

Summary
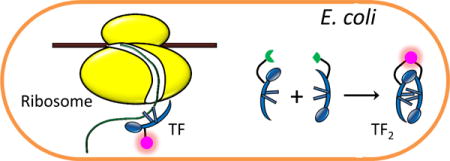
In bacteria, trigger factor (TF) is the molecular chaperone that interacts with the ribosome to assist the folding of nascent polypeptides. Studies in vitro have provided insights into the function and mechanism of TF. Much is to be elucidated, however, about how TF functions in vivo. Here we use single-molecule tracking, in combination with genetic manipulations, to study the dynamics and function of TF in living E. coli cells. We find that TF, besides interacting with the 70S ribosome, may also bind to ribosomal subunits and form TF-polypeptide complexes that may include DnaK/DnaJ proteins. The TF-70S ribosome interactions are highly dynamic inside cells, with an average residence time of ~0.2 s. Our results confirm that the signal recognition particle weakens TF’s interaction with the 70S ribosome, and further identify that this weakening mainly results from a change in TF’s binding to the 70S ribosome, rather than its unbinding. Moreover, using photoconvertible bimolecular fluorescence complementation, we selectively probe TF2 dimers in the cell and show that TF2 does not bind to the 70S ribosome but is involved in the post-translational interactions with polypeptides. These findings contribute to the fundamental understanding of molecular chaperones in assisting protein folding in living cells.
Keywords: molecular chaperone, trigger factor, unbinding kinetics, single-molecule tracking, photoconvertible bimolecular fluorescence complementation
Graphical abstract
In bacteria, trigger factor (TF) is the molecular chaperone that interacts with the ribosome to assist the folding of nascent polypeptides. We use single-molecule tracking, photoconvertible bimolecular fluorescence complementation, and genetic manipulations to probe the function and dynamics of TF and TF2 dimers in live E. coli cells. We gain insights into how TF and TF2 interact with the ribosome, the polypeptides, the DnaK/DnaJ chaperones, or the signal recognition particle in living cells.
Introduction
Upon translation from mRNA by the ribosome, the newly synthesized proteins need to fold properly to become functional. Many proteins fold spontaneously inside cells, but a significant fraction of them need assistance by molecular chaperones to reach their folded states efficiently and timely (Hartl, 1996). In E. coli, trigger factor (TF) is the first molecular chaperone that interacts with emerging nascent polypeptides (Figure 1A) and is responsible for the folding of ~70% proteins in the absence of ATP (Bukau et al., 2000, Hartl & Hayer-Hartl, 2009). Besides TF, the ATP-dependent DnaK/DnaJ/GrpE and GroEL/GroES chaperone systems constitute two major downstream folding pathways in bacterial cells (Hartl & Hayer-Hartl, 2009, Frydman, 2001, Hartl & Hayer-Hartl, 2002). They are responsible for the folding of 9–18% (Deuerling et al., 1999) and 10–15% (Ewalt et al., 1997) proteins, respectively (Figure 1B and C).
Figure 1.
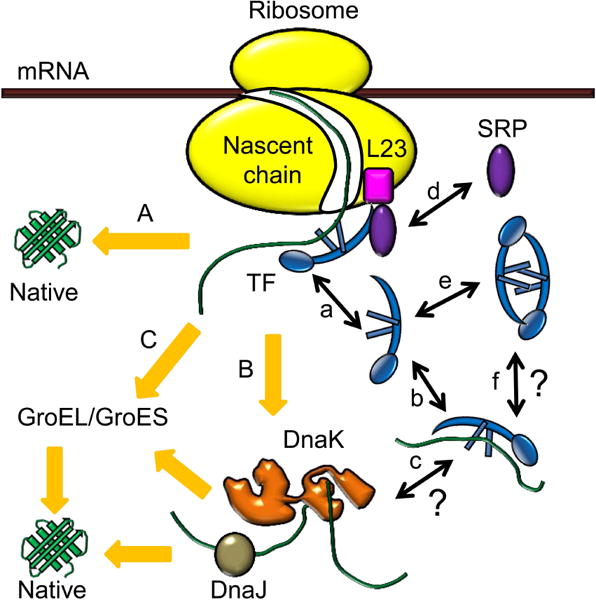
Molecular chaperone systems in E. coli, and functions of trigger factor (TF). Upon emerging from the ribosome, nascent polypeptides first interact with the ribosome-associated TF to fold to native states (pathway A). The polypeptides needing further assistance are transferred to the DnaK/DnaJ system (pathway B) or the GroEL/GroES system (pathway C) to complete folding. When associated with the ribosome, TF binds to the ribosomal protein L23 near the polypeptide exit site and goes through binding-unbinding cycles (a). In the cytoplasm, TF can bind to unfolded or partially folded polypeptides (b), but whether these TF-polypeptide complexes can interact with the DnaK/DnaJ system is unclear (c). Besides TF, another ribosomal associated factor, signal recognition particle (SRP), can also bind to L23 on the ribosome (d). The free TFs in the cytoplasm can exist as both monomers and dimers (e), but the potential role of TF2 dimer in binding to unfolded or partially folded polypeptides remains to be elucidated (f).
As the first chaperone helping nascent polypeptides to fold, TF interacts in 1:1 stoichiometry with the ribosomal protein L23 near the polypeptide exit site of the ribosome (Kramer et al., 2002, Lill et al., 1988). The interaction affinity varies depending on the ribosome’s state: when the ribosome is vacant, the affinity is weak with a dissociation constant of ~1 μM (Patzelt et al., 2002, Raine et al., 2004, Maier et al., 2003); when the ribosome is actively translating, the affinity can increase up to 20 fold, depending on the length and sequence of the nascent peptide chain (Raine et al., 2006). The interaction is also dynamic, in which TF undergoes continual binding-unbinding cycles (Figure 1a) (Kaiser et al., 2006), but there were conflicting results on the kinetics of TF unbinding from the ribosome. Earlier studies reported that TF unbinding was slow with a half-life of ~10 s on vacant ribosomes and up to ~50 s on translating ribosomes (Kaiser et al., 2006, Rutkowska et al., 2008, Maier et al., 2003). A more recent study provided a different range from 60 ms (on vacant ribosomes or ribosomes translating non-TF-binding-polypeptides) to 1.7 s (on ribosomes translating polypeptides with TF-specific sequences) (Bornemann et al., 2014), indicating that unbinding was much faster. This study also showed that the previously reported slow unbinding kinetics could be due to the particular fluorescent probe used to label TF (Bornemann et al., 2014). All these studies were performed in vitro, however. It remains unknown about TF’s unbinding kinetics from the ribosome in a living cell (Figure 1a).
In the cytoplasm, TF’s functions partially overlap with those of DnaK in binding nascent polypeptides or stabilizing and folding unfolded proteins (Figure 1b) (Teter et al., 1999, Agashe et al., 2004, Deuerling et al., 2003). Consequently, TF and DnaK compete for polypeptides, with TF being more competitive (Deuerling et al., 2003). In TF knockout strains, DnaK binds 2–3 fold more nascent polypeptides (Deuerling et al., 1999, Teter et al., 1999). On the other hand, TF and DnaK can also cooperate in folding large multidomain proteins co-translationally (Agashe et al., 2004). Yet, whether TF can cooperate post-translationally in a living cell with DnaK, as well as DnaK’s co-chaperone DnaJ, remains unclear (Figure 1c).
Besides TF, signal recognition particle (SRP), which is the first targeting factor that binds to nascent peptides, also interacts with the ribosomal protein L23 (Figure 1d) (Schaffitzel et al., 2006, Grudnik et al., 2009, Gu et al., 2003) and scans for the emerging hydrophobic signal-anchor sequence for co-translational membrane targeting (Bornemann et al., 2008, Holtkamp et al., 2012). An earlier study presented a model that TF and SRP competed for binding to ribosomes (Ullers et al., 2003). Later studies showed that TF and SRP could concurrently bind to the same vacant ribosome or ribosome-nascent chain complex (RNC), affecting each other’s conformation within the complex (Raine et al., 2004, Buskiewicz et al., 2004). Recent studies also concluded that although TF and SRP could bind simultaneously as ternary complexes with RNCs, the two proteins weakened each other’s interaction with RNC depending on the nascent polypeptides and the translational stages (Ariosa et al., 2015, Bornemann et al., 2014). However, all of these studies were carried out in vitro; how TF and SRP affect each other in interacting with the ribosome in a living cell is not fully defined.
