

ABSTRACT
Gene expression in methanotrophs has been shown to be affected by the availability of a variety of metals, most notably copper-regulating expression of alternative forms of methane monooxygenase. A copper-binding compound, or chalkophore, called methanobactin plays a key role in copper uptake in methanotrophs. Methanobactin is a ribosomally synthesized and posttranslationally modified peptide (RiPP) with two heterocyclic rings with an associated thioamide for each ring, formed from X-Cys dipeptide sequences that bind copper. The gene coding for the precursor polypeptide of methanobactin, mbnA, is part of a gene cluster, but the role of other genes in methanobactin biosynthesis is unclear. To begin to elucidate the function of these genes, we constructed an unmarked deletion of mbnABCMN in Methylosinus trichosporium OB3b and then homologously expressed mbnABCM using a broad-host-range cloning vector to determine the function of mbnN, annotated as coding for an aminotransferase. Methanobactin produced by this strain was found to be substantially different from wild-type methanobactin in that the C-terminal methionine was missing and only one of the two oxazolone rings was formed. Rather, in place of the N-terminal 3-methylbutanoyl-oxazolone-thioamide group, a leucine and a thioamide-containing glycine (Gly-Ψ) were found, indicating that MbnN is used for deamination of the N-terminal leucine of methanobactin and that this posttranslational modification is critical for closure of the N-terminal oxazolone ring in M. trichosporium OB3b. These studies provide new insights into methanobactin biosynthesis and also provide a platform for understanding the function of other genes in the methanobactin gene cluster.
IMPORTANCE Methanotrophs, microbes that play a critical role in the carbon cycle, are influenced by copper, with gene expression and enzyme activity changing as copper levels change. Methanotrophs produce a copper-binding compound, or chalkophore, called methanobactin for copper uptake, and methanobactin plays a key role in controlling methanotrophic activity. Methanobactin has also been shown to be effective in the treatment of Wilson disease, an autosomal recessive disorder where the human body cannot correctly assimilate copper. It is important to characterize the methanobactin biosynthesis pathway to understand how methanotrophs respond to their environment as well as to optimize the use of methanobactin for the treatment of copper-related diseases such as Wilson disease. Here we show that mbnN, encoding an aminotransferase, is involved in the deamination of the N-terminal leucine and necessary for the formation of one but not both of the heterocyclic rings in methanobactin that are responsible for copper binding.
KEYWORDS: methanotrophy, methanobactin, aminotransferase, ribosomally synthesized and posttranslationally modified peptide
INTRODUCTION
Methanotrophs utilize a novel modified peptide termed methanobactin for copper uptake (1–4). Methanobactin is the first example of a chalkophore, or copper-specific chelating agent, and is akin to the siderophores used for iron sequestration by many microbes. Methanobactins are characterized as being small (<1,200 Da) polypeptides with two heterocyclic rings each with an adjacent thioamide group. The N-terminal ring is an oxazolone, while the other is either another oxazolone or an imidazolone or pyrazinedione group (5). Perhaps the best-characterized methanobactin is that from Methylosinus trichosporium OB3b (Fig. 1). Methanobactin from this methanotroph has several interesting features, including that the N-terminal leucine has been deaminated and that the two heterocylic rings are both oxazolones (1, 2, 6).
FIG 1.

Molecular structure of methanobactin from M. trichosporium OB3b. (Modified from reference 8 with permission [copyright 2010 American Chemical Society].) An asterisk (*) indicates a methionine that is observed in some but not all samples.
The genetics underlying methanobactin synthesis are slowly being unraveled. Given the unique structure of methanobactin from M. trichosporium OB3b, it was initially speculated that it was synthesized via a nonribosomal peptide synthase (7). Subsequent investigation, however, suggested that it may be a ribosomally synthesized and posttranslationally modified peptide (RiPP), with a gene—mbnA—possibly coding for the polypeptide precursor of methanobactin identified that includes a 19-amino-acid leader sequence and a 10-amino-acid core sequence (8). When mbnA was knocked out in Methylosinus trichosporium OB3b, this microbe was unable to synthesize methanobactin, providing evidence that methanobactin is a RiPP (4). With these data, the core polypeptide sequence of methanobactin from M. trichosporium OB3b was shown to be Leu-Cys-Gly-Ser-Cys-Tyr-Pro-Cys-Ser-Met, where the heterocyclic rings shown in Fig. 1 are formed from the two X-Cys dipeptide sequences highlighted in bold (5).
mbnA is part of a gene cluster in M. trichosporium OB3b as shown in Fig. 2. Upstream of mbnA is mbnT, coding for TonB-dependent transporter. Recently, mbnT was successfully knocked out in M. trichosporium OB3b, with the resultant mutant able to synthesize methanobactin but unable to take it back up, showing that mbnT encodes the mechanism of methanobactin uptake (9). Immediately upstream of mbnT are mbnR and mbnI, coding for a putative membrane sensor and an extracytoplasmic function sigma factor, respectively. Collectively, these genes appear to be under the control of a σ70-dependent promoter as predicted using BPROM (10). Downstream of mbnA are several other genes, some of which have unknown function but are likely involved in methanobactin biosynthesis (mbnB and mbnC), as well as genes annotated as encoding either an extrusion protein that may be responsible for methanobactin secretion (mbnM) or an aminotransferase that may play a role in methanobactin biosynthesis (mbnN). These genes also appear to be part of an operon under the control of a σ70-dependent promoter as predicted using BPROM. Two additional genes, mbnP and mbnH, encoding a putative diheme cytochrome c peroxidase and its partner, are adjacent to mbnN but look to be under the control of a separate σ70-dependent promoter (again predicted using BPROM). The function(s) of mbnP and mbnH is unclear, although they may be involved in ring formation in methanobactin or may be part of a secondary copper uptake system (5, 9).
FIG 2.

Methanobactin gene cluster of M. trichosporium OB3b.
