

Abstract
The exon junction complex (EJC) is a multiprotein complex integral to mRNA metabolism. Biochemistry and genetic studies have concluded that the EJC is composed of four core proteins, MAGOH, EIF4A3, RBM8A, and CASC3. Yet recent studies in Drosophila indicate divergent physiological functions for Barentsz, the mammalian Casc3 ortholog, raising the question as to whether CASC3 is a constitutive component of the EJC. This issue remains poorly understood, particularly in an in vivo mammalian context. We previously found that haploinsufficiency for Magoh, Eif4a3, or Rbm8a disrupts neuronal viability and neural progenitor proliferation, resulting in severe microcephaly. Here, we use two new Casc3 mouse alleles to demonstrate developmental phenotypes that sharply contrast those of other core EJC components. Homozygosity for either null or hypomorphic Casc3 alleles led to embryonic and perinatal lethality, respectively. Compound embryos lacking Casc3 expression were smaller with proportionately reduced brain size. Mutant brains contained fewer neurons and progenitors, but no apoptosis, all phenotypes explained by developmental delay. This finding, which contrasts with severe neural phenotypes evident in other EJC mutants, indicates Casc3 is largely dispensable for brain development. In the developing brain, CASC3 protein expression is substoichiometric relative to MAGOH, EIF4A3, and RBM8A. Taken together, this argues that CASC3 is not an essential EJC component in brain development and suggests it could function in a tissue-specific manner.
Keywords: Casc3, EJC, brain, development, mouse, peripheral
INTRODUCTION
Post-transcriptional regulation by RNA binding proteins enables a fine-tuned control of gene expression within cells. One such example is the exon junction complex (EJC), which binds mRNA in the nucleus and travels with it into the cytoplasm (Le Hir et al. 2016). Nuclear EJC is implicated in splicing, whereas cytoplasmic EJC influences protein translation, nonsense-mediated decay (NMD), and subcellular mRNA localization. These mRNA metabolism processes are mediated in part via association with peripheral EJC proteins. Biochemical studies in immortalized cells, as well as crystal structures of mammalian and Drosophila proteins, have indicated the core EJC is composed of MAGOH, RBM8A, EIF4A3, and CASC3 (Tange et al. 2005; Andersen et al. 2006; Bono et al. 2006; Stroupe et al. 2006; Buchwald et al. 2010; Wang et al. 2014). Consistent with this biochemistry evidence, genetic studies in the Drosophila oocyte reveal common roles for these four components in mRNA localization (Newmark and Boswell 1994; Hachet and Ephrussi 2001; Mohr et al. 2001; van Eeden et al. 2001; Palacios et al. 2004). There is also emerging evidence to suggest that these components can have distinct functions (Singh et al. 2012, 2015). In the Drosophila eye, Barentsz (Casc3) phenotypes diverge from Mago (Magoh), Y14 (Rbm8a), and Ddx48 (Eif4a3) mutants, three of which each disrupts development and Mapk splicing (Ashton-Beaucage et al. 2010; Roignant and Treisman 2010). These phenotypic differences could be due to different subcellular localization of EJC components (Degot et al. 2004; Baguet et al. 2007), or alternatively could suggest CASC3 is not always a core EJC constituent. This issue remains poorly understood, and in particular has never been addressed in an in vivo mammalian context.
Toward this end, our group recently generated mouse mutants in Magoh, Eif4a3, and Rbm8a, which we used to demonstrate essential requirements of these core EJC components in brain development. Magoh germline mice (MagohMos2/+) are microcephalic (reduced brain size) and perinatal lethal (Silver et al. 2010). Likewise, conditional haploinsufficiency for Magoh, Eif4a3, or Rbm8a within the developing brain causes severe microcephaly (McMahon et al. 2014; Mao et al. 2015, 2016). In these mouse models, reduced brain size is due to aberrant neurogenesis, and specifically altered progenitor proliferation, ectopic neuronal differentiation, and massive apoptosis. Phenotypes from EJC mouse genetics fit with the discovery of human mutations and copy-number variations in EJC components in patients with intellectual disability, autism spectrum disorders, and brain malformations (Brunetti-Pierri et al. 2008; Rosenfeld et al. 2012; Nguyen et al. 2013; Favaro et al. 2014). These data collectively indicate that precise levels of Magoh, Eif4a3, and Rbm8a are essential for mammalian brain development, but beg the question as to whether Casc3 levels are also critical.
Here, we evaluated the in vivo function of Casc3 in embryogenesis using two novel Casc3 mutant mouse models. Analyses of these mutant mice demonstrate that Casc3 is essential for proper developmental progression and animal viability. However, in marked contrast to other core EJC proteins, we observe no significant defects in brain development in Casc3 heterozygous or compound mutant animals. In compound mutant brains in which Casc3 expression was almost entirely eliminated (>99%), neurogenesis defects were evident, but explained by developmental delay. These observed phenotypic differences between Casc3 and other core EJC mutants could be due to substoichiometric protein expression levels of CASC3 in the developing brain, relative to other core EJC components. Together, these data provide mammalian in vivo evidence that Casc3 is critical for proper embryonic development, and shows distinct dosage requirements for brain development relative to other core EJC components.
