

DNA repair is critical for genome integrity. Lawrence et al. reveal a role for a novel complex that links the nucleus to microtubules to promote accurate repair through both inhibition of error-prone nonhomologous end joining and promotion of homologous recombination.
Abstract
The Caenorhabditis elegans SUN domain protein, UNC-84, functions in nuclear migration and anchorage in the soma. We discovered a novel role for UNC-84 in DNA damage repair and meiotic recombination. Loss of UNC-84 leads to defects in the loading and disassembly of the recombinase RAD-51. Similar to mutations in Fanconi anemia (FA) genes, unc-84 mutants and human cells depleted of Sun-1 are sensitive to DNA cross-linking agents, and sensitivity is rescued by the inactivation of nonhomologous end joining (NHEJ). UNC-84 also recruits FA nuclease FAN-1 to the nucleoplasm, suggesting that UNC-84 both alters the extent of repair by NHEJ and promotes the processing of cross-links by FAN-1. UNC-84 interacts with the KASH protein ZYG-12 for DNA damage repair. Furthermore, the microtubule network and interaction with the nucleoskeleton are important for repair, suggesting that a functional linker of nucleoskeleton and cytoskeleton (LINC) complex is required. We propose that LINC complexes serve a conserved role in DNA repair through both the inhibition of NHEJ and the promotion of homologous recombination at sites of chromosomal breaks.
Introduction
Our genomes are under constant assault, accumulating DNA damage that must be repaired before the next cell division. It is especially important to precisely repair DNA in the germline to prevent passing deleterious mutations to the next generation. A large number of pathways sense and repair damaged DNA; these pathways are differentially engaged depending on cell cycle stage, cell type, and the nature of the damage induced (Ciccia and Elledge, 2010). DNA double-strand breaks (DSBs) are primarily repaired by nonhomologous end joining (NHEJ), which ligates breaks without regard for homology, or by homologous recombination (HR), which requires a template for repair (Daley and Sung, 2014). Because NHEJ is more error prone, cells in the germline use HR when possible. However, the mechanisms underlying how germ cells favor HR over NHEJ repair pathways are not well understood. In this study, we report a mechanism conserved from nematodes to mammals in which the linker of nucleoskeleton and cytoskeleton (LINC) functions with the Fanconi anemia (FA) pathway to suppress NHEJ in favor of HR to safeguard the genome.
FA is a complex disorder characterized by genome instability, a predisposition to cancer, and sensitivity to cross-linking agents (Kottemann and Smogorzewska, 2013; Duxin and Walter, 2015). Over 19 genes have been shown to mutate in FA. Critical among these is FANCD2, which, in response to stalled replication forks or interstrand cross-links, is monoubiquinated and recruits repair proteins, such as the nuclease FAN-1 (Kennedy and D’Andrea, 2005; Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010). Interestingly, cross-link sensitivity of fancd2 mutants in Caenorhabditis elegans, human, and DT40 chicken B cells is suppressed by the inactivation of NHEJ, suggesting that the FA pathway ensures HR repair by inhibiting NHEJ (Adamo et al., 2010; Pace et al., 2010). However, in mice, inactivation of NHEJ exacerbates the cross-linking sensitivity of fancd2, indicating that we do not fully understand the interactions between the FA pathway and repair through NHEJ and HR (Bunting et al., 2012).
The conserved LINC complex physically connects the nucleus to the cytoplasm and is essential for nuclear migration, anchorage, centrosome attachment to the outer nuclear membrane, and mechanotransduction (Tapley and Starr, 2013; Luxton and Starr, 2014). LINC consists of inner nuclear membrane SUN domain proteins and outer nuclear membrane KASH domain proteins. Canonical SUN proteins (C. elegans UNC-84; mammalian Sun-1/2) have a nucleoplasmic domain that interacts with lamins, a single transmembrane pass, and the conserved SUN domain (Malone et al., 1999; Crisp et al., 2006; Haque et al., 2006; Jaspersen et al., 2006; Tapley et al., 2011; Bone et al., 2014; Cain et al., 2014). SUN domains interact in the perinuclear space with KASH domain proteins, which span the outer nuclear membrane to connect to the cytoskeleton (Starr and Han, 2002; Padmakumar et al., 2005; Crisp et al., 2006; McGee et al., 2006; Sosa et al., 2012). LINC has been implicated in various aspects of internal nuclear organization and function. Most striking is the conserved requirement for LINC in chromosome movement and pairing during meiosis (Chikashige et al., 2006; Ding et al., 2007; Penkner et al., 2007; Koszul et al., 2008; Prasada Rao et al., 2011; Lee et al., 2012; Christophorou et al., 2015; Varas et al., 2015). LINC has also been implicated in DNA damage signaling and repair (Oza et al., 2009; Lei et al., 2012; Swartz et al., 2014; Lottersberger et al., 2015; Ryu et al., 2015). These studies have suggested that LINC serves as a tether to sequester unrepaired DSBs or dysfunctional telomeres to the nuclear periphery and/or to mediate the mobility of these aberrant ends. However, mechanisms by which LINC facilitates DNA damage repair remain to be elucidated.
To determine how SUN proteins function in DNA repair, we focused on the role of UNC-84 in the adult C. elegans germline. The germline is particularly amenable to these studies, as it is the only actively dividing tissue in the adult worm, and germ cells have a robust response to DNA damage (Bailly and Gartner, 2013; Lawrence et al., 2015). The germline is arranged in a spatiotemporal gradient that includes actively dividing cells in the proliferative zone (PZ) and all stages of meiotic prophase (transition zone, pachytene, diplotene, and diakinesis; Fig. 1 A). We discovered that UNC-84 plays a critical role in the repair of interstrand cross-links by inhibiting NHEJ and recruiting the nuclease FAN-1. We provide evidence that human Sun-1 also functions in the repair of interstrand cross-links. Analysis of SUN and KASH protein mutations and inhibition of microtubule polymerization support a model whereby chromosome motion driven by LINC promotes repair.
Figure 1.

unc-84 PZ nuclei have defects in cell cycle arrest and RAD-51 after HU. (A) Cartoon of the C. elegans germline. TZ, transition zone; EP, early pachytene; MP, mid-pachytene; LP, late pachytene; DP, diplotene; DI, diakinesis. (B) Images of PZ nuclei from wild type (WT), unc-84(n369), unc-84(n369);cku-70(tm1524), cku-70(tm1524), and chk-1(RNAi) worms, minus HU (−HU) and plus 25 mM HU at 0- or 5-h recovery (+HU), stained with DAPI. Bar, 10 µm. (C) The mean number of nuclei in the PZ, which corresponds to the first 100 µm of the gonad 0 h after release from HU. n > 20. The p-value was determined by a two-way analysis of variance. **, P < 0.01; ***, P < 0.0001. (D) Percentages of PZ nuclei smaller than 3.5 µm with (light orange) and without (dark orange) RAD-51 and PZ nuclei larger than 3.5 µm with (light pink) and without (dark pink) RAD-51 5 h after release from HU in wild-type, unc-84(n369), unc-84(n369);cku-70(tm1524), cku-70(tm1524), and chk-1(RNAi) germlines. n > 50. Error bars indicate 95% CI.
Results
NHEJ is engaged at stalled/collapsed replication forks in the absence of UNC-84
C. elegans has two SUN domain–containing proteins: UNC-84 and SUN-1. SUN-1 is essential for germline organization, meiotic chromosome pairing, and early embryonic viability (Malone et al., 2003; Penkner et al., 2007, 2009; Minn et al., 2009; Sato et al., 2009), making it difficult to tease apart a role in DNA damage repair. UNC-84 is essential for nuclear migration and anchorage in the soma, and a role in the germline has not been reported (Malone et al., 1999; Cain et al., 2014). Therefore, we focused on the analysis of unc-84–null mutants (unc-84(n369); Malone et al., 1999; Lee et al., 2002) after challenge with DNA-damaging agents.
We examined the consequences of perturbing DNA replication in PZ germ cells by treating worms with the ribonucleotide reductase inhibitor hydroxyurea (HU). HU depletes nucleotide pools, leading to replication fork stalling/collapse (Petermann et al., 2010). After treatment with 25 mM HU, wild-type PZ nuclei arrest in S/G2 (Fig. S1, C and D) but continue to grow, resulting in enlarged and consequently fewer nuclei (Fig. 1, B and C; MacQueen and Villeneuve, 2001). Inactivation of the DNA damage checkpoint by depletion of checkpoint kinase, CHK1, results in a failure to arrest in response to HU; these animals have smaller and many more nuclei within the PZ (Fig. 1, B and C; Garcia-Muse and Boulton, 2005). Analysis of the PZ in unc-84 mutants after HU revealed an intermediate number of nuclei (Fig. 1, B and C). Thus, unc-84 mutants respond differently to HU than either wild-type or checkpoint-defective mutants, suggesting that UNC-84 is involved in some aspect of DNA damage response (DDR)/repair.
After the release from HU in wild-type worms, germ cells remain arrested (≥3.5 µm in diameter) until repair is completed (Lawrence et al., 2015). 5 h after release from HU, 71.0% of wild-type nuclei were arrested, and 89% of those contained the recombinase RAD-51, a marker of ongoing repair by HR. However, none of the small nuclei (<3.5 µm; 29.0%), which presumably had just divided, had RAD-51 (Fig. 1 D). In contrast, most chk-1(RNAi) germline nuclei challenged with HU bypassed the DNA damage checkpoint and prematurely divided, resulting in significantly more small nuclei (86.4%; Fig. 1 D). A significant fraction (41.5%) of the small nuclei in chk-1(RNAi) contained RAD-51 foci (Fig. 1 D), suggesting that nuclear division occurred despite unrepaired DNA damage. unc-84 PZs had an intermediate number of small nuclei (56.9%), and 28.7% of these nuclei had RAD-51 foci 5 h after HU (Fig. 1 D). These data suggest that a significant percentage of unc-84 mutant nuclei had either completed repair before progressing through the cell cycle or progressed through the cell cycle without completing repair and without loading RAD-51. If repair failed, germ cells damaged in the PZ would be packaged into gametes that, when fertilized, would have decreased viability. We monitored progeny viability 36–72 h after release from HU; this time frame captures progeny derived from PZ germ cells exposed to HU (Jaramillo-Lambert et al., 2007). We observed no difference between wild-type and unc-84 mutants (percentage of progeny inviability mean ± 95% confidence interval [CI]; wild-type: 7.37 ± 2.97%; unc-84: 3.83 ± 1.10%), suggesting that repair was completed in those cells packaged into gametes. As germline apoptosis eliminates defective germ cells and could mask a defect, we also analyzed progeny viability upon inactivation of the essential cell death caspase CED-3 (Ellis and Horvitz, 1986). Although there was a significant difference between ced-3(n717) and unc-84;ced-3 before HU (1.20 ± 0.47 vs. 3.30 ± 1.5; P < 0.01; Fig. S3 D), there was no difference after HU, although brood size was significantly reduced (percentage of progeny inviability [P = 0.06] and brood size [P < 0.0001]; mean ± 95% CI; ced-3: 2.28 ± 1.37 and 167.10 ± 24; unc-84;ced-3: 5.27 ± 2.87 and 57.0 ± 16.70), suggesting that apoptosis does not explain the low progeny inviability of unc84 mutants after HU exposure.
To test the hypothesis that repair proceeds at a subset of lesions in unc-84 cells without loading RAD-51, we examined the consequence of inactivating the major RAD-51–independent pathway, NHEJ, by mutation of cku-70 (C. elegans Ku70 orthologue; Clejan et al., 2006) in wild type and unc-84 mutants. Both unc-84;cku-70(tm1524) and cku-70 mutants were proficient for arrest after HU (Fig. 1, B and C). Furthermore, unc-84;cku-70 and, to a lesser extent, cku-70 mutants had reduced numbers of small nuclei containing RAD-51 (unc-84;cku-70: 9.5%; cku-70: 21%) compared with unc-84 mutants (28.7%), but more than wild type (0%; Fig. 1 D). These results suggest that a subset of stalled/collapsed forks is repaired by NHEJ in the absence of UNC-84.
UNC-84 plays a role in DNA repair independent of tethering breaks to the nuclear periphery
Although the removal of NHEJ improved the response of unc-84 to HU, germ cells were not restored to wild type with respect to RAD-51, suggesting that UNC-84 plays additional roles in DNA repair (Fig. 1 D). To examine the repair of stalled/collapsed replication forks, we treated worms with a 2-h pulse of 5 mM HU and monitored the loading and removal of RAD-51 after release from HU. This dose of HU had no effect on proliferation in either wild-type or unc-84 PZ cells (mitotic index [H3S10-P] 6-h HU recovery: wild type, 5.60 ± 0.30 vs. no HU, 5.00 ± 0.30 [P = 0.12]; unc-84, 4.03 ± 0.40 vs. 3.19 ± 0.40 [P = 0.16]), although we did observe a delay in progression to G2/M in unc-84 in the absence of HU (Fig. S1 B). Nonetheless, labeled nucleotides were incorporated comparably into PZ nuclei (Fig. S1 C), suggesting that S phase is similar in wild type and unc-84. Before HU treatment, there were slightly elevated levels of RAD-51 in unc-84 compared with wild type (P < 0.01); however, immediately after release from HU and 2 h later, unc-84 had significantly fewer RAD-51 foci than wild type (P < 0.001; Fig. 2 A). By 4 h, RAD-51 levels were indistinguishable between unc-84 and wild type. To determine whether the reduced levels of RAD-51 early in the time course were a consequence of channeling repair through NHEJ, we examined RAD-51 in unc-84;cku-70. Immediately after release from HU and 2 h later, the unc-84;cku-70 mutant displayed intermediate levels of RAD-51, and by 4 h there was very little difference between wild type, unc-84, and unc-84;cku-70 (Fig. 2 A). The cku-70 mutant behaved like wild type until 6 h, when more RAD-51 foci were observed (Fig. 2 A). These results suggest that some HU lesions are repaired through NHEJ in the absence of UNC-84.
Figure 2.