Moreover, TF exists in about 2 to 3 fold molar excess over the ribosome inside bacterial cells. The majority of TF is thus free in the cytoplasm (Patzelt et al., 2002), and it can dimerize (Figure 1e) with a dissociation constant of 1–2 μM (Maier et al., 2003). In vitro, the Thermotoga maritima TF can form 2:2 complexes with the ribosomal protein S7 (Martinez-Hackert & Hendrickson, 2009), suggesting that TF dimers (i.e., TF2) may be involved in the ribosome assembly process, while E. coli TF2 can bind denatured proteins (Liu et al., 2005). Inside cells, the function of TF2 is unclear, however (Figure 1f).
To address the above knowledge gaps, we report here a single-molecule tracking (SMT) (Elf et al., 2007, Chen et al., 2015b, English et al., 2011, Javer et al., 2013, Mehta et al., 2013, Bakshi et al., 2011, Mazza et al., 2012, Gahlmann & Moerner, 2014) study of TF dynamics in living E. coli cells, where TF is tagged with a photoconvertible fluorescent protein reporter. This in vivo measurement can potentially uncover TF’s interactions with other proteins inside the cells and provide the actual dynamics of TF in the complex cellular environment rather than a well-controlled in vitro condition. Complementing SMT with photoconvertible bimolecular fluorescence complementation (PC-BiFC) (Nickerson et al., 2014, Liu et al., 2014) further allows us to probe selectively the function and dynamics of TF2 dimers in the cell. By resolving and quantifying the different diffusive behaviors of single TF molecules in combination of genetic manipulations of the cell, we gain insights into how TF and TF2 can interact with the ribosome, the polypeptides, DnaK/DnaJ, or SRP in living bacterial cells.
Results and Discussion
SMT resolves three diffusion states of TF in living E. coli cells; one of them is the freely diffusing state
To visualize TF in a living E. coli cell, we fused to its C-terminus a photoconvertible fluorescent protein mEos3.2 (i.e., mE) (McKinney et al., 2009, Zhang et al., 2012) and encoded this TFmE fusion gene at its chromosomal locus. Protein gel analyses of the cell lysate show that TFmE stays intact in the cell (Figure S1A–B). Cell growth assay under SDS/EDTA stress (Oh et al., 2011) in comparison with the wild-type strain (BW25113) and the TF knockout strain (i.e., Δtig) further shows that this tagged TFmE is as functional as the untagged TF (Figure S2A).
We then used a 405 nm laser to photoconvert the mE tag of TFmE one at a time in a living cell from its naturally green fluorescent form to its red fluorescent form, and subsequently used a 561 nm laser to induce the red fluorescence to image single red fluorescent TFmE via time-lapse stroboscopic imaging (Elf et al., 2007, Chen et al., 2015b, English et al., 2011, Javer et al., 2013, Mehta et al., 2013, Gahlmann & Moerner, 2014, Bakshi et al., 2011, Mazza et al., 2012) (Figure 2A inset; and SI Section S4). By localizing each red TFmE molecule’s position to ~25 nm precision in each image over a time-lapse series, we tracked its position in a living E. coli cell until the mE tag photobleached (Figure 2A).
Figure 2.
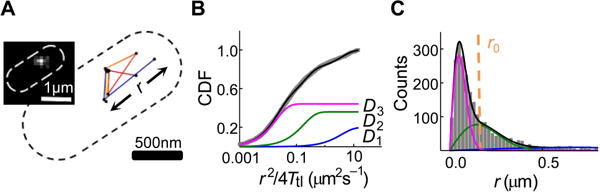
SMT resolves three diffusion states of TF in living E. coli cells. (A) Exemplary tracking position trajectory of a TFmE molecule inside a cell. Dash line is the cell boundary. Inset: a stroboscopic fluorescence image of a TFmE molecule in a cell. (B) Cumulative distribution function (CDF) of displacement length r per time lapse (Ttl = 60 ms) of TFmE in living cells, plotted against r2/4Ttl. Fitting with Eq. S9 (black line; SI Section S6.1) resolves three diffusion components, with their effective diffusion constants (and fractional populations): D1 = 3.85 ± 0.14 μm2 s−1 (20 ± 2%), D2 = 0.18 ± 0.04 μm2 s−1 (36 ± 3%), and D3 = 0.02 ± 0.01 μm2 s−1 (44 ± 1%). The three components are also plotted individually (colored lines). (C) Histogram of displacement length r and the corresponding probability distribution function (PDF, black line) of r from B, with the resolved three individual components (colored lines). The vertical red dashed line indicates the upper threshold r0 = 220 nm that selected dominantly the displacement lengths of the D3 state.
To quantify the diffusive behaviors of TFmE in a cell, we determined the cumulative distribution function (CDF) of its displacement length r per time lapse (Ttl = 60 ms), as well as the corresponding probability distribution function (PDF) of r (Figure 2B and C). The CDF (and PDF) requires minimally three diffusive components to be fitted satisfactorily, each following Brownian diffusion behaviors (Eq. S9 in SI Section S6.1). The effective diffusion constants of the three diffusion states are 3.85 ± 0.14, 0.18 ± 0.04, and 0.02 ± 0.01 μm2 s−1 (referred to as D1, D2, and D3, respectively), with fractional populations of 20 ± 2%, 36 ± 3%, and 44 ± 1%, respectively (Figure 2B and C). These effective diffusion constants are affected by the confinement from the small cell geometry, where fast moving molecules are affected more significantly; and they correspond to intrinsic diffusion constants of ~11, 0.2, and 0.02 μm2 s−1, respectively, on the basis of simulations and control experiments reported previously (Chen et al., 2015b).
We further verified the minimal number of diffusion states of TFmE in the cell via hidden Markov model analysis using the vbSPT software package (Persson et al., 2013), as well as inverse transformation of the confined displacement distributions (ITCDD), which deconvolutes the cell confinement effect (Oswald et al., 2014, Chen et al., 2015a) (SI Sections S6.2–S6.3). The vbSPT analysis gave effective diffusion constants (and fractional populations) of 4.40 ± 0.42 μm2 s−1 (14 ± 1%), 0.19 ± 0.05 μm2 s−1 (31 ± 3%), and 0.02 ± 0.01 μm2 s−1 (54 ± 4%), similar to those of CDF and PDF analyses. The ITCDD analysis directly gave the intrinsic diffusion constants (and their fractional populations) of 7.3 ± 2.4 μm2 s−1 (21 ± 1%), 0.20 ± 0.02 μm2 s−1 (33 ± 3%), and 0.02 ± 0.01 μm2 s−1 (45 ± 2%), again consistent.
We assigned the D1 state as the freely diffusing TFs in the cytoplasm for the following reasons: (1) This freely diffusing state is expected to exist and be the fastest among all possible diffusive behaviors of TF in a cell. (2) Both the effective diffusion constant (3.85 ± 0.14 μm2 s−1) and the intrinsic diffusion constant (7.3 ± 2.4 μm2s−1) of the D1 state are consistent with those of the free mE tag and of the freely diffusing state of a mE-tagged transcription regulator we previously studied in living E. coli cells under the same imaging conditions (i.e., 3.3 to 3.7 μm2 s−1 and 7.0 to 14.9 μm2 s−1 for the effective and intrinsic diffusion constants, respectively) (Chen et al., 2015b).