RESULTS
A Casc3 hypomorphic allele demonstrates expression in the developing brain and requirement for animal viability
To evaluate the requirement of Casc3 in the developing brain, we generated a new Casc3 mouse allele (Casc3Gt(RRU345)Byg, referred to as Casc3RRU345 hereafter). We obtained ES cells (BayGenomics gene-trap resource consortium) (Stryke et al. 2003) containing a gene trap insertion in intron 3 of Casc3. This gene trap has a splice-acceptor sequence upstream of a β-geo reporter gene (a fusion of β-galactosidase and neomycin phosphotransferase II). The resulting insertional mutation is predicted to constitutively produce a fusion transcript of exons 1–3 of Casc3 with the β-galactosidase (LacZ) gene (Fig. 1A). We first took advantage of the LacZ insertion in this gene-trap allele to evaluate reporter expression as a proxy for Casc3 expression, using LacZ activity staining of Casc3RRU345/+ and Casc3RRU345/RRU345 E11.5 and E12.5 whole embryos. This highly sensitive assay detected LacZ activity in Casc3RRU345/+ and Casc3RRU345/RRU345 embryos (Fig. 1B–H). Staining was especially robust in the central nervous system, particularly in the forebrain, midbrain, and neural tube (Fig. 1C,D,F–H). We also noted strong LacZ activity in the developing heart of both heterozygous and homozygous embryos (Fig. 1B,C). Using whole mount in situ hybridization of E11.5 and E12.5 embryos, we confirmed the neuronal expression of endogenous Casc3 mRNA (Fig. 1I–L). These data indicate Casc3 mRNA is expressed in the developing nervous system.
FIGURE 1.

Casc3 expression and phenotypes of a hypomorphic gene trap mutant allele. (A) Schematic representation of the Casc3RRU345 allele with exons (black boxes), introns (thin lines), and gene trap insertion (triangle), not to scale. Genotyping primers (black arrows: WT Forward, WT Reverse; red arrows: LacZ Forward and LacZ reverse) and Taqman probe (green bar: spanning exons 8 and 9). (B–H) Representative images of β-gal staining of whole mount embryos at E11.5 (B–D) and E12.5 (E–H) with indicated genotypes, showing lateral and ventral views. Note staining in the CNS (black arrows) and the heart (white arrows). (H) High-magnification view of an E12.5 embryo. (I–L) Whole mount in situ hybridizations for Casc3 mRNA in E11.5 (I,J) and E12.5 (K,L) wild-type embryos showing lateral and ventral views. Note Casc3 expression in the neocortex at both ages (black arrowheads). (M,N) qRT-PCR of wild-type Casc3 expression in dorsal cortices of E12.5 and E16.5 embryos from indicated genotypes. Casc3 expression in control littermates was set to 1.0, and all values were normalized to Gapdh. n = 3 biological replicates for each genotype for qPCR. (O,P) Quantification of Western analyses for CASC3 in E16.5 dorsal cortical lysates of indicated genotypes. For densitometry analysis (O), CASC3 expression level in control was set to 1.0, and loading was normalized using stain-free detection methods. Representative image (P) of the Western blot and stain-free loading control. n = 2–3 biological replicates for each genotype from two independent Westerns (shown in Supplemental Fig. 1). (Q–T) Representative images of whole mount E18.5 embryos (Q,R) and brains (S,T) from indicated genotypes. (U) Quantification of the ratio of brain size to body length for E18.5 embryos of indicated genotypes, with the ratio of control embryos set to 1.0. Two-tailed Student t-test, (***) P < 0.001, (**) P < 0.01, (*) P < 0.05, (n.s.) not significant. Error bars, SD, n = 6–10 for LacZ staining experiments, n = 3–5 for in situ hybridization. Scale bars, 1 mm.
We next measured Casc3 expression levels in Casc3 mutant heterozygous and homozygous E12.5 and E16.5 brains, at early and late stages of cortical development. qRT-PCR was performed using a Taqman probe spanning exons 8 and 9 of Casc3, which is located after the LacZ insertion and able to detect wild-type but not mutant fusion transcripts. Compared to littermate control embryos, wild-type Casc3 mRNA levels were significantly reduced at both ages. Casc3RRU345/+ showed 52%–54% significant reduction and Casc3RRU345/RRU345 showed 90%–95% reduction in Casc3 mRNA levels (Fig. 1M,N). This result was corroborated by Western analyses of dorsal cortical lysates using CASC3 antibody. By Western analysis, a single protein product was detected. Compared to control, CASC3 protein level was significantly reduced by 72% in E16.5 Casc3RRU345/RRU345 mutant cortices, also apparent at E11.5 and E15.5 (Fig. 1O,P; Supplemental Fig. S1A–C). Together these analyses indicate that Casc3RRU345 is a strong hypomorphic allele in the developing brain.