RAD-51 and RPA are altered after HU treatment in the absence of UNC84. (A) RAD-51–positive nuclei in wild type (WT), unc-84(n369), unc-84(n369);cku-70(tm1524), or cku-70(tm1524) in the absence of or after recovery from 5 mM HU. n > 30. (B) Number of RPA-1::YPF foci per nucleus in wild type, unc-84(n369), unc-84(n369);cku-80(RNAi), or cku-80(RNAi) in the absence of or after recovery from 5 mM HU. n > 30. (C) MAD-2 (red) staining in wild-type or unc-84(n369) germlines after 25 mM HU treatment counterstained with DAPI (blue). (D) High-resolution SIM images of a single nucleus from unc-84(n369) and wild-type worms treated with 25 mM HU and stained with RAD-51 (red), NPC (Mab414; green), and DAPI (blue). Next to each image is the mean number of RAD-51 foci observed in each genotype as well as the mean area of each RAD-51 focus (n = 4 germlines). Images represent a projection of three z sections. The scatterplot indicates the distance in nanometers between NPC and RAD-51 foci in unc-84(n369) and wild-type worms after HU. Bars, 5 µm. Error bars indicate 95% CI. *, P < 0.05; **, P < 0.01; ***, P < 0.0001, when compared with unc-84(n369) single mutants.
We also examined the loading and removal of replication protein A (RPA; RPA-1::YFP; Stergiou et al., 2011), which is loaded onto single-stranded DNA during replication and repair and is exchanged for RAD-51 during HR (Sleeth et al., 2007). In wild-type PZ nuclei, RPA-1::YFP levels peaked 2 h after release from HU and then declined (Fig. 2 A). In unc-84 animals, RPA-1::YFP levels were similar to wild type immediately after release from HU but were slightly higher than wild type at 2 h (P < 0.05) and remained higher than wild type at 4 and 6 h (4 h, P < 0.05; 6 h, P < 0.001; Fig. 2 B). Although RPA-1::YFP was restored to wild-type levels in unc-84;cku-80(RNAi) at 2 h after HU, RPA-1 levels were indistinguishable from unc-84 at 4 and 6 h, suggesting that alteration of RPA levels is not solely a consequence of repair by NHEJ. Surprisingly, depletion of CKU-80 alone led to slightly lower levels of RPA-1 at 0 and 2 h, but these levels were similar to wild type at 4 and 6 h. The subtle alterations in RPA-1 compared with RAD-51 loading suggests that only a subset of RPA-1–marked HU events are channeled into repair through RAD-51.
We recently reported that repair of DNA damage in PZ nuclei is dependent on both the DDR and the spindle assembly checkpoint (SAC) and proposed that the SAC facilitated recruitment of damaged DNA to the nuclear periphery for repair (Lawrence et al., 2015). Because Saccharomyces cerevisiae and Schizosaccharomyces pombe SUN proteins are required for the localization of persistent DSBs to the nuclear periphery (Kalocsay et al., 2009; Oza et al., 2009), we explored the possibility that UNC-84 served as a tether for the SAC and/or damaged DNA. We found that the SAC component MAD-2 was enriched at the nuclear periphery in unc-84 mutants as in wild type (Fig. 2 C). Furthermore, structured illumination microscopy (SIM) revealed that RAD-51 was closely associated with the nuclear periphery after HU in unc-84 mutants as in wild type (Fig. 2 D). Collectively, these results suggest that UNC-84 plays a role in DNA damage repair independent of tethering MAD-2 or RAD-51 to the nuclear periphery.
UNC-84 promotes the repair of interstrand cross-links and irradiation (IR)-induced DSBs
Cisplatin creates DNA interstrand cross-links that block replication fork progression. Repair of interstrand cross-links is regulated by the FA pathway and requires unhooking, trans-lesional synthesis, and HR to repair the resulting replication-dependent DSB (Deans and West, 2011). To determine whether UNC-84 functions in interstrand cross-link repair, we examined the progeny viability, brood size, and developmental progression of unc-84 mutants 36–72 h after cisplatin. We observed a decrease in both progeny viability and brood size as well as an increase in animals with postembryonic developmental defects compared with wild type (Fig. 3 A and Fig. S2, A and B; Adamo et al., 2010). Sensitivity was not specific to cisplatin, as unc-84 mutants were also sensitive to nitrogen mustard (Fig. S2 C).
Figure 3.

UNC-84 plays a role in interstrand cross-link repair. (A) Progeny inviability after 0, 100, 250, or 500 µM cisplatin in wild type (WT) and unc-84(n369) mutants. n > 10. (B) Progeny inviability after 0 or 250 µM cisplatin in wild type, unc-84(n369), cku-70(tm1524), unc-84(n369);cku-70(tm1524), unc-84(n369);lig-4(ok716), and lig-4(ok716). n > 10. (C) Percentages of RAD-51–positive PZ nuclei after recovery for the given times from 100 µM cisplatin for wild type, unc-84(n369), unc-84(n369);cku-70(tm1524), and cku-70(tm1524). The p-value denotes significance compared with wild type at given times, except for unc-84(n369) versus unc-84(n369);cku-70(tm1524) (bracketed). n > 15. (D) Number of RPA-1::YFP foci after recovery for the given times from 100 µM cisplatin for wild type, unc-84(n369), unc-84(n369);cku-80(RNAi), and cku-80(RNAi). n > 15. (E) Progeny viability after 250 µM cisplatin in unc-84(n369), fcd-2(tm1268), fcd-2(tm1268);unc-84(n369), fan-1(tm423), and fan-1(tm423);cku-70(tm1524). n > 10. (F) Percentages of RAD-51–positive nuclei after recovery for the given times from 100 µM cisplatin for 2 h for wild type, unc-84(n369), fcd-2(tm1268), and fcd-2(tm1268);unc-84(n369). n > 15. (G) Viability of HeLa cells with or without mouse Sun-1 (mSun-1) transfected with the indicated siRNA constructs and treated with 0.5 µM DNA-PK inhibitor NU7026 (Veuger et al., 2003) or vehicle before damage induction with 40 ng/ml MMC. Error bars indicate 95% CI. The p-value was determined by two-way analysis of variance. *, P < 0.05; **, P < 0.01; ***, P < 0.0001. cispl, cisplatin.
Inactivation of NHEJ suppresses cross-linking sensitivity in certain FA pathway mutants, suggesting that NHEJ poisons repair of cross-links in the absence of the FA pathway (Adamo et al., 2010; Pace et al., 2010). As NHEJ is differentially engaged after HU-induced damage in the absence of UNC-84, we examined cisplatin sensitivity in mutations in NHEJ, including CKU-70 and LIG-4, a DNA ligase (Clejan et al., 2006), in the presence and absence of UNC-84. cku-70 and lig-4(ok716) mutants displayed wild-type sensitivity to cisplatin. Interestingly, the unc-84;cku-70 and unc-84;lig-4 mutants had elevated levels of progeny inviability in the absence of damage that was not further enhanced by cisplatin (Fig. 3 B), suggesting that although inactivation of NHEJ improved the viability of unc-84 mutant after cisplatin, both NHEJ and UNC-84 play a role in some aspect of meiosis or embryogenesis independent of each other.
To provide insight into why inactivation of NHEJ improved cisplatin sensitivity of unc-84 mutants, we monitored RAD-51 after a 2-h pulse of 100 µM cisplatin in wild-type and unc-84 PZ germ cells. Immediately after release from cisplatin, unc-84 mutants had fewer germ cell nuclei with RAD-51 foci (0 h, P < 0.0001; Fig. 3 C). Over time, RAD-51 levels increased, and by 8 h levels were higher in unc-84 than in wild type (P < 0.01; Fig. 3 C). Furthermore, by 24 h after cisplatin, RAD-51 persisted to a greater extent than wild type in unc-84 mutants (P < 0.05; Fig. 3 C). RAD-51 was restored to wild-type levels immediately after release from cisplatin in unc-84;cku-70 (0 h; Fig. 3 C). However, by 4 h, RAD-51 levels in unc-84;cku-70 were indistinguishable from unc-84. Several hours after release, RAD-51 persisted in a subset of nuclei in the absence of UNC-84 and both UNC-84 and CKU-70.
We also examined RPA loading and removal after cisplatin treatment. In contrast to what was observed in response to HU, RPA-1::YFP levels mirrored RAD-51 and were reduced in the unc-84 mutant; this reduction was partially suppressed by inactivation of NHEJ (unc-84;cku-80). However, over time, RPA-1::YFP levels became elevated in unc-84;cku-80 compared with both wild type and unc-84. These results suggest that a subset of cisplatin-induced lesions is repaired by NHEJ in the absence of UNC-84, and removing NHEJ in unc-84 is sufficient to restore progeny viability. However, the rise in RPA and RAD-51 levels over time in both the presence and absence of NHEJ after cisplatin suggests that UNC-84 also influences repair by impinging on some aspect of HR.
To determine whether UNC-84 contributes to the repair of DSBs, we exposed worms to 10 Gy of γ-IR and monitored progeny viability as well as the loading and disassembly of RAD-51 in wild-type and unc-84 PZ nuclei. Similar to HU, IR had little effect on progeny viability over 36–72 h in unc-84 (wild type, 6.0 ± 1.5% embryo inviability vs. unc-84, 8.6 ± 3.1% [P = 0.5]). However, RAD-51 was perturbed. In wild type, all PZ nuclei had at least a single RAD-51 focus 0.5 h after IR, and foci were gradually disassembled until 16 h when the majority of RAD-51 foci were removed (Fig. 4 A). There was a significant decrease in RAD-51 foci in unc-84–irradiated PZ nuclei at early time points. At 0.5 h after exposure to IR, ∼5% of unc-84 nuclei did not contain any RAD-51 foci, whereas every wild-type nucleus contained at least one (blue in unc-84 column; Fig. 4 A; z test, P < 0.01) and the mean number of RAD-51 foci per nucleus was reduced (unc-84, 4.07 ± 0.16 vs. wild type, 5.86 ± 0.20 [P < 0.0001]). Additionally, there were many fewer nuclei with six or more foci in unc-84 compared with wild type (lack of red/less orange in unc-84 column; Fig. 4 A; z test, P < 0.01). After 2 h, the mean number of RAD-51 was not significantly different between wild type and unc-84 (unc-84, 4.64 ± 0.20 vs. wild type, 4.88 ± 0.26); however, the distribution remained altered, with many fewer nuclei containing more than six foci (less orange/red in unc-84 column compared with wild type; Fig. 4; z test, P < 0.01), and reduction in mean RAD-51 foci was evident at 4 h (unc-84, 2.86 ± 0.26% vs. wild type, 3.70 ± 0.29% [P < 0.0001]). However, at 6 and 16 h, more RAD-51 foci were observed in unc-84 compared with wild type (6 h: unc-84, 3.19 ± 0.26% vs. wild type, 2.54 ± 0.25% [P = 0.004]; 16 h: unc-84, 0.37 ± 0.09% vs. wild type, 0.23 ± 0.07% [P = 0.018]). Thus, RAD-51 was observed at fewer breaks early after IR, but at later time points, RAD-51 persisted in the absence of UNC-84.
Figure 4.
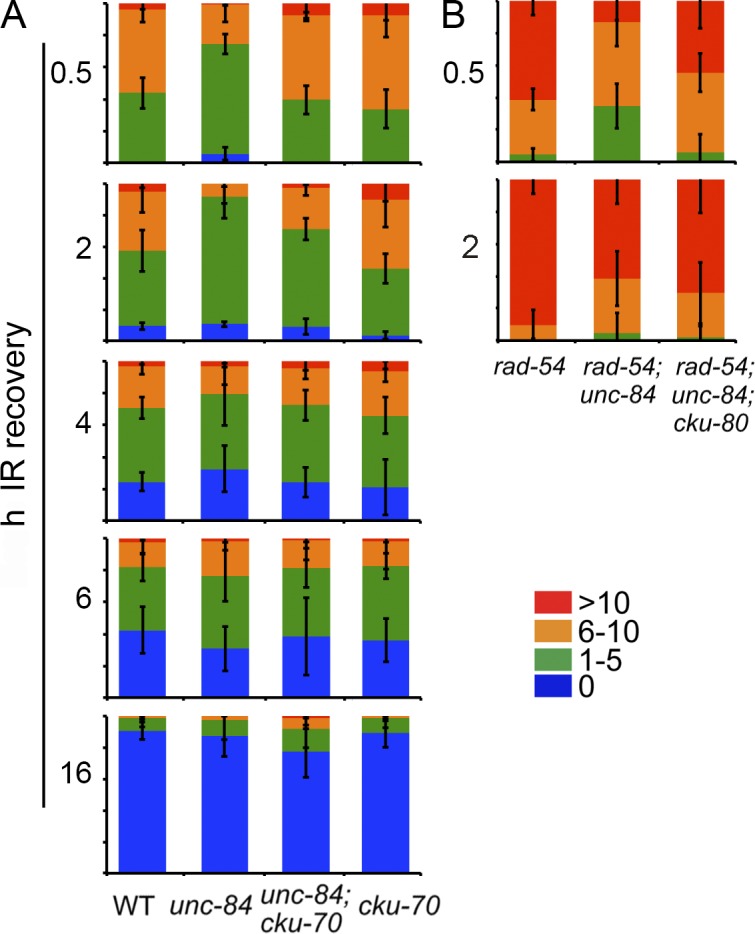
RAD-51 loading and disassembly after IR is altered in the absence of UNC-84. (A and B) Percentages of nuclei with either 0, 1–5, 6–10, or >10 RAD-51 foci per nucleus at indicated times after 10 Gy IR in wild type (WT), unc-84(n369), unc-84(n369);cku-70(tm1524), and cku-70(tm1524) (A); and rad-54(ok615), rad-54(ok615);unc-84(RNAi), and rad-54;unc-84(n369);cku-80(ok861) (B). n > 3 germlines.