There is increasing interest in understanding the methanobactin biosynthesis pathway; e.g., it has been recently shown that methanobactin from M. trichosporium OB3b has the potential to treat individuals afflicted with Wilson disease (11–13). Wilson disease is an autosomal recessive disorder in which the body is unable to correctly assimilate copper, with copper accumulating in the liver and brain, and can result in severe and irreversible damage (14, 15). Current treatment therapies include prescription of chelating agents such as penicillamine and trientine, but it is not uncommon for serious side effects to occur, and, in any case, when using these compounds, copper is excreted through the urine and not the bile, which is the preferred or normal physiological route. Methanobactin, however, was found to bind copper very strongly in a rat model of Wilson disease. Copper was then quickly removed via the bile, and animals treated with methanobactin had sustained clinical recovery (11). Such findings indicate that methanobactin has the potential to be an alternative treatment for Wilson disease, particularly for those patients with acute liver failure (16).
Here we describe the role of mbnN in production of methanobactin from M. trichosporium OB3b by comparing and contrasting methanobactin produced by a mutant where mbnN has been deleted versus that made by M. trichosporium OB3b with the complete methanobactin gene cluster.
RESULTS
Using sucrose counterselection techniques, a markerless deletion of mbnA through mbnN was constructed in M. trichosporium OB3b. This was confirmed by PCR amplification of mbnA through mbnN and verification of the loss of the plasmid backbone (see Fig. S2 in the supplemental material). This was further confirmed by sequencing as well as by the finding that the M. trichosporium OB3b ΔmbnAN strain was kanamycin sensitive and sucrose resistant (data not shown). The construction of this deletion mutant had no effect on expression of mbnPH as confirmed by reverse transcription-PCR (RT-PCR) (Fig. S3). Interestingly, expression of mbnPH was dependent on copper as found earlier for mbnA (4), suggesting that mbnA, mbnP, and mbnH utilize a shared regulatory element for expression.
After verification of the deletion of mbnA through mbnN from the chromosome, methanobactin production was examined in this mutant. The absence of color in the culture medium suggested that no methanobactin was being produced by this mutant even when it was cultured under low-copper conditions (Fig. S4). The lack of methanobactin production in the ΔmbnAN mutant was confirmed via UV/visible light absorption spectral analyses of the spent medium (which was concentrated more than 50-fold in using an HP20 Dianion column; Fig. 3).
FIG 3.
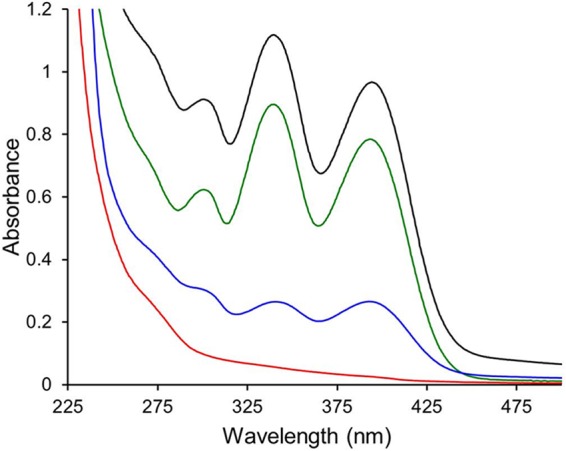
UV-visible light absorption spectra of the spent media from wild-type M. trichosporium OB3b (black trace), the ΔmbnAN mutant (red trace), and the ΔmbnAN + pWG101 mutant (green trace). Cells were cultured on NMS medium amended with 0.2 μM CuCl2 (black and red traces) or on NMS medium amended with 0.2 μM CuCl2 and 20 μg · ml−1 spectinomycin (green trace). The blue trace shows methanobactin production in mutant ΔmbnAN + pWG101 grown on NMS medium amended with 5.0 μM CuCl2 and 20 μg · ml−1 spectinomycin.
The ΔmbnAN strain was then back-complemented with mbnA through mbnN via insertion of these genes into pTJS140 to generate pWG101. Expression of these back-added genes was suggested by the color of the culture medium (Fig. S4) and was confirmed via RT-PCR (Fig. S5). Methanobactin production was clearly visible via UV-visible light absorption spectroscopy (Fig. 3) and was approximately 75% of that observed from wild-type M. trichosporium OB3b. The mass of methanobactin from this back-complemented mutant (M. trichosporium ΔmbnAN + pWG101) was found to be 1,154.27 Da by both liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Fig. S6) and Fourier transform-ion cyclotron resonance-mass spectrometry (FT-ICR-MS) (Fig. S7) and was identical to the mass of methanobactin from wild-type M. trichosporium OB3b, indicating that M. trichosporium ΔmbnAN + pWG101 produced the same form of methanobactin.
The native σ70-dependent promoter upstream of mbnA was also incorporated into pWG101, and, based on earlier findings showing that mbnA expression decreased with increasing copper levels in the M. trichosporium OB3b wild-type strain (4), similar results were expected in M. trichosporium ΔmbnAN + pWG101. Indeed, expression of mbnA and mbnN was visibly reduced in M. trichosporium ΔmbnAN + pWG101 in the presence of copper compared to the results seen in the growth medium in the absence of copper (Fig. S5). Comparable results were seen for methanobactin production; i.e., the level of methanobactin in the spent medium was markedly reduced when M. trichosporium ΔmbnAN + pWG101 was grown in the presence of copper (Fig. 3).