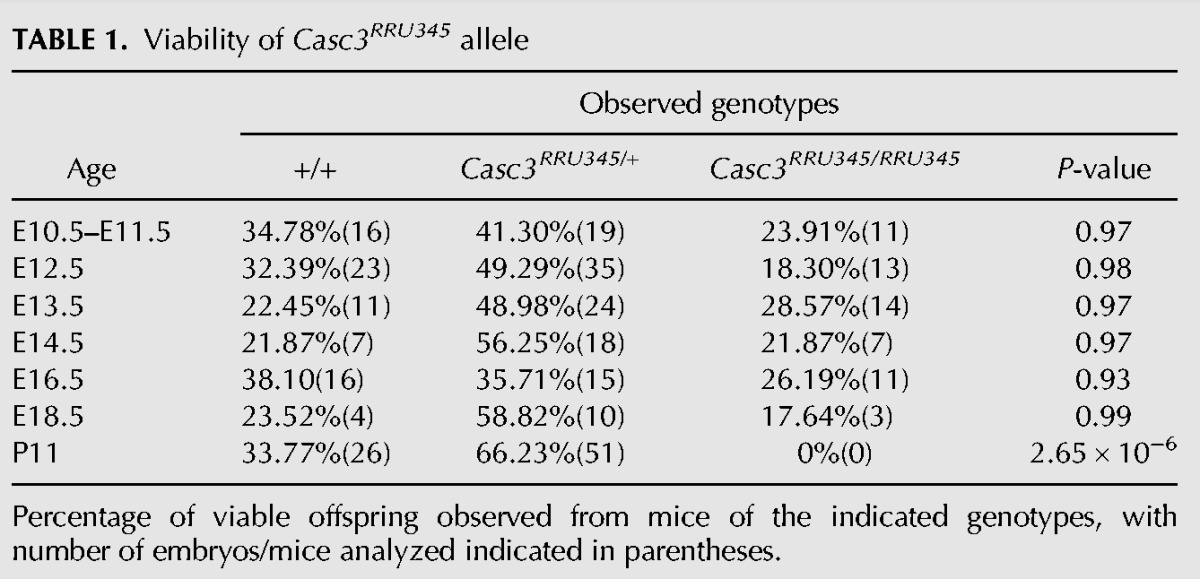
We next examined if reduced Casc3 expression impacted animal viability and development. Casc3RRU345/RRU345 embryos were viable throughout embryogenesis. However, by postnatal day 11, Casc3RRU345/RRU345 mutants were not recovered (P < 0.0001) (Table 1), indicating that Casc3 is essential for animal viability. We then examined mutant embryos at the end of gestation. At E18.5, homozygotes were viable. The Casc3RRU345/RRU345 embryos were markedly smaller than littermate controls. Their brains were also reduced in size. However, measurement of the brain size in whole mount embryos revealed it was proportionally reduced relative to body length (Fig. 1Q–U). This contrasts with MagohMos2/+ heterozygous brains, which are disproportionally reduced by ∼30%, relative to body size (Supplemental Fig. S1D; Silver et al. 2010). Together this indicates that loss of Casc3 expression causes lethality, but may not overtly disrupt brain size.
TABLE 1.
Viability of Casc3RRU345 allele
Depletion of Casc3 in compound embryos results in developmental delay but no striking defect in brain development
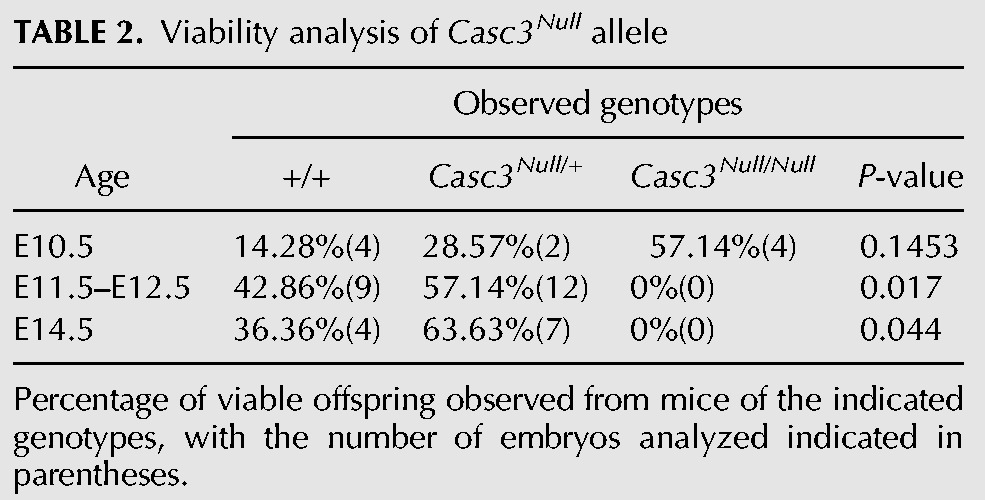
Given that Casc3RRU345/RRU345 animals retain some CASC3 expression in the developing brain, we sought to further challenge the idea that Casc3 is required for brain development. To this end, we generated a germline allele of Casc3 (Casc3tm1(KOMP)Vlcg, referred to as Casc3Null) using ES cells from the KOMP consortium (Fig. 2A). This allele contains a LacZ/Neo insertion that replaces exons 1–11, the entire Casc3 coding sequence. We examined Casc3Null/+ and Casc3Null/Null embryos at E10.5, when the brain is largely composed of neuroepithelial progenitors. At E10.5, Casc3Null/+ embryo size and morphology was comparable to control embryos. In contrast, E10.5 Casc3Null/Null embryos were markedly smaller and dysmorphic relative to control (Fig. 2B–D). One day later, at E11.5 and then E12.5, Casc3Null/+ embryos were phenotypically normal with no overt reductions in brain size compared to control littermates (Supplemental Fig. S2A–D). Thus a 50% depletion of CASC3 does not impair development, which contrasts with Magoh, Eif4a3, and Rbm8a mutants (Silver et al. 2010; McMahon et al. 2014; Mao et al. 2015, 2016). However, at E11.5, we failed to recover Casc3Null/Null embryos, indicating that Casc3Null/Null embryos are embryonic lethal between E10.5 and E11.5 (P = 0.017) (Table 2). This is consistent with a report of embryonic lethality in the same allele by the International Mouse Phenotyping Consortium (Brown and Moore 2012). Embryonic lethality at this stage can be a result of many problems, including defective cardiac development, which is notable given the robust Casc3 expression in the developing heart.
FIGURE 2.

