycan (GAG) participation in PrPSc replication in vitro. Here, we investigated whether GAGs play a role in the faithful replication of PrPSc by using a modified PMCA performed with baculovirus-derived recombinant PrP (Bac-PrP) as a substrate. The addition of heparan sulfate (HS) or its analog heparin (HP) restored the conversion efficiency in PMCA that was inhibited through nucleic acid depletion. Moreover, the PMCA products obtained under these conditions were infectious and preserved the properties of the input PrPSc. These data suggest that HS and HP play the same role as nucleic acids in facilitating faithful replication of prions in PMCA. Furthermore, we showed that HP binds to both Bac-PrP and Bac-PrPSc through the sulfated groups present on HP and that the N-terminal domain of Bac-PrPSc might potentially not be involved in the binding to HP. These results suggest that the interaction of GAGs such as HS and HP with PrPC and/or PrPSc through their sulfate groups is critical for the faithful replication of prions.
Keywords: heparan sulfate, heparin, prion, prion disease, protein folding, baculovirus-recombinant prion protein, protein misfolding cyclic amplification
Introduction
Prions are unique proteinaceous infectious agents that are considered to be the cause of transmissible spongiform encephalopathies, including scrapie in sheep, bovine spongiform encephalopathy (BSE)2 in cattle, and Creutzfeldt-Jakob disease in humans (1). Prions consist primarily of a pathogenic form (PrPSc) of the normal cellular prion protein (PrPC), and PrPSc appears to propagate itself through the autocatalytic conformational conversion of PrPC (2). Although the mechanism of PrPC conversion into PrPSc remains unclear, host cofactors have been suggested to be necessary for efficient replication of PrPSc (3, 4); various biological molecules, such as nucleic acids (5–8), glycosaminoglycans (GAGs) (9–12), lipids (13, 14), and proteins (15–17), have been reported to act as cofactors for PrPC conversion into PrPSc.
Protein misfolding cyclic amplification (PMCA) is an in vitro technique for amplifying undetectable amounts of PrPSc to levels detectable using common methods, such as Western blotting (WB) (18–21). PrPSc propagated using PMCA retains the properties of the input PrPSc, suggesting that PMCA mimics the in vivo replication of PrPSc (22). Therefore, PMCA has been used in studies investigating the PrPSc replication mechanism and the cofactors involved in the replication (23, 24). Deleault et al. (5, 25) demonstrated that RNA and DNA stimulated the in vitro conversion of PrPC into PrPSc, and the Ma group (8) showed that synthetic phospholipids and RNA spontaneously generated infectious PrPSc in PMCA performed using bacterial recombinant PrP (recPrP) as the substrate. Furthermore, we showed that both RNA and DNA were crucial for the faithful replication of mouse-adapted prions in PMCA performed using baculovirus-derived recombinant PrP (Bac-PrP) (26). Thus, available data strongly suggest that nucleic acids promote PrPSc replication in PMCA, although whether nucleic acids are involved in prion replication in vivo is unknown.
GAGs, like nucleic acids, are polyanions, and GAGs function in brain development and are critical determinants of brain structure and function (27–30). The GAG heparan sulfate (HS) localizes at amyloid plaques in the brains of humans and animals with prion disease (31) and Alzheimer's disease (32), and HS is suggested to be involved in the pathogenesis of amyloidosis. Furthermore, sulfated glycans were found to alter PrPC cellular localization and stimulate PrPC endocytosis in cultured cells (33), suggesting that GAGs serve as a cellular receptor for both prion uptake and cell infection. PrPSc accumulation also influences the metabolism of GAGs and results in their accumulation and secretion in urine (34). Furthermore, sulfated GAGs containing HS stimulate the conformational change of PrPC into a PrPSc-like proteinase K (PK)-resistant form even in a cell-free conversion assay (9, 12). Thus, GAG involvement in prion disease pathogenesis and PrPC conversion into PrPSc has been widely reported (35–37), but whether GAGs promote PrPSc replication in PMCA, which mimics in vivo prion replication, is unknown.
We hypothesized that GAGs might play a similar role as nucleic acids in PMCA because GAGs are also polyanions and because GAGs contribute to critical molecular events in the pathogenesis of prion diseases and to both in vivo and in vitro PrPSc replication. Therefore, we investigated whether GAGs were involved in PrPSc replication by a modified PMCA performed using partially purified Bac-PrP as the PrP substrate; we denoted this as insect-cell PMCA (iPMCA). In iPMCA, PK- and heat-treated insect cell lysates were used as the conversion enhancer instead of brain homogenates (BHs), and iPMCA enabled PrPSc propagation that preserved the strain characteristics of the input PrPSc. We showed that HS and its analog heparin (HP) restored the amplification of Bac-PrPSc retaining the strain characteristics of the input PrPSc when amplification was blocked through nucleic acid depletion. Furthermore, HP bound to both Bac-PrP and Bac-PrPSc through its sulfate groups, and the degree of HP sulfation affected the conversion of Bac-PrP into Bac-PrPSc.
Results
GAGs Did Not Affect Bac-PrP Conversion into a PK-resistant Form (Bac-PrPres) in the Presence of Nucleic Acids
To determine whether GAGs were involved in the conversion ofBac-PrP into Bac-PrPSc in iPMCA (26), we first investigated whether treatment of PK- and heat-treated cell lysate (PHCL), which contains the cofactors necessary for Bac-PrPSc conversion, with GAG-degrading enzymes influences the activity inducing Bac-PrP conversion into Bac-PrPres. Electrophoretic separation of PHCL and Alcian blue staining of gels revealed that PHCL contains GAGs (NT; Fig. 1A). Furthermore, we found that most of the GAGs in PHCL were digested following treatment with chondroitinase ABC, which digests chondroitin sulfates (CSs), chondroitins, and hyaluronic acid (HA), but not after treatment with heparinase II, which digests HS and HP (Ch and Hp; Fig. 1A). The results of control experiments demonstrated that chondroitinase ABC and heparinase II efficiently digested purified CSB and HP, respectively, under the iPMCA reaction-buffer condition. These results suggested that PHCL contained CSs, such as CSA, CSB, and CSC, but not HS and HP. Furthermore, treatment of PHCL with both chondroitinase ABC and heparinase II did not affect Bac-PrP conversion into Bac-PrPres in iPMCA in which the reaction mixtures were seeded with ME7 strain prions and mouse-adapted BSE (mBSE) prions (Ch and Hp; Fig. 1B); however, treatment with benzonase, which digests both DNA and RNA, efficiently inhibited the conversion, as reported previously (26). Next, we investigated how the addition of GAGs into the iPMCA reaction mixture affects Bac-PrP conversion into Bac-PrPres (Fig. 1C) and found that none of the seven types of GAGs added independently of the reaction mixtures increased the conversion efficiency. Therefore, we speculated that the presence of a large amount of nucleic acids in PHCL masked the effect of each GAG treatment on Bac-PrPres conversion and that the nucleic acids played a key role in the conversion in iPMCA because the amount of GAGs included in PHCL was insufficient to stimulate the conversion.
FIGURE 1.