The role of mbnN in methanobactin biosynthesis was then investigated by inserting mbnA through mbnM (again under the control of the native σ70-dependent promoter) into pTJS140, creating pWG102, and then conjugating this into M. trichosporium ΔmbnAN. Expression of mbnA and mbnM was evident (Fig. S8), and the methanobactin produced by M. trichosporium ΔmbnAN + pWG102 was isolated and characterized. The molecular mass of methanobactin produced by this strain was found to be 999.47 Da by LC-MS/MS (Fig. S6) and 999.46 Da by FT-ICR MS (Fig. S7), 154.7 Da less than that of methanobactin isolated from the M. trichosporium OB3b wild-type strain (6). The difference suggests that one or both of the heterocyclic rings did not form and that an amino acid residue was missing. Subsequently, methanobactin from M. trichosporium ΔmbnAN + pWG102 was subjected to Edman degradation and was found to have a polypeptide backbone with a sequence consisting of Leu-?-Gly-Ser-?-Tyr-Pro-?-Ser-? (question marks suggest moieties with sulfhydryl groups, e.g., cysteines or non-amino acid moieties). It should be noted that the C-terminal methionine is missing from M. trichosporium ΔmbnAN + pWG102 methanobactin, and this can explain 131.2 Da of the difference in mass in comparing methanobactin from the wild-type strain to that from M. trichosporium ΔmbnAN + pWG102. More importantly, successful amino acid sequencing of methanobactin from M. trichosporium ΔmbnAN + pWG102 was surprising, as previous attempts to sequence wild-type methanobactin via Edman degradation resulted in the nonmethanobactin sequence of Ser-Met-Tyr-Pro-?-Ser-?-Met. The modification of the N-terminal leucine and the presence of the adjacent oxazolone ring A alter or disrupt N-terminal amino acid sequencing in wild-type methanobactin. The successful collection of methanobactin sequence data from M. trichosporium ΔmbnAN + pWG102 indicates that the N-terminal leucine had not been modified and raises the possibility that oxazolone ring A might also have been missing.
To help identify the unknown residues found in the initial amino acid sequence of methanobactin from M. trichosporium ΔmbnAN + pWG102, the compositions of acid-digested methanobactin from M. trichosporium ΔmbnAN + pWG101 and ΔmbnAN + pWG102 were determined and compared. Methanobactin from M. trichosporium ΔmbnAN + pWG101 was found to have 0 leucines, 1.9 serines, 2.1 glycines, 0.8 prolines, 1.8 cysteines, and 0.8 methionines per tyrosine (used as an internal standard). The presence of proline and one more glycine than expected from the structure of wild-type methanobactin (Fig. 1) indicates that under these conditions, oxazolone ring B underwent hydrolysis and decarboxylation to form proline and a glycine (likely containing thioamide or Gly-Ψ) but oxazolone ring A was not degraded. Such a result is not unprecedented as it was shown earlier that when methanobactin from M. trichosporium OB3b was subjected to acid digestion, oxazolone ring B was hydrolyzed and decarboxylated to proline and a thioamide-containing glycine before ring A was converted to an α-oxo-leucine and a thioamide-containing glycine (8).
Methanobactin from M. trichosporium ΔmbnAN + pWG102 was found to have a different composition, i.e., 1.0 leucines, 1.8 serines, 3.3 glycines, 0.7 prolines, and 1.6 cysteines per tyrosine. The absence of a Met was consistent with the N-terminal sequencing results. The presence of leucine and a third glycine is remarkable and is unlikely to be the result of the degradation of oxazolone ring A, as methanobactin from M. trichosporium ΔmbnAN + pWG102 was prepared in the same fashion as that from M. trichosporium ΔmbnAN + pWG101. Rather, the finding of these residues supports the possibility that oxazolone ring A was not present in methanobactin from M. trichosporium ΔmbnAN + pWG102 methanobactin.
To consider this further, UV/visible light absorption spectral analyses of M. trichosporium ΔmbnAN + pWG102 methanobactin were performed and the results were compared to those from methanobactin produced by M. trichosporium ΔmbnAN + pWG101 (Fig. 4). The presence of only one major absorption peak at 337 nm indicates that M. trichosporium ΔmbnAN + pWG102 methanobactin has only oxazolone ring B and not oxazolone ring A, as a second major absorption peak would be expected at 394 nm. Further, copper titration of M. trichosporium ΔmbnAN + pWG102 methanobactin demonstrated copper binding, but the sample saturated at a copper/methanobactin molar ratio of ∼0.5, compared to 1.0 for wild-type M. trichosporium methanobactin (1, 2) and for methanobactin from M. trichosporium ΔmbnAN + pWG101 (Fig. 4). The impaired copper binding by methanobactin from M. trichosporium ΔmbnAN + pWG102 again suggests that oxazolone ring A is missing. Finally, acid digestion of methanobactin produced by M. trichosporium ΔmbnAN + pWG102 followed the hydrolysis pattern observed for oxazolone B from both wild-type M. trichosporium (8) and M. trichosporium ΔmbnAN + pWG101. In wild-type M. trichosporium and M. trichosporium ΔmbnAN + pWG101 methanobactin, oxazolone ring B is hydrolyzed before oxazolone ring A, as evidenced by the decrease in absorption at 340 nm but the minimal change at 394 nm (Fig. S9). The mass changes for methanobactin from M. trichosporium ΔmbnAN + pWG101 (24.98 Da) and ΔmbnAN + pWG102 (25.99, 27, and 28 Da) (Fig. S10) after acid digestion were consistent with the 25.98-Da change predicted following hydrolysis of the oxazolone ring B (8).
FIG 4.

Comparison of the UV/visible light absorption spectra of methanobactin from (A) M. trichosporium OB3b ΔmbnAN + pWG101 and (B) M. trichosporium OB3b ΔmbnAN + pWG102 with the addition of 0.0 to 1.0 Cu(II) per methanobactin in 0.05 increments.