Casc3 compound mutants deplete Casc3 expression in neocortices and exhibit developmental delay. (A) Schematic representation of the wild-type and Casc3Null alleles with exons (black box), introns (line), and null cassette (blue and yellow box), not to scale. Primers for genotyping are shown (black arrows: WT forward and reverse; red arrows: SU forward and LacZ reverse). The insertion was a knock-in of the coding sequence of Casc3 as depicted by homologous sequence in green outside of the exons, not to scale. (B–D) Representative images of E10.5 whole mount embryos with indicated genotypes. Note there was no overt body size or brain size difference of Casc3Null/+ compared to the control, but Casc3Null/Null embryo showed developmental delay. (E–I) Representative images of E13.5 whole mount embryos of indicated genotypes, with neocortices highlighted by dotted lines. Note Casc3RRU345/+ (F) showed no overt difference while Casc3RRU345/RRU345 (G) and Casc3RRU345/Null (H) embryos showed reduced body sizes and developmental delay. MagohMos2/+ (I) mutant showed milder developmental delay but significant brain size reduction. (J–M) Representative images of E14.5 whole mount embryos for control (J,L) and Casc3RRU345/Null (K,M) showing smaller size (J versus K) and developmental delay of limbs (L versus M). Note L is a high-magnification of J, and M is a different embryo than K. (N) qRT-PCR showing the relative Casc3 expression in E14.5 dorsal cortices from indicated genotypes. The Casc3 level in control brains was set to 1.0, and data were normalized to Gapdh. Note that Casc3 expression in Casc3RRU345/Null was virtually eliminated (0.01% that of control). Two-tailed student t-test, (***) P < 0.001. Error bars, SD. n = 3 biological replicates for each genotype. Scale bars: B–D, 500 µm; E–M, 1 mm.
TABLE 2.
Viability analysis of Casc3Null allele
The embryonic lethality associated with Casc3 homozygosity prohibited analysis of brain development, therefore we generated embryos carrying compound Casc3RRU345/Null alleles. Compound embryos were viable at E13.5 and E14.5, critical stages of brain development (Fig. 2E–M). Expression of Casc3 mRNA was measured by qRT-PCR of E14.5 Casc3RRU345/Null and Casc3RRU345/RRU345 dorsal neocortices. Comparison of Casc3 expression to control revealed that Casc3RRU345/RRU345 brains had a 95% reduction, consistent with analyses at younger ages (see Fig. 1M,N). Compound mutant embryos had a significant 99.9% reduction in Casc3, indicating this combination eliminates virtually all Casc3 expression in the developing neocortex (Fig. 2N). Examination of gross embryonic development revealed that E13.5 Casc3RRU345/+ embryos were phenotypically normal with no overt body or brain size differences compared to the littermate controls (Fig. 2E,F). In contrast, E13.5 and E14.5 Casc3RRU345/RRU345 and Casc3RRU345/Null mutants both showed developmental delay (noted by eye pigmentation and limb development) by ∼1 d when compared to the wild-type littermate controls (Fig. 2E,G,H,J–M). In comparison, the Magoh heterozygous germline mutant showed more mild developmental delay, with a disproportionate reduction in brain size (compare Fig. 2I to 2F,G,H).
Due to the developmental delay, we used stage-matched embryos to examine whether Casc3 depletion disrupted neurogenesis similar to other EJC mutants. We previously showed that E13.5 and E14.5 Magoh haploinsufficient embryos have reduced cortical thickness, fewer TBR2+ intermediate progenitors, along with excessive neurons and apoptosis (Silver et al. 2010). These same phenotypes are also evident in conditional models of EJC components (Emx1-Cre;MagohLoxP/+, Rbm8aLoxP/+, and Eif4a3LoxP/+ embryos) (McMahon et al. 2014; Mao et al. 2015, 2016), which delete the respective EJC components in the neocortex beginning at E9.5 (Gorski et al. 2002; Sahara and O'Leary 2009). Thus, we aimed to evaluate these same neurogenesis phenotypes in Casc3 mutants. At E14.5, both Casc3RRU345/RRU345 and Casc3RRU345/Null showed thinner cortical thickness compared to wild-type littermate controls (P = 0.02 and P = 0.002, respectively) (Fig. 3A,E,I,Q). Importantly, this difference was explained by developmental delay, as E13.5 control cortical thickness was similar to the E14.5 mutants (Fig. 3M,Q). We also quantified fewer TBR2+ intermediate progenitors in E14.5 Casc3RRU345/RRU345 and Casc3RRU345/Null brains compared to E14.5 controls. However, again the E14.5 values were similar to those of E13.5 control brains (Fig. 3B,F,J,N,R).
FIGURE 3.

Cortical neurogenesis defects of Casc3 compound mutants due to developmental delay. (A–P) E14.5 (A–L) and E13.5 (M–P) coronal sections of embryonic brains from control (A–D and M–P), Casc3RRU345/RRU345 (E–H), and Casc3RRU345/Null (I–L), stained for PAX6+ radial glia (A,E,I,M), TBR2+ intermediate progenitors (B,F,J,N), TUJ1+ neurons (C,G,K,O), and CC3+ cells (D,H,L,P). Dotted lines denote boundaries of dorsal cortices. (Q–S) Quantification of dorsal cortical thickness (Q) percentage of all cells which are TBR2+ within a 200 µm column (R), and thickness of TUJ1+ layer relative to cortex (S) for indicated genotypes. For cortical thickness, the value of control brains was set to 1.0. (Q–S) Two-tailed Student's t-test, (*) P < 0.05, (n.s.) not significant. Error bars, SD. n = 3–4 biological replicates each. Scale bars: A–P, 50 µm.
In EJC deficient brains, there is an increase in neurons and neuronal layers, and apoptosis of the cortex (Supplemental Fig. S3; Mao et al. 2016). These phenotypes were not evident in Casc3RRU345/RRU345 and Casc3RRU345/Null embryos. Instead, we observed a significant 12%–13% reduction in the thickness of the neuronal layer compared to E14.5 control brains (Fig. 3C,G,K,O,S). Moreover, there was no notable apoptosis in either Casc3RRU345/RRU345 or Casc3RRU345/Null mutants (Fig. 3D,H,L,P). This is in sharp contrast to the Magoh germline haploinsufficiency, which shows marked apoptosis at the same age (Supplemental Fig. 3E). It is also distinct from conditional haploinsufficiency for Magoh, Rbm8a, and Eif4a3 in the developing brain, in which just 50% reduction results in severe neurogenesis defects and microcephaly (McMahon et al. 2014; Mao et al. 2015, 2016). Thus in contrast to other EJC core components, depletion of Casc3 did not significantly affect neuronal differentiation, viability, or microcephaly. Together these analyses argue that neurogenesis defects in Casc3 mutants can be attributed in large part to developmental delay.