Assignment of the D3 state of TF: 70S-ribosome-bound state
The effective and intrinsic diffusion constants of the D3 state of TFmE are very small, ~0.02 μm2 s−1 (they do not differ much because the cell confinement does not affect much the motions of slow-diffusing molecules, which is close to the reported diffusion constant of the fully assembled 70S ribosome (0.04 to 0.06 μm2 s−1) (Bakshi et al., 2012, Sanamrad et al., 2014). Thus we assign it as the 70S-ribosome-bound state. It should be noted this diffusion constant value is close to our localization uncertainty, and therefore the corresponding molecules are effectively immobile in our measurements.
Consistently, when overexpressing TFmE from a plasmid in addition to the chromosomal copy of TFmE, the fractional population (A3) of the D3 state decreases from ~44% to ~16% (Figure 3A), as the increased copy number of TFmE diminishes the percentage of TFmE bound to the 70S ribosome out of the total TFmE in the cell. When TF’s residues 44–46 FRK are mutated to AAA (i.e., ), which is known to reduce TF’s association with the ribosome (Kramer et al., 2002), the fractional population of the D3 state in cells with similar TF expression levels decreases to a negligible ~5% (Figure 3A), further supporting our assignment (note that our experimental uncertainty is ~5% in determining the fractional populations of different diffusion states). For this type of comparisons, the expression level of TFmE in each cell was determined using single-cell quantification of protein concentration (SI Section S4.3), which allowed for sorting individual cells into groups of various protein concentrations and only comparing cells with similar expression levels to avoid any potential concentration-dependent effects.
Figure 3.
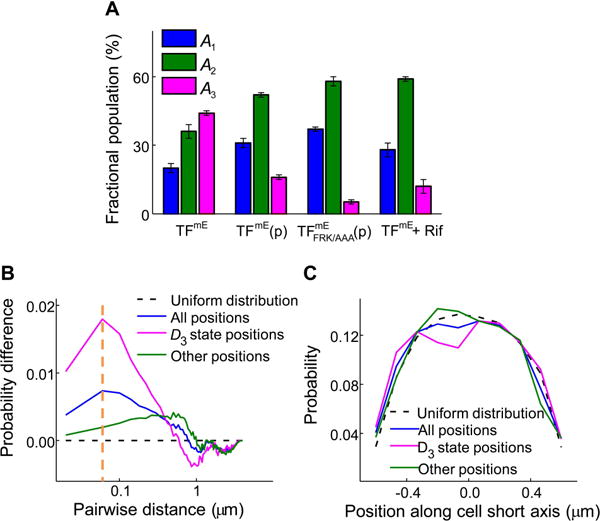
Assignment of the D3 state of TF as the 70S-ribosome-bound state. (A) Comparison of fractional populations of the three diffusion states of TFmE in the chromosomally-tagged strain (i.e., TFmE), further overexpressed from a pBAD24 plasmid (i.e., TFmE(p)), with the FRK/AAA mutations expressed from a pBAD24 plasmid in the tig knockout strain (i.e., (p)), or in the strain treated with 200 μM rifampicin (i.e., TFmE + Rif). (B) Probability distribution difference of pairwise distances of initial TFmE tracking positions relative to the simulated uniform position distributions in the cell. Other positions: the positions excluding those of the thresholded D3 state positions. (C) Probability distribution of the initial TFmE tracking positions along the cell short axis.
To further confirm the D3 state being TFmE bound to the 70S ribosome rather than ribosomal subunits, we treated the cells with the drug rifampicin (Rif) (SI Section S3), which blocks transcription initiation, decreasing the cellular mRNA level and thus the amount of the 70S ribosomes that assemble on mRNA. Consistently, Rif treatment leads to a decrease of the fractional population of the D3 from ~44% to ~12% (Figure 3A), further supporting our assignment.
Past studies have shown that 70S ribosomes often cluster together because of multiple ribosomes actively translating on a single mRNA (Staehelin et al., 1963, Warner et al., 1963), forming so-called polyribosomes whose size could be ~100 nm on the basis of electron microscopy images (Staehelin et al., 1963). Consequently, the 70S-ribosome-bound TFs are expected to cluster spatially as well in the cell. To probe this, we computed the probability distribution of pairwise distances between the initial positions of individual TFmE tracking trajectories and subtract from it the corresponding probability distribution calculated from simulated positions that are uniformly distributed within the cell (SI Section S8.1.1). This probability distribution difference of pairwise distances shows a peak at ~60 nm (blue curve, Figure 3B), supporting that some of TFmE indeed form clusters of comparable size to polyribosomes. It is worth noting that because of the molecular motions during the imaging time, the cluster size determined from the pairwise distance analysis here reflects an upper limit of the actual cluster size.
To more cleanly probe the D3 state of TFmE in the cell, we used the PDF of displacement length r of TFmE in Figure 2C, and thresholded this PDF with an upper limit r0 = 220 nm, below which >99% of the displacement lengths and the corresponding TFmE positions of the D3 state are included. We then calculated the probability distribution of pairwise distances between these thresholded positions and again subtract from it the one from the uniform distribution. This D3-state-dominated probability distribution difference of pairwise distances shows an enhanced peak at ~60 nm (magenta curve, Figure 3B). Concurrently, for the probability distribution difference of pairwise distances from the remaining positions, the peak at ~60 nm vanished (green curve, Figure 3B). Moreover, for , which has weakened association with the 70S ribosome, no significant peak is observed at this pairwise distance (Figure S10A). These analyses thus further support that the D3 state is the 70S-ribosome-bound TFs.
Previous studies (Sanamrad et al., 2014) have also shown that 70S ribosomes, but not ribosomal subunits, are excluded from the bacterial nucleoid, which is located mostly in the middle of the cell. The 70S-ribosome-bound TF is thus expected to be excluded from the nucleoid as well, showing decreased spatial distribution in the middle of the cell. To probe this, we obtained the distribution of all initial positions of TFmE tracking trajectories with respect to the short axis of the rod-shaped E. coli cell (blue curve, Figure 3C), combining and overlaying results from many individual cells (SI Section S8.2). Compared with that from simulated uniformly distributed positions (dashed black line, Figure 3C), this distribution shows a slight dent in the middle, suggesting a decreased localization of TFmE in the middle of the cell. In contrast, the position distribution of , which has weakened association with the 70S ribosome, does not show this dent and has the same shape as that of uniformly distributed positions (magenta curve, Figure S11A). Using the thresholded, D3-state-dominated TFmE positions, this dent in the middle of the cell in the position distribution is even clearer (magenta curve, Figure 3C), beyond the noise level (SI Section S8.2.2). Moreover, upon treating the cells with the drug kanamycin (Kan), which inhibits translation and causes nucleoid contraction (Bakshi et al., 2014, Misumi & Tanaka, 1980, Pestka, 1974, Sohmen et al., 2009), the depth of this dent decreases (Figure S12C vs. B). Altogether, these results further support that the D3 state of TFmE is the 70S-ribosome-bound state, which has a decreased localization in the middle of the cell because of the nucleoid exclusion of the 70S ribosome.
Assignment of the D2 state of TF: a mixture of TFs bound to free ribosomal subunits and TF-polypeptide(-DnaK/DnaJ) complexes
The smaller value of D2 (~0.2 μm2 s−1) of TFmE than the freely diffusing state D1 indicates that TF here must be interacting with other species in the cell. As TF can bind to the protein L23 of the 50S subunit in the assembled 70S ribosome, TF can likely bind to free ribosomal subunit 50S as well, contributing to the D2 state. Consistently, D2 of TFmE is close to the diffusion constant (0.12–0.40 μm2 s−1) (Bakshi et al., 2012, Sanamrad et al., 2014) of free ribosomal subunits (50S and 30S). However, only 15% of ribosomal subunits are free inside cells (Forchhammer & Lindahl, 1971, Sanamrad et al., 2014) and TF is in 2–3 fold molar excess over the ribosome (Patzelt et al., 2002). The fractional population (A2 ~36%) of the TFmE D2 state is thus too large to assign it to be entirely a population bound to free ribosomal subunits. There must be some other component(s) within the D2 state.