Considering the mass spectral, amino acid sequence, composition, and UV/visible light absorption data in toto, we propose that the metal-free form of methanobactin from M. trichosporium ΔmbnAN + pWG102 has the molecular structure shown in Fig. 5. This structure, with a mass of 999.26 Da, agrees well with that measured via mass spectrometry (Fig. S6 and S7). In particular, we predict that leucine is adjacent to a thioamide-containing glycine (Gly-Ψ) and that oxazolone ring B is also present. To verify the presence of this modified glycine in methanobactin, we assayed for the presence of thiol groups before and after reduction performed using tris(2-carboxyethyl)phosphine (TCEP). Before reduction, no thiol groups were measured in methanobactin from the M. trichosporium OB3b wild-type, ΔmbnAN + pWG101, and ΔmbnAN + pWG102 strains (Table 1), suggesting that the thioamide groups associated with the oxazolone rings are not detectable using this methodology and that the cysteine thiol groups were oxidized. After reduction, however, the methanobactins from the M. trichosporium OB3b wild-type and ΔmbnAN + pWG101 strains had 1.7 ± 0.3 and 2.1 ± 0.1 thiols per methanobactin, respectively (Table 1), and were likely created from the reduction of the disulfide bond connecting Cys 3 and Cys 6 (Fig. 1). Methanobactin from M. trichosporium ΔmbnAN + pWG102 had 3.1 ± 0.3 thiols per methanobactin after reduction with TCEP. The finding of an additional thiol after reduction indicates that despite the lack of oxazolone ring A, the sulfur group remains and, as such, supports the conclusion that a thioamide-containing glycine exists in place of oxazolone ring A.
FIG 5.

Proposed structure of methanobactin from M. trichosporium ΔmbnAN + pWG102. Major differences from wild-type methanobactin are shown in red.
TABLE 1.
Quantification of thiol groups in methanobactin from M. trichosporium OB3b wild-type, ΔmbnAN + pWG101, and ΔmbnAN + pWG102 strains as isolated and following reduction by tris(2-carboxyethyl)phosphine under anaerobic conditions
| M. trichosporium OB3b strain | No. of thiol groups per methanobactina |
|
|---|---|---|
| As isolated | Following reduction | |
| Wild type | 3.0 × 10−4 ± 5.1 × 10−4 | 1.7 ± 0.3 |
| ΔmbnAN + pWG101 | 3.7 × 10−3 ± 3.2 × 10−4 | 2.1 ± 0.1 |
| ΔmbnAN + pWG102 | 2.2 × 10−3 ± 1.1 × 10−4 | 3.1 ± 0.3 |
Values are averages ± standard deviations of results from duplicate samples.
DISCUSSION
Since the discovery that the precursor polypeptide of methanobactin is chromosomally encoded and that the gene is part of a gene cluster that includes genes coding for an extrusion protein and for a putative diheme cytochrome c peroxidase and its partner protein, as well genes of unknown function, it has been accepted that methanobactin is a RiPP, but the roles of genes in the methanobactin gene cluster other than mbnT (coding for a TonB-dependent transporter required for uptake of the copper-methanobactin complex [9]) have not been explicitly determined. Here we show through the construction of a markerless deletion of mbnABCMN and expression of mbnABCM from a plasmid that the product of mbnN is necessary for the deamination of the N-terminal leucine. Further, M. trichosporium ΔmbnAN + pWG102 is unable to construct oxazolone ring A in methanobactin, but oxazolone ring B is formed, indicating that these two rings are made independently. At this time, it is unclear if oxazolone rings A and B are formed via the same or different modifying enzyme(s), but from the data collected, it appears that the modifying enzyme(s) responsible for formation of oxazolone ring A can bind and/or modify the N-terminal cysteine only after the deamination of the adjacent leucine.
In our earlier proposal of a pathway for methanobactin biosynthesis (5), we speculated that ring closure occurred before changes in the connectivity of the peptide backbone. On the basis of the findings reported here, however, it appears that deamination of the N-terminal acid must occur before formation of oxazolone ring A in M. trichosporium OB3b, and we present in Fig. 6 an alternative scheme for formation of methanobactin. We stress that we do not know whether, in wild-type methanobactin, rearrangement of the peptide backbone occurs before leucine deamination (or vice versa)—only that both are required for formation of oxazolone ring A.
FIG 6.

Proposed pathway of formation of (A) the N-terminal oxazolone group with associated thioamide group in wild-type methanobactin from M. trichosporium OB3b and (B) the resulting altered pathway in M. trichosporium ΔmbnAN + pWG102.
It should be noted that all methanobactins characterized to date have an oxazolone ring near the C terminus, but the N-terminal heterocyclic ring is an oxazolone ring, a pyrazinedione ring, or an imidazolone ring. In our earlier proposal of a pathway for methanobactin biosynthesis (5), we speculated that pyrazinedione and imidazolone rings are formed from modification of an oxazolone ring, but further consideration of available bioinformatic data juxtaposed with the findings reported here suggests that pyrazinedione and imidazolone rings can be created without an oxazolone ring platform. That is, in our original model of ring formation, deamination of the amino acid residue adjacent to the cysteine converted to an oxazolone ring was not necessary for ring formation. The data presented here, however, show such deamination is required for the formation of oxazolone ring A. Methanobactin of Methylocystis rosea and methanobactin of Methylocystis strain SB2 have an N-terminal pyrazinedione ring and imidazolone ring, respectively, but the methanobactin gene clusters of these two strains do not show any clear evidence of an encoded aminotransferase (5). Given that the N-terminal oxazolone ring in methanobactin of M. trichosporium OB3b is formed only when mbnN is expressed, it is possible that the creation of the alternative rings found in other forms of methanobactin does not require an oxazolone ring to be formed first. Rather, the formation of pyrazinedione and imidazolone rings may be due to the concerted activity of a suite of modifying enzymes, including a putative flavin adenine dinucleotide (FAD)-dependent oxidoreductase found in the methanobactin gene clusters of Methylocystis rosea and Methylocystis strain SB2 but not in M. trichosporium OB3b (5). As noted earlier, however, formation of imidazolone or pyrazinedione rings without an oxazolone intermediate would likely necessitate multiple changes in the connectivity of the peptide backbone (5).