We tested whether removal of NHEJ by mutation of cku-70 altered the pattern of RAD-51 after IR treatment in unc-84 mutants. At 0.5 h after IR, RAD-51 foci were significantly elevated in unc-84;cku-70 mutants compared with unc-84 (6.29 ± 0.22 vs. 4.07 ± 0.16 [P < 0.0001]; Fig. 4 A) but were similar to cku-70 (6.72 ± 0.21 [P = 0.07]; Fig. 4 A). However, at 2 h, there were fewer RAD-51 foci in unc-84;cku-70 (3.4 ± 0.20) compared with unc-84 (4.63 ± 0.20) and even higher levels of RAD-51 in cku-70 alone (6.04 ± 0.25 [P < 0.0001]). These results suggest that in addition to some breaks being repaired by NHEJ, unc-84 mutants have a defect in loading RAD-51 and/or in processing breaks. In the absence of CKU-70, the higher levels of RAD-51 presumably reflect more breaks loaded with RAD-51 because repair by NHEJ is blocked. Over time, the difference diminished, and by 16 h there were more RAD-51 foci in the double mutant than in either single mutant (unc-84;cku-70, 0.95 ± 0.19 vs. unc-84, 0.37 ± 0.09 and cku-70, 0.28 ± 0.08), which could reflect a defect in RAD-51 disassembly or an overall slower resolution of breaks without perturbing RAD-51 removal. Thus, UNC-84 plays multiple roles in DSB repair.
As break repair is dynamic, cytological analysis of RAD-51 in fixed germlines reflects both break formation and ongoing repair. To trap RAD-51 repair intermediates, we analyzed RAD-51 levels upon inactivation of RAD-54 (Solinger et al., 2002; Mets and Meyer, 2009). In rad-54(ok615) mutants, RAD-51 levels remain high, as RAD-54 is essential for RAD-51–mediated strand exchange during HR and is required for RAD-51 disassembly (Fig. 4, compare A with B). Although overall levels of RAD-51 were increased as a result of the absence of RAD-54, significantly fewer RAD-51 foci were observed in rad-54;unc-84 compared with rad-54 alone after IR (P < 0.0001; Fig. 4 B). These data suggest that in the absence of UNC-84, some breaks fail to load RAD-51 and/or load RAD-51 slowly. To determine whether reduction in RAD-51 was caused by the repair of a subset of breaks by NHEJ, we examined RAD-51 levels in rad-54;unc-84;cku-80(ok861) and found that RAD-51 levels were similar to rad-54 and significantly different than rad-54;unc-84 (0.5 h, P < 0.0001; 2 h, P = 0.001; Fig. 4 B). At 2 h after IR, rad-54;unc-84;cku-80 had intermediate levels of RAD-51 compared with rad-54 and rad-54;unc-84, consistent with the hypothesis that UNC-84 also plays a role in RAD-51 loading. Thus, our data are consistent with a model whereby UNC-84 functions to promote repair by HR, and in its absence, NHEJ is inappropriately engaged.
Human Sun-1 facilitates repair of interstrand cross-links
To determine whether human Sun-1 or Sun-2 plays a role in interstrand cross-link repair, we examined cross-link sensitivity in HeLa cells depleted for Sun-1/2. Inactivation of Sun-1 alone by siRNAs resulted in sensitivity to cross-linking agent mitomycin C (MMC), similar to what has been reported for the inactivation of FANCD2 (Fig. 3 F; Adamo et al., 2010). Sun-2 was not efficiently depleted by siRNA, and as Sun-1 depletion alone resulted in MMC sensitivity, Sun-2 was not examined further. To determine the extent to which sensitivity to MMC is a result of a role for Sun-1 in inhibiting NHEJ, we used a potent and specific DNA-dependent protein kinase (PK) inhibitor NU7026 (Veuger et al., 2003). HeLa cells treated with Sun-1 siRNAs resulted in hypersensitivity to MMC, whereas blocking NHEJ by addition of NU7026 suppressed the MMC sensitivity of Sun-1–depleted cells to levels comparable to the control (Fig. 3 F). We also examined cross-link sensitivity in HeLa cells expressing mouse Sun-1 (Poser et al., 2008) in the presence and absence of human Sun-1 siRNAs. Consistent with Sun-1 siRNAs specifically inactivating human Sun-1, we observed similar levels of sensitivity as in control-treated cells (Fig. 3 F). Therefore, as in C. elegans, human Sun-1 also functions to suppress NHEJ to facilitate repair of DNA cross-links.
UNC-84 plays a role in repair of meiotic DSBs
In addition to DNA damage, DSBs are intentionally induced during meiosis. Meiotic DSBs are processed to promote crossovers in the context of paired and synapsed homologous chromosomes. In C. elegans, pairing is not dependent on recombination, and each of the six homologous chromosome pairs is connected by a single crossover. To determine whether UNC-84 plays a role in the processing of meiotic DSBs or in the pairing of homologous chromosomes, which indirectly affects meiotic DSB repair, chromosome pairing was analyzed in germlines of worms harboring LacO sequences on chromosome V (LacO-V) with LacI-GFP (Severson and Meyer, 2014). unc-84 mutants paired LacO-V as efficiently as wild type; in the majority of PZ germ cells, two LacI-GFP spots were detected (wild type, 88.48%; unc-84, 86.61%), indicating that homologues were not paired before meiosis. In the transition zone, 51.5% of wild-type and 55.26% of unc-84 nuclei, and by early pachytene, 93.93% of wild-type and 97.64% of unc-84 nuclei, had one spot, indicative of paired chromosome Vs. SYP-1, a central region component of the synaptonemal complex, loaded onto chromosomes between axes at the same time during meiotic prophase as wild type (Fig. S3 A) and colocalized with the axial component HTP3 (Fig. S3 B). Thus, UNC-84 is not important for chromosome pairing and synapsis.
To determine whether UNC-84 plays a role in meiotic DSB repair, we examined loading and removal of RAD-51 throughout meiotic prophase in unc-84 germlines. In wild type, RAD-51 foci peak in abundance in early to mid-pachytene and are largely disassembled by late pachytene (Colaiácovo et al., 2003). In unc-84 mutants, there was a small but significant reduction of RAD-51 foci in early pachytene (P < 0.0001; Mann-Whitney U test; Fig. 5 A), suggesting that fewer DSBs are formed, that some DSBs are processed independently of HR, and/or that kinetics of the loading/disassembly of RAD-51 are altered in the absence of UNC-84. However, by mid-pachytene, more RAD-51 foci were observed in the unc-84 mutant (P < 0.0001; Fig. 5 A), suggesting that RAD-51 loading is slower or delayed in the absence of UNC-84. We next examined the consequence of inactivating NHEJ in the unc-84 mutant and found that the reduced levels of RAD-51 were partially suppressed at early pachytene; however, by mid- and late pachytene, RAD-51 levels were elevated in the double mutant compared with either wild type or unc-84 (Fig. 5 A). Inactivation of NHEJ alone resulted in elevated RAD-51 in mid- and late pachytene, suggesting that NHEJ functions independently of UNC-84 and that some meiotic DSBs are repaired by NHEJ beginning in mid-pachytene.
Figure 5.

UNC-84 facilitates repair at a subset of meiotic DSBs. (A and B) Mean numbers of RAD-51 foci per nucleus in early pachytene (EP), mid-pachytene (MP), and late pachytene (LP) in wild type (WT), unc-84(n369), unc-84(n3690);cku-70(tm1524), cku-70(tm1524) (A); and rad-54(ok615), rad-54(ok615);unc-84(RNAi), rad-54(ok615);unc-84(n369);cku-80(ok861), and rad-54(ok615);cku-80(ok861) (B). n > 3 germlines per genotype. (C) Number of DAPI-stained bodies in diakinesis in wild type, unc-84(n369), cku-70(tm1524), syp-1(me17), syp-1(me17);unc-84(n369), syp-1(me17);unc-84(n369);cku-70(tm1524), and syp-1(me17);cku-70(tm1524). n > 60. (D) Images of DAPI-stained diakinesis chromosomes for the given genotypes; the arrow marks a misshapen DAPI body that likely represents a chromosome fusion. Bar, 5 µm. Error bars indicate 95% CI. *, P < 0.05; **, P < 0.01; ***, P < 0.0001.
Errors in meiotic recombination activate a checkpoint, which leads to elevated apoptosis to cull defective germ cells (MacQueen and Hochwagen, 2011). Consistent with a defect in meiotic recombination, germline apoptosis was increased in unc-84, unc-84;cku-70, and cku-70 mutants; elevated apoptosis was largely abrogated in the absence of meiotic DSBs (spo-11; Fig. S3 C), suggesting the increased apoptosis is dependent on meiotic recombination. Although unc-84 mutants have slightly elevated levels of progeny inviability, removal of the apoptotic machinery resulted in significantly more inviable progeny (ced-3; Fig. S3 D), suggesting that unc-84 defects in recombination compromise germ cell function. Together, these results suggest that UNC-84 plays a role in meiotic DSB repair.
To elucidate the mechanism underlying the alteration in RAD-51 in the absence of UNC-84, we examined RAD-51 in rad-54 mutant germlines. During meiosis, inactivation of RAD-54 not only blocks RAD-51 turnover, but, unlike IR, it also extends the time for which DSBs are formed (Mets and Meyer, 2009; Rosu et al., 2013; Stamper et al., 2013). Although levels of RAD-51 foci were higher in rad-54;unc-84 compared with unc-84 germlines as expected, there was a significant reduction in RAD-51 foci in early pachytene in the double mutant compared with rad-54 alone (P < 0.0001; Fig. 5 B). The decrease in RAD-51 foci in rad-54;unc-84 doubles in early pachytene was alleviated by removal of NHEJ (rad-54;unc-84;cku-80 triple mutant; Fig. 5 B). However, in mid- and late pachytene, RAD-51 levels were not statistically different between rad-54 and rad-54;unc-84. Furthermore, the rad-54;cku-80 double had elevated RAD-51 beginning in mid-pachytene, and this increase was only partially alleviated by the removal of UNC-84, suggesting that UNC-84 facilitates RAD-51 loading independently of NHEJ. Collectively, these data are consistent with a subset of breaks being repaired by NHEJ in the absence of UNC-84 in early prophase and that UNC-84 plays additional roles in HR.
The subtle defect in RAD-51 processing in unc-84 mutants suggests that UNC-84 only operates on a subset of meiotic DSBs. Consistent with this, COSA-1, a crossover-promoting protein that concentrates at nascent crossover sites (Yokoo et al., 2012), was observed at six foci in unc-84 mutants as in wild type in late pachytene (wild type, 67.5% nuclei and six foci; unc-84(RNAi), 70.5% nuclei and six foci [P = 0.89]), suggesting that DSBs are efficiently processed into crossovers in the absence of UNC-84. However, during meiosis, many more DSBs are induced than will become crossovers. The contribution and interplay between pathways responsible for the repair of DSBs not destined to become crossovers is not well understood. When homologue interactions are blocked by mutation of the synaptonemal complex (e.g., syp-1 or syp-2 mutants), additional requirements for repair can be uncovered. For example, in fcd-2 (C. elegans FANCD2) mutants, chromosome fusions as analyzed by FISH were observed in the absence of SYP-2 and were dependent on NHEJ (Adamo et al., 2010). In contrast, inactivation of SYP-2 in brc-1 (C. elegans BRCA1) mutants resulted in chromosome fragments caused by an inability to repair a subset of breaks (Adamo et al., 2008). Diakinesis nuclei from wild-type and unc-84 mutants contained six bivalents (Fig. 5, C and D). In syp-1(me17) mutants, 12 univalents were observed (Fig. 5, C and D), as DSBs are efficiently repaired when homologues are not connected in an otherwise wild-type worm (MacQueen et al., 2002). syp-1;unc-84 double mutants had significantly fewer DAPI-stained bodies as compared with syp-1 alone (syp-1;unc-84, 11.25 ± 0.13; syp-1, 11.80 ± 0.06 [P < 0.0001]; Fig. 5, C and D). Furthermore, some of the DAPI bodies were abnormal in shape (Fig. 5 D, arrow), consistent with formation of chromosomal fusions. The presence of fewer DAPI bodies is similar to fcd-2 mutants, although in fcd-2 mutants ∼70% of diakinesis nuclei contained <12 DAPI bodies (Adamo et al., 2010), whereas 41.2% of unc-84 nuclei had <12 DAPI bodies. To test the hypothesis that the aberrant chromosomes arose because of inappropriate repair by NHEJ, we examined diakinesis nuclei in the unc-84;syp-1;cku-70 triple mutant and observed an increase in DAPI bodies compared with syp-1;unc-84 (syp-1;unc-84, 11.25 ± 1.16; syp-1;unc-84;cku-70, 12 ± 0.69; syp-1;cku-70, 12 ± 0.32 DAPI bodies [P < 0.0001]; Fig. 5, C and D). The presence of nuclei with >12 DAPI bodies suggests that some breaks are not repaired in the absence of UNC-84 even when NHEJ is not present. Collectively, these results are consistent with a role for UNC-84 in meiotic DSB repair.
UNC-84 protein is expressed in proliferating germ cells in response to DNA damage
UNC-84 localizes to the nuclear envelope in the proximal gonad and most somatic cells during embryogenesis, larval, and adult stages (Malone et al., 1999; Lee et al., 2002; Cain et al., 2014). To examine UNC-84 in germ cell nuclei, we engineered a transgenic line expressing UNC-84::GFP from the endogenous unc-84 locus. Homozygous UNC-84::GFP worms were functional for embryonic nuclear positioning (mispositioned hyp7 nuclei in L1 animals: unc-84::gfp, 0.02 ± 0.14 [n = 53]; wild type, 0 ± 0 [n = 11]; unc-84, 14.3 ± 1.5 [n = 25]; Tapley et al., 2011). Furthermore, immunofluorescence using antibodies against GFP revealed localization of UNC-84::GFP at the periphery of nuclei in the proximal germline beginning at late pachytene stage (Figs. 6 A and S4 A) and in most somatic cells (Fig. 6 A, distal tip cells/sheath cells). No UNC-84 was detected in the male germline (Fig. S4 B), suggesting that expression of UNC-84 in late pachytene was specific to oogenesis and most likely important for early embryonic development (Xiong et al., 2011).
Figure 6.