When overexpressing TFmE from a plasmid in addition to the chromosomal copy of TFmE, the value of D2 increases from ~0.2 to ~0.7 μm2 s−1 (Figure 4A), again suggesting that the D2 state contains at least another component than those bound to free ribosomal subunits, and that this component has faster diffusion than the ribosomal subunits. Moreover, for the mutant with reduced association with the ribosome (and thus the ribosomal subunit as well), D2 further increases to ~0.9 μm2 s−1 (Figure 4A), which is consistent with that the D2 state now has less contribution from the relatively slower population bound to free ribosomal subunits. Taken together, these results support that the D2 state of TF contains at least two major components: one slower component that is bound to free ribosomal subunit, the other faster component in which TF interacts with other proteins that are smaller than free ribosomal subunits; and these two components are not directly resolved in our analysis of CDF and PDF of TF’s displacement lengths as in Figure 2B–C. Considering TF’s role as a chaperone and that many proteins that are known to interact with TF are inactive proteins (e.g., polypeptides) (Lill et al., 1988, Kandror et al., 1999, Crooke & Wickner, 1987, Lecker et al., 1989, Kandror et al., 1995), we are inclined to that the second major component of the D2 state is TF’s interaction complexes with polypeptides in the cytoplasm; these complexes might contain some other proteins (e.g., DnaK or DnaJ; see below) and could contain multiple copies of the involved proteins to be significant in molecular mass.
Figure 4.
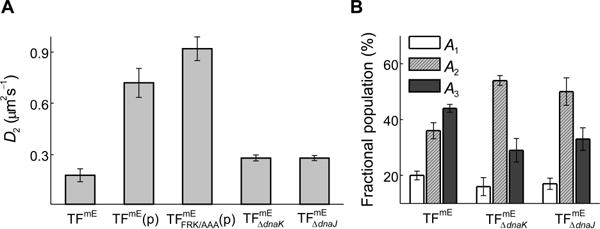
Assignment of the D2 state of TF. (A) Comparison of the value of diffusion constant D2 of TFmE in the chromosomally-tagged strain (TFmE), further overexpressed from a pBAD24 plasmid (i.e., TFmE(p)), containing the FRK/AAA mutations expressed from a pBAD24 plasmid in the tig strain ( (p)), chromosomally-tagged in dnaK strain ( ), and chromosomally-tagged in dnaJ strain ( ). (B) Comparison of fractional populations of the three diffusion states of TFmE, , and .
To further support the presence of the two major components of the D2 state, we examined chromosomally expressed TFmE in strains whose dnaK or dnaJ gene was deleted (i.e., or ). In both cases, the value of D2 does not change much (~0.3 μm2 s−1 for and , compared with ~0.2 μm2 s−1 for TFmE, Figure 4A), but the fractional population (A2) of the D2 state increases from ~36% to ~54% ( ) or ~50% ( ) (Figure 4B), suggesting that both DnaK and DnaJ are involved, directly or indirectly, in the functions of TF’s D2 state. This involvement likely affects both components of the D2 state approximately equally, so that when DnaK or DnaJ is deleted, the fractional populations of both components in the D2 state increase by comparable extents, leading to no significant changes in the value of the diffusion constant D2. Upon deleting dnaK or dnaJ, the population increase of TF bound to the free ribosomal subunits can be rationalized by the possibility that TF takes over DnaK’s role as a complement in facilitating ribosome assembly (Alix & Guerin, 1993, Sbai & Alix, 1998), while the population increase of the polypeptide-bound TF can be rationalized by the interruption of the pathway in which TF-polypeptide complexes interact with DnaK/DnaJ to transfer the unfolded polypeptides to complete folding (Liu et al., 2005).
Altogether, we assign the D2 state of TF as most likely a mixture that contains two unresolved major components: (1) a slower-diffusing TF population bound to free ribosomal subunits, and (2) a relatively faster diffusing TF-polypeptide complexes, which may include DnaK/DnaJ proteins and which could contain multiple copies of TF and/or DnaK/J.
TF-70S ribosome interaction is transient in living cells
From the single-molecule tracking trajectories (Figure 2A), we obtained displacement length r per time-lapse vs. time trajectories, which sometimes show clear transitions between large and small r values (Figure 5A). The smaller r values (e.g., r ≤ 220 nm) are dominated by the D3 state, i.e., TFmE bound to the 70S ribosome (Figure 2C), while also having contributions from TFmE in the D2 state (~35%). Thresholding this r-vs.-time trajectory with an upper displacement limit r0 = 220 nm (see Figure 2C) selects out the small displacements as well as the individual time durations (i.e., microscopic residence time τ) dominated by a single TFmE protein molecule bound to a 70S ribosome. Each microscopic residence time τ starts when r drops below r0 and ends when r increases above r0 (e.g., τ1 in Figure 5A) or when the mE-tag photobleaches or photoblinks (e.g., τ2 in Figure 5A). Combining the individual τ values from many single-molecule r-vs.-time trajectories, we obtained the distribution of τ (Figure 5B). Using a simple kinetic model that accounts for the photobleaching/photoblinking kinetics of the mE tag, we analyzed the distribution of τ to obtain an estimate of the unbinding rate constant kd (= 5.0 ± 0.8 s−1) of TFmE from the 70S ribosome (see analysis details in SI Sections S7.1–S7.2). We further validated the analysis of this r0-thresholded residence time by varying the r0 value and by analysis using the hidden Markov model (SI Section S7.3).
Figure 5.
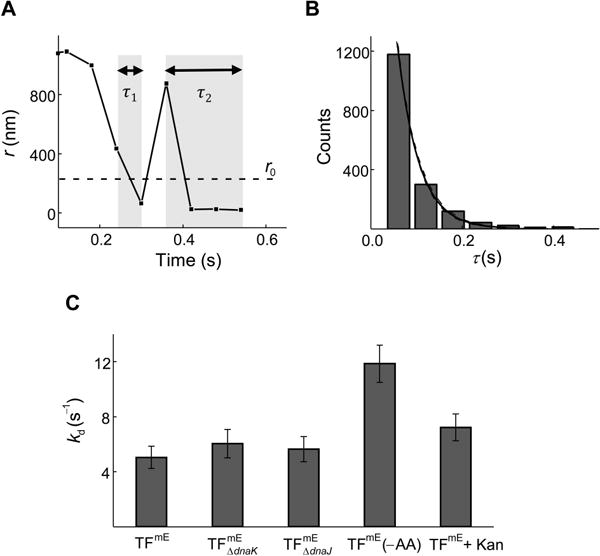
Unbinding kinetics of TF from the 70S ribosome. (A) Exemplary single-molecule displacement length r per time lapse (Ttl = 60 ms) vs. time trajectory of TFmE in the chromosomally-tagged strain (TFmE) The dashed line denotes the r0 =220 nm threshold as in Figure 2C, and the shaded regions represent two of the microscopic residence time τ. (B) Histogram of residence time τ of TFmE. The black solid line is a fit with (Tint is the laser exposure time during each image and Ttl is the time lapse, Eq. S11), where the photobleaching/blinking rate constant is kbl = 248 s−1 (Figure S8B). The dashed lines indicate the 95% confidence bounds of the fit. (C) Comparison of the unbinding rate constant kd of TFmE , , TFmE (−AA), and TFmE + Kan.