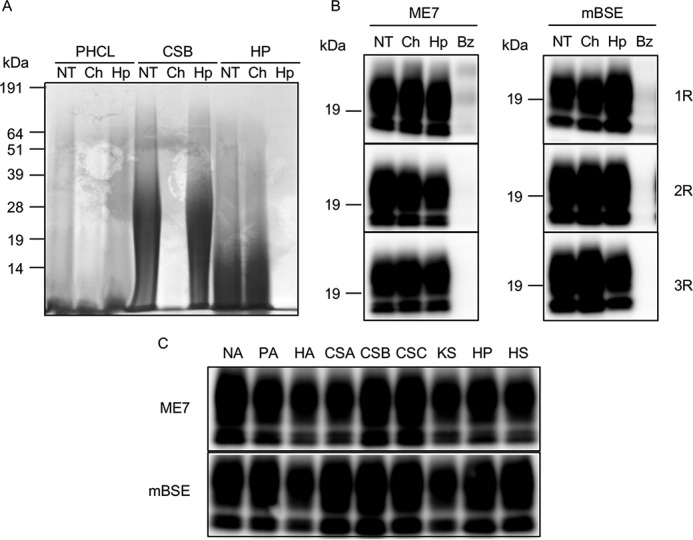
Effects of treatment with GAG-degrading enzymes and the addition of GAGs on Bac-PrPres amplification. A, PK- and heat-treated cell lysate (PHCL), chondroitin sulfate B (CSB), and heparin (HP) were digested with chondroitinase ABC (Ch) or heparinase II (Hp). CSB and HP were digested in 1× PBS containing 4 mm EDTA and 0.25% Triton X-100. NT, non-treated sample. Samples were separated using SDS-PAGE, and the gel was stained with Alcian blue to visualize GAGs. B, ME7- and mBSE-seeded PMCA reactions were performed using PHCL digested with chondroitinase ABC, heparinase II, or benzonase (Bz). C, ME7- and mBSE-seeded iPMCA reactions in the presence of nucleic acids were performed by adding polyA (PA), hyaluronic acid (HA), chondroitin sulfate A (CSA), CSB, chondroitin sulfate C (CSC), keratan sulfate (KS), HP, or HS at 50 μg/ml final concentration. NA, no additive.
Certain GAGs Enhanced the Conversion Efficiency Suppressed by Nucleic Acid Depletion
To directly test whether the nucleic acids present in the iPMCA reaction mixtures masked the effects of the GAGs, we examined how GAGs influenced Bac-PrPres conversion in iPMCA under nucleic acid depletion by using three prion strains, ME7, mBSE, and Chandler. In accordance with our previous work (26), benzonase treatment lowered the conversion efficiency and precluded sequential amplification (NA; Fig. 2), and the addition of synthetic poly(A) (PA) to the reaction mixtures completely restored the conversion (Fig. 2). When HP and GAGs except for HP were added at 10 and 100 μg/ml to the nucleic acid-depleted reaction mixtures, all of the GAGs enhanced the efficiencies of first round conversion relative to the basal level (NA; Fig. 2), although the effect of HA and keratan sulfate was very weak. The addition of HS or HP enabled sequential amplification of Bac-PrPres under nucleic acid-depleted conditions in the case of all three prion strains, but for the sequential amplification, HS was required at a 10-fold higher concentration than HP (50-fold in the case of ME7). This quantitative difference in the effects on conversion could be attributed to the distinct degrees of sulfation.
FIGURE 2.

Recovery effects of GAGs on Bac-PrPres amplification inhibited through nucleic acid depletion. ME7-, mBSE-, and Chandler-seeded iPMCA reactions were performed under conditions of nucleic acid depletion by adding PA, HA, CSA, CSB, CSC, keratan sulfate (KS), HP, or HS. PA, HP, and GAGs except for HP were added at 40, 10, and 100 μg/ml, respectively. Moreover, HS and CSB were added to a final concentration of 500 μg/ml for ME7- and Chandler-seeded PMCA (500HS and 500CSB). Ten rounds of serial amplification were conducted. The results of the first, second, third, fifth, and tenth rounds of PMCA are shown. ND, not done (amplification not performed). NA, no additive.
All CSs tested were incapable of maintaining ME7-seeded sequential amplification; however, in the case of mBSE, CSA promoted at least five rounds of Bac-PrPres amplification but did not maintain the amplification until the 10th round, and, by contrast, CSB and CSC enabled sequential amplification. Last, for Chandler, all CSs facilitated Bac-PrPres amplification until the fifth round but could not maintain the amplification until the tenth round, although CSB added at 500 μg/ml enabled 10 rounds of amplification of the Chandler-seeded iPMCA products (500CSB). These results suggest that the effects of CSA, CSB, and CSC on conversion depended on the prion strains.
Sulfate Groups on HP Are Critical for Bac-PrPres Amplification
GAG length and degree of sulfation were previously reported to affect the conversion of PrPC or recPrP into PrPSc (9, 10, 35–38). Therefore, to examine the relative effects of GAG length and sulfation degree on Bac-PrP conversion into Bac-PrPres, we added chemically modified HPs to iPMCA reaction mixtures under nucleic acid-depleted conditions (Fig. 3A). We selected HP for this experiment because structurally modified forms are obtained from only HP. Low molecular mass HPs (lane 5; <3 kDa), which are composed of ∼8 monosaccharide units, induced conversion to the same extent as normal HP (lane 4; 17–19 kDa), but disaccharided HP (lane 6; 0.7 kDa) had lost this ability. These results suggested that a chain length of ∼8 saccharide units is adequate for stimulating the conversion, but a disaccharide unit cannot induce Bac-PrP conversion into Bac-PrPres. Next, we investigated the effects of sulfate groups on HP. De-N-sulfated (DeN) HP, which completely lacks N-sulfation, and de-O-sulfated (DeO) HP, which completely lacks O-sulfation, exhibited a weak but clearly detectable conversion-inducing activity (lanes 7 and 8) in both ME7 and mBSE strains. The converting activity of DeN HP was slightly stronger than that of DeO HP, which suggested that O-sulfation is more effective than N-sulfation in promoting Bac-PrP conversion into Bac-PrPres. Conversely, N-acetyl-de-O-sulfated (NaDO) HP, which completely lacks O-sulfation and in which N-sulfation is markedly diminished, was less active than DeN HP and DeO HP (lane 9), and the activity measured was equivalent to that obtained with the addition of either no HP (lane 2) or heparinase II-treated normal HP (lane 10). To quantify the effects of desulfated HPs on the conversion, PMCA was performed with various amounts of desulfated HPs added under the nucleic acid-depleted condition (Fig. 3B). In the case of normal HP, the activity peaked when it was added at 10–20 μg/ml in the case of both ME7 and mBSE strains, and the conversion rates were higher than those measured in the PMCA performed in the presence of nucleic acids (open circles); however, the conversion activity of normal HP was decreased when it was added at >50 μg/ml. By contrast, the activity of DeN HP in the ME7 and mBSE strains peaked at 500 and 100 μg/ml, respectively, although the conversion rates measured at these concentrations were also higher than the rates of the PMCA performed in the presence of nucleic acids (filled triangles). In the case of DeO HP, the highest activities were observed at 1000 μg/ml in both ME7 and mBSE strains, but the conversion rates were ∼65% of the rate in PMCA performed in the presence of nucleic acids (filled circles); however, the conversion rates measured with DeO HP might be increased at concentrations >1000 μg/ml. Last, with NaDO HP, the conversion rates were almost equivalent to those of PMCA performed with no added HPs under nucleic acid-depleted conditions (dotted line, 24.5% for ME7 and 18.1% for mBSE), even when the input amounts were increased to 1000 μg/ml (filled squares). These results suggested that the sulfate groups on HP are critical for the conversion of Bac-PrP into Bac-PrPres. Furthermore, the diminished ability of DeN HP and DeO HP to induce Bac-PrPres conversion could be compensated for by increasing the amounts of the input. This finding suggested that the adequate amount of sulfate groups required for inducing the conversion to Bac-PrPSc does not have to be present on a single HP molecule and that the conversion can be promoted by the interaction of multiple low sulfated HP molecules with Bac-PrP and/or Bac-PrPSc through the sulfate groups from each molecule.