UNC-84 and ZYG-12 are up-regulated in PZ germ cells in response to DNA damage and can physically associate in vitro. (A) Dissected germline from an UNC-84::GFP worm stained with anti–UNC-84 (N-terminal epitope; red) and anti-GFP (C-terminal epitope; green). Insets show staining in all zones of the germline from the PZ, transition zone (TZ), early pachytene (EP), mid-pachytene (MP), late pachytene (LP), diplotene (DP), and diakinesis (DI), as well as the somatic distal tip cell (DTC) and sheath cell (Sh). Bars: (main image) 100 µm; (insets) 2 μm. The arrow indicates a somatic sheath cell. The asterisk indicates a distal tip cell. (B) UNC-84::GFP is detected at the nuclear periphery in arrested PZ germ cells after cisplatin treatment. The UNC-84::GFP germline after 48 h of 250 µM cisplatin recovery, stained for GFP, RAD-51, and DAPI. (C) ZYG-12::GFP is present in patches in proliferating germ cells in the absence of cisplatin and shows higher intensity in RAD-51–positive nuclei 48 h after removal from 250 µM cisplatin (left). Quantification of pixel intensity plotted as fold GFP fluorescence at the nuclear envelope in RAD-51–positive nuclei compared with RAD-51–negative nuclei in ZYG-12::GFP and ZYG-12::GFP;unc-84(n369) worms (right); n > 90. Error bars indicate 95% CI. ***, P < 0.0001. (D) Image of ZYG-12::GFP (green), UNC-84 (red), RAD-51 (white), and DAPI (blue) localization in the PZ 48 h after 250 µM cisplatin. The arrow indicates a somatic distal tip cell. Bars, 10 µm. (E) MBP::ZYG-12 and UNC-84 coelute as a single complex upon size-exclusion chromatography (Superose 6 column; top). The asterisk represents a degradation product. Fractions were collected and analyzed by SDS-PAGE and Coomassie blue staining (bottom).
To determine whether UNC-84 was regulated in response to DNA damage, we treated worms with cisplatin and examined UNC-84::GFP over time. We observed UNC-84::GFP at the nuclear periphery at 16, 24, and 48 h after release from cisplatin in a subset of PZ cells in hermaphrodite (48 h shown; Fig. 6 B) and male germlines (Fig. S4 C). These cells were large and contained RAD-51, suggesting that they were arrested and were undergoing DNA repair. Treatment with HU or IR also resulted in UNC-84::GFP at the nuclear envelope of PZ germ cells; however, fewer cells were labeled under these conditions (Fig. S4, D and E). We hypothesize that we are not detecting all UNC-84 but are capturing cells in prolonged arrest where UNC-84 accumulates. As the male germline is not surrounded by sheath cells in the distal gonad (Kimble and Hirsh, 1979) and a defect in RAD-51 was also observed in unc-84 mutant males (Fig. S4 F), these results suggest that UNC-84 is expressed in at least a subset of cells and is required cell autonomously in PZ germ cells to promote accurate repair in response to DNA-damaging agents.
Nuclear localization of FAN-1 is altered in the absence of UNC-84 or FCD-2
Our data show that both C. elegans UNC-84 and human Sun-1 are required to repair interstrand cross-links when NHEJ is present. As the FA pathway is critical for repair of interstrand cross-links and the key FA component, FANCD2, is also required in the presence of NHEJ (Adamo et al., 2010; Pace et al., 2010), we explored the relationship between UNC-84 and FCD-2. The fcd-2(tm1268) mutant is sensitive to cross-linking agents (Collis et al., 2006), displaying sensitivity similar to unc-84 (P = 0.08; Fig. 3 D). The fcd-2;unc-84 double mutant looked similar to fcd-2 (P = 0.89) but was elevated compared with unc-84 (P < 0.03; Fig. 3 D). These results suggest that UNC-84 likely functions in the same pathway as FCD-2 in the repair of interstrand cross-links.
We next examined RAD-51 loading after exposure to cross-linking agents in unc-84;fcd-2 and corresponding single mutants. Surprisingly, fcd-2 mutants had higher levels of RAD-51 foci in PZ germ cells immediately after cisplatin treatment than wild type (Adamo et al., 2010), whereas unc-84 mutants had reduced RAD-51 (Fig. 3 E). Analysis of fcd-2;unc-84 double mutants revealed RAD-51 levels were similar to fcd-2 (Fig. 3 E). Thus, fcd-2 is epistatic to unc-84 for RAD-51 and likely functions downstream of UNC-84.
Repair of interstrand cross-links involves the recruitment and coordination of multiple nucleases. One of these, FAN-1, interacts with FANCD2 in mammalian cells and is important for DNA interstrand cross-link repair (Kennedy and D’Andrea, 2005; Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010). C. elegans fan-1(tm423) mutants were more sensitive to cisplatin treatment than either fcd-2 (MacKay et al., 2010) or unc-84 mutants (P < 0.0001; Fig. 3 D). However, cisplatin sensitivity of fan-1 mutants was not suppressed by the inactivation of NHEJ (Fig. 3 D), suggesting that, unlike FCD-2 and UNC-84, FAN-1 does not function to inhibit NHEJ during processing of cross-links.
In mammalian cells, FAN-1 recruitment to cross-links is dependent on FANCD2 (Smogorzewska et al., 2010); however, recruitment is dispensable for cross-link repair (Lachaud et al., 2016). Using a FAN-1::GFP transgene (Kratz et al., 2010), we found that recruitment of FAN-1 after cisplatin was abrogated in the absence of either FCD-2 or UNC-84 (Fig. 7). Thus, UNC-84 and FCD-2 are both important for recruiting FAN-1.
Figure 7.

UNC-84 and FCD-2 are required for the efficient recruitment of FAN-1 after cisplatin treatment. (Left) Dissected germlines from FAN-1::GFP, FAN-1::GFP;unc-84(n369), FAN-1::GFP;fcd-2(tm1268), and FAN-1::GFP;zyg-12(or577) worms in the absence (−) and presence (+) of 250 µM cisplatin. (Right) Quantification of GFP intensity in the given genotypes where the ratio of nucleoplasmic to cytoplasmic signal was determined in the absence and presence of 250 µM cisplatin. n > 50 nuclei. Bar, 10 μm. Error bars indicate 95% CI. ***, P < 0.0001.
A novel LINC complex mediates repair of interstrand cross-links
SUN proteins in the inner nuclear membrane recruit KASH partners to the outer nuclear membrane to create LINC complexes for microtubule-based motion. Alternatively, N-terminal domains of SUN proteins could function in the nucleoplasm independently of KASH partners. To distinguish between these two hypotheses for the role of SUN proteins in the repair of DNA damage, we analyzed the cisplatin sensitivity of unc-84 alleles. These mutants were grouped into classes based on phenotype, identity of molecular lesion, and intragenic complementation (Fig. 8 A; Malone et al., 1999). One class of mutants maps to the nucleoplasmic N terminus, including unc-84(P91S), and disrupts the interaction between UNC-84 and lamin, leading to a partial defect in nuclear migration (Bone et al., 2014). A second, complementary class of missense mutants map to the perinuclear SUN domain and disrupts interactions between SUN and canonical KASH proteins (Malone et al., 1999; Lee et al., 2002; McGee et al., 2006; Sosa et al., 2012). unc-84(P91S) mutants had a cisplatin-sensitive phenotype similar to unc-84(null) (Fig. 8 A), suggesting that the nucleoplasmic domain of UNC-84 is critical for the repair of interstrand cross-links. Surprisingly, the three SUN domain missense mutants revealed differences in sensitivity, even though they all behave as nulls with respect to nuclear positioning (Malone et al., 1999). Two of the mutants, unc-84(C994Y) and unc-84(G1002D), showed sensitivity at the highest cisplatin concentration, whereas the third mutant, unc-84(S988F), was not sensitive to cisplatin at any concentration (Fig. 8 A). The differences in cisplatin sensitivity in the mutants are not likely caused by changes in protein expression, as the levels of UNC-84 are similar in the P91S, G1002D, and S988F alleles (Lee et al., 2002; Bone et al., 2014) and all of the mutant proteins were detected after DNA damage in the PZ, as well as in late prophase (Fig. S4 H). Furthermore, we observed elevated germline apoptosis (P91S and C994Y) and rescue of the cisplatin sensitivity upon inactivation of NHEJ in P91S, G1002D, and C994Y mutants (Fig. S5). These results suggest that changes to the luminal domain that interfere with KASH interactions for nuclear migration and anchorage do not completely abrogate DNA repair function. The differential effect of SUN mutations suggests that UNC-84 partners with a different KASH protein to promote DNA repair.
Figure 8.

UNC-84, ZYG-12, and the microtubule network are required for efficient repair of DNA cross-links. (A) Schematic of UNC-84 domains and mutations (top). TM, transmembrane domain. Percentages of progeny inviability of wild-type (WT) worms and unc-84(n369), unc-84(P91S), unc-84(S988F), unc-84(C994Y), and unc-84(G1002D) after exposure to the given concentrations of cisplatin (bottom). n > 10. cispl, cisplatin. (B) Percentages of progeny inviability after exposure to the given concentrations of cisplatin of wild-type worms and worms harboring mutations in KASH domain–containing proteins unc-83(e1408), anc-1(e1873), kdp-1(RNAi), and zyg-12(RNAi) at 20°C, as well as wild type, zyg-12(or577), and zyg-12(or577);cku-70(tm1524) at the semipermissive temperature of 16°C. n > 10. (C) Percentages of RAD-51–positive nuclei after treatment with 100 µM cisplatin in wild type, unc-84(n369), kdp-1(RNAi), zyg-12(or577), and zyg-12(or577);cku-70(tm1524) at 16°C. n > 30. (D) Percentages of RAD-51–positive nuclei after treatment with 100 µM cisplatin in wild-type and cku-70(tm1524) worms in the presence of the given concentrations of colchicine. n > 30. Error bars indicate 95% CI. P-values were determined by two-way analyses of variance. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
There are four known KASH proteins in C. elegans (McGee et al., 2009). UNC-83 and ANC-1 are the canonical UNC-84 partners that mediate nuclear migration and anchorage, respectively (Starr and Han, 2001; Starr and Han, 2002). We found that both unc-83(e1408)– and anc-1(e1873)–null mutants had wild-type levels of cisplatin sensitivity (Fig. 8 B), suggesting that UNC-84 does not mediate DNA repair through ANC-1 or UNC-83. The two other C. elegans KASH proteins, ZYG-12 and KDP-1, are both expressed in germ cells. ZYG-12 mediates centrosome attachment in early embryos and ensures proper homologous chromosome pairing during meiosis. Both of these previously described functions of ZYG-12 require the second, divergent C. elegans SUN protein SUN-1 (Malone et al., 2003; Minn et al., 2009; Sato et al., 2009). KDP-1 can interact with either UNC-84 or SUN-1 and is important for cell cycle kinetics in the germline and early embryo (McGee et al., 2009). The KASH domains of ZYG-12 and KDP-1 are poorly conserved when compared with UNC-83 and ANC-1, Drosophila melanogaster Klarsicht and MSP-300, or the mammalian Nesprins (McGee et al., 2009). To explore the possibility that ZYG-12 or KDP-1 functions with UNC-84 to promote DNA repair, we analyzed cisplatin sensitivity in worms partially depleted of these proteins, as both are essential for viability and organization of the gonad. zyg-12(RNAi), but not kdp-1(RNAi), displayed sensitivity to cisplatin (zyg-12(RNAi) vs. wild type; P < 0.05; Fig. 8 B). We also observed a significant increase in progeny inviability in a temperature-sensitive mutant (zyg-12(or577)) treated with cisplatin (P < 0.0001; Fig. 8 B), suggesting that ZYG-12 plays a role in cross-link repair.
ZYG-12 localizes to all germ cells, including in the PZ (Fig. 6 C; Sato et al., 2009). After cisplatin treatment, we observed significantly more ZYG-12::GFP at the nuclear periphery of arrested (>3.5 µm) PZ germ cells; these nuclei also contained RAD-51, suggesting that they were engaged in DNA repair (Fig. 6 C). The enrichment of ZYG-12::GFP in these cells was dependent on UNC-84, as the increase in fluorescence intensity was abrogated in unc-84 mutants (Fig. 6 C). We also examined the colocalization of ZYG-12::GFP and UNC-84 after cisplatin treatment. Although every cell that contained UNC-84 also had enhanced ZYG-12::GFP (Fig. 6 D), not all cells with enhanced ZYG-12::GFP were UNC-84 positive, which we attribute to inefficient detection with UNC-84 antibody. Finally, to determine whether UNC-84 and ZYG-12 interact in vitro, tagged versions of the SUN domain of UNC-84 and the KASH domain of ZYG-12 were coexpressed in Escherichia coli and purified on size-exclusion and ion exchange columns. 6×His::UNC-84(912–1,111) and MBP::ZYG-12(749–777) coeluted as a single complex (Fig. 6 E), showing that they directly interact. Together, these data are consistent with a novel LINC complex consisting of UNC-84 and ZYG-12 mediating the repair of interstrand cross-links.
We next monitored RAD-51 and found that, similar to unc-84 mutants (Figs. 3 C and 8 A), zyg-12 PZ germ cells had low levels of RAD-51 immediately after release from cisplatin (Fig. 8 C). Inactivation of both zyg-12 and cku-70 resulted in increased levels of RAD-51 (Fig. 8 C), consistent with ZYG-12 playing a similar role in repair as UNC-84. However, progeny inviability was exacerbated, not rescued, in the zyg-12;cku-70 mutant (Fig. 8 B; compare with Fig. 3 B for unc-84). These results suggest that whereas ZYG-12 plays a role in intrastrand cross-link repair, it is not identical to UNC-84. Because mutations in KASH proteins do not disrupt the localization of SUN proteins (Lee et al., 2002), it is likely that UNC-84 still localizes to the inner nuclear membrane in zyg-12 mutants, where it mediates the recruitment of FAN-1, altering response to cisplatin. To test this, we examined FAN-1::GFP in zyg-12 and found that it was localized as in wild type (Fig. 7, left and right). Thus, our data are consistent with an UNC-84–ZYG-12 complex mediating repair, but only UNC-84 is required for FAN-1 recruitment.