This kd corresponds to an average residence time of ~0.2 s for TFmE bound to the 70S ribosome (including both vacant ribosomes and RNCs), close to that of a recent in vitro study (0.09 to 2.4 s, corresponding to a half-life of 0.06 to 1.7 s; the variability here comes from TF binding to vacant ribosomes or different RNCs; the largest value is for TF’s interaction with ribosomes translating TF-specific sequences (Bornemann et al., 2014)). Alternatively, a hidden Markov model analysis of the single-molecule tracking trajectories using the vbSPT software package (Persson et al., 2013) gave an estimated residence time of ~1.4 s (SI Section S7.3.2), within an order of magnitude of ~0.2 s (the difference here could be due to that our r0-thresholded residence times contain contributions from the D2 state; SI Section S7.3.2) and also close to the recent in vitro study (Bornemann et al., 2014). Considering that the average ribosome translation speed in E. coli is about 12 to 21 amino acids per second (Young & Bremer, 1976, Dennis & Bremer, 1974) and a typical protein contains about 50–1000 amino acids, the average translation time for a protein in an E. coli cell is about seconds to minutes, significantly longer than the average residence time of TFmE on the 70S ribosome determined here. Therefore, during the translation of one polypeptide chain in a living E. coli cell, the interactions of TF with the translating 70S ribosome is very dynamic: within each TF binding event, the TF molecule only stays bound to the 70S ribosome for a small portion of the entire translation period and unbinds from the ribosome-nascent chain complex (RNC) promptly. This transient interaction could allow each TF molecule to sample multiple 70S ribosomes in a cell during a short time period and facilitate the folding of the nascent proteins efficiently (Bornemann et al., 2014).
Moreover, in the dnaK and dnaJ strains, kd does not show any significant changes (Figure 5C), suggesting that DnaK and DnaJ do not affect the stability of the TF-70S ribosome complex and that DnaK and DnaJ likely do not interact with TF directly on the 70S ribosome. When using cells grown under amino acid deficiency (TFmE (−AA)) or treated with the translation inhibitor Kan (TFmE + Kan), the unbinding rate constant kd increases appreciably (Figure 5C), consistent with that these cells have inactive protein translation so that TF molecules’ residence time is shorter on the 70S ribosome. This trend agrees with previous in vitro results that TF stays shorter on non-translating ribosomes than on translating ribosomes (except for ribosomes translating polypeptides without TF-specific sequences) (Kaiser et al., 2006, Rutkowska et al., 2008, Bornemann et al., 2014, Maier et al., 2003).
SRP weakens TF’s interaction with RNC, but does not affect the kinetics of TF’s unbinding from it
Despite the many in vitro studies, how TF and SRP affect each other’s interaction with the 70S ribosome remains to be better defined in living cells. To probe their relations, we overexpressed the protein component Ffh of SRP in the TFmE strain, generating the TFmE + SRP strain (SI Section S1.5 and Figure S1D). As SRP’s RNA component, 4.5S RNA, exists in the cell in about four-fold excess over Ffh (Jensen & Pedersen, 1994), this Ffh overexpression should increase the cellular SRP level, where we can examine its effect on TF-70S ribosome interactions.
While not affecting the diffusion constants of all diffusion states of TFmE (Table S4), this SRP overexpression decreases the fractional population of TF’s 70S-ribosome-bound state (A3 drops from 44 ± 1% to 36 ± 3%; Figure 6A), indicating that SRP weakens TF’s interaction with the 70S ribosome, which agrees with the in vitro observation that SRP decreases the binding affinity of TF to the ribosome (Bornemann et al., 2014). On the other hand, the unbinding rate constant kd of TFmE from the 70S ribosome does not show significant changes (Figure 6B). Therefore, the SRP’s influence mainly results from slowing TF’s binding to the 70S ribosome; once TF is bound to the 70S ribosome, SRP either hardly bind to the TF-ribosome complex or co-bind to the ribosome L23 site without increasing the unbinding kinetics of TF.
Figure 6.
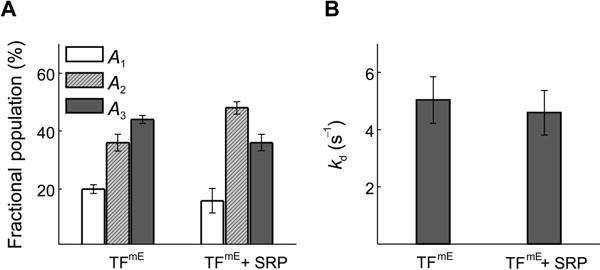
Effects of SRP on TF’s interaction with the 70S ribosome. (A) Comparison of fractional populations of the three diffusion states of TFmE and TFmE + SRP. (B) Comparison of the unbinding rate constant kd of TFmE and TFmE + SRP.
In normal cells without overexpression, cellular SRP concentration is much lower, where it would be even less likely for SRP to affect the unbinding of TF from the ribosome. Assuming the influence between SRP and TF for binding to the ribosome is bilateral (i.e., TF can also affect SRP’s binding to ribosome), then once a ribosome is occupied by TF, the timing for SRP to bind to the same ribosome for effective targeting would be interfered. Therefore, the facile unbinding of TF from the 70S ribosome discussed in the previous section, as well as the fact that SRP is recruited to translating ribosomes even before the nascent peptide emerges from the ribosome exit tunnel (Bornemann et al., 2008), would both be important to ensure SRP to have sufficient chances to interact with ribosomes timely.
Photoconvertible bimolecular fluorescence complementation (PC-BiFC) probes the single-molecule dynamics of TF2 dimer
Since free TFs exist in a monomer-dimer equilibrium in the cytoplasm, the fast-moving freely diffusing state D1 contains also TF2 dimers besides the monomers. Regarding the D2 state that involves TF interacting with free-ribosomal-subunits and polypeptides, it remains unclear if TF2 dimers would also be involved. To probe specifically the function and dynamics of TF2 dimers, we explored a PC-BiFC approach to trap TF2 dimers in the cell (Nickerson et al., 2014, Liu et al., 2014). In this approach, the photoconvertible fluorescent protein mE is split into two fragments, a larger N-terminal fragment mEN (residue 1–164) and the other smaller C-terminal fragment mEC (residue 165–225); each is used to fuse to TF’s C- or N-terminus via a flexible linker, creating TF-mEN and mEC-TF, respectively (see details in SI Section S1.6). When the two fragment-tagged TFs are co-expressed in a cell, the dimerization of TF brings together the two fragments, which complement irreversibly to form a functional mE, generating (Figure 7A). Subsequent controlled photoconversion and single-molecule stroboscopic imaging then allows for tracking single molecules in a living cell. In this complex, the two TF can transiently separate but will rapidly dimerize again “intramolecularly” due to the tethering by the complemented mE. On the basis of TF’s natural dimerization affinity, this tethered complex should spend >98% of its presence in the dimerized form rather than two monomers tethered by mE (SI Section S9.2).
Figure 7.
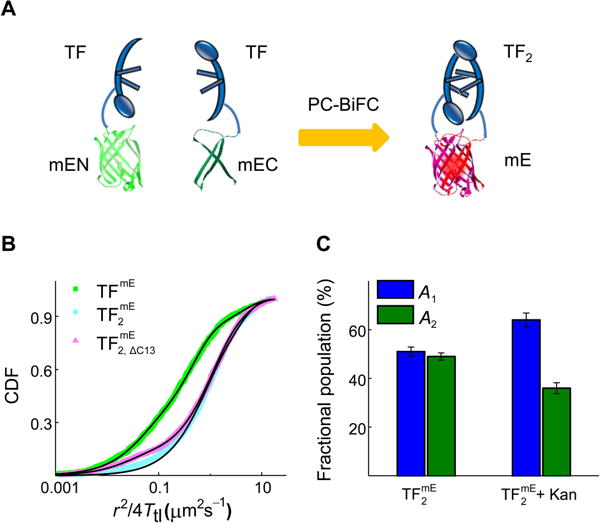
PC-BiFC probes the dynamics of TF2 dimer. (A) Design of probing TF2 using the PC-BiFC method. (B) Cumulative distribution function (CDF) of displacement length r per time lapse (Ttl = 60 ms) of TFmE overexpressed from a plasmid (TFmE), mE-fragment-tagged TFs expressed from a plasmid and complemented ( ), and the complementation of mE-fragment-tagged truncated TF and mE-fragment-tagged regular TF ( ) in living BL21(DE3) cells. The black lines are fittings by Eqs. S8–S9 (SI Section S6). The CDF analysis of TFmE resolves three diffusion components, with their effective diffusion constants (and fractional populations): D1 = 3.96 ± 0.27 μm2 s−1 (20 ± 1%), D2 = 0.42 ± 0.06 μm2 s−1 (58 ± 2%), and D3 = 0.04 ± 0.01 μm2 s−1 (22 ± 2%). The CDF analysis of resolves two diffusion components, with effective diffusion constants (and fractional populations) of D1 = 3.04 ± 0.10 μm2 s−1 (51 ± 2%) and D2 = 0.62 ± 0.06 μm2 s−1 (49 ± 1%). The CDF analysis of resolves three diffusion components, with effective diffusion constants (and fractional populations): D1 = 3.75 ± 0.15 μm2 s−1 (32 ± 1%), D2 = 0.94 ± 0.10 μm2 s−1 (58 ± 2%), and D3 = 0.02 ± 0.01 μm2 s−1 (10 ± 2%). (C) Comparison of fractional populations of the two diffusion states of and + Kan.