FIGURE 3.

Recovery effects of various HP derivatives on Bac-PrPres amplification under nucleic acid-depleted conditions. A, three rounds of ME7- and mBSE-seeded iPMCA were performed, under conditions of nucleic acid depletion, in the presence of HP and various HP derivatives: normal HP (HP, 17–18 kDa; lane 4), low molecular mass HP (<3 kDa; lane 5), disaccharided HP (0.7 kDa; lane 6), de-N-sulfated HP (lane 7), de-O-sulfated HP (lane 8), N-acetyl-de-O-sulfated HP (lane 9), and heparinase II-treated normal HP (lane 10). Lanes 1–3, non-treated sample, benzonase-treated sample, and benzonase-treated sample with added PA, respectively. All additives were used at a final concentration of 10 μg/ml. B, one round of ME7- and mBSE-seeded iPMCA performed using benzonase-treated PHCL; the amplification was conducted with various concentrations of normal HP (open circles), de-N-sulfated HP (closed triangles), de-O-sulfated HP (closed circles), and N-acetyl-de-O-sulfated HP (closed squares) (2.5–1000 μg/ml). The experiments were repeated at least three times. The conversion efficiencies for each sample are expressed as a percentage change (mean ± S.D. (error bars)) relative to the control value (control = 100). The conversion value of PMCA performed using non-treated PHCL served as a control. The conversion rates measured with the use of N-acetyl-de-O-sulfated HP were almost equivalent to those of PMCA performed with no addition of HPs under nucleic acid-depleted conditions (dotted line, 24.5% for ME7 and 18.1% for mBSE).
HP Binds to Both Bac-PrP and Bac-PrPSc
We investigated whether GAGs bound to Bac-PrP and/or Bac-PrPres by employing a binding assay that was performed using HP-conjugated agarose beads. The iPMCA reaction mixtures before and after the reaction were incubated with HP-conjugated beads, and after centrifuging the samples, the distributions of Bac-PrP or Bac-PrPres between the pellet (HP-bound fraction) and the supernatant (unbound fraction) were determined through densitometric quantification of their signals in WB analysis. When the sample tested was the iPMCA reaction mixture before the reaction, which contained only Bac-PrP, 40% of the input Bac-PrP was located in the HP-bound fraction, but only 10% of the input Bac-PrP was in the control FLAG-bead fraction (left columns; Fig. 4), and the difference was statistically significant. Notably, the amount of Bac-PrP in the HP-bound fraction was decreased to 20% when an excess amount of free normal HP (NM; Fig. 4) was added. By contrast, when NaDO HP was added to the mixture, the binding rate of Bac-PrP with HP was 38%, and no significant difference was observed between the NaDO HP addition and no addition. These results strongly suggested that Bac-PrP specifically bound to HP. Next, we performed the HP-binding assay by using the PK-digested reaction mixture after ME7-seeded iPMCA (right columns; Fig. 4). This sample did not contain Bac-PrP because of the digestion with PK and contained only the PK-resistant core of Bac-PrPres. Approximately 60% of the PK-resistant core of Bac-PrPres was contained in the HP-bound fraction, but only ∼10% of it was in the FLAG-bead fraction. The amount of the PK-resistant core of Bac-PrPres in the HP-bound faction was reduced substantially, to ∼10%, when free HP was added, but NaDO HP addition did not markedly affect the binding of the PK-resistant core to HP-agarose beads. These results suggested that both the PK-resistant core of Bac-PrPres and Bac-PrP specifically bound to HP and that of the two, the truncated Bac-PrPres bound more efficiently to HP. Furthermore, in the case of the non-treated iPMCA mixtures after the reaction, which included both Bac-PrP and full-length Bac-PrPres, the binding rates measured for each experimental group were between those of Bac-PrP and the PK-resistant core of Bac-PrPres (middle columns; Fig. 4), which suggested that full-length Bac-PrPres also specifically bound to HP.
FIGURE 4.
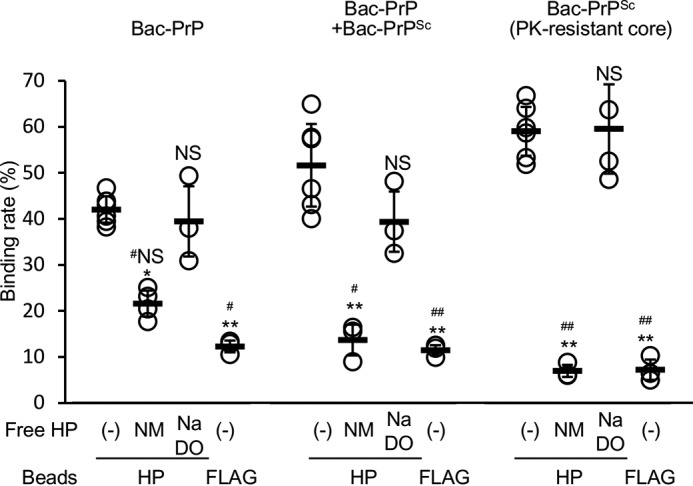
Specific binding of HP to Bac-PrP and Bac-PrPSc. HP-agarose was incubated with the non-seeded and non-reacted iPMCA reaction solution (Bac-PrP), the non-PK-digested reaction solution after PMCA performed using ME7 PrPSc as the seed (Bac-PrP + Bac-PrPSc), and the PK-digested reaction solution after PMCA (Bac-PrPSc, PK-resistant core). After centrifugation, WB was performed to analyze the HP-agarose beads and supernatants, and the band intensities of each fraction were quantified using a ChemiImager. The HP-binding rates are shown as the ratio of Bac-PrP and/or Bac-PrPSc in the bead fraction to total Bac-PrP and/or Bac-PrPSc. Free normal HP (NM) or NaDO was added into the mixtures of each iPMCA reaction solution and the HP-agarose beads during the incubation. Each iPMCA reaction solution was incubated with FLAG-beads (FLAG) as a control. Open circles indicate the binding rates from individual experiments; bars, mean of the binding rates of independent experiments; error bars, S.D. * and #, p < 0.01 (versus without any addition and with addition of free NaDO HP, respectively, in HP-agarose beads). ** and ##, p < 0.001 (versus without any addition and with the addition of free NaDO HP, respectively, in HP-agarose beads). NS and #NS, no significant difference between non-addition and the addition of free NaDO HP and between the addition of free normal HP and the addition of NaDO HP, respectively.
