

Abstract
Sepsis is a poorly understood syndrome of systemic inflammation responsible for hundreds of thousands of deaths every year. The integrity of the gut epithelium and competence of adaptive immune responses are notoriously compromised during sepsis, and the prevalent assumption in the scientific and medical community is that intestinal commensals have a detrimental role in the systemic inflammation and susceptibility to nosocomial infections seen in critically ill, septic patients. However, breakthroughs in the last decade provide strong credence to the idea that our mucosal microbiome plays an essential role in adaptive immunity, where a human host and its prokaryotic colonists seem to exist in a carefully negotiated armistice with compromises and benefits that go both ways. In this review, we re-examine the notion that intestinal contents are the driving force of critical illness. An overview of the interaction between the microbiome and the immune system is provided, with a special focus on the impact of commensals in priming and the careful balance between normal intestinal flora and pathogenic organisms residing in the gut microbiome. Based on the data in hand, we hypothesize that sepsis induces imbalances in microbial populations residing in the gut, along with compromises in epithelial integrity. As a result, normal antigen sampling becomes impaired, and proliferative cues are intermixed with inhibitory signals. This situates the microbiome, the gut, and its complex immune network of cells and bacteria, at the center of aberrant immune responses during and after sepsis.
Keywords: Sepsis, T cell, immune suppression, gut microbiome, infection, apoptosis
Introduction
Human beings—among other mammalian life forms on this planet—are colonized by microbes at all epithelial barriers as soon as they are born.1 As the largest body surface directly exposed to the environment, the human intestine is populated with an enormous number of microorganisms from predominantly bacterial origin.2 A mind-boggling 400 m2 of the intestinal epithelial surface is colonized by ∼100 trillion organisms, which outnumber the cells in the human body by 10–100 fold.3,4 While recent data have proposed a revision to the ratio of commensal bacteria to human cells to be closer to 1:1,5 the vast array of bacteria that colonize the typical human still translates to each person carrying about 100 times more bacterial genes than human genes.6 The diversity and density of this microbial colony within the gut can be altered by antibiotics, disease co-morbidity, diet, and other environmental factors.7,8 Through advances in genomic sequencing technology during the last decade, we now know alterations in the membership of the gut microbiome are associated with immunopathology in several autoimmune, inflammatory, and allergic diseases, as well as altered susceptibility to infectious diseases.9–13 The symbiotic relationship between host and microbiome can be best appreciated within our own bodies in the context of health and disease.
Sepsis is a poorly understood syndrome of systemic inflammation responsible for thousands of deaths every year, with no specific therapies on the current market. Since the 1990s, sepsis researchers have reported many patients surviving the acute stages of sepsis exhibit clinically relevant immunosuppression.14 As part of this immunosuppression, acutely septic patients show reduced lymphocyte function and a temporal decrease in circulating lymphocyte numbers.15,16 This is particularly evident for CD4 T cells, which orchestrate cell-mediated immunity in the body.17 Traditionally, researchers and physicians have assumed the intestinal commensals have a detrimental role in promoting systemic inflammation and infection in the critically ill septic patient, an idea termed the “toxic gut lymph.” Advances in the understanding of microbial commensalism, however, compel us to re-examine the notion that the intestinal contents are the driving force of critical illness.18 Paradoxically, extensive evidence from the last decade supports the theory that our mucosal microbiome—the collective symbiotic and pathobiotic commensals in the gut—plays an essential role in the establishment of tolerogenic as well as immunocompetent CD4 T-cell responses, homeostatic proliferation, and repertoire determination. Although these bacteria are oftentimes beneficial, imbalances between pathogenic and symbiotic commensal species (dysbiosis) can be seen in a wide variety of diseases.19