To test and validate this PC-BiFC approach for trapping and visualizing protein dimers (SI Section S9.1), we co-expressed mEN- or mEC-tagged leucine zippers CZ and NZ (i.e., CZ-mEN, mEC-NZ); the two leucine zippers are well-known to assemble into complexes in E. coli (Ghosh et al., 2000). In addition, we also co-expressed Tsr-CZ-mEN (where Tsr is an inner membrane protein (Kim et al., 1999, Kim et al., 2002)) and mEC-NZ, so that the complementation complex is targeted to the cell membrane. For both systems we observed complementation in the expected cellular locations (i.e., in the cytoplasm and on the membrane, respectively; SI Figs. S13 and S14), demonstrating the effectiveness of this PC-BiFC approach. It happened that our choice of mE and the split position to make the mEN and mEC fragments as well as using the leucine zippers for validation are almost identical to the recent work by Sun et al (Liu et al., 2014). and similar to that by Nan et al. who used the split fragments of the photoactivatable fluorescent protein PAmCherry1 (Nickerson et al., 2014).
To overexpress the two fragment-tagged TFs (i.e., TF-mEN and mEC-TF) as equally as possible in E. coli, we encoded them in a pET vector (i.e., pETDuet-1 vector) that has two multiple-cloning sites, and transformed it into the BL21(DE3) strain (see details in SI Section S1.6)—in the BW25113 strain, which was used for all our other studies here, pET vectors cannot be expressed due to the strain’s lack of T7 RNA polymerase. SDS-PAGE and Western blot analyses show that both TF-mEN and mEC-TF are intact in the cell, and the proteolytic cleavage of the fragment tags is minimal (<5%; SI Section S2). Cell growth assay under SDS/EDTA stress shows that both TF-mEN and mEC-TF are comparably functional as the untagged TF (Figure S2B).
We then performed SMT on and determined the CDF of its displacement length r per time lapse (Ttl = 60 ms) (Figure 7B). This CDF merely needs two diffusion components to be fitted satisfactorily (Eq. S8 in SI Section S6.1), with effective diffusion constants (and fractional populations) of D1 = 3.04 ± 0.10 μm2 s−1 (51 ± 2%) and D2 = 0.62 ± 0.06 μm2 s−1 (49 ± 1%), respectively. We further verified this minimal two-state diffusion of via hidden Markov model analysis (Persson et al., 2013) and ITCDD analysis (Oswald et al., 2014, Chen et al., 2015a) (Table S5, SI Sections S6.2–S6.3).
In parallel, we also studied TFmE encoded in a pET vector (i.e., pET-21b) under the same expression condition as in BL21(DE3) cells (Figure 7B). The CDF of TFmE requires minimally three diffusion components to be fitted satisfactorily; their effective diffusion constants (and fractional population) are: D1 = 3.96 ± 0.27 μm2 s−1 (20 ± 1%), D2 = 0.42 ± 0.06 μm2 s−1 (58 ± 2%), and D3 = 0.04 ± 0.01 μm2 s−1 (22 ± 2%), consistent with those of TFmE in BW25113 cells.
Comparing with TFmE, the diffusive behaviors of are missing the 70S-ribosome-bound D3 state, directly supporting that TF2 dimer does not interact with the 70S ribosome. Consistently, the pairwise distance analysis of positions does not support the existence of clustering that is associated with polyribosomes (Fig. S10B). The spatial distribution of does not show decreased probability in the middle of the cell, either, consistent with the lack of the nucleoid exclusion effect experienced by 70S ribosomes (Fig. S11B).
As a control, we truncated the C-terminal 13 amino acids of TF within mEC-TF (i.e., giving mEC-TF C13), which reduces its dimerization capability (Zeng et al., 2006). Co-expressing mEC-TFΔC13 with TF-mEN in the same cell also leads to the complemented , although the complementation efficiency is much lower than that of (SI Section 9.3 and Fig. S16). In this , the two TF should spend a significant amount of time non-dimerized, thus behaving like two monomers but tethered together. Consistently, CDF analysis of the SMT results of gave three diffusion components, in which the 70S-ribosome-bound D3 state reappeared compared with those of (Figure 7B). Therefore, the absence of the D3 state for provides the direct in vivo experimental evidence—for the first time to our knowledge—that TF2 dimers cannot bind to the 70S ribosome.
To learn more about the D2 state of , we treated the cells expressing with the translation inhibitor Kan and found the fractional population (A2) of ’s D2 state decreases significantly (Figure 7C). This decrease suggests that the D2 state of contains a polypeptide-bound population so that the decrease in its fractional population could be explained by the decreased protein synthesis in the cell under Kan treatment. In addition, the value of D2 for (~0.6 μm2 s−1) is consistent with that of TFmE (0.2 to 0.9 μm2 s−1, depending on the strain; Figure 4A). Therefore, both TF2 dimers and TF monomers can interact with polypeptides. Considering TF2 dimers cannot bind to the 70S ribosome, it is possible that TF2 dimers and TF monomers have different functions or different substrate pools in binding polypeptides. TF2 dimers might provide an enhanced protecting environment due to its geometric characteristics for chaperoning polypeptides with particular shapes or properties, so that they are only involved in interacting with polypeptides post-translationally, rather than co-translationally inside the cells.
Conclusion
Using a combination of SMT, PC-BiFC, and genetic manipulations, we have studied the diffusive behaviors of the molecular chaperone TF, as well as its dimeric form TF2 selectively, in living E. coli cells. Besides its freely diffusing state and the 70S-ribosome-bound state, our results suggest that TF can bind to free ribosomal subunits, and form TF-polypeptide complexes that could include DnaK/DnaJ proteins. TF-70S ribosome interactions were found to be transient in the cell, in which the TF’s average residence time on the 70S ribosome is ~0.2 s, much shorter than the average protein translation time. This transient interaction allows TF to sample multiple 70S ribosomes within a short time period as well as sufficient chances for other ribosome-binding proteins to interact with the 70S ribosome timely. Our results further indicate that SRP weakens TF’s interaction with the 70S ribosome in the cell, but this weakening mainly comes from affecting TF’s binding to the 70S ribosome, rather than its unbinding. For the TF2 dimer specifically, our results directly support that TF2 does not interact with the 70S ribosome in the cell, but can be involved in the post-translational interactions with polypeptides. Our findings here contribute to the fundamental understanding of the various functions of TF inside cells, which is an important step toward understanding chaperone-assisted protein folding in cells.
Experimental Procedures
The supporting information (SI) presents: (1) Materials and sample preparation (Sections S1–S3), including the design and construction of mEos3.2 and mEos3.2-fragment tagged TFs, the protein expression and cell growth conditions, and the functional assay. (2) Single-molecule tracking and cellular protein concentration quantification (Section S4). (3) Details of data analysis (Sections S5–S8), including single-molecule localization, diffusion analysis, unbinding kinetics analysis, and spatial distribution analysis. (4) Validation and additional results of photoconvertible bimolecular fluorescence complementation (Section S9). (5) Additional references (Section S10).
