

ABSTRACT
Some life-threatening, foodborne, and zoonotic infections are transmitted through poultry birds. Inappropriate and indiscriminate use of antimicrobials in the livestock industry has led to an increased prevalence of multidrug-resistant bacteria with epidemic potential. Here, we present a functional molecular epidemiological analysis entailing the phenotypic and whole-genome sequence-based characterization of 11 H. pullorum isolates from broiler and free-range chickens sampled from retail wet markets in Hyderabad City, India. Antimicrobial susceptibility tests revealed all of the isolates to be resistant to multiple antibiotic classes such as fluoroquinolones, cephalosporins, sulfonamides, and macrolides. The isolates were also found to be extended-spectrum β-lactamase producers and were even resistant to clavulanic acid. Whole-genome sequencing and comparative genomic analysis of these isolates revealed the presence of five or six well-characterized antimicrobial resistance genes, including those encoding a resistance-nodulation-division efflux pump(s). Phylogenetic analysis combined with pan-genome analysis revealed a remarkable degree of genetic diversity among the isolates from free-range chickens; in contrast, a high degree of genetic similarity was observed among broiler chicken isolates. Comparative genomic analysis of all publicly available H. pullorum genomes, including our isolates (n = 16), together with the genomes of 17 other Helicobacter species, revealed a high number (8,560) of H. pullorum-specific protein-encoding genes, with an average of 535 such genes per isolate. In silico virulence screening identified 182 important virulence genes and also revealed high strain-specific gene content in isolates from free-range chickens (average, 34) compared to broiler chicken isolates. A significant prevalence of prophages (ranging from 1 to 9) and a significant presence of genomic islands (0 to 4) were observed in free-range and broiler chicken isolates. Taken together, these observations provide significant baseline data for functional molecular infection epidemiology of nonpyloric Helicobacter species such as H. pullorum by unraveling their evolution in chickens and their possible zoonotic transmission to humans.
IMPORTANCE Globally, the poultry industry is expanding with an ever-growing consumer base for chicken meat. Given this, food-associated transmission of multidrug-resistant bacteria represents an important health care issue. Our study involves a critical baseline approach directed at genome sequence-based epidemiology and transmission dynamics of H. pullorum, a poultry pathogen having established zoonotic potential. We believe our studies would facilitate the development of surveillance systems that ensure the safety of food for humans and guide public health policies related to the use of antibiotics in animal feed in countries such as India. We sequenced 11 new genomes of H. pullorum as a part of this study. These genomes would provide much value in addition to the ongoing comparative genomic studies of helicobacters.
KEYWORDS: Helicobacters, genomics, molecular epidemiology, poultry
INTRODUCTION
Helicobacter pullorum is a urease-negative, bile-resistant bacterium that was first isolated from the ceca of asymptomatic poultry and was also recovered from the intestinal contents and livers of laying hens with enteritis and vibrionic hepatitis (1). This nonpyloric Helicobacter species has also been isolated from people suffering from gastroenteritis (1–4) and is likely to be associated with inflammatory bowel disease (5, 6) and chronic liver disease (7–12). Laharie et al. (13) also detected DNA of H. pullorum and H. canadensis in Crohn's disease patients, as well as in controls. Moreover, several reports suggest that some enterohepatic Helicobacter species, such as Helicobacter hepaticus, Helicobacter bilis, Helicobacter canis, Helicobacter cinaedi, and H. pullorum, produce a common, well-characterized bacterial virulence factor, the cytolethal distending toxin (CDT) (14–18). This bacterial protein triggers G2/M cell cycle arrest in a wide range of mammalian cell lines, leading to enlarged or distended cells dying with apoptosis (19). A recent finding also highlighted that CDT in H. pullorum is responsible for major cytopathogenic effects in vitro, reinforcing its role as a major virulence factor in pathogenesis (20).
The infectious disease burden is very high globally, and inappropriate and irrational use of antimicrobial agents against these infectious diseases has already resulted in the emergence of multidrug-resistant (MDR) bacteria. The prevalence of MDR organisms in livestock and their products has traditionally been linked to the disproportionate use of antimicrobials in veterinary practice and animal husbandry settings in the form of prophylactic agents and growth promoters (21). These drug-resistant bacteria greatly reduce treatment efficacy and cause increased morbidity and mortality in livestock, as well as in humans (22). The problem of antimicrobial resistance (AMR) is even more complex in developing countries because of high population density, poor sanitation, and less stringent antibiotic policies. In addition to this, there are only a few novel antimicrobial agents that are expected to be available for use in the next few years (23).
The problem of AMR is one of the greatest threats to public health. The resistant bacteria and their associated genes can move within and between populations of humans and animals, which makes AMR a contentious issue. As this problem continues to grow, it is imperative to characterize organisms that are resistant to multiple antimicrobial agents so that epidemiological surveillance data can be reliably obtained.
Chickens are used as a major source of food proteins worldwide. In India, consumption of poultry meat is growing at about 12% a year, making it one of the fastest-growing markets in the world (National Sample Survey Office of India report, 2014). In order to fulfill the high demand for chicken meat, poultry farmers or feed manufacturers may resort to the use of growth promoters, including various antibiotics, sometimes indiscriminately, for faster weight gain by chickens. Moreover, epidemiological investigations suggest that the majority of the foodborne bacterial infections caused by different enteric pathogens majorly spread through foods of animal origin (24, 25). The mainstream medical literature lacks any report from India or other developing countries on systematic epidemiological tracking of bacterial pathogens from poultry, in particular, H. pullorum. In poultry, H. pullorum was found to colonize predominantly the cecum (26). Recently, Borges et al. isolated H. pullorum directly from chicken meat (27). The presence of H. pullorum on poultry carcasses has also been reported, which might be due to contamination during the poultry rearing, handling, and slaughtering processes (28). Therefore, with the risk of zoonosis together with its ability to spread AMR, H. pullorum could be an emerging foodborne human pathogen that needs to be deciphered at the genomic and molecular levels, particularly by elucidating its zoonotic potential.
In this study, we attempted to rigorously characterize a modest set of H. pullorum isolates with respect to their multidrug resistance potential and their genome sequence-based phylogeny and epidemiology by analyzing their core and pan-genomes, mobilome, and virulence gene repertoire.
RESULTS
Isolation, characterization, and genome sequencing of H. pullorum isolates.