Inhibition of microtubule polymerization leads to defects in RAD-51 loading at cisplatin-induced lesions
Our data support a model whereby UNC-84–ZYG-12 connects the nucleoskeleton to the cytoskeleton to mediate repair of interstrand cross-links. As ZYG-12 interacts with microtubule motors to drive nuclear, centrosome, and chromosome movements (Malone et al., 2003; Sato et al., 2009; Zhou et al., 2009; Rog and Dernburg, 2015), we hypothesized that similar mechanisms were required for DNA repair. To test this hypothesis, we disrupted microtubule polymerization by treating worms with colchicine, induced interstrand cross-links, and monitored RAD-51. Inhibition of microtubule polymerization in wild-type germlines resulted in low levels of RAD-51 after cisplatin treatment at all concentrations of colchicine (Fig. 8 D), even though cytological disruption of the microtubule networks was only observed at higher concentrations (Fig. S6 A). The reduced levels of RAD-51 were unlikely to be caused by alteration of the cell cycle, as the G2 marker, phospho–CDK-1/NCC-1 (Moser et al., 2009), was similar to the untreated controls in the presence of the lowest concentration of colchicine (Fig. S6 D). Reduced RAD-51 after colchicine treatment was comparable to what we observed in unc-84 and zyg-12 mutants (Fig. 8, compare D with C), suggesting that the microtubule network is important for repair. By 4 h after treatment, there was no difference between RAD-51 in the presence or absence of colchicine (Fig. S6 B). To determine whether the reduction in RAD-51 observed by preventing microtubule polymerization was relieved by inactivating NHEJ, we monitored RAD-51 loading after treatment with colchicine in the cku-70 mutant and found that RAD-51 levels were similar to those found in wild type (Fig. 8 D). Inhibition of microtubule polymerization by nocodazole had a similar effect (Fig. S6 C). Collectively, these data suggest that microtubules play a role in the inhibition of NHEJ after DNA damage, which we propose is mediated through a UNC-84–ZYG-12 LINC complex.
Discussion
In this study, we provide evidence that a novel LINC complex plays a role in DNA damage repair. This function is conserved from C. elegans to humans. LINC contributes to the repair of DSBs associated with IR, the removal of interstrand cross-links, and the resolution of stalled replication forks in mitotically dividing cells and induced DSBs during meiosis. LINC was previously implicated in the repair of DNA damage (Oza et al., 2009; Lei et al., 2012; Swartz et al., 2014; Lottersberger et al., 2015; Ryu et al., 2015), but the mechanisms for how they do so were not clear. Our data indicate that LINC functions to promote HR and, in the presence of interstrand cross-links, recruits FAN-1 nuclease to the nucleoplasm. Collectively, our data highlight the importance of communication between the nucleus and cytoskeleton in the critical transactions of DNA repair and recombination.
Models for LINC complex function in DNA repair
We discuss four nonmutually exclusive mechanistic models suggested by our data for how SUN and KASH proteins favor HR. First, LINC could recruit DSBs to the periphery of the nucleus where the environment favors repair by HR. Second, LINC could function in mechanotransduction of a signal across the nuclear envelope to facilitate DNA repair. Third, SUN proteins could inhibit NHEJ by directly interacting with the DNA-PK/Ku70/Ku80 complex. Fourth, LINC could mediate motion along cytoplasmic microtubules to license DNA repair by HR. The data presented here argue against the first and support the latter two models for LINC function in DNA repair in C. elegans germline nuclei and HeLa cells.
Given that LINC complexes are embedded in the nuclear envelope, it is attractive to propose that DSB recruitment to the nuclear periphery through LINC facilitates repair by HR. Consistent with this, in both budding and fission yeast, SUN proteins recruit DSBs to the nuclear envelope. However, these DSBs are unrepairable (Oza et al., 2009; Swartz et al., 2014), and thus recruitment may represent a mechanism to sequester these breaks rather than to promote repair. We show that damaged DNA marked by RAD-51 is recruited to the periphery in the absence of UNC-84 in C. elegans (Fig. 2 D), suggesting that the primary function of LINC is not the recruitment of DSBs to the periphery. Furthermore, other groups have shown that DSBs are not recruited to the periphery in mammalian cells but rather that DSB repair is compartmentalized at the periphery (Soutoglou et al., 2007; Lemaître et al., 2014). Thus, we propose that SUN proteins influence repair independently of recruitment of damaged DNA to the periphery.
An alternative model posits that LINC is critical in maintaining the integrity of the nuclear envelope to promote DDR signaling. The checkpoint kinases ATR/ATM are upstream activators of the DDR (Ciccia and Elledge, 2010), and ATM signaling is decreased in the absence of SUN proteins (Lei et al., 2012). We show that unc-84 mutants respond differently to DNA damage than either wild-type or checkpoint-defective mutants (Fig. 1), suggesting that loss of SUN is not solely explained by defective ATR/ATM signaling. ATR is critical for nuclear envelope integrity and for signaling mechanical stress (Kumar et al., 2014). Given the established role of LINC in mechanotransduction (Chambliss et al., 2013), it is intriguing to hypothesize that in the absence of SUN or KASH, mechanical stress mediated by DNA damage fails to fully activate ATR/ATM, leading to inappropriate use of NHEJ.
We favor a model whereby LINC both directly inhibits the Ku70/Ku80/LIG-4 complex through SUN proteins and licenses repair by HR through microtubules (Fig. 9). Lei et al. (2012) identified an interaction between DNA-PK (C. elegans does not contain DNA-PK), Ku, and the nucleoplasmic domains of Sun-1/2. In this study, we show that inactivation of NHEJ by mutation of Ku70/80 in C. elegans or inhibition of DNA-PK in HeLa cells suppress DNA damage-sensitivity phenotypes of unc-84 mutant animals or Sun-1 siRNA–treated HeLa cells. We propose that SUN proteins interact with the (DNA-PK/)Ku70/Ku80 complex, thereby inhibiting NHEJ.
Figure 9.

LINC complexes facilitate DNA repair through both the inhibition of NHEJ and the promotion of HR. (A) In response to DSBs, wild-type UNC-84 interacts with ZYG-12 at the nuclear envelope to form a LINC complex that connects the cytoskeleton with the nucleoplasm. Motors that associate with microtubules are unknown. In the nucleoplasm, UNC-84 (human Sun-1) interacts with NHEJ machinery (KU70/KU80/LIG-4 and DNA-PK in mammals) to drive microtubule-dependent inhibition of NHEJ and the promotion of RAD-51 loading/HR. (B) After DNA cross-links, UNC-84 also recruits FAN-1 to the nucleus to facilitate processing of interstrand cross-links; additional nucleases are involved in processing cross-links. (C) In the absence of UNC-84, NHEJ is not inhibited, and HR is also impaired in response to DSBs. Additionally, FAN-1 is not recruited to the nucleus upon induction of cross-links, and other nucleases may be inappropriately engaged. Blue, ZYG-12; purple, UNC-84; green, DNA-PK/KU70/KU80; orange, FAN-1. MT, microtubule; Ce, C. elegans; WT, wild-type.
LINC could promote HR through motion-dependent licensing, whereby chromosomal motion prevents NHEJ from operating on DSBs (Fig. 9). Consistent with this model, inactivation of microtubule dynamics or LINC components led to a defect in RAD-51. The RAD-51 defect was partially suppressed by the removal of NHEJ, suggesting that SUN proteins normally act to inhibit NHEJ. Additionally, we observed both slower accumulation of RAD-51 and persistent RAD-51 in the absence of a functional LINC, suggesting that LINC also facilitates processing of DSBs by HR. This model is attractive, as it is similar to the proposed role for LINC-mediated chromosomal movement in licensing pairing/synapsis during meiosis (Sato et al., 2009). An opposite conclusion was made when it was reported that a LINC complex was important for promoting motion-dependent repair of naked telomeres by NHEJ (Lottersberger et al., 2015). In this study, we show that LINC and microtubules are important for inhibiting NHEJ in response to interstrand cross-links, DNA damage generated by HU, IR, and meiotic DSBs. The disparity between this study and Lottersberger et al. (2015) could be a consequence of the damage examined (i.e., naked telomeres vs. induced damage/meiotic DSBs). Alternatively, there could be inherent differences between humans and mice consistent with the reported suppression (in humans) versus enhancement (in mice) of the FA pathway by inactivation of NHEJ (Adamo et al., 2010; Bunting et al., 2012). Our genetic data show that inhibiting NHEJ machinery partially suppresses DNA damage–sensitive phenotypes of LINC or microtubule disruption. We therefore conclude that motion acts to inhibit NHEJ and license HR repair of DNA damage.
In our model, UNC-84 interacts with ZYG-12 to form a novel LINC complex, and in support of this, we show that these two proteins interact in vitro (Fig. 6 E). Insights into SUN–KASH interactions have been revealed by the crystal structure of human Sun-2 (Sosa et al., 2012), yet our analysis of SUN domain mutants suggests that not all SUN and KASH domains interact in the same way. This could reflect the requirement for different resistance in tension in different physiological processes (i.e., moving nuclei vs. inhibiting NHEJ; Cain et al., 2014). Furthermore, the microtubule motor that generates motion mediated by this novel LINC complex is unknown (Fig. 9). ZYG-12 interacts with dynein through the dynein light chain (Malone et al., 2003). Other KASH proteins interact with kinesin 1 or kinesin 2 (Fan and Beck, 2004; Meyerzon et al., 2009; Roux et al., 2009; Zhang et al., 2009). In mouse embryonic fibroblasts, kinesin 2 generates the forces to move naked telomeres (Lottersberger et al., 2015). Thus, any of these motors could couple with LINC to mediate DNA repair.
Roles of LINC complexes and nucleases in interstrand cross-link repair
Unlike other DNA-damaging agents, interstrand cross-links led to embryonic lethality upon inactivation of UNC-84, suggesting that LINC has a more important role in processing these lesions. Previous work has uncovered unique requirements for the repair of interstrand cross-links. The RAD-51 paralogue RFS-1 is required for cross-link repair but not for repair of HU, IR, or meiotic DSBs, suggesting that the nature of the HR substrate is distinct when replication is blocked (Ward et al., 2007). Interestingly, like unc-84, rfs-1 mutants show a reduction of RAD-51 after treatment with cross-linking agents; however, the extent of similarities between unc-84 and rfs-1 is unknown, as only a single time point was examined, and we observed a complex pattern of RAD-51 with cross-linking agents. Additionally, although RFS-1 is relatively specific to the repair of replication-blocking lesions, UNC-84 also plays a role in processing lesions induced by HU, IR, and meiotic DSBs.
One possible explanation for UNC-84’s unique role in cross-link repair is that FAN-1, which has a specific requirement in cross-link repair (Kennedy and D’Andrea, 2005; Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010), is not efficiently recruited to the nucleoplasm in the absence of UNC-84 (Fig. 7). Unlike inhibition of NHEJ, FAN-1 localization to the nucleus does not require a functional LINC, suggesting that UNC-84 serves as a tether for FAN-1. Although multiple mechanisms have been described to target SUN proteins to the inner nuclear membrane (Turgay et al., 2010; Gardner et al., 2011; Tapley et al., 2011; Ungricht et al., 2015), how UNC-84 promotes FAN-1 recruitment is not clear. Once in the nucleus, the presence of FAN-1 leads to unproductive repair when NHEJ is not inhibited by LINC (Fig. 8). The role of FAN-1 has been enigmatic; it was identified by several groups as interacting with the FA pathway through FANCD2 (Kennedy and D’Andrea, 2005; Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010), yet it is not a classic FA gene. Adding to its significance, FAN-1 has been postulated to be a tumor suppressor through its role in stabilizing replication forks, and FAN-1 mutations have been identified in pancreatic, colorectal, and breast cancer (Seguí et al., 2015; Lachaud et al., 2016; Smith et al., 2016). Biochemically, FAN-1 has several activities, including unhooking interstrand cross-links and strand incision (Gwon et al., 2014; Pennell et al., 2014; Takahashi et al., 2015). Further, multiple other nucleases participate in interstrand cross-link repair (Kottemann and Smogorzewska, 2013; Duxin and Walter, 2015). Consequently, loss of FAN-1 may lead to the inappropriate use of other nucleases, resulting in unproductive and potentially lethal repair products (Figs. 3 and 9). Whether SUN proteins coordinate the activity of these additional nucleases remains an open question.
The complexity of DNA repair pathways
DNA repair pathways are critical for maintaining genome integrity, and, consequently, repair can be abrogated in cancer and neurological diseases (Suberbielle et al., 2015; Ceccaldi et al., 2016). A challenge is in understanding how the large numbers of repair pathways are integrated in different cellular contexts. This is evident in our analyses of FCD-2 and UNC-84. Although many of the phenotypes of fcd-2 and unc-84 are similar, RAD-51 foci are elevated in fcd-2 mutants after cross-linking agents and in meiosis (Adamo et al., 2010), whereas they are reduced in the absence of UNC-84 (Figs. 3 and 5). These results suggest that DSBs formed in repairing cross-links or during meiosis can be processed by many different pathways. Furthermore, it is likely that when one pathway is inactivated, different pathways are engaged and make distinct contributions to the repair profile of the organism. These results highlight our gap in understanding the interplay and complexities between different repair pathways, particularly under pathological conditions.
Our studies also highlight the importance of robustly inhibiting NHEJ in meiosis. Meiotic DSB repair must be tightly controlled to ensure that DSBs are repaired by HR to promote chiasma formation. After cutting by the meiosis-specific topoisomerase Spo-11, the MRN complex processes the break, thereby preventing resealing by NHEJ (Yin and Smolikove, 2013). In contrast to MRN, where very few RAD-51 foci are observed, inactivation of UNC-84 has subtle defects, as it appears that only a subset of breaks in early prophase are repaired through NHEJ in the absence of UNC-84 (Fig. 5). A previous study had revealed that RAD-51 levels were only perturbed in lig-4 mutants when both synapsis and sister chromatid cohesion were blocked (Smolikov et al., 2007). Our analysis of cku-70 mutants revealed elevated RAD-51 in mid- and late pachytene in an otherwise wild-type worm. The difference is most likely a result of ends being bound by CKU-70/80 in the absence of LIG-4. Thus, our results suggest that NHEJ is active throughout meiotic prophase and multiple pathways have evolved to ensure it is not inappropriately used.