Supplementary Material
Acknowledgments
We acknowledge the National Institute of Health (GM106420, GM109993, and AI117295), Army Research Office (66998-LS/W911NF-15-1-0268) and National Science Foundation (CMMI-1463084) for funding, and Yimon Aye and Cynthia Kinsland for helping with molecular biology.
Footnotes
The authors do not have any conflict of interest to declare.
References
- Agashe VR, Guha S, Chang HC, Genevaux P, Hayer-Hartl M, Stemp M, Georgopoulos C, Hartl FU, Barral JM. Function of trigger factor and DnaK in multidomain protein folding: increase in yield at the expense of folding speed. Cell. 2004;117:199–209. doi: 10.1016/s0092-8674(04)00299-5. [DOI] [PubMed] [Google Scholar]
- Alix JH, Guerin MF. Mutant DnaK chaperones cause ribosome assembly defects in Escherichia coli. Proc Natl Acad Sci U S A. 1993;90:9725–9729. doi: 10.1073/pnas.90.20.9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariosa A, Lee JH, Wang S, Saraogi I, Shan SO. Regulation by a chaperone improves substrate selectivity during cotranslational protein targeting. Proc Natl Acad Sci U S A. 2015;112:E3169–3178. doi: 10.1073/pnas.1422594112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi S, Bratton BP, Weisshaar JC. Subdiffraction-limit study of Kaede diffusion and spatial distribution in live Escherichia coli. Biophys J. 2011;101:2535–2544. doi: 10.1016/j.bpj.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi S, Choi H, Mondal J, Weisshaar JC. Time-dependent effects of transcription- and translation-halting drugs on the spatial distributions of the Escherichia coli chromosome and ribosomes. Mol Microbiol. 2014;94:871–887. doi: 10.1111/mmi.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi S, Siryaporn A, Goulian M, Weisshaar JC. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol Microbiol. 2012;85:21–38. doi: 10.1111/j.1365-2958.2012.08081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornemann T, Holtkamp W, Wintermeyer W. Interplay between trigger factor and other protein biogenesis factors on the ribosome. Nat Commun. 2014;5:4180. doi: 10.1038/ncomms5180. [DOI] [PubMed] [Google Scholar]
- Bornemann T, Jockel J, Rodnina MV, Wintermeyer W. Signal sequence-independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat Struct Mol Biol. 2008;15:494–499. doi: 10.1038/nsmb.1402. [DOI] [PubMed] [Google Scholar]
- Bukau B, Deuerling E, Pfund C, Craig EA. Getting newly synthesized proteins into shape. Cell. 2000;101:119–122. doi: 10.1016/S0092-8674(00)80806-5. [DOI] [PubMed] [Google Scholar]
- Buskiewicz I, Deuerling E, Gu SQ, Jockel J, Rodnina MV, Bukau B, Wintermeyer W. Trigger factor binds to ribosome-signal-recognition particle (SRP) complexes and is excluded by binding of the SRP receptor. Proc Natl Acad Sci U S A. 2004;101:7902–7906. doi: 10.1073/pnas.0402231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Jung W, Santiago AG, Yang F, Krzeminski L, Chen P. Quantifying Multistate Cytoplasmic Molecular Diffusion in Bacterial Cells via Inverse Transform of Confined Displacement Distribution. J Phys Chem B. 2015a;119:14451–14459. doi: 10.1021/acs.jpcb.5b08654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Santiago AG, Jung W, Krzeminski L, Yang F, Martell DJ, Helmann JD, Chen P. Concentration- and chromosome-organization-dependent regulator unbinding from DNA for transcription regulation in living cells. Nat Commun. 2015b;6:7445. doi: 10.1038/ncomms8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke E, Wickner W. Trigger factor: a soluble protein that folds pro-OmpA into a membrane-assembly-competent form. Proc Natl Acad Sci U S A. 1987;84:5216–5220. doi: 10.1073/pnas.84.15.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PP, Bremer H. Differential rate of ribosomal protein synthesis in Escherichia coli B/r. J Mol Biol. 1974;84:407–422. doi: 10.1016/0022-2836(74)90449-5. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Patzelt H, Vorderwulbecke S, Rauch T, Kramer G, Schaffitzel E, Mogk A, Schulze-Specking A, Langen H, Bukau B. Trigger Factor and DnaK possess overlapping substrate pools and binding specificities. Mol Microbiol. 2003;47:1317–1328. doi: 10.1046/j.1365-2958.2003.03370.x. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–696. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science (New York, NY) 2007;316:1191–1194. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English BP, Hauryliuk V, Sanamrad A, Tankov S, Dekker NH, Elf J. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A. 2011;108:E365–373. doi: 10.1073/pnas.1102255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewalt KL, Hendrick JP, Houry WA, Hartl FU. In vivo observation of polypeptide flux through the bacterial chaperonin system. Cell. 1997;90:491–500. doi: 10.1016/s0092-8674(00)80509-7. [DOI] [PubMed] [Google Scholar]
- Forchhammer J, Lindahl L. Growth rate of polypeptide chains as a function of the cell growth rate in a mutant of Escherichia coli 15. J Mol Biol. 1971;55:563–568. doi: 10.1016/0022-2836(71)90337-8. [DOI] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Gahlmann A, Moerner WE. Exploring bacterial cell biology with single-molecule tracking and super-resolution imaging. Nat Rev Microbiol. 2014;12:9–22. doi: 10.1038/nrmicro3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh I, Hamilton AD, Regan L. Antiparallel Leucine Zipper-Directed Protein Reassembly: Application to the Green Fluorescent Protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- Grudnik P, Bange G, Sinning I. Protein targeting by the signal recognition particle. Biol Chem. 2009;390:775–782. doi: 10.1515/BC.2009.102. [DOI] [PubMed] [Google Scholar]
- Gu SQ, Peske F, Wieden HJ, Rodnina MV, Wintermeyer W. The signal recognition particle binds to protein L23 at the peptide exit of the Escherichia coli ribosome. RNA (New York, NY) 2003;9:566–573. doi: 10.1261/rna.2196403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science (New York, NY) 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- Holtkamp W, Lee S, Bornemann T, Senyushkina T, Rodnina MV, Wintermeyer W. Dynamic switch of the signal recognition particle from scanning to targeting. Nat Struct Mol Biol. 2012;19:1332–1337. doi: 10.1038/nsmb.2421. [DOI] [PubMed] [Google Scholar]
- Javer A, Long Z, Nugent E, Grisi M, Siriwatwetchakul K, Dorfman KD, Cicuta P, Cosentino Lagomarsino M. Short-time movement of E. coli chromosomal loci depends on coordinate and subcellular localization. Nat Commun. 2013;4:3003. doi: 10.1038/ncomms3003. [DOI] [PubMed] [Google Scholar]
- Jensen CG, Pedersen S. Concentrations of 4.5S RNA and Ffh protein in Escherichia coli: the stability of Ffh protein is dependent on the concentration of 4.5S RNA. Journal of bacteriology. 1994;176:7148–7154. doi: 10.1128/jb.176.23.7148-7154.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser CM, Chang HC, Agashe VR, Lakshmipathy SK, Etchells SA, Hayer-Hartl M, Hartl FU, Barral JM. Real-time observation of trigger factor function on translating ribosomes. Nature. 2006;444:455–460. doi: 10.1038/nature05225. [DOI] [PubMed] [Google Scholar]
- Kandror O, Sherman M, Goldberg A. Rapid degradation of an abnormal protein in Escherichia coli proceeds through repeated cycles of association with GroEL. J Biol Chem. 1999;274:37743–37749. doi: 10.1074/jbc.274.53.37743. [DOI] [PubMed] [Google Scholar]