CASC3 protein levels and expression are distinct from MAGOH, RBM8A, and EIF4A3 in the developing brain
The absence of a striking developmental defect in neurogenesis in Casc3 mutants could be due to at least two possibilities. MAGOH, EIF4A3, and RBM8A could influence brain development primarily via non-EJC functions. Alternatively, perhaps CASC3 is not a core EJC protein in the developing brain. To further evaluate these possibilities, we examined expression patterns of CASC3 in the developing brain. We previously showed that MAGOH, EIF4A3, and RBM8A are all highly expressed in neural progenitors and enriched in nuclei (Mao et al. 2016). Consistent with the whole mount in situ and LacZ analyses (see Fig. 1), we detected CASC3 in the neocortex. Immunofluorescence against CASC3 showed its expression throughout the cortex, with enrichment in the cortical plate where neurons reside (Fig. 4A). CASC3 was primarily cytoplasmic, as has been previously reported in immortalized cells (Degot et al. 2004; Baguet et al. 2007). We also assessed expression of EJC components in isolated progenitors and neurons. Using E12.5 Dcx-dsRed dorsal cortices, neurons (Dcx positive), and progenitors (Dcx negative) were FACs sorted. qRT-PCR showed that Magoh, Rbm8a, and Eif4a3 were enriched in progenitors relative to the post-mitotic neurons (6.6-fold in Magoh, P = 0.004; 3.3-fold in Rbm8a, P = 0.002; and 3.8-fold in Eif4a3, P = 0.002). In contrast, Casc3 levels were similar between progenitors and neurons (Fig. 4B). These analyses suggest that Casc3 is distinct from the other EJC core components in its expression pattern and localization in the developing brain.
FIGURE 4.
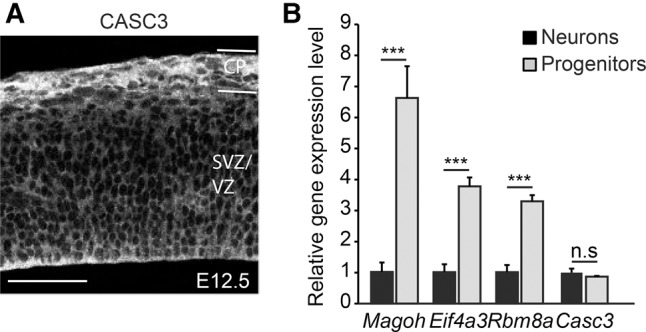
CASC3 expression in the developing cortex differs from other EJC core components. (A) Coronal section of E12.5 wild-type embryos stained for CASC3. Note that cytoplasmic localized CASC3 is expressed throughout the dorsal cortex with enrichment in the cortical plate (CP). (B) Quantification of gene expression for EJC components in sorted E12.5 progenitors (Dcx-dsRed negative, gray) and neurons (Dcx-dsRed positive, black). Note, Casc3 levels were comparable in progenitors and neurons while the other EJC components showed enrichment in progenitors. The levels of individual genes in the neurons (Dcx positive) were set to 1.0. Two-tailed Student's t-test, (***) P < 0.001. Error bars, SD. n = 2 biological replicates each for B. Scale bar, 50 µm.
In addition to these expression patterns we also examined CASC3 protein levels relative to MAGOH, EIF4A3, and RBM8A, using unbiased quantitative proteomic analyses of E11.5 cortices (n = 3 biological replicates) (Mao et al. 2016). Among 3587 proteins detected, MAGOH, RBM8A, and EIF4A3 were abundant (ranked 966, 2554, and 360, respectively). In contrast, CASC3 was not detected, suggesting that its protein levels are significantly lower than other core EJC components. This reinforces findings from HEK293 cells, in which CASC3 pull-down with MAGOH or EIF4A3 is significantly lower relative to other EJC proteins (Singh et al. 2012) and in which Casc3 mRNA levels are lower (Singh et al. 2015). Altogether, this indicates that in the developing brain, CASC3 exhibits distinct expression and localization patterns and protein levels relative to MAGOH, RBM8A, and EIF4A3. Taken together with our genetic analyses, this suggests that CASC3 is not a core EJC component in the developing neocortex.
DISCUSSION
The exon junction complex is an essential node for post-transcriptional regulation. Four components, MAGOH, EIF4A3, RBM8A, and CASC3, are thought to compose the core machinery of this complex. Yet this idea has not been challenged in a mammalian system in vivo. Here we demonstrate that unlike the other core components, Casc3 is largely dispensable for brain development. Casc3 depletion disrupts developmental progression resulting in smaller brain size and ultimately animal lethality. However, Casc3 depleted brains are not disproportionately reduced relative to body size, indicating that brain development is not nearly as sensitive to Casc3 dosage as it is for other components. We discover that CASC3 protein expression and levels are distinct from those of other core components in the developing brain. Together, this suggests that CASC3 may act as a peripheral EJC component in the developing brain and/or have tissue-specific functions. Our findings have implications for interpreting EJC function in development both within and outside of the brain.