HS and HP Can Substitute for Nucleic Acids in Promoting the Propagation of Infectious Bac-PrPSc in iPMCA
We investigated whether the products generated from iPMCA performed using HS and HP instead of nucleic acids were infectious. We collected the products generated from 13 rounds of ME7- and mBSE-seeded iPMCA performed with HP or HS added under nucleic acid-depleted conditions and intracerebrally inoculated the products into Tga20 mice. This resulted in the development of clinical symptoms and death in the case of all inoculated mice (Table 1). However, the mice inoculated with the products from both ME7- and mBSE-seeded iPMCA performed with HP or HS under nucleic acid-depleted conditions survived significantly longer than did the mice inoculated with the product from the non-treated iPMCA, although the Bac-PrPres amounts in each inoculum did not differ considerably (data not shown). By contrast, survival times did not differ in a statistically significant manner between mice that were inoculated with the products of iPMCA performed with PA or non-treated iPMCA in the case of both of ME7 and mBSE. Last, we investigated the biochemical properties of the PrPSc accumulated in the brain of mice inoculated with each PMCA product. The banding patterns in WB analysis (Fig. 5A) and the PK susceptibility (Fig. 5B) of PrPSc from the mice inoculated with each iPMCA product were identical or nearly identical to each other in the case of both ME7 and mBSE. These results suggested that HS and HP functioned similarly as the nucleic acids did in promoting faithful prion replication in vitro.
TABLE 1.
Mean survival times of Tga20 mice following intracerebral inoculation
The final dilutions of the ME7 and mBSE seeds in the “seed” samples were 1 × 10−15.
| Inoculum | Transmission rate (total deaths/total number) | Mean survival time ± S.D. |
|---|---|---|
| ME7 seed | % | days |
| Non-treated | 100 (6/6) | 133.8 ± 8.0 |
| Benzonase-treated | ||
| +PA | 100 (5/5) | 157.2 ± 16.0 |
| +HP | 100 (6/6) | 182.2 ± 27.5 |
| +HS | 100 (7/7) | 163.6 ± 15.6 |
| Non-additive | 0 (0/6) | >600 |
| mBSE seed | ||
| Non-treated | 100 (7/7) | 139.4 ± 4.5 |
| Benzonase-treated | ||
| +PA | 100 (6/6) | 135.7 ± 4.2 |
| +HP | 100 (7/7) | 196.1 ± 14.0 |
| +HS | 100 (6/6) | 188.3 ± 15.7 |
| Non-additive | 0 (0/5) | >600 |
FIGURE 5.
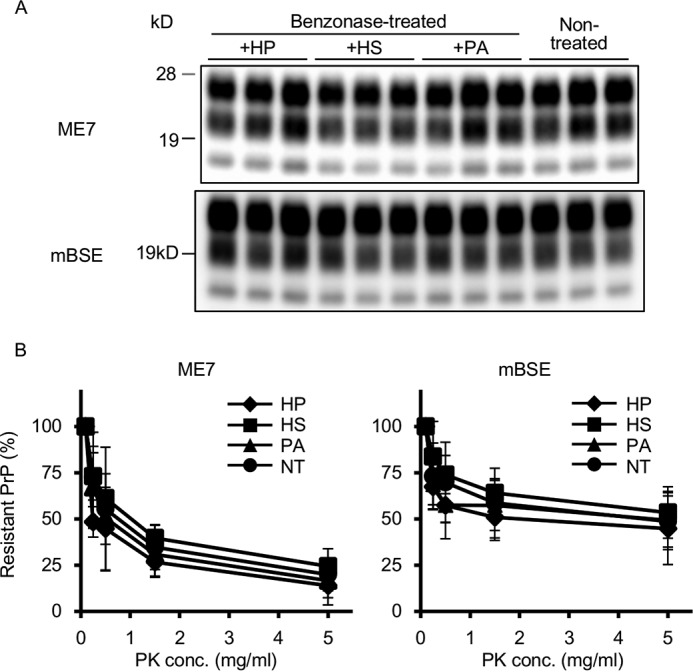
Biochemical properties of PrPSc accumulated in the brain of mice inoculated with the products of PMCA performed with HS or HP under nucleic acid-depleted conditions. A, WB was performed to analyze the PrPSc accumulated in the brain of mice inoculated with ME7- or mBSE-seeded PMCA products. The inoculums were the products of benzonase-treated iPMCA performed with the addition of HP (+HP), HS (+HS), or PA (+PA) and the non-treated normal iPMCA products (Non-treated). B, PK sensitivities of the PrPSc from mice inoculated with each PMCA product were measured by performing the PK degradation assay. WB was performed to analyze the samples from at least three independent experiments. The graphs show the average relative intensities of the PrPSc signal together with the S.D. (error bars) at each tested PK concentration.
Discussion
Nucleic acids play a crucial role in prion replication in PMCA (5, 25, 26, 39). Here, we showed that GAG addition into the reaction mixtures improved the Bac-PrPres amplification that was blocked through nucleic acid depletion in iPMCA. However, all GAGs did not equally influence prion replication, and HS and its analog HP were found to completely restore the replication of three prion strains, ME7, Chandler, and mBSE. This result suggests that HS and HP perform the same function as nucleic acids in prion replication in vitro. HS and HP have been known to promote PrP conversion into a PrPSc-like form in in vitro cell-free conversion assays (10, 12); however, we have demonstrated for the first time that HS and HP are critical for the replication of infectious prions. Furthermore, our study revealed that the HS and HP concentrations required to induce the conversion differed between prion strains. We also showed that HP bound, through its sulfate groups, to both Bac-PrP and Bac-PrPSc (whose N-terminal region was truncated) and that the sulfation was crucial for the conversion to Bac-PrPSc. These results suggest that HS and HP function in prion replication by binding to PrPC and/or PrPSc through the sulfate groups in HS and HP.
HS is present in the brain and is colocalized with amyloid plaques in the brain in prion diseases (31) and Alzheimer's disease (32, 40). Furthermore, HS localizes on the plasma membrane and in the extracellular matrix, and HS chains that exist as membrane-anchored proteoglycans enter the endocytic pathway following incorporation into a cell through endocytosis. PrPC conversion into PrPSc is considered to occur either on the cell surface or in an endocytic pathway compartment, such as the lysosome or recycling endosome, mainly in the CNS (41, 42). Consequently, the histological localization and subcellular localization of HS coincide with the sites where PrPSc conversion occurs. These findings strongly suggest that HS plays a key role in PrPSc replication in vivo as a cofactor in the cell membrane environment of the CNS. This suggestion is supported by recent data showing that following scrapie infection, heparanase-overexpressing transgenic mice survive considerably longer than control mice (43). Horonchik et al. (44) have previously shown that HS can serve as a cellular receptor for prion uptake, and, therefore, HS might play a dual role, as a cofactor for PrPSc conversion and as a receptor for prion uptake into cells.