We isolated 11 H. pullorum isolates from 100 chicken samples that were collected from seven different retail wet markets in Hyderabad City, India. These 11 isolates originated from four different markets that were spread out in different places in Hyderabad City. Of these 11 strains, 6 were isolated from free-range chickens and were designated NAP1W4, NAP2W5, NAP3W17, NAP5W19, NAP6W24, and NAP8W25 and 5 were from broiler chickens and named NAP10B8, NAP11B31, NAP12B32, NAP13B35, and NAP14B36. The broiler chickens we sampled were reared on farms that employ formula feed supplemented with antibiotics, while the free-range chickens originated in rural areas. Gram staining and urease testing revealed small, curved, Gram-negative, and urease-negative bacilli that were confirmed as H. pullorum by species-specific PCR and 16S rRNA gene sequencing. Whole-genome sequencing (WGS) of 11 H. pullorum isolates produced high-quality genomic assemblies. The detailed genome characteristics of the 11 sequenced H. pullorum isolates are shown in Table 1, and an alignment of their whole genome sequences with the H. pullorum MIT985489 genome as a reference is shown in Fig. 1. The result of BLAST ring image generator (BRIG) analysis of this whole-genome alignment demonstrated that the 11 H. pullorum isolates of free-range and broiler chickens together with 1 H. pullorum isolate from a human were homogeneous in their genetic architecture, with no significant genomic divergence despite their geographic and host differences.
TABLE 1.
Genome statistics of the whole-genome sequences of the 11 H. pullorum isolates in this study
| Strain | Avg fold genome coverage | No. of contigs | Genome size (bp) | No. of CDSs | Avg CDS length (bp) | % coding capacity | % G+C | No. of genes |
|
|---|---|---|---|---|---|---|---|---|---|
| rRNA | tRNA | ||||||||
| NAP1W4 | 202 | 162 | 1,889,077 | 1,947 | 885 | 88.2 | 34.16 | 3 | 37 |
| NAP2W5 | 212 | 140 | 1,804,031 | 1,814 | 886 | 89.1 | 34.38 | 3 | 36 |
| NAP3W17 | 286 | 74 | 1,733,930 | 1,746 | 906 | 91.2 | 34.44 | 3 | 37 |
| NAP5W19 | 166 | 127 | 1,731,995 | 1,719 | 893 | 88.6 | 34.45 | 3 | 36 |
| NAP6W24 | 171 | 108 | 1,691,767 | 1,688 | 904 | 90.2 | 34.56 | 5 | 38 |
| NAP8W25 | 110 | 134 | 1,783,256 | 1,819 | 883 | 90 | 34.25 | 3 | 37 |
| NAP10B8 | 119 | 105 | 1,717,485 | 1,705 | 907 | 90.1 | 34.46 | 4 | 36 |
| NAP11B31 | 80 | 139 | 1,862,853 | 1,886 | 876 | 88.7 | 34.08 | 3 | 37 |
| NAP12B32 | 97 | 130 | 1,856,360 | 1,883 | 877 | 88.9 | 34.11 | 2 | 36 |
| NAP13B35 | 83 | 123 | 1,856,511 | 1,881 | 885 | 89.6 | 34.11 | 2 | 36 |
| NAP14B36 | 76 | 101 | 1,855,374 | 1,888 | 879 | 89.4 | 34.11 | 3 | 36 |
FIG 1.

Whole-genome alignment of H. pullorum comprising six free-range (green) and five broiler (blue) chicken isolates generated by BRIG with the H. pullorum MIT985489 genome positioned as a reference. The key to the right lists the isolates from top to bottom as they appear from the inside of the circle toward the outside.
Antimicrobial susceptibility profiles and identification of AMR genes.
All of the isolates from both broiler and free-range chickens were found to be MDR, as all 11 isolates were resistant to at least three different antibiotic classes. The isolates from broiler and free-range chickens did not show any significant difference in their antibiotic resistance profiles, as shown in Table 2. The AMR rates of broiler and free-range chicken isolates were found to be 100% for nalidixic acid, enrofloxacin, co-trimoxazole, and cefotaxime, while 80% of the broiler and 83% of the free-range chicken isolates were resistant to ciprofloxacin. Also, 80% of the broiler and 67% of the free-range chicken isolates were resistant to clarithromycin. However, all of the isolates were susceptible to tetracycline, neomycin, chloramphenicol, and colistin. Phenotypically, the rate of extended-spectrum β-lactamase (ESBL) production in broiler and free-range chicken isolates was found to be 100%. In addition, all of the ESBL producers were resistant to the beta-lactamase inhibitor clavulanic acid. Correlating with the phenotypic observations, the genomic data of all of the isolates showed the presence of five or six well-characterized AMR genes linked to resistance to various antibiotic classes (see Table S2 in the supplemental material). All of the strains, irrespective of their isolation source, were found to harbor common AMR-associated genes such as the elfamycin resistance gene tufA, a quinolone/fluoroquinolone resistance-related gene encoding DNA gyrase subunit A, and a multidrug resistance-nodulation-division (RND) family transporter efflux pump gene (cmeB). All of the isolates carried aminoglycoside resistance gene aph(3″)-Ib, except for one free-range chicken isolate, NAP8W25. The broiler chicken isolates carried one or two additional genes encoding aminoglycoside resistance (see Table S2 in the supplemental material).
TABLE 2.
Prevalence of antimicrobial resistance in H. pullorum isolates obtained from free-range and broiler chickens
| Phenotype | Overall prevalence of resistance phenotype in population (%) | Estimated fraction due to isolates from: |
|
|---|---|---|---|
| Free-range chickens | Broiler chickens | ||
| Quinolone/fluoroquinolone resistance | |||
| Ciprofloxacin | 82 | 0.55 | 0.44 |
| Nalidixic acid | 100 | 0.54 | 0.45 |
| Enrofloxacin | 100 | 0.54 | 0.45 |
| Sulfonamide/trimethoprim (co-trimoxazole) resistance | 100 | 0.54 | 0.45 |
| Cephalosporin (cefotaxime) resistance | 100 | 0.54 | 0.45 |
| Macrolide (clarithromycin) resistance | 73 | 0.50 | 0.50 |
| Aminoglycoside (neomycin) resistance | 0 | 0 | 0 |
| Phenicol (chloramphenicol) resistance | 0 | 0 | 0 |
| Tetracycline resistance | |||
| Oxytetracycline | 0 | 0 | 0 |
| Chlortetracycline | 0 | 0 | 0 |
| Doxycycline hydrochloride | 0 | 0 | 0 |
| Miscellaneous (colistin) resistance | 0 | 0 | 0 |
| ESBL production | 100 | 0.54 | 0.45 |
| Clavulanic acid resistance | 100 | 0.54 | 0.45 |
| Multidrug resistance | 100 | 0.54 | 0.45 |
Phylogenetic analysis.
To determine the genetic relatedness of heterogeneous Helicobacter species and H. pullorum strains, we performed a consensus bootstrapped core genome maximum-likelihood-based phylogenetic analysis. The results showed that all 33 Helicobacter, Wolinella, and Campylobacter genomes could be broadly separated into two distinct clusters (Fig. 2). H. canadensis and H. rodentium clustered closely with H. pullorum isolates, revealing high similarity to H. pullorum among all of the other Helicobacter species analyzed. This is in agreement with the previous reports of 16S rRNA gene-based phylogenetic analyses (29, 30). Interestingly, the genomes of Wolinella succinogenes and Campylobacter jejuni, although they belong to different genera, coclustered with H. pullorum, exhibiting their genetic relatedness to the latter.
FIG 2.