Our analyses have exposed vastly different phenotypes between Casc3 and the other EJC components. Haploinsufficiency of Magoh, Rbm8a, and Eif4a3 each cause severe microcephaly due to massive apoptosis and altered progenitor proliferation (McMahon et al. 2014; Mao et al. 2015, 2016). In contrast, using compound embryos we show that a 99% reduction of Casc3 during brain development causes neither microcephaly nor apoptosis. Additionally, MAGOH, EIF4A3, and RBM8A are each essential for mitosis, whereas CASC3 is not (Silver et al. 2010; Ishigaki et al. 2013). This is relevant because mitotic defects in neural progenitors may underlie microcephaly in Magoh mutants (Pilaz et al. 2016). Moreover, if CASC3 works with MAGOH, EIF4A3, and RBM8A to influence neurogenesis, we predicted it should show similar expression patterns in the developing brain; however, our data suggest otherwise.
How can we reconcile these findings to understand CASC3 function? Given its expression in the developing brain, we hypothesize that in this tissue, CASC3 is primarily a peripheral EJC component, only interacting with a subset of EJCs for specific functions. The question of whether CASC3 functions as a peripheral EJC component in all tissues is fascinating and will require detailed molecular study. Another plausible interpretation is that CASC3 has distinct functions in different cells/tissues. Our current results do not differentiate between these two possibilities, and importantly, they are not necessarily mutually exclusive.
Based on our data and those of others, we posit that Casc3 functions in the nervous system primarily in post-mitotic neurons. Indeed, CASC3 was enriched in post-mitotic neurons of the cortical plate. In contrast, other core EJC components were more highly expressed in progenitors, and Magoh depletion from post-mitotic neurons has no overt impact on neuron number (Pilaz et al. 2016). This suggests that, instead of regulating neural progenitor differentiation, Casc3 might primarily influence neural maturation and synaptic function. CASC3 is implicated in neurons as a downstream target of miR-128 (Bruno et al. 2011). CASC3 is also important for mRNA transport in neurons, independent of MAGOH, RBM8A, and EIF4A3. This was demonstrated in E17 rat brains, in which only CASC3 interacted with STAUFEN2, a double-stranded RNA binding protein known to repress transport and translation in neurons (Köhrmann et al. 1999; Fritzsche et al. 2013). Future analyses of the role of Casc3 in post-mitotic neurons will be valuable for exposing EJC-dependent and possibly independent functions.
There may also be a differential requirement of CASC3 in other organs in the developing embryos. We showed that Casc3 expression is enriched in the developing heart, in which the enrichment of other EJC core components has not been reported. Depletion of Casc3 in early embryogenesis might cause cardiovascular defects and eventually lead to embryonic lethality of Casc3Null/Null embryos prior to E11.5. Thus it will also be of interest in future studies to evaluate heart development in CASC3 deficient embryos.
To conclude, the present work shows that Casc3 is required for proper embryogenesis and viability in mouse models. A recent study using iCLIP in HeLa cells showed that CASC3 binds to canonical EJC sites, which the authors argue supports the idea that CASC3 is a core protein (Hauer et al. 2016). This may be the case for many cell types; however, the work presented here, together with research in Drosophila models, also argues that CASC3 can have organ- and cell-specific requirements in vivo. This idea may extend to other EJC components as well, given that human mutations of RBM8A and EIF4A3 cause developmental disorders affecting distinct organs (bone and blood defects versus craniofacial). Altogether these genetic studies challenge assumptions of EJC composition in every cell.
MATERIALS AND METHODS
Mouse husbandry and generation of Casc3 mutant alleles
All experiments were performed in agreement with the guidelines from the Division of Laboratory Animal Resources from Duke University School of Medicine and IACUC. Plug dates were defined as embryonic day (E) 0.5 on the morning the plug was identified. The Casc3Gt(RRU345)Byg (Casc3RRU345) gene trap allele ES cells were acquired from MMRRC. The Casc3tm1(KOMP)Vlcg (Casc3Null) allele was acquired from the KOMP Repository Knockout mouse project from UC Davis. All experiments to analyze these lines were done on an inbred C57BL/6J background. For genotyping mice, the following conditions were used: 94°C × 2 min (1×); 94°C × 15 sec, 65°C × 30 sec, 72°C × 30 sec (30×); 72°C × 7 min (1×). For the gene trap allele: LacZ Forward: GATCCGCGCTGGCTACCGGC; LacZ Reverse: GGATACTGACGAAACGCCTGCC with internal control primers; Rapmycin Forward: AGCTTCTCATTGCTGCGCGCCAGGTTCAGG; Rapmycin Reverse: AGGACTGGGTGGCTTCCAACTCCCAGACAC. For the wild-type allele: Casc3 WT Forward: CAGCCACTTGACTGGCTCCTCA; Casc3 WT Reverse: GGGCTAAGGCAGAACACTCCGC. For genotyping Casc3Null mutant allele, SU Forward: AGAAGAGCTCGAAACCCTCC; LacZRev Reverse: GTCTGTCCTAGCTTCCTCACTG. Dcx-dsRed (TgDcx-DsRed)14Qlu mice were acquired from Jackson Laboratory.
Western blots and quantification
E16.5 embryonic dorsal cortices and bodies were collected from C57BL/6J (wild-type), Casc3RRU345/+, and Casc3RRU345/RRU345 mice and lysed in IP lysis buffer with protease inhibitors (Pierce). Cortical lysates were run on 4%–20% precasted Stain-Free SDS–Polyacrylamide gels (Bio-Rad). Gels were transferred onto nitrocellulose membranes and blotted using the following primary antibodies: rabbit anti-CASC3 (1:500, kind gift from Dr. Michael Kiebler, Germany). Blots were developed using ECL reagent (Pierce). Densitometry was performed using ImageJ. Loading control was determined through UV-induced fluorescence of separated protein bands, imaged by ChemiDoc MP Systems (Bio-Rad), and quantified by Image Lab software (Bio-Rad). Final values were quantified by normalizing CASC3 levels to loading controls and analyzed for significance using a two-tailed Student's t-test.