LINC and cancer
The finding that LINC functions in DNA repair opens up exciting possibilities for the treatment of human diseases such as FA and cancer. FA mutants exist for which the gene has not yet been identified, and it is possible that LINC inactivation could be the responsible lesion. Furthermore, the recent finding that SUN expression levels alter sensitivity to cisplatin treatment in lung cancers (Lv et al., 2015) suggests that manipulation of SUN may be an effective chemotherapy.
Materials and methods
Genetics
Worms were maintained at 20°C under standard conditions except the unc-84 alleles: unc-83(e1408) and zyg-12(or577) worms were maintained at 15°C. The wild-type strain was N2 Bristol. Strains were obtained from the Caenorhabditis Genetics Center unless otherwise noted. We used the following strains in this study. I: anc-1(1753); rad-54–snx-3(ok615) I/hT2 [bli-4(e973) let-(q782 qIs48)](I:III). III: cku-70(tm1524); cku-80(ok861); lig-4(ok716); brc-1(tm1145). IV: ced-3(n717); fcd-2(tm1268); fan-1(tm423); spo-11(ok79). V: unc-83(e1408); syp-1(me17) V/nT1 [unc(n754) let qIs50] (IV;V). X: unc-84(n369); unc-84(P91S); unc-84(C994Y); unc-84(G1002D); unc-84(S988F) (Malone et al., 1999). RPA-1::YFP(opls263) and FAN-1::GFP(opls406) were obtained from M. Hengartner (University of Zurich, Zurich, Switzerland; Kratz et al., 2010; Stergiou et al., 2011). ZYG-12::GFP was obtained from C. Malone (The Pennsylvania State University, University Park, PA; Malone et al., 2003).
Clustered regularly interspaced short palindromic repeats
The knock-in UNC-84::GFP strain was created using the methods described by Dickinson et al. (2013). In brief, GFP was tagged to the C terminus of UNC-84 by PCR gene splicing by overlapping extension with five overlapping fragments (1.6 kb of the 3′ end of the unc-84 coding region, gfp, 373 bp of the unc-84 3′ UTR, the unc-119(+) cassette, and a 1.5-kb homologous genomic region downstream of unc-84) and cloning into TOPO-XL to create pSL696. The unc-84 single guide RNA + Cas9 vector (pSL695) was made with the Q5 Site-Directed Mutagenesis kit (New England Biolabs, Inc.). unc-119(ed3) III animals were injected with 10 ng/µl pSL696, 50 ng/µl pSL695, 10 ng/µl pMA122, 10 ng/µl pGH8, 2.5 ng/µl pCFJ90, and 5 ng/µl pCFJ104 (all markers from Addgene) as described previously (Dickinson et al., 2013). To excise the unc-119(+) cassette, the clustered regularly interspaced short palindromic repeat strain was injected with 50 ng/µl pDD104 and 100 ng/µl podr1::RFP and then outcrossed to N2 males three times to eliminate off-target mutations.
Cytological analyses
Immunostaining of germlines was performed as described by Jaramillo-Lambert and Engebrecht (2010). Germlines were fixed in 1% PFA for 5 min and freeze cracked in liquid nitrogen followed by 1 min of cold (−20°C) methanol. Slides were blocked in 0.7% BSA for 1 h before primary antibodies were incubated at room temperature overnight. Secondary antibodies were incubated for 2 h at room temperature. Specificity of antibody staining was verified by examining the absence of staining in RNAi-depleted or mutant worms.
The following primary antibodies were purchased and used at the indicated dilutions: rabbit anti–RAD-51 (1:10,000; Novus Biologicals), rabbit anti–H3S10-P (1:200; EMD Millipore), mouse anti–nuclear pore complex (NPC) proteins (Mab414; 1:100; Abcam), rabbit anti–GFP (1:500; Novus Biologicals), mouse anti–α-tubulin (1:500; Sigma-Aldrich), and rabbit anti–Cdk-1(pTyr15) (1:100; EMD Millipore). Mouse anti–UNC-84 IgM (1:100) has been described previously (Cain et al., 2014). Rabbit anti–SYP-1 (1:200), guinea pig anti–HTP-3 (1:1,000), and rabbit anti–MAD-2 (1:2,000) were gifts from A. Villeneuve (Stanford University, Palo Alto, CA), A. Dernburg (University of California, Berkeley, Berkeley, CA), and R. Kitagawa (University of Texas Health Science Center at San Antonio, San Antonio, TX), respectively. The following secondary antibodies from Thermo Fisher Scientific were used at 1:500 dilutions: Alexa Fluor 555 goat anti–rabbit IgG, Alexa Fluor 488 goat anti–rabbit IgG, Alexa Fluor 546 goat anti–mouse IgG, Alexa Fluor 488 goat anti–mouse IgG, Alexa Fluor 555 goat anti–mouse IgM, and Alexa Fluor 488 goat anti–guinea pig IgG. 2 µg/ml DAPI (Sigma-Aldrich) was used to counterstain DNA.
LacI-His6-GFP, purified as described previously (Checchi et al., 2014), was added directly to dissected gonads after standard fixation at a concentration of 5–25 ng/ml and incubated overnight at 4°C (Severson and Meyer, 2014).
Germlines from COSA-1::GFP, FAN-1::GFP, and RPA-1::YFP worms were dissected and fixed for 1 min in cold 95% ethanol after PFA fixation, and endogenous GFP fluorescence was imaged. Quantification of COSA-1 was performed by counting COSA-1::GFP foci in late pachytene nuclei, excluding the first two rows of nuclei containing COSA-1::GFP foci and the last two rows of late pachytene. Quantification of RPA-1 was performed by counting RPA::YFP foci in all PZ nuclei. Quantification of FAN-1 was performed by acquiring the integrated density of FAN-1::GFP fluorescence in ImageJ (National Institutes of Health), which is the product of pixel intensity and area of the region of interest, in nuclei and cytoplasm within the PZ, and a nucleoplasmic to cytoplasmic signal ratio was calculated.
The fluorescence intensity of ZYG-12::GFP in nuclei was calculated as for FAN-1::GFP using a GFP antibody and was normalized by dividing by cytoplasmic signal. The intensity of normalized ZYG-12::GFP in nuclei containing RAD-51 foci was compared with nuclei lacking RAD-51 in the given genotypes.
Collection of images was performed using a deconvolution microscope (API DeltaVision; GE Healthcare). Images were deconvolved using Applied Precision SoftWoRx image analysis software (GE Healthcare) and were subsequently processed and analyzed using ImageJ Fiji (a gift from W. Rasband, National Institutes of Health, Bethesda, MD). All images are projections through approximately half of the germline unless otherwise stated.
SIM analysis was performed using an N-SIM superresolution microscope (Nikon) and NIS-Elements 2 image processing software (Nikon). Images were further processed using ImageJ.
RAD-51 measurements in SIM images
Distances between RAD-51 and the NPC were determined by obtaining fluorescence intensity plots with line scans in ImageJ. The number of pixels between the peaks of each signal was determined and converted to nanometers.
RNA-mediated interference (RNAi) analysis
RNAi experiments were performed using the feeding method (Kamath and Ahringer, 2003) at 20°C. Unless otherwise noted, gravid hermaphrodites were fed RNAi-inducing HT115(DE3) bacteria strains or the same bacteria transformed with the empty feeding vector, L4440. chk-1(RNAi) was performed on L1 larvae; kdp-1(RNAi) and zyg-12(RNAi) were performed on L4 larvae for 24 h. All feeding strains were obtained from a genomic RNAi feeding library (Kamath and Ahringer, 2003). Cultures were plated onto nematode growth medium (NGM) plates containing 25 µg/ml carbenicillin and 1 mM IPTG and were used within 2 wk.
IR experiments
Young adult worms were irradiated with 10 Gy (1,000 rad) from a Cs-137 source. Worms were dissected at 0.5, 2, 4, 6, and 16 h.
HU experiments
For high-dose experiments, L4s were placed on NGM plates containing 25 mM HU (Sigma-Aldrich) for 16 h before either dissection, transfer for recovery, or progeny viability assays. Cell cycle arrest was assayed by counting DAPI-stained nuclei with 100 µm of the distal tip of the gonad. For low-dose HU experiments, staged young adults were exposed to 5 mM HU for 2 h before being moved to a plate without HU (−HU) for dissection, recovery, or progeny viability assays. HU was allowed to dissipate into plates for at least 3 h before worms were introduced. For low-dose HU exposure, cell cycle kinetics were assayed by staining with anti–H3S10-P and counting positive nuclei. Progeny viability was monitored for 72 h after release from HU.
Cross-linking agents
For cisplatin sensitivity, young adults were placed on NGM plates containing 0, 100, 250, or 500 µM cisplatin (Sigma-Aldrich) for 16 h. Worms were moved to new NGM plates and allowed to lay progeny for 36 h, after which the parent was moved to a new plate for 36 h. Progeny were counted 24 h after removal of the parent; progeny inviability was recorded for the 36–72-h brood. Nitrogen mustard sensitivity was measured by placing young adults on NGM plates containing 0, 50, or 100 µM nitrogen mustard (Sigma-Aldrich) and assayed as with cisplatin. For RAD-51 assays, worms were placed on 100 µM cisplatin plates for 2 h and allowed to recover for the given times before dissection and staining with RAD-51 antibody. Cross-linking agents were allowed to dissipate into plates for at least 3 h before worms were introduced.
Microtubule inhibition
Young adult worms were incubated in a liquid M9 containing E. coli OP50 and the indicated concentration of colchicine for 16 h with shaking. Worms were recovered on NGM plates and germlines processed as desribed in the Cytological analyses section. Nocodazole treatment was performed on NGM medium containing the indicated concentrations. The extent of microtubule deploymerization was monitored by α-tubulin staining.
Meiotic RAD-51
Germlines were divided by PZ, transition zone, and pachytene. Pachytene was partitioned into early, mid, and late by dividing pachytene into three equal parts.
EdU time courses
EdU (Click-it kit; Thermo Fisher Scientific) experiments were performed as in Fox et al. (2011). 10-mm M9 plates were spotted with bacteria that had previously incorporated EdU. For the pulse-chase experiment, wild-type and unc-84(n369) worms were fed EdU bacteria for 30 min, moved to OP50 plates, and dissected at given time points. Germlines were stained for H3S10-P, and the Click-it reaction was performed, followed by DAPI staining. Time 0 is when we put worms onto the EdU plate. To analyze cell cycle kinetics, the percentage of H3S10-P–positive germlines that were also EdU labeled was determined for each time point. For the 2-h EdU exposure, worms were fed EdU bacteria for 2 h and were immediately dissected, and the Click-it reaction was performed.
Apoptosis
Apoptotic germline nuclei were scored in indicated worms ∼48 h after L4 larvae by acridine orange (AO; Invitrogen) with fluorescence microscopy. 0.5 ml of 50-µg/ml AO in M9 was added to a 60-mm plate containing adult worms and incubated at room temperature for 1 h. Worms were moved to new 60-mm plates, allowed to recover, and then mounted under coverslips in M9 on 3% agarose pads containing 0.2 mM tetramisole (Sigma-Aldrich; Jaramillo-Lambert and Engebrecht, 2010).
Mammalian experiments
HeLa Kyoto cells expressing a localization and affinity purification (LAP)–tagged mouse Sun-1 from a stable bacterial artificial chromosome pool (mSun-1–LAP; ENSMUSG00000036817, MCP_ky_2083) were a gift from A. Hyman (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany; Poser et al., 2008). HeLa Kyoto cells, also from A. Hyman, were used as controls (Poser et al., 2008). Cells were grown in high-glucose DMEM supplemented with 10% FBS and cultured at 37°C and 5% CO2. 20 µM siRNA constructs (DharmaFECT Smartpool siRNAs; GE Healthcare) were transfected with 8 µl/well DharmaFECT 1 transfection reagent (GE Healthcare), as suggested by the manufacturer. For survival assays, cells were transfected with siRNA for 6 h, split 24 h later into 6-well dishes, and allowed to recover for 24 h before being treated with drugs. Cells were treated with 0.5 µM DNA-PK inhibitor (NU7026; Tocris Bioscience) or vehicle control for 2 h before damage induction with 40 ng/ml MMC (Sigma-Aldrich; Adamo et al., 2010). Cells were then harvested 24 h later, and cell survival was assessed with trypan blue.
UNC-84–ZYG-12 protein complex purification
6×Histidine-tagged UNC-84912–1,111 and maltose-binding protein (MBP)–fused ZYG-12749–777 from C. elegans were cloned into a modified bicistronic expression plasmid (pETDuet-1) and were coexpressed in E. coli LOBSTR-RIL(DE3) (Kerafast; Esra Demircioglu et al., 2016). Transformed cells were grown at 37°C to an OD600 of 0.6 and then shifted to 18°C, and protein expression was induced by the addition of IPTG to a final concentration of 0.2 mM for 16 h. Cells were harvested by centrifugation at 6,000 g, resuspended in lysis buffer (50 mM potassium phosphate, pH 8.0, 400 mM NaCl, and 40 mM imidazole), and lysed using an LM20 Microfluidizer processor (Microfluidics). The lysate was cleared by centrifugation at 10,000 g for 25 min. The soluble fraction was incubated with Ni-Sepharose 6 Fast Flow beads (GE Healthcare) for 30 min at 4°C. The beads were washed with lysis buffer, and the protein was eluted (250 mM imidazole, 10 mM Tris/HCl, pH 8.0, 150 mM NaCl). Samples were first purified via size-exclusion chromatography on a Superdex S200 16/60 column (GE Healthcare) and equilibrated in running buffer (10 mM Tris/HCl, pH 8.0, and 150 mM NaCl, and 0.2 mM EDTA). Subsequently, the protein complex was further purified by ion exchange chromatography on a MonoQ 5/50 column (GE Healthcare). Finally, the purified UNC-84–ZYG-12 complex was analyzed on a Superose 6 10/300 column (GE Healthcare) equilibrated in running buffer. Elution fractions were analyzed by SDS-PAGE using Coomassie blue staining.