Core genome-based consensus maximum-likelihood phylogenetic tree of 11 in-house and 5 publicly available H. pullorum genomes with 1 C. jejuni, 1 W. succinogenes, and 17 other Helicobacter species genomes. The tree was generated after 1,000 replicates/bootstraps with RAxML. The output of RAxML was visualized with the iTOL tool.
The phylogenetic diversity within H. pullorum isolates is depicted by a tree in Fig. 3. From the phylogenetic tree, we could glean that all of our H. pullorum isolates were intermixed with each other, forming mixed clusters, irrespective of their geographic origins and isolation sources. Hence, our results suggest that our H. pullorum strains did not show geographically restricted lineages, unlike H. pylori. Additionally, H. pullorum strains from broiler and free-range chickens and a single human isolate showed mixed clusters, indicating that H. pullorum strains from different hosts and geographical locations share significant genetic features with each other and they particularly did not exhibit a host-specific genomic architecture. The results also demonstrated that the H. pullorum isolates from broiler chickens were genetically more similar than H. pullorum isolates from free-range chickens (Fig. 3).
FIG 3.

Core genome-based consensus maximum-likelihood phylogenetic tree of only H. pullorum isolates comprising 11 in-house and 5 publicly available H. pullorum genomes. The tree was generated after 1,000 replicates/bootstraps with RAxML and visualized by the iTOL tool. FRC, free-range chicken isolates; BC, broiler chicken isolates.
Core and pan-genome analysis.
The core genome contents of 11 sequenced H. pullorum strains, H. pullorum strain 229313, and human-derived strain MIT985489 were analyzed by OrthoMCL. A total of 25,438 proteins were predicted from the 13 strains. These predicted proteins formed 2,188 orthologous gene clusters. Of these, around 1,359 orthologous gene clusters were found to constitute the core genome. Of the 1,359 core gene clusters, 1,041 could be characterized into various functional categories according to the Clusters of Orthologous Groups of proteins (COG) database, as shown in Fig. 4. The COG functional classification revealed a large proportion of genes belonging to the J, E, and M functional classes. Class J contains genes having functions related to translation, ribosomal structure, and biogenesis, while classes E and M consist of genes involved in amino acid transport/metabolism, and cell wall/envelope biogenesis, respectively. The core genome comprised genes encoding multidrug efflux pumps, which contribute to intrinsic drug resistance in bacteria (31). The genes encoding metallo-β-lactamase enzymes that confer resistance to β-lactam antibiotics, including carbapenems, were also identified in the core genome. H. pullorum-specific gene content analysis revealed a total of 8,560 H. pullorum-specific proteins among all 16 H. pullorum isolates, with an average of 535 specific proteins per isolate. Of the total of 8,560 H. pullorum-specific genes, 83% (7,136) belong to a hypothetical category whereas the remaining 17% (1,424) were classified into different COG categories, with the majority of them being assigned to cell wall/membrane/envelope biogenesis, transcription, defense mechanisms, and other multiple classes.
FIG 4.

COG functional classification of 1,041 core genes obtained by comparing 13 H. pullorum isolates (11 in-house and 2 publicly available genomes). On the x axis is the number of genes in each functional category on the y axis.
Further analysis of our H. pullorum isolates for the presence of strain-specific content revealed a total of 327 strain-specific genes. H. pullorum strains from free-range chickens were found to contain 204 strain-specific genes, with an average of 34 genes per genome, while H. pullorum from broiler chicken strains contained only 14 strain-specific genes. The genomes of H. pullorum strains 229313 and MIT985489 carried 59 and 50 strain-specific genes, respectively (Fig. 5). Analysis of these strain-specific genes revealed that a majority of them were predicted to code for hypothetical proteins, prophages, transposons, and functions related to cell wall/envelope biogenesis and transcription. In addition, some of these genes also code for type IV DNA secretion systems and type I restriction-modification systems. Moreover, a few strain-specific genes were also found to be associated with bacterial defense mechanisms, replication, recombination, and signal transduction.
FIG 5.
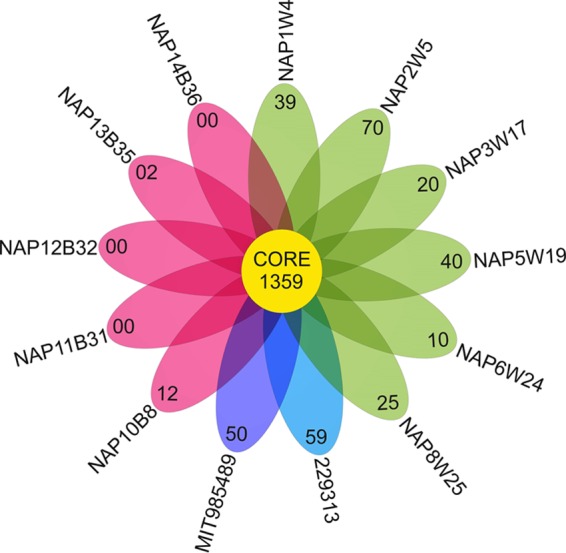
Diagrammatic representation of core and specific gene contents of free-range and broiler chicken isolates and two other H. pullorum strains (MIT985489 and 229313).
Identification of virulence factors and mobile genetic elements.
To understand the pathogenicity of H. pullorum and the most prevalent virulence profiles, we investigated the virulence gene contents of all of our isolates. A total of 182 different virulence factors were predicted from among our H. pullorum isolates (see Table S3 in the supplemental material). The results revealed that around 116 virulence genes were conserved in all of the H. pullorum isolates; the other 66 genes were present in some but absent from other strains of H. pullorum. It was found that about 40 such virulence genes encode functions related to bacterial motility that could be essential for the colonization of chicken ceca by bacteria. The presence of a histidine kinase (FlgS) and a response regulator protein (FlgR) also indicated the existence of bacterial two-component regulatory (TCR) systems. A total of 20 genes were found to encode adhesion function proteins with the unique characteristic of being heat stable. Other virulence factors identified in H. pullorum strains included 18 genes that are part of a glycosylation system and 7 genes coding for hypothetical lipopolysaccharide endotoxins. Most importantly, all of the isolates encoded the CdtB subunit of a common cell cycle-arresting cytotoxic protein named CDT.
The draft genome sequences of H. pullorum strains from both broiler and free-range chickens were also screened for the presence of putative phage sequences, genomic islands, and insertion elements. The genomes of broiler chicken isolates NAP11B31, NAP12B32, and NAP13B35 carried various phage remnants along with one intact phage region. While NAP14B36 carried only one intact phage sequence, NAP10B8 harbored four incomplete phage regions. The sizes of the intact phage regions present in NAP11B31, NAP12B32, NAP13B35, and NAP14B36 were found to be 8.7, 6.4, 7.7, and 19.8 kb, respectively, containing 18, 12, 15, and 24 coding sequences (CDS), respectively. All intact phage regions contained Escherichia phage vB_EcoM-ep3, except for the intact region of NAP11B31, which contained Stenotrophomonas phage Smp131 of Stenotrophomonas maltophilia.