qRT-PCR analysis
Embryonic cortices were collected from E12.5, E14.5, and E16.5 C57BL/6J (wild-type), Casc3RRU345/+, Casc3RRU345/RRU345, and E14.5 Casc3RRU345/Null embryos, and RNA was extracted using the RNeasy Micro kit (QIAGEN). cDNA was prepared according to the iScript kit (Bio-Rad). qPCR was performed in triplicates using Taqman probes (Life Technologies): Casc3(Mm01296308_m1), and Gapdh (4352339E) with Taqman Real-Time PCR master mix (Thermo Fisher Scientific). Values were normalized to Gapdh loading control and the values of wild-type were set to 1.0 at each age. Student's t-test was used to determine the significance of mRNA level changes. For each genotype, three embryos were examined. For Dcx-dsRed mice, dorsal cortices from two E12.5 embryos were dissociated and FACS sorted. RNA was extracted from both Dcx-positive and Dcx-negative cells.
Staining and quantification of tissue sections
Brains were fixed overnight in 4% paraformaldehyde (PFA) at 4°C, followed by submersion in 30% sucrose until sinking, as previously described (Silver et al. 2010). Brain cryostat sections (20 µm) were prepared and stored at −80°C until use. Sections were permeabilized with 0.25% Triton X-100 for 10 min and blocked with MOM block reagent (Vector laboratories) for 1 h at room temperature (RT). Sections were incubated with primary antibodies for 2 h at RT or overnight at 4°C. Sections were then incubated in species appropriate secondary antibodies and Hoechst for 15 min at room temperature. The following primary antibodies were used: rabbit anti-CC3 (diluted 1:200; Cell Signaling); rabbit anti-TBR2 (1:1,000; Abcam); rabbit anti-PAX6 (1:1,000; Millipore); mouse anti-TUJ1 (1:400; Covance). The following secondary antibodies were used: Alex Fluor 488 and Alex Fluor 594 (1:400; Invitrogen). Hoechst (Thermo Fisher Scientific) was used for nucleus counterstain. High-magnification images were captured using a Zeiss Axio Observer Z.1 microscope coupled with an apotome. Cortical thickness was measured with Zen software. Cell quantification was performed with ImageJ/FIJI. Three sections from anatomically comparable regions per embryo and three biological replicates from control (wild-type) and mutant alleles (Casc3RRU345/+, Casc3RRU345/RRU345, and Casc3RRU345/Null) were quantified.
Whole mount LacZ staining and in situ hybridization
LacZ staining was done as previously described (Boyd et al. 2015). Whole mount in situ hybridization was performed as previously described (Silver et al. 2013). For in situ hybridizations, Casc3 sense and antisense probes were generated from a cDNA construct obtained from Open Biosystems (EMM1002-5827453). For antisense probes, T7 polymerase and EcoR1 digestion were used to generate a 700 nt probe. For sense probes, SP6 polymerase and HindIII digestion were used to generate a 700 nt probe. Only antisense data are included in the paper as sense showed no signal.
Mass spectrometry
We performed relative quantitation proteomic study of E11.5 dorsal cortices using the Duke Proteomics Core Facility. This methodology and the complete data set are described in Mao et al. (2016).
Prenatal and postnatal viability test
Yolk sacs from embryos and ear clips from P12 animals were collected for genomic DNA preparation. Genotypes were carried out as aforementioned. χ2 test was carried out to determine the viability.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Michael Kiebler for the generous gift of anti-Casc3 antibody. We thank members of the Silver laboratory for helpful discussions. We thank Jeremy Rouanet and Lei Shi for technical assistance, and the Duke Mouse Transgenic facility, Genome Sequencing facility, and Proteomics facility. Funding was provided by National Institutes of Health (NIH) grants R00-NS064197 and R01-NS083897.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.058826.116.
REFERENCES
- Andersen CBF, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CLP, Pedersen JS, Séraphin B, Le Hir HE, Andersen GR. 2006. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 313: 1968–1972. [DOI] [PubMed] [Google Scholar]
- Ashton-Beaucage D, Udell CM, Lavoie H, Baril C, Lefrançois M, Chagnon P, Gendron P, Caron-Lizotte O, Bonneil E, Thibault P, et al. 2010. The exon junction complex controls the splicing of MAPK and other long intron-containing transcripts in Drosophila. Cell 143: 251–262. [DOI] [PubMed] [Google Scholar]
- Baguet A, Degot S, Cougot N, Bertrand E, Chenard M-P, Wendling C, Kessler P, Le Hir HE, Rio M-C, Tomasetto C. 2007. The exon-junction-complex-component metastatic lymph node 51 functions in stress-granule assembly. J Cell Sci 120: 2774–2784. [DOI] [PubMed] [Google Scholar]
- Bono F, Ebert J, Lorentzen E, Conti E. 2006. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126: 713–725. [DOI] [PubMed] [Google Scholar]
- Boyd JL, Skove SL, Rouanet JP, Pilaz L-J, Bepler T, Gordân R, Wray GA, Silver DL. 2015. Human-chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr Biol 25: 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SDM, Moore MW. 2012. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome 23: 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, Lalani SR, Graham B, Lee B, Shinawi M, et al. 2008. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 40: 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, Song H-W, Corbett MA, Gifford WD, Gécz J, et al. 2011. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell 42: 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald G, Ebert J, Basquin C, Sauliere J, Jayachandran U, Bono F, Le Hir HE, Conti E. 2010. Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc Natl Acad Sci 107: 10050–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degot S, Le Hir HE, Alpy F, Kedinger V, Stoll I, Wendling C, Séraphin B, Rio M-C, Tomasetto C. 2004. Association of the breast cancer protein MLN51 with the exon junction complex via its speckle localizer and RNA binding module. J Biol Chem 279: 33702–33715. [DOI] [PubMed] [Google Scholar]