It thus appears that different strategies have been developed by different methanotrophs for the formation of methanobactin, and creation and characterization of more mutants are clearly required to fully reconstruct these different biosynthetic pathways. That M. trichosporium OB3b is the only methanotroph known to produce methanobactin with a validated system for generation of mutants represents a challenge, however. Similar approaches may be possible in other methanotrophs that produce methanobactin, but in the event that working genetic systems are difficult to construct in these strains, it may be useful to pursue efforts whereby methanobactin genes from other strains are heterologously expressed in either Escherichia coli or methanobactin-defective mutants of M. trichosporium OB3b.
In conclusion, through the construction and back-complementation of a deletion mutant defective in methanobactin production, we have elucidated the function of mbnN and can now utilize the deletion mutant platform to unravel the role of other genes in the methanobactin gene cluster. With such data, we can develop a better mechanistic understanding of the methanobactin biosynthesis pathway and also create strategies to modify methanobactin for enhanced treatment of copper-related diseases such as Wilson disease.
MATERIALS AND METHODS
Bacterial strains, growth media, and culture conditions.
Plasmid construction was accomplished using Escherichia coli strain TOP10 (Invitrogen, Carlsbad, CA). Plasmids used and constructed during this study are shown in Table 2. The donor strain for conjugation of plasmids into Methylosinus trichosporium OB3b was E. coli S17-1 (17). E. coli strains were cultivated at 37°C in Luria broth medium (Dot Scientific, Burton, MI). Strains of M. trichosporium OB3b were cultivated at 30°C on nitrate mineral salts (NMS) medium (20) either in 250-ml flasks with side arms at 200 rpm or in a 15-liter New Brunswick Bioflow and Cellgen 310 fermentor (Eppendorf, Hauppauge, NY, USA) using methane as the sole carbon and energy source. Where necessary, filter-sterilized solutions of copper, as CuCl2, or antibiotics (Sigma-Aldrich, St. Louis, MO) were added to culture media aseptically. The working concentrations of antibiotics were as follows: ampicillin, 100 μg · ml−1; kanamycin, 25 μg · ml−1 (for E. coli) or 5 μg · ml−1 (for M. trichosporium OB3b transconjugants); nalidixic acid, 15 μg · ml−1; spectinomycin, 20 μg · ml−1. Chemicals were purchased from Fisher Scientific (Waltham, MA) or Sigma-Aldrich (St. Louis, MO) and were of American Chemical Society reagent grade or better.
TABLE 2.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Genotype, description, or sequence (5′–3′)a | Restriction site(s) | Source or reference |
|---|---|---|---|
| E. coli TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu) 7697 galU galK rpsL (Strr) endA1 nupG | Invitrogen | |
| E. coli S17.1 | recA1 thi pro hsdR-RP4-2Tc::Mu Km::Tn7 | 17 | |
| M. trichosporium OB3b | Wild type | ||
| M. trichosporium ΔmbnAN | mbnABCMN deleted | This study | |
| M. trichosporium ΔmbnAN + pWG101 | M. trichosporium ΔmbnAN back-complemented by pWG101 | This study | |
| M. trichosporium ΔmbnAN + pWG102 | mbnN deleted, constructed by the use of M. trichosporium ΔmbnAN carrying pWG102 | This study | |
| Plasmids | |||
| pK18mobsacB | Mobilizable suicide vector; oriT Kmr lacZ sacB | 18 | |
| pWG012 | pK18mobsacB carrying 2-kb ligated arms used to knock out mbnABCMN | This study | |
| pTJS140 | Broad-host-range cloning vector; Mob Apr Spr Smr lacZ | 19 | |
| pWG101 | pTJS140 carrying 5-kb mbnABCMN with its native promoter | This study | |
| pWG102 | pTJS140 carrying 4-kb mbnABCM with its native promoter | This study | |
| Primers | |||
| dmbnaF | ATTTTTggatccCGAAGGACAATAACAAGGCG | BamHI | This study |
| dmbnaR | ATTTTAggtaccACTCCAAACAgcatgcGATA | KpnI/SphI | This study |
| dmbnbF | ATTTTAggtaccATCCTTCTATGTCTGCAGCC | KpnI | This study |
| dmbnbR | ATTTTTaagcttGATCCTCCTCGAATTCCCTC | HindIII | This study |
| mbn21 | GACGTTCGGGTCTTCTTCGC | This study | |
| mbn22 | CGCCTCTAGATCATTCCGAC | This study | |
| pK18-bb-F | CTCTGGTAAGGTTGGGAAGC | This study | |
| pK18-bb-R | GCAATATCACGGGTAGCCAA | This study | |
| mbnANf | ATTTTTggtaccGACGTTCGGGTCTTCTTCGC | KpnI | This study |
| mbnANr | ATTTTTggtaccCGCCTCTAGATCATTCCGAC | KpnI | This study |
| mbnAMr | ATTTTTggtaccTTCGTTTCACATGGGATCGC | KpnI | This study |
| mbnPH-F | TTCGTGACGATCGAGGTC | This study | |
| mbnPH-R | GGTGCGTCCGTCGGTAAA | This study | |
| qmbnA_FO | TGGAAACTCCCTTAGGAGGAA | 4 | |
| qmbnA_RO | CTGCACGGATAGCACGAAC | 4 | |
| mbnN-F | GCTCGGAATTCTCGCTTTCC | This study | |
| mbnN-R | CGCCTCTAGATCATTCCGAC | This study | |
| mbnM-F | GTTCGGCTATTTCCTGACGC | This study | |
| mbnM-R | CTAGGCGCATCATCATCACA | This study |
Restriction sites are shown in lowercase letters. Apr, ampicillin resistance; Kmr, kanamycin resistance; Smr or Strr, streptomycin resistance; Spr, spectinomycin resistance.
General DNA methods, transformation, and conjugation.