Statistical analyses
Statistics were determined with an unpaired Student’s t test, z test, two-way analysis of variance, or Mann-Whitney U test, as indicated; 95% CI are included.
Online supplemental material
Fig. S1 shows cell cycle effects. Fig. S2 shows that cross-linking agents reduce unc-84(n369) brood size and progeny viability and increase developmental abnormalities. Fig. S3 shows that unc-84 mutants do not display defects in chromosome synapsis but have elevated germline apoptosis. Fig. S4 shows UNC-84::GFP in male germlines and in response to different DNA-damaging agents. Fig. S5 shows the embryonic lethality of UNC-84 point mutants after cisplatin is also suppressed by loss of NHEJ. Fig. S6 shows that microtubule destabilizers affect the microtubule network and RAD-51 levels.
Supplementary Material
Acknowledgments
We thank Anne Villeneuve, Rita Kitagawa, Abby Dernburg, and Didier Hodzic for generously providing antibodies and the Caenorhabditis Genetic Center for providing strains. We also thank Courtney Bone, Natalie Cain, and Venecia Valdez for help with analysis of the unc-84 hyp-7 phenotype, Eugenel Espiritu for the germline diagram, Rachel Merenda and Bailey Firth for the generation of syp-1;unc-84 and syp-1;unc-84;cku-70 worms, respectively, and Adam Mitner for initial analysis of zyg-12(RNAi).
This work was supported by National Institutes of Health grant GM103860 and Agricultural Experimental Station California-Davis grant *MCB-7237-H to J. Engebrecht, National Institutes of Health grant T32GM0070377 to K.S. Lawrence, National Institutes of Health grant R01GM073874 to D.A. Starr, National Institutes of Health grant AR065484 to T.U. Schwartz, and National Institutes of Health grant T32GM007287 to V.E. Cruz. We thank Ken Burtis and the Fanconi Anemia Research Fund for additional support.
The authors declare no competing financial interests.
Author contributions: K.S. Lawrence: conceptualization, formal analysis, investigation, methodology, review, and editing; E.C. Tapley: conceptualization, formal analysis, investigation, review, and editing; V.E. Cruz: formal analysis, investigation, review, and editing; Q. Li: investigation; K. Aung: investigation; K.C. Hart: investigation; T.U. Schwartz: conceptualization, formal analysis, funding acquisition, supervision, review, and editing; D.A. Starr: conceptualization, formal analysis, funding acquisition, supervision, review, and editing; J. Engebrecht: conceptualization, formal analysis, funding acquisition, investigation, supervision, and original draft.
Footnotes
Abbreviations used:
- AO
- acridine orange
- CI
- confidence interval
- DDR
- DNA damage response
- DSB
- double-strand break
- FA
- Fanconi anemia
- HR
- homologous recombination
- HU
- hydroxyurea
- IR
- irradiation
- LINC
- linker of nucleoskeleton and cytoskeleton
- MBP
- maltose-binding protein
- MMC
- mitomycin C
- NGM
- nematode growth medium
- NHEJ
- nonhomologous end joining
- NPC
- nuclear pore complex
- PK
- protein kinase
- PZ
- proliferative zone
- SAC
- spindle assembly checkpoint
- SIM
- structured illumination microscopy
References
- Adamo A., Montemauri P., Silva N., Ward J.D., Boulton S.J., and La Volpe A.. 2008. BRC-1 acts in the inter-sister pathway of meiotic double-strand break repair. EMBO Rep. 9:287–292. 10.1038/sj.embor.7401167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamo A., Collis S.J., Adelman C.A., Silva N., Horejsi Z., Ward J.D., Martinez-Perez E., Boulton S.J., and La Volpe A.. 2010. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell. 39:25–35. 10.1016/j.molcel.2010.06.026 [DOI] [PubMed] [Google Scholar]
- Bailly A., and Gartner A.. 2013. Germ cell apoptosis and DNA damage responses. Adv. Exp. Med. Biol. 757:249–276. 10.1007/978-1-4614-4015-4_9 [DOI] [PubMed] [Google Scholar]
- Bone C.R., Tapley E.C., Gorjánácz M., and Starr D.A.. 2014. The Caenorhabditis elegans SUN protein UNC-84 interacts with lamin to transfer forces from the cytoplasm to the nucleoskeleton during nuclear migration. Mol. Biol. Cell. 25:2853–2865. 10.1091/mbc.E14-05-0971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting S.F., Callén E., Kozak M.L., Kim J.M., Wong N., López-Contreras A.J., Ludwig T., Baer R., Faryabi R.B., Malhowski A., et al. 2012. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell. 46:125–135. 10.1016/j.molcel.2012.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain N.E., Tapley E.C., McDonald K.L., Cain B.M., and Starr D.A.. 2014. The SUN protein UNC-84 is required only in force-bearing cells to maintain nuclear envelope architecture. J. Cell Biol. 206:163–172. 10.1083/jcb.201405081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R., Rondinelli B., and D’Andrea A.D.. 2016. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26:52–64. 10.1016/j.tcb.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss A.B., Khatau S.B., Erdenberger N., Robinson D.K., Hodzic D., Longmore G.D., and Wirtz D.. 2013. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci. Rep. 3 10.1038/srep01087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checchi P.M., Lawrence K.S., Van M.V., Larson B.J., and Engebrecht J.. 2014. Pseudosynapsis and decreased stringency of meiotic repair pathway choice on the hemizygous sex chromosome of Caenorhabditis elegans males. Genetics. 197:543–560. 10.1534/genetics.114.164152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikashige Y., Tsutsumi C., Yamane M., Okamasa K., Haraguchi T., and Hiraoka Y.. 2006. Meiotic proteins bqt1 and bqt2 tether telomeres to form the bouquet arrangement of chromosomes. Cell. 125:59–69. 10.1016/j.cell.2006.01.048 [DOI] [PubMed] [Google Scholar]
- Christophorou N., Rubin T., Bonnet I., Piolot T., Arnaud M., and Huynh J.R.. 2015. Microtubule-driven nuclear rotations promote meiotic chromosome dynamics. Nat. Cell Biol. 17:1388–1400. 10.1038/ncb3249 [DOI] [PubMed] [Google Scholar]
- Ciccia A., and Elledge S.J.. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell. 40:179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clejan I., Boerckel J., and Ahmed S.. 2006. Developmental modulation of nonhomologous end joining in Caenorhabditis elegans. Genetics. 173:1301–1317. 10.1534/genetics.106.058628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaiácovo M.P., MacQueen A.J., Martinez-Perez E., McDonald K., Adamo A., La Volpe A., and Villeneuve A.M.. 2003. Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Dev. Cell. 5:463–474. 10.1016/S1534-5807(03)00232-6 [DOI] [PubMed] [Google Scholar]
- Collis S.J., Barber L.J., Ward J.D., Martin J.S., and Boulton S.J.. 2006. C. elegans FANCD2 responds to replication stress and functions in interstrand cross-link repair. DNA Repair (Amst.). 5:1398–1406. 10.1016/j.dnarep.2006.06.010 [DOI] [PubMed] [Google Scholar]
- Crisp M., Liu Q., Roux K., Rattner J.B., Shanahan C., Burke B., Stahl P.D., and Hodzic D.. 2006. Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 172:41–53. 10.1083/jcb.200509124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley J.M., and Sung P.. 2014. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 34:1380–1388. 10.1128/MCB.01639-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans A.J., and West S.C.. 2011. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 11:467–480. 10.1038/nrc3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson D.J., Ward J.D., Reiner D.J., and Goldstein B.. 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods. 10:1028–1034. 10.1038/nmeth.2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X., Xu R., Yu J., Xu T., Zhuang Y., and Han M.. 2007. SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev. Cell. 12:863–872. 10.1016/j.devcel.2007.03.018 [DOI] [PubMed] [Google Scholar]
- Duxin J.P., and Walter J.C.. 2015. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 37:49–60. 10.1016/j.ceb.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis H.M., and Horvitz H.R.. 1986. Genetic control of programmed cell death in the nematode C. elegans. Cell. 44:817–829. 10.1016/0092-8674(86)90004-8 [DOI] [PubMed] [Google Scholar]
- Esra Demircioglu F., Cruz V.E., and Schwartz T.U.. 2016. Purification and structural analysis of SUN and KASH domain proteins. Methods Enzymol. 569:63–78. 10.1016/bs.mie.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., and Beck K.A.. 2004. A role for the spectrin superfamily member Syne-1 and kinesin II in cytokinesis. J. Cell Sci. 117:619–629. 10.1242/jcs.00892 [DOI] [PubMed] [Google Scholar]
- Fox P.M., Vought V.E., Hanazawa M., Lee M.H., Maine E.M., and Schedl T.. 2011. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development. 138:2223–2234. 10.1242/dev.059535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Muse T., and Boulton S.J.. 2005. Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J. 24:4345–4355. 10.1038/sj.emboj.7600896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner J.M., Smoyer C.J., Stensrud E.S., Alexander R., Gogol M., Wiegraebe W., and Jaspersen S.L.. 2011. Targeting of the SUN protein Mps3 to the inner nuclear membrane by the histone variant H2A.Z. J. Cell Biol. 193:489–507. 10.1083/jcb.201011017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwon G.H., Kim Y., Liu Y., Watson A.T., Jo A., Etheridge T.J., Yuan F., Zhang Y., Kim Y., Carr A.M., and Cho Y.. 2014. Crystal structure of a Fanconi anemia-associated nuclease homolog bound to 5′ flap DNA: basis of interstrand cross-link repair by FAN1. Genes Dev. 28:2276–2290. 10.1101/gad.248492.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque F., Lloyd D.J., Smallwood D.T., Dent C.L., Shanahan C.M., Fry A.M., Trembath R.C., and Shackleton S.. 2006. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 26:3738–3751. 10.1128/MCB.26.10.3738-3751.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Lambert A., and Engebrecht J.. 2010. A single unpaired and transcriptionally silenced X chromosome locally precludes checkpoint signaling in the Caenorhabditis elegans germ line. Genetics. 184:613–628. 10.1534/genetics.109.110338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo-Lambert A., Ellefson M., Villeneuve A.M., and Engebrecht J.. 2007. Differential timing of S phases, X chromosome replication, and meiotic prophase in the C. elegans germ line. Dev. Biol. 308:206–221. 10.1016/j.ydbio.2007.05.019 [DOI] [PubMed] [Google Scholar]
- Jaspersen S.L., Martin A.E., Glazko G., Giddings T.H. Jr., Morgan G., Mushegian A., and Winey M.. 2006. The Sad1-UNC-84 homology domain in Mps3 interacts with Mps2 to connect the spindle pole body with the nuclear envelope. J. Cell Biol. 174:665–675. 10.1083/jcb.200601062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalocsay M., Hiller N.J., and Jentsch S.. 2009. Chromosome-wide RAD51 spreading and SUMO-H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol. Cell. 33:335–343. 10.1016/j.molcel.2009.01.016 [DOI] [PubMed] [Google Scholar]
- Kamath R.S., and Ahringer J.. 2003. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 30:313–321. 10.1016/S1046-2023(03)00050-1 [DOI] [PubMed] [Google Scholar]
- Kennedy R.D., and D’Andrea A.D.. 2005. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 19:2925–2940. 10.1101/gad.1370505 [DOI] [PubMed] [Google Scholar]
- Kimble J., and Hirsh D.. 1979. The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev. Biol. 70:396–417. 10.1016/0012-1606(79)90035-6 [DOI] [PubMed] [Google Scholar]
- Koszul R., Kim K.P., Prentiss M., Kleckner N., and Kameoka S.. 2008. Meiotic chromosomes move by linkage to dynamic actin cables with transduction of force through the nuclear envelope. Cell. 133:1188–1201. 10.1016/j.cell.2008.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann M.C., and Smogorzewska A.. 2013. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 493:356–363. 10.1038/nature11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz K., Schöpf B., Kaden S., Sendoel A., Eberhard R., Lademann C., Cannavó E., Sartori A.A., Hengartner M.O., and Jiricny J.. 2010. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell. 142:77–88. 10.1016/j.cell.2010.06.022 [DOI] [PubMed] [Google Scholar]
- Kumar A., Mazzanti M., Mistrik M., Kosar M., Beznoussenko G.V., Mironov A.A., Garrè M., Parazzoli D., Shivashankar G.V., Scita G., et al. 2014. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell. 158:633–646. 10.1016/j.cell.2014.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaud C., Moreno A., Marchesi F., Toth R., Blow J.J., and Rouse J.. 2016. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science. 351:846–849. 10.1126/science.aad5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence K.S., Chau T., and Engebrecht J.. 2015. DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity. PLoS Genet. 11:e1005150 10.1371/journal.pgen.1005150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.Y., Conrad M.N., and Dresser M.E.. 2012. Meiotic chromosome pairing is promoted by telomere-led chromosome movements independent of bouquet formation. PLoS Genet. 8:e1002730 10.1371/journal.pgen.1002730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.K., Starr D., Cohen M., Liu J., Han M., Wilson K.L., and Gruenbaum Y.. 2002. Lamin-dependent localization of UNC-84, a protein required for nuclear migration in Caenorhabditis elegans. Mol. Biol. Cell. 13:892–901. 10.1091/mbc.01-06-0294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K., Zhu X., Xu R., Shao C., Xu T., Zhuang Y., and Han M.. 2012. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr. Biol. 22:1609–1615. 10.1016/j.cub.2012.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaître C., Grabarz A., Tsouroula K., Andronov L., Furst A., Pankotai T., Heyer V., Rogier M., Attwood K.M., Kessler P., et al. 2014. Nuclear position dictates DNA repair pathway choice. Genes Dev. 28:2450–2463. 10.1101/gad.248369.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Ghosal G., Yuan J., Chen J., and Huang J.. 2010. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science. 329:693–696. 10.1126/science.1192656 [DOI] [PubMed] [Google Scholar]