All of the H. pullorum isolates from free-range chickens contained incomplete phage-associated regions of various sizes, ranging from 5.6 to 15.8 kb. Cronobacter sakazakii phage GAP32 and Cafeteria roenbergensis phage BV-PW1 were the common phage-associated regions found in these isolates. These phage-associated regions consisted of genes encoding putative functions relevant to bacterial metabolism, membrane-associated protein functions, restriction-modification systems, and metalloproteases together with phage-related tail and base plate assembly proteins and some hypothetical proteins. For detailed descriptions of the phages identified in the genomes of H. pullorum and their gene contents, see Table S4 in the supplemental material.
Several genomic islands ranging in size from 4 to 67 kb were identified in 7 of the 11 H. pullorum genomes sequenced. No genomic island was detected in the NAP2W5, NAP3W17, NAP6W24, or NAP10B8 isolate. These islands mainly contain genes associated with bacterial outer membrane proteins, type II/IV and VI secretory systems, restriction-modification system(s), and putative hypothetical proteins. Table S5 in the supplemental material presents a detailed account of all of the genomic islands identified in seven isolates along with their major predicted functions.
DISCUSSION
From the above observations, it is evident that our study exhaustively unraveled, as a pioneering effort, the genotypic, phenotypic, chromosomal, and molecular characteristics of H. pullorum isolates from the intestines of broiler and free-range chickens in India. Other studies (26, 32) previously reported on the prevalence of H. pullorum in the raw meat, cecum, and liver of poultry from different countries, which could possibly pose a high health risk to humans via its transmission through food similar to that of Campylobacter species. However, the prevalence and molecular characteristics of H. pullorum from chicken carcasses have not been studied in greater detail, including the genomic and epidemiological attributes of field strains. Our study also gains significance in the Indian context, as there is currently no effective implementation of antibiotic use guidelines in the poultry industry in this vast country that has a predominantly agriculture-based and rural-area-focused economy. On the other hand, the rate of consumption of chicken meat is increasing exponentially. The reason for the paucity of work in this area could be that H. pullorum is a fastidious organism and its isolation is troublesome; this has, in many ways, hindered the establishment of this organism as a foodborne pathogen. In this study, we validated and reassessed the usefulness of a convenient method for isolation of H. pullorum from the poultry cecum, a membrane filter technique described earlier (26).
Further, we have comprehensively characterized the AMR profiles of H. pullorum isolates from both broiler and free-range chickens by testing them against several different classes of antibiotics and also compared their genomic features. Previous studies involving antimicrobial susceptibilities of H. pullorum by Zanoni et al. (26) and Ceelen et al. (33) did not detect resistance to the macrolide and fluoroquinolone classes. Although Borges et al. (27) reported H. pullorum resistance against fluoroquinolones, they only included four strains isolated from a single poultry source and did not examine the ESBL production of H. pullorum strains. In our study, we found that all of the isolates were MDR, irrespective of their isolation source. Our findings showed that all of our Indian isolates were prominent in harboring resistance to various classes of antibiotics (quinolones/fluoroquinolones, cephalosporins, sulfonamides, and macrolides), unlike previous reports (26, 33). Moreover, all of our isolates were also resistant to clavulanic acid. We were expecting a lower prevalence of AMR in free-range chicken isolates because there is no intensive use of antibiotics in their rearing process, unlike that of broiler chickens. Surprisingly, our broiler and free-range chicken isolates showed similar resistance patterns, which might be due to the ability of these bacteria to acquire genes horizontally (34) from resistant microorganisms in the surrounding environment as the exposure of free-range chickens to environmental contamination is much greater than that of farm-reared chickens (35, 36). This could also be due to the indiscriminate use of antibiotics in humans and animals, which results in high selection pressure. Free-range chickens have high exposure to human and animal excreta and their wastes. Because they inhabit human communities, these birds pick up antibiotic residues/resistance determinants, thus making their gut microbiota resistant to different antibiotics. We believe that this is how the free-range and broiler chicken isolates in this study came to have similar AMR profiles.
Resistance to clavulanate might be due to hyperproduction of the enzyme beta-lactamase, which overwhelms the action of this enzyme inhibitor (37). However, the presence of an RND family efflux pump(s) and its involvement in clavulanate resistance cannot be ruled out because of its wide range of efflux substrates (38, 39). In addition to performing phenotypic antimicrobial susceptibility tests, we also genotypically confirmed the presence of molecular determinants of AMR toward elfamycin, quinolone/fluoroquinolone resistance genes, and a multidrug RND family transporter efflux pump in all of our isolates, with the aminoglycoside resistance genes being predominant (92% of our isolates). In particular, more aminoglycoside resistance genes were detected in H. pullorum from broiler chickens than in H. pullorum from free-range chickens.
The core genome is the part of a genome that is present in all of the isolates of a species and is crucial for survival in a particular habitat. Core genome analysis plays a key role in determining population structure, which in turn sheds light on the evolutionary trajectories of strains (40). Core genome-based phylogenetic analysis clearly demonstrated an interspecies genetic relatedness of Helicobacter isolates in cluster 1 of Fig. 2. High intraspecies genomic diversity of H. pullorum from free-range chickens was observed, as shown in Fig. 3. Analysis of phylogenetic trees revealed that the genomes of W. succinogenes, C. jejuni, H. canadensis, H. rodentium, and H. pullorum clustered together, showing high conservation of their core genome content compared to that of other Helicobacter species, which indicates their common ancestry and suggests that H. pullorum could have descended from or shared lineages with any of the above species, particularly H. canadensis. The habitat of an organism also plays an important role in the evolution of bacterial species, as some of them, such as H. canadensis (29), C. jejuni, and H. pullorum, have the same host, and this might be the reason for shared core genome contents compared to others. The presence of a remarkable number of genomic islands and a large inventory of prophages in H. pullorum isolates points toward plausibility of its ability to acquire genes via horizontal gene transfer, which has a potential impact on the evolutionary adaptation of bacterial species (41).
Our analyses also made it clear that the genomes of H. pullorum isolates exhibited some degree of genomic diversity, which is consistent with a previous study based on two fingerprinting techniques, amplified fragment length polymorphism and pulsed-field gel electrophoresis (42). We found that strains from free-range chickens were genetically more diverse than broiler chicken isolates, which could possibly be due to the unrestricted movement of free-range chickens in their surroundings, where continuous exposure to a large number of microorganisms from a variety of foods and contaminated water and air shapes their gut microbiota composition. In contrast, broiler chickens were restricted at a specific place and fed formula feed with a clean water supply and compulsory exposure to antibiotics used as anabolics and administered also for prophylaxis and treatment. Because of this restricted environment and the continuous selection pressure of antibiotics, it was obvious that the guts of broiler chickens on poultry farms would contain a largely clonal and identical repertoire of microbial species that would be very different from that of their free-ranging counterparts. Hence, we saw less diversity in H. pullorum isolates obtained from broiler chickens.