- Kandror O, Sherman M, Rhode M, Goldberg AL. Trigger factor is involved in GroEL-dependent protein degradation in Escherichia coli and promotes binding of GroEL to unfolded proteins. EMBO J. 1995;14:6021–6027. doi: 10.1002/j.1460-2075.1995.tb00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- Kim SH, Wang W, Kim KK. Dynamic and clustering model of bacterial chemotaxis receptors: structural basis for signaling and high sensitivity. Proc Natl Acad Sci U S A. 2002;99:11611–11615. doi: 10.1073/pnas.132376499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer G, Rauch T, Rist W, Vorderwulbecke S, Patzelt H, Schulze-Specking A, Ban N, Deuerling E, Bukau B. L23 protein functions as a chaperone docking site on the ribosome. Nature. 2002;419:171–174. doi: 10.1038/nature01047. [DOI] [PubMed] [Google Scholar]
- Lecker S, Lill R, Ziegelhoffer T, Georgopoulos C, Bassford PJ, Jr, Kumamoto CA, Wickner W. Three pure chaperone proteins of Escherichia coli–SecB, trigger factor and GroEL–form soluble complexes with precursor proteins in vitro. EMBO J. 1989;8:2703–2709. doi: 10.1002/j.1460-2075.1989.tb08411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Crooke E, Guthrie B, Wickner W. The “trigger factor cycle” includes ribosomes, presecretory proteins, and the plasma membrane. Cell. 1988;54:1013–1018. doi: 10.1016/0092-8674(88)90116-x. [DOI] [PubMed] [Google Scholar]
- Liu CP, Perrett S, Zhou JM. Dimeric trigger factor stably binds folding-competent intermediates and cooperates with the DnaK-DnaJ-GrpE chaperone system to allow refolding. J Biol Chem. 2005;280:13315–13320. doi: 10.1074/jbc.M414151200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xing D, Su QP, Zhu Y, Zhang J, Kong X, Xue B, Wang S, Sun H, Tao Y, Sun Y. Super-resolution imaging and tracking of protein-protein interactions in sub-diffraction cellular space. Nat Commun. 2014;5:4443. doi: 10.1038/ncomms5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier R, Eckert B, Scholz C, Lilie H, Schmid FX. Interaction of trigger factor with the ribosome. J Mol Biol. 2003;326:585–592. doi: 10.1016/s0022-2836(02)01427-4. [DOI] [PubMed] [Google Scholar]
- Martinez-Hackert E, Hendrickson WA. Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone. Cell. 2009;138:923–934. doi: 10.1016/j.cell.2009.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza D, Abernathy A, Golob N, Morisaki T, McNally JG. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 2012;40:e119. doi: 10.1093/nar/gks701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P, Jovanovic G, Lenn T, Bruckbauer A, Engl C, Ying L, Buck M. Dynamics and stoichiometry of a regulated enhancer-binding protein in live Escherichia coli cells. Nat Commun. 2013;4:1997. doi: 10.1038/ncomms2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi M, Tanaka N. Mechanism of inhibition of translocation by kanamycin and viomycin: a comparative study with fusidic acid. Biochem Biophys Res Commun. 1980;92:647–654. doi: 10.1016/0006-291x(80)90382-4. [DOI] [PubMed] [Google Scholar]
- Nickerson A, Huang T, Lin LJ, Nan X. Photoactivated localization microscopy with bimolecular fluorescence complementation (BiFC-PALM) for nanoscale imaging of protein-protein interactions in cells. PloS one. 2014;9:e100589. doi: 10.1371/journal.pone.0100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G, Weissman JS, Bukau B. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 2011;147:1295–1308. doi: 10.1016/j.cell.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald F, BE LM, Bollen YJ, Peterman EJ. Imaging and quantification of trans-membrane protein diffusion in living bacteria. Phys Chem Chem Phys. 2014;16:12625–12634. doi: 10.1039/c4cp00299g. [DOI] [PubMed] [Google Scholar]
- Patzelt H, Kramer G, Rauch T, Schonfeld HJ, Bukau B, Deuerling E. Three-state equilibrium of Escherichia coli trigger factor. Biol Chem. 2002;383:1611–1619. doi: 10.1515/BC.2002.182. [DOI] [PubMed] [Google Scholar]
- Persson F, Linden M, Unoson C, Elf J. Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nat Methods. 2013;10:265–269. doi: 10.1038/nmeth.2367. [DOI] [PubMed] [Google Scholar]
- Pestka S. The use of inhibitors in studies on protein synthesis. Methods Enzymol. 1974;30:261–282. doi: 10.1016/0076-6879(74)30030-4. [DOI] [PubMed] [Google Scholar]
- Raine A, Ivanova N, Wikberg JE, Ehrenberg M. Simultaneous binding of trigger factor and signal recognition particle to the E. coli ribosome. Biochimie. 2004;86:495–500. doi: 10.1016/j.biochi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Raine A, Lovmar M, Wikberg J, Ehrenberg M. Trigger factor binding to ribosomes with nascent peptide chains of varying lengths and sequences. J Biol Chem. 2006;281:28033–28038. doi: 10.1074/jbc.M605753200. [DOI] [PubMed] [Google Scholar]
- Rutkowska A, Mayer MP, Hoffmann A, Merz F, Zachmann-Brand B, Schaffitzel C, Ban N, Deuerling E, Bukau B. Dynamics of trigger factor interaction with translating ribosomes. J Biol Chem. 2008;283:4124–4132. doi: 10.1074/jbc.M708294200. [DOI] [PubMed] [Google Scholar]
- Sanamrad A, Persson F, Lundius EG, Fange D, Gynna AH, Elf J. Single-particle tracking reveals that free ribosomal subunits are not excluded from the Escherichia coli nucleoid. Proc Natl Acad Sci U S A. 2014;111:11413–11418. doi: 10.1073/pnas.1411558111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbai M, Alix JH. DnaK-dependent ribosome biogenesis in Escherichia coli: competition for dominance between the alleles dnaK756 and dnaK+ Mol Gen Genet. 1998;260:199–206. doi: 10.1007/s004380050886. [DOI] [PubMed] [Google Scholar]
- Schaffitzel C, Oswald M, Berger I, Ishikawa T, Abrahams JP, Koerten HK, Koning RI, Ban N. Structure of the E. coli signal recognition particle bound to a translating ribosome. Nature. 2006;444:503–506. doi: 10.1038/nature05182. [DOI] [PubMed] [Google Scholar]
- Sohmen D, Harms JM, Schlunzen F, Wilson DN. SnapShot: Antibiotic inhibition of protein synthesis I. Cell. 2009;138:1248 e1241. doi: 10.1016/j.cell.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Staehelin T, Brinton CC, Wettstein FO, Noll H. STRUCTURE AND FUNCTION OF E. COLI ERGOSOMES. Nature. 1963;199:865–870. doi: 10.1038/199865a0. [DOI] [PubMed] [Google Scholar]
- Teter SA, Houry WA, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C, Hartl FU. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 1999;97:755–765. doi: 10.1016/s0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Ullers RS, Houben EN, Raine A, ten Hagen-Jongman CM, Ehrenberg M, Brunner J, Oudega B, Harms N, Luirink J. Interplay of signal recognition particle and trigger factor at L23 near the nascent chain exit site on the Escherichia coli ribosome. J Cell Biol. 2003;161:679–684. doi: 10.1083/jcb.200302130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR, Knopf PM, Rich A. A multiple ribosomal structure in protein synthesis. Proc Natl Acad Sci U S A. 1963;49:122–129. doi: 10.1073/pnas.49.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R, Bremer H. Polypeptide-chain-elongation rate in Escherichia coli B/r as a function of growth rate. Biochem J. 1976;160:185–194. doi: 10.1042/bj1600185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LL, Yu L, Li ZY, Perrett S, Zhou JM. Effect of C-terminal truncation on the molecular chaperone function and dimerization of Escherichia coli trigger factor. Biochimie. 2006;88:613–619. doi: 10.1016/j.biochi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, Zhang J, Xu P, Xu T. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.