- Lottersberger F., Karssemeijer R.A., Dimitrova N., and de Lange T.. 2015. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell. 163:880–893. 10.1016/j.cell.2015.09.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton G.W., and Starr D.A.. 2014. KASHing up with the nucleus: novel functional roles of KASH proteins at the cytoplasmic surface of the nucleus. Curr. Opin. Cell Biol. 28:69–75. 10.1016/j.ceb.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X.B., Liu L., Cheng C., Yu B., Xiong L., Hu K., Tang J., Zeng L., and Sang Y.. 2015. SUN2 exerts tumor suppressor functions by suppressing the Warburg effect in lung cancer. Sci. Rep. 5 10.1038/srep17940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay C., Déclais A.C., Lundin C., Agostinho A., Deans A.J., MacArtney T.J., Hofmann K., Gartner A., West S.C., Helleday T., et al. 2010. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. 142:65–76. 10.1016/j.cell.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen A.J., and Hochwagen A.. 2011. Checkpoint mechanisms: the puppet masters of meiotic prophase. Trends Cell Biol. 21:393–400. 10.1016/j.tcb.2011.03.004 [DOI] [PubMed] [Google Scholar]
- MacQueen A.J., and Villeneuve A.M.. 2001. Nuclear reorganization and homologous chromosome pairing during meiotic prophase require C. elegans chk-2. Genes Dev. 15:1674–1687. 10.1101/gad.902601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen A.J., Colaiácovo M.P., McDonald K., and Villeneuve A.M.. 2002. Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev. 16:2428–2442. 10.1101/gad.1011602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone C.J., Fixsen W.D., Horvitz H.R., and Han M.. 1999. UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development. 126:3171–3181. [DOI] [PubMed] [Google Scholar]
- Malone C.J., Misner L., Le Bot N., Tsai M.C., Campbell J.M., Ahringer J., and White J.G.. 2003. The C. elegans hook protein, ZYG-12, mediates the essential attachment between the centrosome and nucleus. Cell. 115:825–836. 10.1016/S0092-8674(03)00985-1 [DOI] [PubMed] [Google Scholar]
- McGee M.D., Rillo R., Anderson A.S., and Starr D.A.. 2006. UNC-83 IS a KASH protein required for nuclear migration and is recruited to the outer nuclear membrane by a physical interaction with the SUN protein UNC-84. Mol. Biol. Cell. 17:1790–1801. 10.1091/mbc.E05-09-0894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee M.D., Stagljar I., and Starr D.A.. 2009. KDP-1 is a nuclear envelope KASH protein required for cell-cycle progression. J. Cell Sci. 122:2895–2905. 10.1242/jcs.051607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mets D.G., and Meyer B.J.. 2009. Condensins regulate meiotic DNA break distribution, thus crossover frequency, by controlling chromosome structure. Cell. 139:73–86. 10.1016/j.cell.2009.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerzon M., Fridolfsson H.N., Ly N., McNally F.J., and Starr D.A.. 2009. UNC-83 is a nuclear-specific cargo adaptor for kinesin-1-mediated nuclear migration. Development. 136:2725–2733. 10.1242/dev.038596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn I.L., Rolls M.M., Hanna-Rose W., and Malone C.J.. 2009. SUN-1 and ZYG-12, mediators of centrosome-nucleus attachment, are a functional SUN/KASH pair in Caenorhabditis elegans. Mol. Biol. Cell. 20:4586–4595. 10.1091/mbc.E08-10-1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser S.C., von Elsner S., Büssing I., Alpi A., Schnabel R., and Gartner A.. 2009. Functional dissection of Caenorhabditis elegans CLK-2/TEL2 cell cycle defects during embryogenesis and germline development. PLoS Genet. 5 (published erratum appears in PLoS Genet. 2011. 5) 10.1371/journal.pgen.1000451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P., Jaspersen S.L., Miele A., Dekker J., and Peterson C.L.. 2009. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 23:912–927. 10.1101/gad.1782209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace P., Mosedale G., Hodskinson M.R., Rosado I.V., Sivasubramaniam M., and Patel K.J.. 2010. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 329:219–223. 10.1126/science.1192277 [DOI] [PubMed] [Google Scholar]
- Padmakumar V.C., Libotte T., Lu W., Zaim H., Abraham S., Noegel A.A., Gotzmann J., Foisner R., and Karakesisoglou I.. 2005. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J. Cell Sci. 118:3419–3430. 10.1242/jcs.02471 [DOI] [PubMed] [Google Scholar]
- Penkner A., Tang L., Novatchkova M., Ladurner M., Fridkin A., Gruenbaum Y., Schweizer D., Loidl J., and Jantsch V.. 2007. The nuclear envelope protein Matefin/SUN-1 is required for homologous pairing in C. elegans meiosis. Dev. Cell. 12:873–885. 10.1016/j.devcel.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Penkner A.M., Fridkin A., Gloggnitzer J., Baudrimont A., Machacek T., Woglar A., Csaszar E., Pasierbek P., Ammerer G., Gruenbaum Y., and Jantsch V.. 2009. Meiotic chromosome homology search involves modifications of the nuclear envelope protein Matefin/SUN-1. Cell. 139:920–933. 10.1016/j.cell.2009.10.045 [DOI] [PubMed] [Google Scholar]
- Pennell S., Déclais A.C., Li J., Haire L.F., Berg W., Saldanha J.W., Taylor I.A., Rouse J., Lilley D.M., and Smerdon S.J.. 2014. FAN1 activity on asymmetric repair intermediates is mediated by an atypical monomeric virus-type replication-repair nuclease domain. Cell Reports. 8:84–93. 10.1016/j.celrep.2014.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., and Helleday T.. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 37:492–502. 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser I., Sarov M., Hutchins J.R., Hériché J.K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A.W., et al. 2008. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods. 5:409–415. 10.1038/nmeth.1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasada Rao H.B., Shinohara M., and Shinohara A.. 2011. Mps3 SUN domain is important for chromosome motion and juxtaposition of homologous chromosomes during meiosis. Genes Cells. 16:1081–1096. 10.1111/j.1365-2443.2011.01554.x [DOI] [PubMed] [Google Scholar]
- Rog O., and Dernburg A.F.. 2015. Direct visualization reveals kinetics of meiotic chromosome synapsis. Cell Reports. 10:1639–1645. 10.1016/j.celrep.2015.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosu S., Zawadzki K.A., Stamper E.L., Libuda D.E., Reese A.L., Dernburg A.F., and Villeneuve A.M.. 2013. The C. elegans DSB-2 protein reveals a regulatory network that controls competence for meiotic DSB formation and promotes crossover assurance. PLoS Genet. 9:e1003674 10.1371/journal.pgen.1003674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux K.J., Crisp M.L., Liu Q., Kim D., Kozlov S., Stewart C.L., and Burke B.. 2009. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc. Natl. Acad. Sci. USA. 106:2194–2199. 10.1073/pnas.0808602106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu T., Spatola B., Delabaere L., Bowlin K., Hopp H., Kunitake R., Karpen G.H., and Chiolo I.. 2015. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 17:1401–1411. 10.1038/ncb3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A., Isaac B., Phillips C.M., Rillo R., Carlton P.M., Wynne D.J., Kasad R.A., and Dernburg A.F.. 2009. Cytoskeletal forces span the nuclear envelope to coordinate meiotic chromosome pairing and synapsis. Cell. 139:907–919. 10.1016/j.cell.2009.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguí N., Mina L.B., Lázaro C., Sanz-Pamplona R., Pons T., Navarro M., Bellido F., López-Doriga A., Valdés-Mas R., Pineda M., et al. 2015. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology. 149:563–566. 10.1053/j.gastro.2015.05.056 [DOI] [PubMed] [Google Scholar]
- Severson A.F., and Meyer B.J.. 2014. Divergent kleisin subunits of cohesin specify mechanisms to tether and release meiotic chromosomes. eLife. 3:e03467 10.7554/eLife.03467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeth K.M., Sørensen C.S., Issaeva N., Dziegielewski J., Bartek J., and Helleday T.. 2007. RPA mediates recombination repair during replication stress and is displaced from DNA by checkpoint signalling in human cells. J. Mol. Biol. 373:38–47. 10.1016/j.jmb.2007.07.068 [DOI] [PubMed] [Google Scholar]
- Smith A.L., Alirezaie N., Connor A., Chan-Seng-Yue M., Grant R., Selander I., Bascuñana C., Borgida A., Hall A., Whelan T., et al. 2016. Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett. 370:302–312. 10.1016/j.canlet.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A., Desetty R., Saito T.T., Schlabach M., Lach F.P., Sowa M.E., Clark A.B., Kunkel T.A., Harper J.W., Colaiácovo M.P., and Elledge S.J.. 2010. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell. 39:36–47. 10.1016/j.molcel.2010.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolikov S., Eizinger A., Hurlburt A., Rogers E., Villeneuve A.M., and Colaiácovo M.P.. 2007. Synapsis-defective mutants reveal a correlation between chromosome conformation and the mode of double-strand break repair during Caenorhabditis elegans meiosis. Genetics. 176:2027–2033. 10.1534/genetics.107.076968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinger J.A., Kiianitsa K., and Heyer W.D.. 2002. Rad54, a Swi2/Snf2-like recombinational repair protein, disassembles RAD51:dsDNA filaments. Mol. Cell. 10:1175–1188. 10.1016/S1097-2765(02)00743-8 [DOI] [PubMed] [Google Scholar]
- Sosa B.A., Rothballer A., Kutay U., and Schwartz T.U.. 2012. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell. 149:1035–1047. 10.1016/j.cell.2012.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E., Dorn J.F., Sengupta K., Jasin M., Nussenzweig A., Ried T., Danuser G., and Misteli T.. 2007. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 9:675–682. 10.1038/ncb1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamper E.L., Rodenbusch S.E., Rosu S., Ahringer J., Villeneuve A.M., and Dernburg A.F.. 2013. Identification of DSB-1, a protein required for initiation of meiotic recombination in Caenorhabditis elegans, illuminates a crossover assurance checkpoint. PLoS Genet. 9 (published erratum appears in PLoS Genet. 2013. 9) 10.1371/journal.pgen.1003679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr D.A., and Han M.. 2002. Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science. 298:406–409. 10.1126/science.1075119 [DOI] [PubMed] [Google Scholar]
- Stergiou L., Eberhard R., Doukoumetzidis K., and Hengartner M.O.. 2011. NER and HR pathways act sequentially to promote UV-C-induced germ cell apoptosis in Caenorhabditis elegans. Cell Death Differ. 18:897–906. 10.1038/cdd.2010.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E., Djukic B., Evans M., Kim D.H., Taneja P., Wang X., Finucane M., Knox J., Ho K., Devidze N., et al. 2015. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 6 10.1038/ncomms9897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz R.K., Rodriguez E.C., and King M.C.. 2014. A role for nuclear envelope-bridging complexes in homology-directed repair. Mol. Biol. Cell. 25:2461–2471. 10.1091/mbc.E13-10-0569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi D., Sato K., Hirayama E., Takata M., and Kurumizaka H.. 2015. Human FAN1 promotes strand incision in 5′-flapped DNA complexed with RPA. J. Biochem. 158:263–270. 10.1093/jb/mvv043 [DOI] [PubMed] [Google Scholar]
- Tapley E.C., and Starr D.A.. 2013. Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Curr. Opin. Cell Biol. 25:57–62. 10.1016/j.ceb.2012.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapley E.C., Ly N., and Starr D.A.. 2011. Multiple mechanisms actively target the SUN protein UNC-84 to the inner nuclear membrane. Mol. Biol. Cell. 22:1739–1752. 10.1091/mbc.E10-08-0733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay Y., Ungricht R., Rothballer A., Kiss A., Csucs G., Horvath P., and Kutay U.. 2010. A classical NLS and the SUN domain contribute to the targeting of SUN2 to the inner nuclear membrane. EMBO J. 29:2262–2275. 10.1038/emboj.2010.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungricht R., Klann M., Horvath P., and Kutay U.. 2015. Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J. Cell Biol. 209:687–703. 10.1083/jcb.201409127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varas J., Graumann K., Osman K., Pradillo M., Evans D.E., Santos J.L., and Armstrong S.J.. 2015. Absence of SUN1 and SUN2 proteins in Arabidopsis thaliana leads to a delay in meiotic progression and defects in synapsis and recombination. Plant J. 81:329–346. 10.1111/tpj.12730 [DOI] [PubMed] [Google Scholar]
- Veuger S.J., Curtin N.J., Richardson C.J., Smith G.C., and Durkacz B.W.. 2003. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 63:6008–6015. [PubMed] [Google Scholar]
- Ward J.D., Barber L.J., Petalcorin M.I., Yanowitz J., and Boulton S.J.. 2007. Replication blocking lesions present a unique substrate for homologous recombination. EMBO J. 26:3384–3396. 10.1038/sj.emboj.7601766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H., Mohler W.A., and Soto M.C.. 2011. The branched actin nucleator Arp2/3 promotes nuclear migrations and cell polarity in the C. elegans zygote. Dev. Biol. 357:356–369. 10.1016/j.ydbio.2011.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., and Smolikove S.. 2013. Impaired resection of meiotic double-strand breaks channels repair to nonhomologous end joining in Caenorhabditis elegans. Mol. Cell. Biol. 33:2732–2747. 10.1128/MCB.00055-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoo R., Zawadzki K.A., Nabeshima K., Drake M., Arur S., and Villeneuve A.M.. 2012. COSA-1 reveals robust homeostasis and separable licensing and reinforcement steps governing meiotic crossovers. Cell. 149:75–87. 10.1016/j.cell.2012.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lei K., Yuan X., Wu X., Zhuang Y., Xu T., Xu R., and Han M.. 2009. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron. 64:173–187. 10.1016/j.neuron.2009.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K., Rolls M.M., Hall D.H., Malone C.J., and Hanna-Rose W.. 2009. A ZYG-12-dynein interaction at the nuclear envelope defines cytoskeletal architecture in the C. elegans gonad. J. Cell Biol. 186:229–241. 10.1083/jcb.200902101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.