HP, the hypersulfated form of HS, also stimulates faithful prion replication in iPMCA. However, we do not consider HP to be involved in PrPSc replication in the brain because almost no HP is detected in the brain (45). By contrast, CSA, CSB, and CSC, which stimulate the conversion of Bac-PrP into a PrPSc-like form, might be involved in PrPSc replication in the brain. CSA and CSC are mainly present in cartilage and in the cornea, and much of CSB is contained in the skin. However, these CSs are also contained in the mammalian brain and are mainly detected in the brain extracellular matrix. In the nervous system, CSs are known to be involved in mediating various physiological effects (29, 46) and are suggested to be involved in prion infection in the brain in cattle (45). These findings suggest that CSs might also facilitate PrPC conversion into PrPSc in the CNS. Furthermore, our results showed that the abilities of CSA, CSB, and CSC to stimulate Bac-PrPSc conversion differed according to prion strains; therefore, this strain-dependent difference in CS ability to promote PrPC conversion to PrPSc might represent one of the underlying causes of strain-specific differences in neuropathology.
The binding of HP and HS has been reported to increase the thermal stability, β-sheet structure, or PK resistance of recPrP (12, 47, 48) and alter the solubility of recPrP (38); these findings suggest that PrPC binding to HS and HP induces a conformational change in PrPC. Therefore, Bac-PrP binding to HP and HS might also induce a conformational change in Bac-PrP and contribute to the conversion of Bac-PrP into Bac-PrPSc. Moreover, because HP also bound to Bac-PrPSc, this binding to Bac-PrPSc might further promote the conversion to Bac-PrPSc by affecting the structure of the template PrPSc. Low molecular weight HP (3 kDa) was shown to bind to recPrP in a 1:1 ratio at pH 7.4 (49), and we found that low molecular weight HP stimulated the conversion to Bac-PrPres (Fig. 3A); these results suggest that the binding of Bac-PrP and HP at a ratio of at least 1:1 might be required for the conversion to Bac-PrPSc. The average molecular masses of HS and HP are 10–70 and 10–20 kDa, respectively, and thus the linear chain structure of these GAGs might enable PrPC and PrPSc to be efficiently positioned near each other on the GAGs; such proximity between the molecules might further enhance the conversion efficiencies.
PrPC and recPrP were reported to bind to HP through the octarepeat region (50), the 120–130-amino acid region, or the lysine-rich region. Here, we found that, relative to Bac-PrP, N-terminally truncated Bac-PrPSc bound to HP as efficiently or more efficiently. This result suggests that the N-terminal region of Bac-PrPSc might not be necessary for HP binding and that Bac-PrPSc could potentially be capable of efficiently binding to HP through other regions. A previous study showed that murine recPrP lacking residues 23–99 bound to HP at pH 5.5 but not pH 7.4 (51), whereas we found that N-terminal-truncated Bac-PrPSc bound to HP at pH 7.4; thus, the HP-binding site of N-terminally truncated Bac-PrPSc could be the same as the binding region of murine recPrP at pH 5.5, which might be buried inside the molecule at pH 7.4. The HP-binding region of N-terminally truncated Bac-PrPSc is considered to be exposed to the outside through a conformational change from Bac-PrP, although the region is normally buried in the protein core in Bac-PrP. This region might be the 120–130-amino acid region or the lysine-rich region. Alternatively, the HP-binding regions or the HP-binding modes of Bac-PrPSc might differ from those of PrPC or recPrP, because the conformation of PrPSc is completely different from the conformations of those proteins. Identification of the binding regions of PrPSc is considered to be critical for elucidating the role of PrPSc in the conversion. Conversely, PrPC and recPrP were reported to directly bind to GAGs containing HP (12, 47, 52), but whether Bac-PrPSc directly binds to HP is unknown because iPMCA reaction mixtures contain numerous biomolecules. Thus, the mode of PrPSc binding to HP must be investigated to clarify the conversion mechanism. However, we cannot exclude the possibility that Bac-PrPSc interacts with GAGs, such as HP, in a manner distinct from the aforementioned manner of binding. Bac-PrPSc can form a multimeric complex and therefore holds the potential for multivalency, which would probably increase the affinity even if the individual binding sites in the PK-resistant core exhibit low avidity.
Phosphatidylethanolamine was shown to be adequate by itself for amplifying infectious mouse PrPSc in PMCA in the presence of PrPSc seeds (14). However, the PrPSc amplified through PMCA performed using phosphatidylethanolamine alone did not preserve the prion strain characteristics of the input PrPSc (53). Synthetic phospholipids and RNA induced the spontaneous generation of two types of prions in Escherichia coli recPrP-based PMCA, and their prion strain characteristics appeared to be unchanged during the serial amplification (8, 54, 55). Our study suggests that polyanions such as GAGs and nucleic acids were essential for accurate in vitro replication of prions in PMCA performed using Bac-PrP. These findings suggest that phosphatidylethanolamine alone is inadequate for faithful in vitro replication of prions and that this replication requires polyanions such as nucleic acids and GAGs.
In conclusion, the findings of this study show that HS and HP stimulate faithful prion replication in PMCA performed using Bac-PrP and that this is mediated by the binding of HS and HP to PrPC and/or PrPSc through the sulfate groups in the GAGs. These results suggest that HS might play a critical role in PrPC conversion into PrPSc in the in vivo cell membrane environment. However, Bac-PrP conversion into Bac-PrPSc in PMCA is not induced with the use of GAGs alone (data not shown), and thus additional cofactors are probably required for the conversion. The Bac-PrP-based PMCA system used here could facilitate the identification of these other cofactors involved in prion replication. If all of the cofactors involved in prion replication in PMCA can be identified, considerable progress would be made in the elucidation of the mechanism underlying the in vivo conversion of PrPC into PrPSc.
Experimental Procedures
Ethics Statement
The animal experiments were approved by the Committee of Animal Experiments of the National Institute of Animal Health (approval numbers 11-008 and 13-005) and were performed in accordance with the Guideline for Animal Experiments at the Ministry of Agriculture, Forestry, and Fisheries of Japan.
Reagents
The following reagents were from commercial sources: synthetic PA (P9403), HA (H1504), CSA (C9819), CSB (C3788), HP (H3393), and HS (H7640) from Sigma; CSC (08815-84) from Nacalai Tesque (Kyoto, Japan); keratan sulfate (400760) from Seikagaku Biobusiness (Tokyo, Japan); HP disaccharide I-S, sodium salt (H1001), DeN HP (heparin I-H; H3001), and NaDO HP (heparin IV-A; H3004) from Dextra Laboratories (Reading, UK); DeO HP (GT6011) from Neoparin (Alameda, CA); and low molecular weight HP (194114) from MP Biomedicals (Santa Ana, CA).