Corroborating the results of whole-genome phylogeny, the results of strain-specific gene content analysis confirmed the increased genomic diversity of H. pullorum from free-range chickens. Consequently, we identified a higher number of specific genes in all of our free-range chicken isolates than in our broiler chicken isolates, three of which were completely lacking specific genes, while only 12 and 2 specific genes were observed in two such isolates. According to our restricted/unrestricted-movement assumption, free-range chickens have an opportunity to make their gut microbiota more dynamic and more prone to acquire genetically diverse bacteria from their surroundings than broiler chickens.
Although the pathogenic mechanisms of H. pullorum are not well understood, it is known that adherence, cell invasion, and intracellular resistance, along with toxin-producing factors, are prerequisites for establishing a bacterial infection and for its outcome. As motility is an essential process involved in direct colonization (43), the presence of a notable number of virulence genes associated with bacterial motility and signal transduction indicates H. pullorum's potential to colonize chickens, as well as humans. Also, the presence of genes for a TCR system in H. pullorum may suggest its importance in supporting bacterial growth at various temperatures in different hosts (44).
A noteworthy number of genes were found to be associated with bacterial adherence, immune evasion, and glycosylation systems. These genes mostly correspond to bacterial membranous lipopolysaccharides and capsule biosynthesis and are considered heat-stable antigens and therefore may be required for the pathogen's stability and proper function at the high body temperature of chickens. In addition, the presence of about 20 glycosylation system-related genes makes it highly probable that they are required to change the carbohydrate structure and thus orient carbohydrates to mimic surface-exposed host carbohydrate molecules, as in Guillain-Barre syndrome caused by C. jejuni (45).
In our virulence factor analysis, we found genes for synthesizing a common subunit of CDT, CdtB. We identified it as a C. jejuni-like toxin subunit; CdtB in H. pullorum was found to possess characteristics of type I DNases. Brief exposure of cultured cells to this subunit results in remarkable chromatin disruption, induction of cytoplasmic distention, and eventually cell cycle arrest (46). So, the existence of this protein in H. pullorum might play an important role in several complications associated with human, as well as animal, health.
Besides these virulence factors, the core genomes of all of our H. pullorum isolates were found to have an RND family efflux pump, which is not restricted only to drugs but could also play an important role in bacterial pathogenicity. In addition to their wide range of efflux substrates, these multiple drug resistance pumps also facilitate bacterial adaptation to different niches (47). The presence of AcrAB-TolC and CmeABC RND efflux pumps suggests H. pullorum's extraordinary defense against a wide range of host-derived substrates, and these pumps could also play an important role in bacterial adherence to and invasion of host epithelial cells, as observed in Salmonella Typhimurium (48) and C. jejuni (49) infections, respectively. Because of the wide range of antibiotic resistances associated with them, multiple-drug efflux pumps may also serve as potential antibacterial targets, as their inhibition may help to achieve the maximum effectiveness of current and future antibiotics (50).
In conclusion, our study suggests that chickens could be a major source for transmission of the emerging MDR pathogen H. pullorum from poultry to humans. Additionally, our study provides a strong argument that H. pullorum is an emerging foodborne pathogen because of its high genomic relatedness to some seasoned pathogens such as C. jejuni. Given that India has the world's fifth largest poultry industry and a large consumer base of approximately 500 million customers for chicken meat, food-associated acquisition of AMR and MDR genes and virulence genotypes and phenotypes represents an alarming situation, and our study constitutes a baseline pioneering effort at dissecting this situation from a pathogen biology and epidemiology point of view. This would likely underpin increased and augmented surveillance systems for the safety of food for humans and also for monitoring animal health. Further, the genomes newly sequenced in this study would be an important resource for future studies involving the comparative genomics and molecular epidemiology of pyloric and nonpyloric Helicobacter isolates in different hosts.
MATERIALS AND METHODS
Sampling, isolation, and identification of strains.
Samples from the gastrointestinal tracts of 55 broiler chickens and 45 free-range chickens from commercial retail poultry outlets were collected in separate sterilized plastic tubes and processed within 3 to 4 h. The ceca were aseptically severed, and their surfaces were washed with 1× phosphate-buffered saline to minimize contamination. The cecal contents were removed, and tissues were cut into small pieces and put into a sterile tissue homogenizer containing 5 ml of brain heart infusion (BHI) broth to get a homogeneous suspension. Tissue homogenates (100 μl) were diluted in 300 μl of BHI broth. Diluted samples were inoculated onto BHI agar supplemented with 10% horse serum and antibiotics (vancomycin [10 mg/liter], amphotericin B [3 mg/liter], and cefoperazone [32 mg/liter]) by the modified filter technique of Steele and McDermott (51). In brief, sterile cellulose acetate membrane filters (0.65-μm pore size) were applied with sterile tweezers onto the surface of a BHI agar plate and 100 μl of diluted homogenate was placed in the middle of each membrane filter. The plate was then incubated for 1 to 2 h in a microaerobic atmosphere (90% N2, 5% O2, 5% CO2) at 37°C. The membrane filters were then removed with sterile tweezers, and the filtrate was streaked onto the agar with a sterile loop. The plates were then incubated under the same conditions for 3 to 4 days. Approximately 5 to 10 grayish white bacterial colonies were observed on the plates in each case.
Colony PCR assays for the H. pullorum 16S rRNA gene with specific primers (forward, 5′ ATG AAT GCTAGT TGT TGT CAG 3′; reverse, 5′ GAT TGG CTC CACTTC ACA 3′) were performed for confirmation to the species level. Amplified products were run on 1.2% agarose gel and visualized with a gel documentation system (Major Science, USA). Amplified products were also sequenced for further confirmation.
Antimicrobial susceptibility testing and identification of AMR genes.
The standard Kirby-Bauer disk diffusion method was used to test the antimicrobial susceptibility of all isolates. The antimicrobial drugs (HiMedia, Mumbai, India) used for this experiment included oxytetracycline (30 μg), doxycycline hydrochloride (30 μg), chlortetracycline (30 μg), ciprofloxacin (5 μg), nalidixic acid (30 μg), enrofloxacin (5 μg), chloramphenicol (30 μg), neomycin (10 μg), co-trimoxazole (25 μg), cefotaxime (30 μg), clarithromycin (15 μg), and colistin (10 μg). To standardize the bacterial suspension (in BHI broth medium), the bacterial density was adjusted to a 0.5 McFarland standard and spread over the entire surface of each BHI agar plate with a sterile cotton swab. Antimicrobial discs were placed on the agar surface with sterile tweezers, and then the plates were incubated at 37°C for 3 to 4 days under microaerobic conditions as described before. A double-disc synergy test was used to check all isolates for ESBL production (52). Thereafter, both results were interpreted according to Clinical and Laboratory Standards Institute recommendations (73). In addition, the amino acid sequences encoded by the predicted genes of 11 H. pullorum strains were compared with those in the comprehensive antibiotic resistance database (http://arpcard.mcmaster.ca) by using BLASTp with identity and query coverage of >50% and >80%, respectively.
Isolation of genomic DNA and WGS.