DNA purification and plasmid extraction were performed using QIAquick and QIAprep kits from Qiagen following the manufacturer's instructions. DNA cloning, preparation of chemically competent cells, and plasmid transformation with E. coli were performed according to Sambrook and Russell (21). Enzymes used for restriction digestion and ligation were purchased from New England BioLabs (Ipswich, MA). PCR of DNA for cloning purposes was accomplished using iProof-High Fidelity polymerase (Bio-Rad, Hercules, CA). PCR for general purposes was accomplished using GoTaq DNA polymerase (Promega, Fitchburg, WI). PCR programs were set according to manufacturers' suggestions. Plasmids were conjugated into methanotrophic strains with E. coli S17.1 as the donor strain as described by Martin and Murrell (22).
Construction of a new methanobactin-defective methanotrophic strain.
A M. trichosporium mbnA::Gmr mutant was constructed previously where mbnA was knocked out via marker-exchange mutagenesis (4). While this mutant was unable to synthesize methanobactin, it does not allow easy mutation of other genes in the mbn operon. We therefore constructed a new M. trichosporium ΔmbnAN mutant, in which mbnABCMN was deleted by counterselection technique. Specifically, a 1-kb DNA fragment from upstream of mbnA and a 1-kb fragment spanning the 3′ region of mbnN were amplified using dmbnaF/dmbnaR and dmbnbF/dmbnbR (Table 2) primer sets, respectively. The amplified fragments were ligated together using the KpnI site and then cloned into pK18mobsacB at BamHI and HindIII sites to create plasmid pWG012. pWG012 was then conjugated into the M. trichosporium OB3b wild-type strain. Counterselection of mutants followed the procedure described by Puri et al. (23). First, transconjugants were selected by resistance to kanamycin for successful single homologous recombination and nalidixic acid to remove donor E. coli. Second, selected transconjugants were grown on NMS medium with no added copper and with 2.5% (mass/vol) sucrose for counterselection of double homologous recombination. Resulting colonies were then checked for kanamycin sensitivity and genotype via sequencing. A 3.5-kb region containing all of mbnABCM and 490 bp of the 5′ region of mbnN (>40%) was thus deleted.
Back-complementation of the M. trichosporium ΔmbnAN mutant and construction of the ΔmbnN mutant.
The suitability of mutant ΔmbnAN as a host for expression of recombinant methanobactin-synthesizing genes was established by introducing pWG101, an expression vector containing the wild-type mbn operon (mbnABCMN) with its native promoter, into mutant ΔmbnAN for back-complementation. pWG101 was constructed by cloning a 4.8-kb DNA fragment amplified by mbnANf/mbnANr primers (Table 2) into the pTJS140 vector (19) at the KpnI site.
To characterize the function of mbnN, a ΔmbnN mutant was constructed by introducing pWG102 expression vector into the ΔmbnAN mutant. pWG102 was constructed by cloning a 3.7-kb DNA fragment of mbnABCM, leaving out mbnN, amplified by mbnANf/mbnAMr primers (Table 2) into the pTJS140 vector (19) at the KpnI site. Both the ΔmbnAN + pWG101 and ΔmbnAN + pWG102 M. trichosporium strains were maintained on NMS medium containing 20 μg · ml−1 spectinomycin and with no added copper. Figure S1 in the supplemental material graphically illustrates the composition of the ΔmbnAN strain as well as the insertions into pWG101 and pWG102.
Extraction of RNA and reverse transcription-PCR (RT-PCR).
To check the expression of various genes from the plasmids and chromosomes of different M. trichosporium OB3b strains and their response to copper, RNAs from different cultures were collected, purified, and reverse transcribed to cDNA to perform RT-PCR. Total RNA was isolated from cells grown to exponential phase using a phenol-chloroform method modified from that of Griffiths et al. (24). Details have been described previously (4, 9). Following isolation, RNA was purified using a Zymo RNA Clean & Concentrator kit (Zymo Research, Irvine, CA) with a combination of at least two DNase treatments using Qiagen RNase-free DNase (Qiagen, Germany). Removal of DNA was confirmed by the absence of 16S rRNA PCR product from PCRs. The purified RNA was quantified using a NanoDrop instrument (NanoDrop ND-1000; NanoDrop Technologies, Inc., Wilmington, DE). The same amount of RNA (500 ng) was used for reverse transcription with SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA) for all reactions. RT-PCR analyses were performed to confirm expression of mbnPH (primers mbnPH-F/mbnPH-R) in the M. trichosporium ΔmbnAN mutant and of mbnA (qmbnA_FO/qmbnA_RO), mbnM (mbnM-F/mbnM-R), and mbnN (mbnN-F/mbnN-R) in M. trichosporium strains ΔmbnAN + pWG101 and ΔmbnAN + pWG102. To compare the expression levels of select genes in response to the copper level, the same amount of cDNA was used as a PCR template, and the same amount of PCR mix was loaded for gel analysis.
Isolation of methanobactin.
Methanobactin was isolated from the spent media of wild-type M. trichosporium OB3b, ΔmbnAN + pWG101, and ΔmbnAN + pWG102 strains as previously described by Bandow et al. (25) with the following modifications. Following freeze-drying, the samples were dissolved in prefiltered (0.22-μm-pore-size filter) H2O (>18 M Ω · cm) and loaded onto a 2.2-cm-by-25-cm Vydac Protein and Peptide C18 column (The Separations Group, CA, USA). The sample was washed with one column volume of H2O (>18 M Ω · cm) and one column volume of 5% methanol/95% H2O (>18 M Ω · cm). Methanobactin from M. trichosporium ΔmbnAN + pWG102 was eluted in one column volume of 10% methanol/90% H2O (>18 M Ω · cm), and methanobactin from the M. trichosporium OB3b wild-type and ΔmbnAN + pWG101 strains was eluted with 15% methanol/85% H2O (>18 M Ω · cm).
Mass spectroscopy.