Preparation of PrPC Source
Immobilized metal affinity chromatography (IMAC)-purified Bac-PrP was used as the PrPC source for PMCA. IMAC purification was performed as described in our previous report (56). After washing in 1× PBS, insect cells expressing Bac-PrP (1 × 108 cells) were suspended in 36 ml of lysis buffer (1× PBS, 300 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate) containing an EDTA-free protease/inhibitor mixture (Nacalai Tesque). Cells were disrupted by sonication for 2 min, using an ice-cold ultrasonic bath sonicator (Bioruptor; Cosmo Bio, Tokyo, Japan), incubated on ice for 60 min, and then centrifuged at 12,000 × g for 90 min at 4 °C. The supernatant fraction was filtered through a 0.45-μm filter. The clarified supernatant was loaded onto a His TALONTM superflow cartridge (Clontech, Mountain View, CA) equilibrated with 10 ml of equilibration buffer (1× PBS, 300 mm NaCl, 1% Triton X-100), after which the cartridge was washed with 10 ml of equilibration buffer and 20 ml of wash buffer (1× PBS, 300 mm NaCl, 2 mm imidazole, 1% Triton X-100). Last, Bac-PrP was eluted in 5 ml of elution buffer (1× PBS, 300 mm NaCl, 500 mm imidazole, 1% Triton X-100) containing the EDTA-free protease/inhibitor mixture, and the concentration of eluted Bac-PrP was adjusted to 100 ng/μl with elution buffer. The IMAC-purified Bac-PrP was ∼37% pure (56).
PrPSc Seeds
The mouse-adapted scrapie strains ME7 and Chandler and mouse-adapted typical BSE (mBSE) were used as PrPSc seeds. These prion strains were propagated in ICR mice. The brains of mice at the terminal stage of the disease were homogenized at 10% concentration (w/v) in 1× PBS, and the 10% BHs were also further diluted to 1% (w/v) with 1× PBS; both homogenates were stored at −80 °C after aliquoting.
Insect Cell PMCA
We performed iPMCA by using an automatic cross-ultrasonic protein-activating apparatus (ELESTEIN 070-GOT, Elekon Science Corp., Chiba, Japan), as reported previously (26). Amplification was performed with 32 cycles of sonication (pulse oscillation for 3 s, repeated five times at intervals of 0.1 s), and this was followed by incubation at 37 °C for 30 min with gentle agitation. The PK- and heat-treated High Five cell lysate (Life Technologies, Inc., Carlsbad, CA) was used as a conversion inducer. We mixed 500 μl of the High Five cell suspension (5 × 104/μl in distilled water) with 500 μl of 2× PMCA buffer (2× PBS, 8 mm EDTA, 2% Triton X-100) and added PK to the mixtures to a final concentration of 100 μg/ml. Following sonication for 1 min, the cell lysate was incubated at 37 °C for 1 h and then heated at 100 °C for 2.5 h to inactivate PK; the resultant cell lysate was designated as PHCL. The reaction mixture was prepared by mixing 2 μl of IMAC-purified Bac-PrP (∼100 ng/μl), 10 μl of PHCL, and 90 μl of 1× PBS containing 4 mm EDTA and 0.25% Triton X-100. To each iPMCA reaction mixture, we added 1 μl of the 1% infected BHs as a PrPSc seed. The amplified products obtained after the first round of amplification were diluted 1:10 with each PrPC substrate, and a second round of amplification was performed. This process was repeated when necessary.
Western Blotting
The PMCA products were digested with 50 μg/ml PK at 37 °C for 1 h, following which an equal volume of 2× SDS sample buffer was added, and the samples were boiled for 5 min. The samples were separated by performing SDS-PAGE in NuPAGE 12% BisTris gels (Life Technologies) and then transferred onto PVDF membranes. The membranes were probed with the anti-PrP HRP-conjugated monoclonal antibody T2 (57) and developed with SuperSignal West Dura Extended Duration Substrate (Pierce), and the chemiluminescence signals were detected using a ChemiImager (Alpha InnoTec, San Leandro, CA).
Enzyme Treatment of PHCL
Chondroitinase ABC (Sigma) was added to PHCL at a final concentration of 1 unit/ml, and heparinase II (New England Biolabs, Ipswich, MA) was added together with the supplied reaction buffer at a final concentration of 160 units/ml. Benzonase (Novagen, Madison, WI) was added together with 2 mm MgCl2 at a final concentration of 1000 units/ml. After these enzymes were added, the PHCLs were incubated at 37 or 30 °C (for heparinase II) overnight with shaking (850 rpm) and then heat-treated at 100 °C for 10 min to inactivate the enzymes. Subsequently, each enzyme-treated PHCL was used for PMCA. To detect the GAGs contained in the PHCLs, an equal volume of 2× SDS sample buffer was added to the samples and boiled for 5 min, and the samples were separated by performing SDS-PAGE in NuPAGE 12% BisTris gels. The GAGs in the gels were visualized by staining (1 h, room temperature) with a solution of 0.125% (v/v) Alcian blue dissolved in 25% (v/v) ethanol and 10% (v/v) acetic acid. The same solution without the dye was used for destaining.
HP-binding Assay
We mixed 20 μl of HP-agarose resin (Sigma), which was prewashed with PBS, with 40 μl of non-treated or PK-digested iPMCA reaction mixtures and incubated the samples at room temperature for 1 h with gentle shaking. After incubation, empty microspin columns (Hoefer, Holliston, MA) were filled with the agarose suspension, and the columns were centrifuged at 1500 × g for 5 min, and the flow-through was recovered. Next, 40 μl of a solution containing 0.1% Triton X-100, 1× PBS, and 4 mm EDTA was added into the columns, and the agarose suspension was collected into microtubes by centrifuging the columns upside down. Last, 40 μl of 2× SDS sample buffer was added to the flow-through and to the agarose suspension, and 15 μl of each mixture was subjected to WB analysis. The blots were analyzed densitometrically using ChemiImager software.
Bioassay
The PMCA products subjected to the bioassay were prepared as follows. We added 1 μl of 1% ME7- and mBSE-infected BHs to each reaction mixture of iPMCA together with 40 μg/ml PA, 25 μg/ml HP, or 500 μg/ml HS under nucleic acid-depleted conditions and then performed one round of PMCA. Next, the process of 10-fold dilution of the PMCA product and its subsequent amplification was repeated 12 times. The PMCA products were diluted 1:10 with 1× PBS, and 20 μl of each diluted sample was injected intracerebrally into 3–5-week-old Tga20 mice overexpressing murine PrP; the mice were injected under sevoflurane anesthesia. All mice were then housed in a biosafety level 3 room of our animal facility, and their clinical status was monitored at least 3 times/week. The animals were euthanized by using a sevoflurane overdose either following evidence of progressive neurologic dysfunction or >600 days after inoculation in the case of mice that showed no neurological disease symptoms, and then the brains were removed. The right hemisphere of the brain was fixed in formalin for histopathological analysis, and the left hemisphere was stored at −80 °C for biochemical analysis. The average survival times of the experimental groups were analyzed using one-way analysis of variance and the Tukey-Kramer multiple comparison test.
PK Degradation Assay
To compare the resistance of infected mouse brain-derived PrPSc to PK digestion, the concentrations of each infected BH were adjusted with 10% Prnp0/0 BH to obtain similar signal intensities of PrPSc in WB analysis. These samples were then digested with various concentrations of PK (50–5000 μg/ml) at 37 °C for 1 h, following which the enzymatic reaction was stopped by boiling in 1× SDS sample buffer. WB was used to analyze the samples in several independent experiments, and the average PrPSc signal intensity measured with each sample was expressed as a percentage relative to that in the sample digested with 50 μg/ml PK.
Author Contributions
M. I. designed and performed experiments, analyzed the data, and wrote the paper. N. T. and N. K. conducted experiments together with M. I. Y. M. provided mice. Y. I., T. Y., and Y. M. discussed the results and the implications and wrote the paper together with M. I.