Genomic DNA was extracted with the Qiagen DNeasy blood and tissue kit (Qiagen, Germany). The yield and quality of DNA were assessed with a NanoDrop spectrophotometer (Thermo Scientific). WGS of all isolates was carried out with the Illumina MiSeq sequencer, which generated approximately 0.5 to 1.2 million paired-end reads of 300 bp, with an average insert size of 400 to 500 bp. Filtering and trimming of the paired-end reads were done with the help of NGS QC Toolkit (53) and FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), respectively. To determine the optimum hash length and the best assembly, VelevetOptimiser.pl script and Velvet (v1.2.10), respectively, were employed (54). Gene prediction and functional annotation of the draft genomes were carried out with RAST (Rapid Annotation Using Subsystem Technology) (55–57), followed by validation with GeneMarkS (58) and Glimmer (59). Genome statistics of these strains were gleaned with ARTEMIS (60). The numbers of tRNA and rRNA genes were identified with tRNAscan-SE (61) and RNAmmer (62), respectively.
Phylogenetic analysis.
The predicted amino acid sequences of 11 H. pullorum isolates along with 5 other strains of H. pullorum, 17 other Helicobacter species, 1 C. jejuni strain, and 1 Wolinella succinogenes strain from the National Center for Biotechnology Information (see Table S1 in the supplemental material) were compared all versus all with BLASTp, followed by the Markov clustering algorithm to identify orthologs (63). Here, the percent identity threshold and E value were adjusted to 70% and 0.00001, respectively. Only genes comprising a minimum of 50 amino acids were included in ortholog analysis. The orthologous gene clusters that did not have any paralogs were considered for further core genome-based phylogeny. Core orthologous genes were aligned with Mafft by using default parameters, followed by removal of gaps with TrimAL (64, 65). The processed alignments were concatenated by using an in-house perl script and used as the input for RAxML (66). The program was run by using the general time-reversible nucleotide substitution model with gamma correction. A consensus maximum-likelihood-based phylogenetic tree was generated after 1,000 replicates/bootstraps. Similarly, we constructed a separate phylogenetic tree exclusively for H. pullorum isolates of free-range and broiler chickens to get a better understanding of their genotypic resemblance. The output of RAxML was visualized with the interactive tree of life (iTOL) tool (67). Further, the whole genomes of free-range and broiler chicken isolates of H. pullorum were also compared by using BRIG (68) to visualize the approximate similarity within their genomes with BLASTn.
Core and pan-genome analysis.
The sequenced H. pullorum isolates along with two others (human H. pullorum isolate MIT985489 and poultry isolate 229313) were analyzed to calculate their core and accessory genome contents. The orthologs were determined on the basis of the threshold values as mentioned in the above section. The orthologous gene clusters that carried orthologs in all of the genomes constituted the core content, whereas others were considered accessory gene content.
The specific gene content of H. pullorum was analyzed by pooling the predicted amino acid sequences of 17 different species of Helicobacter (see Table S1 in the supplemental material) together with all of our H. pullorum isolates (n = 16). Core and accessory gene contents were identified with the OrthoMCL program. H. pullorum accessory genes were extracted from the pooled accessory gene content of all Helicobacter species. BLASTp analysis was performed by using H. pullorum accessory proteins as the query with Helicobacter accessory proteins as the database. The best hits of BLASTp output for each protein were obtained, and proteins <70% identical were considered H. pullorum-specific proteins. Further, H. pullorum strain-specific genes were also identified by using an in-house perl script. The functional categories of the strain-specific genes were predicted by using the COG database (69). The genes were classified into multiple classes that comprise more than one domain of a COG functional category, while those genes that did not show any significant hit against the database were identified as hypothetical genes.
Identification of virulence genes and detection of mobile genetic elements.
The whole genomes of all of our isolates were screened with the BLASTp program for the presence of virulence genes listed in the Virulence Factor Database (VFDB, http://www.mgc.ac.cn/VFs/). The amino acid sequences of the predicted genes from all of the 11 strains were compared with the database of virulence-related genes of C. jejuni obtained from the VFDB. The identity cutoff and query coverage values were kept at >50% and >80%, respectively.
In addition, all of these strains were screened for the presence of mobile genetic elements. Phages or phage-like elements were identified by the PHAST tool (70). Genomic islands in these strains were determined with IslandViewer (71), which makes use of three algorithms (IslandPick, IslandPath-DIMOB, and SIGI-HMM) for prediction and facilitates the visualization and analysis of these islands. Insertion elements were predicted with the ISfinder web tool (72), which compares the query sequences against a database of insertion sequence elements.
Accession number(s).
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under accession numbers MAOZ00000000 (H. pullorum NAP10B8), MAPA00000000 (H. pullorum NAP8W25), MAPB00000000 (H. pullorum NAP6W24), MAPC00000000 (H. pullorum NAP5W19), MAPD00000000 (H. pullorum NAP3W17), MAPE00000000 (H. pullorum NAP2W5), MANJ00000000 (H. pullorum NAP13B35), MANK00000000 (H. pullorum NAP14B36), MAJF00000000 (H. pullorum NAP11B31), MAJG00000000 (H. pullorum NAP12B32), and LXWI00000000 (H. pullorum NAP1W4), including all 11 of the isolates sequenced in this study.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funding received from the Department of Biotechnology, Government of India (BT/PR6921/MED/29/699/2013), and support from the Indo-German International Research Training Group, Internationales Graduiertenkolleg (GRK1673)—functional molecular infection epidemiology, an initiative of the German Research Foundation (DFG) and the University of Hyderabad (India), for which N.A. served as a speaker from the Indian side. N.A. is an adjunct professor at the Academy of Scientific and Innovative Research (ACSIR), India. S.Q. acknowledges the award of a junior research fellowship from the University Grants Commission of India.
We thank Lothar H. Wieler for his advice and discussions.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02305-16.