Molecular masses of methanobactin from the M. trichosporium ΔmbnAN + pWG101 and ΔmbnAN + pWG102 strains were initially screened using a Voyager DE Pro matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometer (JBI Scientific, Huntsville, TX, USA). Samples (10 to 20 μg · μl−1) were mixed in a 1:1 ratio with matrix Super dihydroxybenzoic acid (Super DHB), and 1 μl of this mixture was loaded on a V700666 plate (from JBI Scientific) and allowed to dry at room temperature. Super DHB was prepared from 9 parts 2,5-dihydroxybenzoic acid (DHB) and 1 part 2-hydroxy-5-methoxybenzoic acid (Sigma-Aldrich, St. Louis, MO), both prepared in 70% acetonitrile–29.9% H2O–0.1% trifluoroacetic acid.
Exact molecular masses were determined on a Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled with an 1260 Infinity liquid chromatography system (Agilent Technologies, Santa Clara, CA, USA) in either the MS mode (LC-MS) or the MS/MS mode (LC-MS/MS). Samples were separated on a 0.5-mm-by-150-mm Zorbax SB C18 column (Agilent Technologies, Santa Clara, CA, USA) with a linear gradient of buffer A (0.1% formic acid, 99.9% H2O) and buffer B (0.1% formic acid, 99.9% acetonitrile).
Molecular masses of methanobactin from M. trichosporium ΔmbnAN + pWG101 and ΔmbnAN + pWG102 were also determined by direct injection on a Bruker SolariX Fourier transform-ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker, MA, USA) in positive electron spray ionization mode. Masses of methanobactin samples digested with 100 mM HCl for 12 h were also measured using LC-MS.
Amino acid sequence and composition analyses.
The N-terminal amino acid sequence of methanobactin from M. trichosporium ΔmbnAN + pWG101 and ΔmbnAN + pWG102 was determined by Edman degradation on a model 494 protein/peptide sequencer (Applied Biosystems, Carlsbad, CA, USA). The amino acid composition of methanobactin from M. trichosporium ΔmbnAN + pWG101 and ΔmbnAN + pWG102 was also determined by the Experiment Station Chemical Laboratories University of Missouri—Columbia using acid hydrolysis and performic acid oxidation methods as previously described (26). Amino acids were separated by cation exchange chromatography–high-performance liquid chromatography (cIEC-HPLC) coupled with postcolumn ninhydrin derivatization and quantified on an L8900 amino acid analyzer (Hitachi, Tokyo, Japan).
UV-visible light absorption spectroscopy.
UV-visible light absorption spectroscopy, acid hydrolysis of oxazolone rings, and copper titration experiments were performed as described earlier (1, 2, 8).
Thiol determination.
The concentration of thiol groups in methanobactin from M. trichosporium OB3b wild-type, ΔmbnAN + pWG101, and ΔmbnAN + pWG102 strains [as isolated and following reduction with tris(2-carboxyethyl)phosphine (TCEP)] was determined using a thiol fluorometric detection kit from Creative BioMart (Shirley, NY, USA). For detection of thiols after reduction, methanobactin was first placed in a Coy anaerobic chamber (95% argon/5% hydrogen atmosphere). Reaction mixtures for reduction contained 100 μl of 1 mM methanobactin from each strain and 200 μl of 8 mM TCEP and were incubated for 30 min at room temperature. Following reduction, samples were transferred to a 96-well microplate and sealed with microplate adhesive film (USA Scientific Inc., Ocala, FL, USA). Microplates were then removed from the anaerobic chamber and read immediately on a Tecan Safire fluorescence, absorbance, and luminescence reader (Tecan Group, Ltd., Männedorf, Switzerland).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Office of Science (Biological and Environmental Research), U.S. Department of Energy (grant no. DE-SC0006630 to J.D.S. and A.A.D.). Use of the Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer was made possible through a generous gift from the Roy J. Carver Charitable Trust (Muscatine, IA). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02619-16.
REFERENCES
- 1.Choi DW, Do YS, Zea CJ, McEllistrem MT, Lee SW, Semrau JD, Pohl NL, Kisting CJ, Scardino LL, Hartsel SC, Boyd ES, Geesey GG, Riedel TP, Shafe PH, Kranski KA, Tritsch JR, Antholine WE, DiSpirito AA. 2006. Spectral and thermodynamic properties of Ag(I), Au(III), Cd(II), Co(II), Fe(III), Hg(II), Mn(II), Ni(II), Pb(II), U(IV), and Zn(II) binding by methanobactin from Methylosinus trichosporium OB3b. J Inorg Biochem 100:2150–2161. [DOI] [PubMed] [Google Scholar]
- 2.Choi DW, Zea CJ, Do YS, Semrau JD, Antholine WE, Hargrove MS, Pohl NL, Boyd ES, Geesey GG, Hartsel SC, Shafe PH, McEllistrem MT, Kisting CJ, Campbell D, Rao V, de la Mora AM, DiSpirito AA. 2006. Spectral, kinetic, and thermodynamic properties of Cu(I) and Cu(II) binding by methanobactin from Methylosinus trichosporium OB3b. Biochemistry 45:1442–1453. [DOI] [PubMed] [Google Scholar]
- 3.Kim HJ, Graham DW, DiSpirito AA, Alterman MA, Galeva N, Larive CK, Asunskis D, Sherwood PM. 2004. Methanobactin, a copper-acquisition compound from methane-oxidizing bacteria. Science 305:1612–1615. doi: 10.1126/science.1098322. [DOI] [PubMed] [Google Scholar]
- 4.Semrau JD, Jagadevan S, DiSpirito AA, Khalifa A, Scanlan J, Bergman BH, Freemeier BC, Baral BS, Bandow NS, Vorobev A, Haft DH, Vuilleumier S, Murrell JC. 2013. Methanobactin and MmoD work in concert to act as the ‘copper-switch’ in methanotrophs. Environ Microbiol 15:3077–3086. [DOI] [PubMed] [Google Scholar]