Acknowledgments
We thank Noriko Shimozaki, Tomoaki Yamamura, and Junko Yamada for technical assistance. We are also grateful to the animal caretakers for their contribution to the study.
This study was supported by JSPS KAKENHI Grant 25460575 and a grant-in-aid from the BSE Control Project of the Ministry of Agriculture, Forestry, and Fisheries of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
- BSE
- bovine spongiform encephalitis
- mBSE
- mouse-adapted BSE
- PrPC
- normal cellular prion protein
- PrPSc
- abnormal pathogenic PrP
- recPrP
- recombinant PrP
- Bac-PrP
- baculovirus-derived recPrP
- PMCA
- protein misfolding cyclic amplification
- iPMCA
- insect cell PMCA
- GAG
- glycosaminoglycan
- HA
- hyaluronic acid
- HP
- heparin
- HS
- heparan sulfate
- DeN
- de-N-sulfated
- DeO
- de-O-sulfated
- NaDO
- N-acetyl-de-O-sulfated
- CS
- chondroitin sulfate
- PA
- poly(A)
- PK
- proteinase K
- PHCL
- PK- and heat-treated cell lysate
- BH
- brain homogenate
- WB
- Westernblotting
- IMAC
- immobilized metal affinity chromatography
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1.Collinge J. (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24, 519–550 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S. B. (1998) The prion diseases. Brain Pathol. 8, 499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soto C. (2011) Prion hypothesis: the end of the controversy? Trends Biochem. Sci. 36, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma J. (2012) The role of cofactors in prion propagation and infectivity. PLoS Pathog. 8, e1002589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deleault N. R., Lucassen R. W., and Supattapone S. (2003) RNA molecules stimulate prion protein conversion. Nature 425, 717–720 [DOI] [PubMed] [Google Scholar]
- 6.Cordeiro Y., Machado F., Juliano L., Juliano M. A., Brentani R. R., Foguel D., and Silva J. L. (2001) DNA converts cellular prion protein into the β-sheet conformation and inhibits prion peptide aggregation. J. Biol. Chem. 276, 49400–49409 [DOI] [PubMed] [Google Scholar]
- 7.Deleault N. R., Harris B. T., Rees J. R., and Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang F., Wang X., Yuan C. G., and Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lawson V. A., Lumicisi B., Welton J., Machalek D., Gouramanis K., Klemm H. M., Stewart J. D., Masters C. L., Hoke D. E., Collins S. J., and Hill A. F. (2010) Glycosaminoglycan sulphation affects the seeded misfolding of a mutant prion protein. PLoS One 5, e12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoyama T., Takeuchi A., Yamamoto M., Kitamoto T., Ironside J. W., and Morita M. (2011) Heparin enhances the cell-protein misfolding cyclic amplification efficiency of variant Creutzfeldt-Jakob disease. Neurosci. Lett. 498, 119–123 [DOI] [PubMed] [Google Scholar]
- 11.Klajnert B., Cortijo-Arellano M., Bryszewska M., and Cladera J. (2006) Influence of heparin and dendrimers on the aggregation of two amyloid peptides related to Alzheimer's and prion diseases. Biochem. Biophys. Res. Commun. 339, 577–582 [DOI] [PubMed] [Google Scholar]
- 12.Wong C., Xiong L. W., Horiuchi M., Raymond L., Wehrly K., Chesebro B., and Caughey B. (2001) Sulfated glycans and elevated temperature stimulate PrP(Sc)-dependent cell-free formation of protease-resistant prion protein. EMBO J. 20, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazlauskaite J., Sanghera N., Sylvester I., Vénien-Bryan C., and Pinheiro T. J. (2003) Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 42, 3295–3304 [DOI] [PubMed] [Google Scholar]
- 14.Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., and Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DebBurman S. K., Raymond G. J., Caughey B., and Lindquist S. (1997) Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc. Natl. Acad. Sci. U.S.A. 94, 13938–13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leucht C., Simoneau S., Rey C., Vana K., Rieger R., Lasmézas C. I., and Weiss S. (2003) The 37 kDa/67 kDa laminin receptor is required for PrP(Sc) propagation in scrapie-infected neuronal cells. EMBO Rep. 4, 290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mays C. E., and Ryou C. (2010) Plasminogen stimulates propagation of protease-resistant prion protein in vitro. FASEB J. 24, 5102–5112 [DOI] [PubMed] [Google Scholar]
- 18.Soto C., Anderes L., Suardi S., Cardone F., Castilla J., Frossard M. J., Peano S., Saa P., Limido L., Carbonatto M., Ironside J., Torres J. M., Pocchiari M., and Tagliavini F. (2005) Pre-symptomatic detection of prions by cyclic amplification of protein misfolding. FEBS Lett. 579, 638–642 [DOI] [PubMed] [Google Scholar]
- 19.Saá P., Castilla J., and Soto C. (2006) Presymptomatic detection of prions in blood. Science 313, 92–94 [DOI] [PubMed] [Google Scholar]
- 20.Murayama Y., Yoshioka M., Okada H., Takata M., Yokoyama T., and Mohri S. (2007) Urinary excretion and blood level of prions in scrapie-infected hamsters. J. Gen. Virol. 88, 2890–2898 [DOI] [PubMed] [Google Scholar]
- 21.Murayama Y., Ono F., Shimozaki N., and Shibata H. (2016) l-Arginine ethylester enhances in vitro amplification of PrP in macaques with atypical L-type bovine spongiform encephalopathy and enables presymptomatic detection of PrP in the bodily fluids. Biochem. Biophys. Res. Commun. 470, 563–568 [DOI] [PubMed] [Google Scholar]
- 22.Castilla J., Saá P., Hetz C., and Soto C. (2005) In vitro generation of infectious scrapie prions. Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 23.Deleault N. R., Kascsak R., Geoghegan J. C., and Supattapone S. (2010) Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49, 3928–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abid K., Morales R., and Soto C. (2010) Cellular factors implicated in prion replication. FEBS Lett. 584, 2409–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deleault N. R., Geoghegan J. C., Nishina K., Kascsak R., Williamson R. A., and Supattapone S. (2005) Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J. Biol. Chem. 280, 26873–26879 [DOI] [PubMed] [Google Scholar]
- 26.Imamura M., Kato N., Okada H., Yoshioka M., Iwamaru Y., Shimizu Y., Mohri S., Yokoyama T., and Murayama Y. (2013) Insect cell-derived cofactors become fully functional after proteinase K and heat treatment for high-fidelity amplification of glycosylphosphatidylinositol-anchored recombinant scrapie and BSE prion proteins. PLoS One 8, e82538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aono S., and Oohira A. (2006) Chondroitin sulfate proteoglycans in the brain. Adv. Pharmacol. 53, 323–336 [DOI] [PubMed] [Google Scholar]
- 28.Avram S., Shaposhnikov S., Buiu C., and Mernea M. (2014) Chondroitin sulfate proteoglycans: structure-function relationship with implication in neural development and brain disorders. Biomed. Res. Int. 2014, 642798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwok J. C., Warren P., and Fawcett J. W. (2012) Chondroitin sulfate: a key molecule in the brain matrix. Int. J. Biochem. Cell Biol. 44, 582–586 [DOI] [PubMed] [Google Scholar]