REFERENCES
- 1.Stanley J, Linton D, Burnens AP, Dewhirst FE, On SL, Porter A, Owen RJ, Costas M. 1994. Helicobacter pullorum sp. nov.-genotype and phenotype of a new species isolated from poultry and from human patients with gastroenteritis. Microbiology 140(Pt 12):3441–3449. doi: 10.1099/13500872-140-12-3441. [DOI] [PubMed] [Google Scholar]
- 2.Steinbrueckner B, Haerter G, Pelz K, Weiner S, Rump JA, Deissler W, Bereswill S, Kist M. 1997. Isolation of Helicobacter pullorum from patients with enteritis. Scand J Infect Dis 29:315–318. doi: 10.3109/00365549709019053. [DOI] [PubMed] [Google Scholar]
- 3.Burnens AP, Stanley J, Morgenstern R, Nicolet J. 1994. Gastroenteritis associated with Helicobacter pullorum. Lancet 344:1569–1570. [DOI] [PubMed] [Google Scholar]
- 4.Bascuñana P, Padrones I, Picazo JJ, Velasco AC. 2011. Diarrhea associated with Helicobacter pullorum in a child. Rev Esp Quimioter 24:50–51. (In Spanish.) [PubMed] [Google Scholar]
- 5.Bohr UR, Glasbrenner B, Primus A, Zagoura A, Wex T, Malfertheiner P. 2004. Identification of enterohepatic Helicobacter species in patients suffering from inflammatory bowel disease. J Clin Microbiol 42:2766–2768. doi: 10.1128/JCM.42.6.2766-2768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veijola L, Nilsson I, Halme L, Al-Soud WA, Makinen J, Ljungh A, Rautelin H. 2007. Detection of Helicobacter species in chronic liver disease and chronic inflammatory bowel disease. Ann Med 39:554–560. doi: 10.1080/07853890701545714. [DOI] [PubMed] [Google Scholar]
- 7.Karagin PH, Stenram U, Wadström T, Ljungh A. 2010. Helicobacter species and common gut bacterial DNA in gallbladder with cholecystitis. World J Gastroenterol 16:4817–4822. doi: 10.3748/wjg.v16.i38.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponzetto A, Pellicano R, Leone N, Cutufia MA, Turrini F, Grigioni WF, D'Errico A, Mortimer P, Rizzetto M, Silengo L. 2000. Helicobacter infection and cirrhosis in hepatitis C virus carriage: is it an innocent bystander or a troublemaker? Med Hypotheses 54:275–277. doi: 10.1054/mehy.1999.0987. [DOI] [PubMed] [Google Scholar]
- 9.Rocha M, Avenaud P, Menard A, Le Bail B, Balabaud C, Bioulac-Sage P, de Magalhaes Queiroz DM, Megraud F. 2005. Association of Helicobacter species with hepatitis C cirrhosis with or without hepatocellular carcinoma. Gut 54:396–401. doi: 10.1136/gut.2004.042168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pellicano R, Menard A, Rizzetto M, Megraud F. 2008. Helicobacter species and liver diseases: association or causation? Lancet Infect Dis 8:254–260. doi: 10.1016/S1473-3099(08)70066-5. [DOI] [PubMed] [Google Scholar]
- 11.Casswall TH, Nemeth A, Nilsson I, Wadstrom T, Nilsson HO. 2010. Helicobacter species DNA in liver and gastric tissues in children and adolescents with chronic liver disease. Scand J Gastroenterol 45:160–167. doi: 10.3109/00365520903426915. [DOI] [PubMed] [Google Scholar]
- 12.Fox JG, Dewhirst FE, Shen Z, Feng Y, Taylor NS, Paster BJ, Ericson RL, Lau CN, Correa P, Araya JC, Roa I. 1998. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology 114:755–763. doi: 10.1016/S0016-5085(98)70589-X. [DOI] [PubMed] [Google Scholar]
- 13.Laharie D, Asencio C, Asselineau J, Bulois P, Bourreille A, Moreau J, Bonjean P, Lamarque D, Pariente A, Soule JC, Charachon A, Coffin B, Perez P, Megraud F, Zerbib F. 2009. Association between entero-hepatic Helicobacter species and Crohn's disease: a prospective cross-sectional study. Aliment Pharmacol Ther 30:283–293. doi: 10.1111/j.1365-2036.2009.04034.x. [DOI] [PubMed] [Google Scholar]
- 14.Chien CC, Taylor NS, Ge Z, Schauer DB, Young VB, Fox JG. 2000. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J Med Microbiol 49:525–534. doi: 10.1099/0022-1317-49-6-525. [DOI] [PubMed] [Google Scholar]
- 15.Young VB, Knox KA, Schauer DB. 2000. Cytolethal distending toxin sequence and activity in the enterohepatic pathogen Helicobacter hepaticus. Infect Immun 68:184–191. doi: 10.1128/IAI.68.1.184-191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor NS, Ge Z, Shen Z, Dewhirst FE, Fox JG. 2003. Cytolethal distending toxin: a potential virulence factor for Helicobacter cinaedi. J Infect Dis 188:1892–1897. doi: 10.1086/379837. [DOI] [PubMed] [Google Scholar]
- 17.Young VB, Knox KA, Pratt JS, Cortez JS, Mansfield LS, Rogers AB, Fox JG, Schauer DB. 2004. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect Immun 72:2521–2527. doi: 10.1128/IAI.72.5.2521-2527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young VB, Chien CC, Knox KA, Taylor NS, Schauer DB, Fox JG. 2000. Cytolethal distending toxin in avian and human isolates of Helicobacter pullorum. J Infect Dis 182:620–623. doi: 10.1086/315705. [DOI] [PubMed] [Google Scholar]
- 19.Jinadasa RN, Bloom SE, Weiss RS, Duhamel GE. 2011. Cytolethal distending toxin: a conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 157:1851–1875. doi: 10.1099/mic.0.049536-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varon C, Mocan I, Mihi B, Pere-Vedrenne C, Aboubacar A, Morate C, Oleastro M, Doignon F, Laharie D, Megraud F, Menard A. 2014. Helicobacter pullorum cytolethal distending toxin targets vinculin and cortactin and triggers formation of lamellipodia in intestinal epithelial cells. J Infect Dis 209:588–599. doi: 10.1093/infdis/jit539. [DOI] [PubMed] [Google Scholar]
- 21.Shah PM, Schafer V, Knothe H. 1993. Medical and veterinary use of antimicrobial agents: implications for public health. A clinician's view on antimicrobial resistance. Vet Microbiol 35:269–274. [DOI] [PubMed] [Google Scholar]
- 22.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core Surveillance (ABCs) MRSA Investigators. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 23.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 24.Schroeder CM, Zhao C, DebRoy C, Torcolini J, Zhao S, White DG, Wagner DD, McDermott PF, Walker RD, Meng J. 2002. Antimicrobial resistance of Escherichia coli O157 isolated from humans, cattle, swine, and food. Appl Environ Microbiol 68:576–581. doi: 10.1128/AEM.68.2.576-581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angulo FJ, Johnson KR, Tauxe RV, Cohen ML. 2000. Origins and consequences of antimicrobial-resistant nontyphoidal Salmonella: implications for the use of fluoroquinolones in food animals. Microb Drug Resist 6:77–83. doi: 10.1089/mdr.2000.6.77. [DOI] [PubMed] [Google Scholar]
- 26.Zanoni RG, Rossi M, Giacomucci D, Sanguinetti V, Manfreda G. 2007. Occurrence and antibiotic susceptibility of Helicobacter pullorum from broiler chickens and commercial laying hens in Italy. Int J Food Microbiol 116:168–173. doi: 10.1016/j.ijfoodmicro.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Borges V, Santos A, Correia CB, Saraiva M, Menard A, Vieira L, Sampaio DA, Pinheiro M, Gomes JP, Oleastro M. 2015. Helicobacter pullorum isolated from fresh chicken meat: antibiotic resistance and genomic traits of an emerging foodborne pathogen. Appl Environ Microbiol 81:8155–8163. doi: 10.1128/AEM.02394-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González A, Piqueres P, Moreno Y, Canigral I, Owen RJ, Hernandez J, Ferrus MA. 2008. A novel real-time PCR assay for the detection of Helicobacter pullorum-like organisms in chicken products. Int Microbiol 11:203–208. [PubMed] [Google Scholar]