- 5.DiSpirito AA, Semrau JD, Murrell JC, Gallagher WH, Dennison C, Vuilleumier S. 2016. Methanobactin and the link between copper and bacterial methane oxidation. Microbiol Mol Biol Rev 80:387–409. doi: 10.1128/MMBR.00058-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behling LA, Hartsel SC, Lewis DE, DiSpirito AA, Choi DW, Masterson LR, Veglia G, Gallagher WH. 2008. NMR, mass spectrometry and chemical evidence reveal a different chemical structure for methanobactin that contains oxazolone rings. J Am Chem Soc 130:12604–12605. doi: 10.1021/ja804747d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ, Galeva N, Larive CK, Alterman M, Graham DW. 2005. Purification and physical-chemical properties of methanobactin: a chalkophore from Methylosinus trichosporium OB3b. Biochemistry 44:5140–5148. doi: 10.1021/bi047367r. [DOI] [PubMed] [Google Scholar]
- 8.Krentz BD, Mulheron HJ, Semrau JD, Di Spirito AA, Bandow NL, Haft DH, Vuilleumier S, Murrell JC, McEllistrem MT, Hartsel SC, Gallagher WH. 2010. A comparison of methanobactins from Methylosinus trichosporium OB3b and Methylocystis strain SB2 predicts methanobactins are synthesized from diverse peptide precursors modified to create a common core for binding and reducing copper ions. Biochemistry 49:10117–10130. doi: 10.1021/bi1014375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu W, Haque MF, Baral BS, Turpin EA, Bandow NL, Kremmer E, Flatley A, Zischka H, DiSpirito AA, Semrau JD. 2016. A TonB-dependent transporter is responsible for methanobactin uptake by Methylosinus trichosporium OB3b. Appl Environ Microbiol 82:1917–1923. doi: 10.1128/AEM.03884-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solovyev V, Salamov A. 2011. Automatic annotation of microbial genomes and metagenomic sequences, p 61–78. In Li RW. (ed), Metagenomics and its applications in agriculture, biomedicine and environmental studies. Nova Science Publishers, Hauppauge, NY. [Google Scholar]
- 11.Lichtmannegger J, Leitzinger C, Wimmer R, Schmitt S, Schulz S, Kabiri Y, Eberhagen C, Rieder T, Janik D, Neff F, Straub BK, Schirmacher P, DiSpirito AA, Bandow N, Baral BS, Flatley A, Kremmer E, Denk G, Reiter FP, Hohenester S, Eckardt-Schupp F, Dencher NA, Adamski J, Sauer V, Niemietz C, Schmidt HHJ, Merle U, Gotthardt DN, Kroemer G, Weiss KH, Zischka H. 2016. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J Clin Invest 126:2721–2735. doi: 10.1172/JCI85226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Summer KH, Lichtmannegger J, Bandow N, Choi DW, DiSpirito AA, Michalke B. 2011. The biogenic methanobactin is an effective chelator for copper in a rat model for Wilson disease. J Trace Elem Med Biol 25:36–41. doi: 10.1016/j.jtemb.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Zischka H, Lichtmannegger J, Schmitt S, Jagemann N, Schulz S, Wartini D, Jennen L, Rust C, Larochette N, Galluzzi L, Chajes V, Bandow N, Gilles VS, DiSpirito AA, Esposito I, Goettlicher M, Summer KH, Kroemer G. 2011. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest 121:1508–1518. doi: 10.1172/JCI45401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ala A, Walker AP, Ashkan A, Dooley JS, Schilsky M. 2007. Wilson's disease. Lancet 369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 15.Roberts EA. 2011. Wilson's disease. Medicine 39:602–604. doi: 10.1016/j.mpmed.2011.08.006. [DOI] [Google Scholar]
- 16.Kaler SG. 2016. Microbial peptide de-coppers mitochondria: implications for Wilson disease. J Clin Invest 126:2412–2414. doi: 10.1172/JCI88617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simon R. 1984. High frequency mobilization of gram-negative bacterial replicons by the in vitro constructed Tn5-Mob transposon. Mol Gen Genet 196:413–420. doi: 10.1007/BF00436188. [DOI] [PubMed] [Google Scholar]
- 18.Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A. 1994. Small mobilizable multipurpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 19.Smith TJ, Slade SE, Burton NP, Murrell JC, Dalton H. 2002. Improved system for protein engineering of the hydroxylase component of soluble methane monooxygenase. Appl Environ Microbiol 68:5265–5273. doi: 10.1128/AEM.68.11.5265-5273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whittenbury R, Phillips KC, Wilkinson JF. 1970. Enrichment, isolation and some properties of methane-utilizing bacteria. J Gen Microbiol 61:205–218. doi: 10.1099/00221287-61-2-205. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 22.Martin H, Murrell JC. 1995. Methane monooxygenase mutants of Methylosinus trichosporium constructed by marker-exchange mutagenesis. FEMS Microbiol Lett 127:243–248. doi: 10.1111/j.1574-6968.1995.tb07480.x. [DOI] [Google Scholar]
- 23.Puri AW, Owen S, Chu F, Chavkin T, Beck DA, Kalyuzhnaya MG, Lidstrom ME. 2015. Genetic tools for the industrially promising methanotroph Methylomicrobium buryatense. Appl Environ Microbiol 81:1775–1781. doi: 10.1128/AEM.03795-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffiths RI, Whiteley AS, O'Donnell AG, Bailey MJ. 2000. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol 66:5488–5491. doi: 10.1128/AEM.66.12.5488-5491.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bandow NL, Gallagher WH, Behling L, Choi DW, Semrau JD, Hartsel SC, Gilles VS, Dispirito AA. 2011. Isolation of methanobactin from the spent media of methane-oxidizing bacteria. Methods Enzymol 495:259–269. doi: 10.1016/B978-0-12-386905-0.00017-6. [DOI] [PubMed] [Google Scholar]
- 26.AOAC International. 1994. Official methods of analysis: 77, 1362. Official method 994.12. AOAC International, Gaithersburg, MD. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.