- 30.Maeda N., Ishii M., Nishimura K., and Kamimura K. (2011) Functions of chondroitin sulfate and heparan sulfate in the developing brain. Neurochem. Res. 36, 1228–1240 [DOI] [PubMed] [Google Scholar]
- 31.Snow A. D., Wight T. N., Nochlin D., Koike Y., Kimata K., DeArmond S. J., and Prusiner S. B. (1990) Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab. Invest. 63, 601–611 [PubMed] [Google Scholar]
- 32.Perlmutter L. S., Barrón E., Saperia D., and Chui H. C. (1991) Association between vascular basement membrane components and the lesions of Alzheimer's disease. J. Neurosci. Res. 30, 673–681 [DOI] [PubMed] [Google Scholar]
- 33.Shyng S. L., Lehmann S., Moulder K. L., and Harris D. A. (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J. Biol. Chem. 270, 30221–30229 [DOI] [PubMed] [Google Scholar]
- 34.Mayer-Sonnenfeld T., Zeigler M., Halimi M., Dayan Y., Herzog C., Lasmezas C. I., and Gabizon R. (2005) The metabolism of glycosaminoglycans is impaired in prion diseases. Neurobiol. Dis. 20, 738–743 [DOI] [PubMed] [Google Scholar]
- 35.Caughey B., and Raymond G. J. (1993) Sulfate polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J. Virol. 67, 643–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabizon R., Meiner Z., Halimi M., and Ben-Sasson S. A. (1993) Heparin-like molecules bind differentially to prion-proteins and change their intracellular metabolic fate. J. Cell. Physiol. 157, 319–325 [DOI] [PubMed] [Google Scholar]
- 37.Caughey B., Brown K., Raymond G. J., Katzenstein G. E., and Thresher W. (1994) Binding of the protease-sensitive form of PrP (prion protein) to sulfated glycosaminoglycan and congo red. J. Virol. 68, 2135–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellett L. J., Coleman B. M., Shambrook M. C., Johanssen V. A., Collins S. J., Masters C. L., Hill A. F., and Lawson V. A. (2015) Glycosaminoglycan sulfation determines the biochemical properties of prion protein aggregates. Glycobiology 25, 745–755 [DOI] [PubMed] [Google Scholar]
- 39.Saá P., Sferrazza G. F., Ottenberg G., Oelschlegel A. M., Dorsey K., and Lasmézas C. I. (2012) Strain-specific role of RNAs in prion replication. J. Virol. 86, 10494–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snow A. D., Sekiguchi R. T., Nochlin D., Kalaria R. N., and Kimata K. (1994) Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer's disease brain. Am. J. Pathol. 144, 337–347 [PMC free article] [PubMed] [Google Scholar]
- 41.Caughey B., Raymond G. J., Ernst D., and Race R. E. (1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 65, 6597–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borchelt D. R., Taraboulos A., and Prusiner S. B. (1992) Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 267, 16188–16199 [PubMed] [Google Scholar]
- 43.Kovalchuk Ben-Zaken O., Nissan I., Tzaban S., Taraboulos A., Zcharia E., Matzger S., Shafat I., Vlodavsky I., and Tal Y. (2015) Transgenic over-expression of mammalian heparanase delays prion disease onset and progression. Biochem. Biophys. Res. Commun. 464, 698–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horonchik L., Tzaban S., Ben-Zaken O., Yedidia Y., Rouvinski A., Papy-Garcia D., Barritault D., Vlodavsky I., and Taraboulos A. (2005) Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 280, 17062–17067 [DOI] [PubMed] [Google Scholar]
- 45.Papakonstantinou E., Karakiulakis G., Roth M., Verghese-Nikolakaki S., Dawson M., Papadopoulos O., and Sklaviadis T. (1999) Glycosaminoglycan analysis in brain stems from animals infected with the bovine spongiform encephalopathy agent. Arch. Biochem. Biophys. 370, 250–257 [DOI] [PubMed] [Google Scholar]
- 46.Dyck S. M., and Karimi-Abdolrezaee S. (2015) Chondroitin sulfate proteoglycans: key modulators in the developing and pathologic central nervous system. Exp. Neurol. 269, 169–187 [DOI] [PubMed] [Google Scholar]
- 47.Andrievskaia O., Potetinova Z., Balachandran A., and Nielsen K. (2007) Binding of bovine prion protein to heparin: a fluorescence polarization study. Arch. Biochem. Biophys. 460, 10–16 [DOI] [PubMed] [Google Scholar]
- 48.Boshuizen R. S., Morbin M., Mazzoleni G., Tagliavini F., Meloen R. H., and Langedijk J. P. (2007) Polyanion induced fibril growth enables the development of a reproducible assay in solution for the screening of fibril interfering compounds, and the investigation of the prion nucleation site. Amyloid 14, 205–219 [DOI] [PubMed] [Google Scholar]
- 49.Vieira T. C., Reynaldo D. P., Gomes M. P., Almeida M. S., Cordeiro Y., and Silva J. L. (2011) Heparin binding by murine recombinant prion protein leads to transient aggregation and formation of RNA-resistant species. J. Am. Chem. Soc. 133, 334–344 [DOI] [PubMed] [Google Scholar]
- 50.Taubner L. M., Bienkiewicz E. A., Copié V., and Caughey B. (2010) Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J. Mol. Biol. 395, 475–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vieira T. C., Cordeiro Y., Caughey B., and Silva J. L. (2014) Heparin binding confers prion stability and impairs its aggregation. FASEB J. 28, 2667–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.González-Iglesias R., Pajares M. A., Ocal C., Espinosa J. C., Oesch B., and Gasset M. (2002) Prion protein interaction with glycosaminoglycan occurs with the formation of oligomeric complexes stabilized by Cu(II) bridges. J. Mol. Biol. 319, 527–540 [DOI] [PubMed] [Google Scholar]
- 53.Deleault N. R., Walsh D. J., Piro J. R., Wang F., Wang X., Ma J., Rees J. R., and Supattapone S. (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U.S.A. 109, E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z., Zhang Y., Wang F., Wang X., Xu Y., Yang H., Yu G., Yuan C., and Ma J. (2013) De novo generation of infectious prions with bacterially expressed recombinant prion protein. FASEB J. 27, 4768–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y., Wang F., Wang X., Zhang Z., Xu Y., Yu G., Yuan C., and Ma J. (2014) Comparison of 2 synthetically generated recombinant prions. Prion 8, 28669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Imamura M., Kato N., Iwamaru Y., Mohri S., Yokoyama T., and Murayama Y. (2016) Multiple-affinity purification of a baculovirus-derived recombinant prion protein with in vitro ability to convert to its pathogenic form. Prep. Biochem. Biotechnol. 26, 1–7 [DOI] [PubMed] [Google Scholar]
- 57.Hayashi H., Takata M., Iwamaru Y., Ushiki Y., Kimura K. M., Tagawa Y., Shinagawa M., and Yokoyama T. (2004) Effect of tissue deterioration on postmortem BSE diagnosis by immunobiochemical detection of an abnormal isoform of prion protein. J. Vet. Med. Sci. 66, 515–520 [DOI] [PubMed] [Google Scholar]