- 29.Fox JG, Chien CC, Dewhirst FE, Paster BJ, Shen Z, Melito PL, Woodward DL, Rodgers FG. 2000. Helicobacter canadensis sp. nov. isolated from humans with diarrhea as an example of an emerging pathogen. J Clin Microbiol 38:2546–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waldenström J, On SL, Ottvall R, Hasselquist D, Harrington CS, Olsen B. 2003. Avian reservoirs and zoonotic potential of the emerging human pathogen Helicobacter canadensis. Appl Environ Microbiol 69:7523–7526. doi: 10.1128/AEM.69.12.7523-7526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olivares J, Bernardini A, Garcia-Leon G, Corona F, Sanchez MB, Martinez JL. 2013. The intrinsic resistome of bacterial pathogens. Front Microbiol 4:103. doi: 10.3389/fmicb.2013.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manfreda G, Parisi A, Lucchi A, Zanoni RG, De Cesare A. 2011. Prevalence of Helicobacter pullorum in conventional, organic, and free-range broilers and typing of isolates. Appl Environ Microbiol 77:479–484. doi: 10.1128/AEM.01712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ceelen L, Decostere A, Devriese LA, Ducatelle R, Haesebrouck F. 2005. In vitro susceptibility of Helicobacter pullorum strains to different antimicrobial agents. Microb Drug Resist 11:122–126. doi: 10.1089/mdr.2005.11.122. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed N, Dobrindt U, Hacker J, Hasnain SE. 2008. Genomic fluidity and pathogenic bacteria: applications in diagnostics, epidemiology and intervention. Nat Rev Microbiol 6:387–394. doi: 10.1038/nrmicro1889. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz T, Kohnen W, Jansen B, Obst U. 2003. Detection of antibiotic-resistant bacteria and their resistance genes in wastewater, surface water, and drinking water biofilms. FEMS Microbiol Ecol 43:325–335. doi: 10.1111/j.1574-6941.2003.tb01073.x. [DOI] [PubMed] [Google Scholar]
- 36.Oluyege JO, Dada AC, Odeyemi AT. 2009. Incidence of multiple antibiotic resistant Gram-negative bacteria isolated from surface and underground water sources in south western region of Nigeria. Water Sci Technol 59:1929–1936. doi: 10.2166/wst.2009.219. [DOI] [PubMed] [Google Scholar]
- 37.Gulay Z, Bicmen M, Amyes SG, Yulug N. 2000. Beta-lactamase patterns and betalactam/clavulanic acid resistance in Escherichia coli isolated from fecal samples from healthy volunteers. J Chemother 12:208–215. doi: 10.1179/joc.2000.12.3.208. [DOI] [PubMed] [Google Scholar]
- 38.Pumbwe L, Randall LP, Woodward MJ, Piddock LJ. 2004. Expression of the efflux pump genes cmeB, cmeF and the porin gene porA in multiple-antibiotic-resistant Campylobacter jejuni. J Antimicrob Chemother 54:341–347. doi: 10.1093/jac/dkh331. [DOI] [PubMed] [Google Scholar]
- 39.Elkins CA, Mullis LB. 2006. Mammalian steroid hormones are substrates for the major RND- and MFS-type tripartite multidrug efflux pumps of Escherichia coli. J Bacteriol 188:1191–1195. doi: 10.1128/JB.188.3.1191-1195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. 2003. The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- 41.Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev 33:376–393. doi: 10.1111/j.1574-6976.2008.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibson JR, Ferrus MA, Woodward D, Xerry J, Owen RJ. 1999. Genetic diversity in Helicobacter pullorum from human and poultry sources identified by an amplified fragment length polymorphism technique and pulsed-field gel electrophoresis. J Appl Microbiol 87:602–610. [DOI] [PubMed] [Google Scholar]
- 43.Ottemann KM, Lowenthal AC. 2002. Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect Immun 70:1984–1990. doi: 10.1128/IAI.70.4.1984-1990.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brás AM, Chatterjee S, Wren BW, Newell DG, Ketley JM. 1999. A novel Campylobacter jejuni two-component regulatory system important for temperature-dependent growth and colonization. J Bacteriol 181:3298–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allos BM. 1998. Campylobacter jejuni infection as a cause of the Guillain-Barre syndrome. Infect Dis Clin North Am 12:173–184. [DOI] [PubMed] [Google Scholar]
- 46.Lara-Tejero M, Galan JE. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290:354–357. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- 47.Blanco P, Hernando-Amado S, Reales-Calderon J, Corona F, Lira F, Alcalde-Rico M, Bernardini A, Sanchez M, Martinez J. 2016. Bacterial multidrug efflux pumps: much more than antibiotic resistance determinants. Microorganisms 4:E14. doi: 10.3390/microorganisms4010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buckley AM, Webber MA, Cooles S, Randall LP, La Ragione RM, Woodward MJ, Piddock LJ. 2006. The AcrAB-TolC efflux system of Salmonella enterica serovar Typhimurium plays a role in pathogenesis. Cell Microbiol 8:847–856. doi: 10.1111/j.1462-5822.2005.00671.x. [DOI] [PubMed] [Google Scholar]
- 49.Lin J, Sahin O, Michel LO, Zhang Q. 2003. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect Immun 71:4250–4259. doi: 10.1128/IAI.71.8.4250-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vila J, Martinez JL. 2008. Clinical impact of the over-expression of efflux pump in nonfermentative Gram-negative bacilli, development of efflux pump inhibitors. Curr Drug Targets 9:797–807. doi: 10.2174/138945008785747806. [DOI] [PubMed] [Google Scholar]
- 51.Steele TW, McDermott SN. 1984. The use of membrane filters applied directly to the surface of agar plates for the isolation of Campylobacter jejuni from feces. Pathology 16:263–265. [DOI] [PubMed] [Google Scholar]
- 52.Hussain A, Ranjan A, Nandanwar N, Babbar A, Jadhav S, Ahmed N. 2014. Genotypic and phenotypic profiles of Escherichia coli isolates belonging to clinical sequence type 131 (ST131), clinical non-ST131, and fecal non-ST131 lineages from India. Antimicrob Agents Chemother 58:7240–7249. doi: 10.1128/AAC.03320-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomason JA III, Stevens R, Vonstein V, Wattam AR, Xia F. 2015. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5:8365. doi: 10.1038/srep08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mural RJ. 2000. ARTEMIS: a tool for displaying and annotating DNA sequence. Brief Bioinform 1:199–200. doi: 10.1093/bib/1.2.199. [DOI] [PubMed] [Google Scholar]
- 61.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L, Stoeckert CJ Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Capella-Gutiérrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langille MG, Brinkman FS. 2009. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25:664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Clinical and Laboratory Standards Institute. 2009. Performance standards for antimicrobial disk susceptibility tests; approved standard—10th edition. CLSI document M02-A10. CLSI, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.