- Favaro FP, Alvizi L, Zechi-Ceide RM, Bertola D, Felix TM, de Souza J, Raskin S, Twigg SRF, Weiner AMJ, Armas P, et al. 2014. A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am J Hum Genet 94: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsche R, Karra D, Bennett KL, Ang FY, Heraud-Farlow JE, Tolino M, Doyle M, Bauer KE, Thomas S, Planyavsky M, et al. 2013. Interactome of two diverse RNA granules links mRNA localization to translational repression in neurons. Cell Rep 5: 1749–1762. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, Jones KR. 2002. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22: 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachet O, Ephrussi A. 2001. Drosophila Y14 shuttles to the posterior of the oocyte and is required for oskar mRNA transport. Curr Biol 11: 1666–1674. [DOI] [PubMed] [Google Scholar]
- Hauer C, Sieber J, Schwarzl T, Hollerer I, Curk T, Alleaume A-M, Hentze MW, Kulozik AE. 2016. Exon junction complexes show a distributional bias toward alternatively spliced mRNAs and against mRNAs coding for ribosomal proteins. Cell Rep 16: 1588–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y, Nakamura Y, Tatsuno T, Hashimoto M, Shimasaki T, Iwabuchi K, Tomosugi N. 2013. Depletion of RNA-binding protein RBM8A (Y14) causes cell cycle deficiency and apoptosis in human cells. Exp Biol Med 238: 889–897. [DOI] [PubMed] [Google Scholar]
- Köhrmann M, Luo M, Kaether C, DesGroseillers L, Dotti CG, Kiebler MA. 1999. Microtubule-dependent recruitment of Staufen-green fluorescent protein into large RNA-containing granules and subsequent dendritic transport in living hippocampal neurons. Mol Biol Cell 10: 2945–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir HE, Sauliere J, Wang Z. 2016. The exon junction complex as a node of post-transcriptional networks. Nat Rev Mol Cell Biol 17: 41–54. [DOI] [PubMed] [Google Scholar]
- Mao H, Pilaz LJ, McMahon JJ, Golzio C, Wu D, Shi L, Katsanis N, Silver DL. 2015. Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J Neurosci 35: 7003–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, McMahon JJ, Tsai YH, Wang Z, Silver DL. 2016. Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53-mediated microcephaly. PLoS Genet 12: e1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon JJ, Shi L, Silver DL. 2014. Generation of a Magoh conditional allele in mice. Genesis 52: 752–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Dillon ST, Boswell RE. 2001. The RNA-binding protein Tsunagi interacts with Mago Nashi to establish polarity and localize oskar mRNA during Drosophila oogenesis. Genes Dev 15: 2886–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmark PA, Boswell RE. 1994. The mago nashi locus encodes an essential product required for germ plasm assembly in Drosophila. Development 120: 1303–1313. [DOI] [PubMed] [Google Scholar]
- Nguyen LS, Kim H-G, Rosenfeld JA, Shen Y, Gusella JF, Lacassie Y, Layman LC, Shaffer LG, Gécz J. 2013. Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet 22: 1816–1825. [DOI] [PubMed] [Google Scholar]
- Palacios IM, Gatfield D, St Johnston D, Izaurralde E. 2004. An eIF4AIII-containing complex required for mRNA localization and nonsense-mediated mRNA decay. Nature 427: 753–757. [DOI] [PubMed] [Google Scholar]
- Pilaz L-J, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, Silver DL. 2016. Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron 89: 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roignant J-Y, Treisman JE. 2010. Exon junction complex subunits are required to splice Drosophila MAP kinase, a large heterochromatic gene. Cell 143: 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Traylor RN, Schaefer GB, McPherson EW, Ballif BC, Klopocki E, Mundlos S, Shaffer LG, Aylsworth AS; 1q21.1 Study Group. 2012. Proximal microdeletions and microduplications of 1q21.1 contribute to variable abnormal phenotypes. Eur J Hum Genet 20: 754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S, O'Leary DDM. 2009. Fgf10 regulates transition period of cortical stem cell differentiation to radial glia controlling generation of neurons and basal progenitors. Neuron 63: 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DL, Watkins-Chow DE, Schreck KC, Pierfelice TJ, Larson DM, Burnetti AJ, Liaw H-J, Myung K, Walsh CA, Gaiano N, et al. 2010. The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nat Neurosci 13: 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver DL, Leeds KE, Hwang H-W, Miller EE, Pavan WJ. 2013. The EJC component Magoh regulates proliferation and expansion of neural crest-derived melanocytes. Dev Biol 375: 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ. 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151: 750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Pratt G, Yeo GW, Moore MJ. 2015. The clothes make the mRNA: past and present trends in mRNP fashion. Annu Rev Biochem 84: 325–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroupe ME, Tange TØ, Thomas DR, Moore MJ, Grigorieff N. 2006. The three-dimensional arcitecture of the EJC core. J Mol Biol 360: 743–749. [DOI] [PubMed] [Google Scholar]
- Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, Harper CA, Meng EC, Lee RE, Yee A, L'Italien L, et al. 2003. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res 31: 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tange TØ, Shibuya T, Jurica MS, Moore MJ. 2005. Biochemical analysis of the EJC reveals two new factors and a stable tetrameric protein core. RNA 11: 1869–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeden FJ, Palacios IM, Petronczki M, Weston MJ, St Johnston D. 2001. Barentsz is essential for the posterior localization of oskar mRNA and colocalizes with it to the posterior pole. J Cell Biol 154: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Murigneux V, Le Hir HE. 2014. Transcriptome-wide modulation of splicing by the exon junction complex. Genome Biol 15: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.