In this review, we provide an overview of the interaction between the microbiome and the immune system, with a special focus on the impact of commensals in priming, as well as the development of competent CD4 T-cell responses. Within the immune system, we will be focusing our discussion herein primarily on T cells, but we acknowledge that innate immunity is also strongly affected during a sepsis with the quantitative and qualitative alterations observed in NK cells and neutrophils. We direct the reader to other commentary centered on changes in these other immune system components during sepsis elsewhere.20,21 The polarization and diversity of Ag-specific CD4 T cell responses are linked to the integrity of epithelial barrier function and the careful balance between normal intestinal flora and pathogenic organisms residing in the gut microbiome. Both of these elements—the integrity of the epithelium and balance between helpful and harmful commensals—are notoriously compromised in sepsis. Patients who survive sepsis often suffer severely compromised immune function, and studies in animals and humans suggest the immune injuries that occur during sepsis may be critical to the pathogenesis and subsequent mortality.22 We believe sepsis induces dysbiosis and compromises epithelial integrity. As a result, normal Ag sampling is impaired, and proliferative cues are intermixed with inhibitory signals, suggesting the microbiome, the gut, and its complex immune network of cells and bacteria is situated at the center of aberrant immunity during and after sepsis.18
Rethinking the germ theory of disease
Bacteria populated the earth 2 billion years before the first signs of eukaryotic life, and they inhabit virtually every terrestrial and aquatic niche on the planet, even some of the most extreme. It should be of no surprise that humans (as an ecological niche) have also been colonized, and bacteria have even integrated themselves into our evolution as mitochondria, the descendants of ancient bacterial organisms.23 In spite of their long-standing claim to this world, we were unaware of their existence until very recently (∼150 years), when bacteriology was placed at the center of our understanding of infection and illness. Pioneers like Lister, Koch, Pasteur, and Metchnikoff valiantly propelled the germ theory of disease forward at the turn of the 20th century.24 Before this paradigm shift in medicine, patients would develop overwhelming sepsis as a direct result of gut leakage and complete ignorance regarding the transfer of pathogens by healthcare workers.25 Aseptic technique was unheard of: surgeons seldom washed their hands and students commonly put their hands in the operative field for didactic purposes.26 The discovery that enemies invisible to the naked eye were the cause of many ailments radically changed the course of medicine,27,28 and microbial pathogenesis became the recurrent theme of 20th-century medicine. Yet, despite the tremendous advances this theory has created, it is interesting that voices within the research community warned for many years that the focus on pathogenic microbes would distract from important research into the role of commensals in health and disease.29 In fact, Metchnikoff was among the first to also propose that modification of the gut flora might improve human health.30,31 A century later, we have come to understand that rather than waging an endless war, the host and its prokaryotic colonists exist in a carefully negotiated armistice, with compromises and benefits that go both ways.
Despite the evolutionary co-existence between the microbiome and host, the high load of bacterially derived antigen (Ag) found in the gut lumen is a challenge to mucosal lymphocytes, and one that is still very much actively investigated in settings of health and disease. Work with animal models indicates the gut is innately predisposed to inflammation, and the influence of balanced symbiotic and pathobiotic components of the microbiome in the immune system is immunosuppressive or tolerogenic.32 These findings are supportive of the central paradox in the “Hygiene Hypothesis”: human societies that adopt a certain level of hygiene to decrease the prevalence of many infections observe a concurrent increase in non-infectious immune disorders. The rise in allergy and autoimmunity in hygienic environments is as dramatic as the drop in infection rates. The “non-self” nature of bacteria (regardless of symbiont or pathobiont status) makes Ag presented and processed by gut antigen-presenting cells (APCs) likely to be recognized by some or many gut-tropic T cells. Therefore, in situations where genetic predisposition is a factor (e.g. patients with autoimmune diseases), microbial modulation would be key to dampening potentially self-reactive or cross-reactive T cells. Although genetic differences clearly affect the development of autoimmunity, hyper-hygienic living environments may cull or limit commensal diversity, as well as affect the microbiome's ability to steer the maturation of the immune system toward a state of homeostatic tolerance.
Composition of the enteric microbiome
Four phyla dominate the adult human microbiome. Of those, 80% of all intestinal bacterial species fall under the most heavily studied: Firmicutes and Bacteroidetes.33 Firmicutes are Gram-positive bacterial taxa composed of many familiar-sounding genera (such as Clostridia, Streptococcaceae, Staphylococcaceae, Enterococcaceae, and Lactobacillae)34; many of these occupants are facultative anaerobes. Bacteroidetes, on the other hand, are Gram-negative bacteria composed largely of Bacteroides species, which are obligate anaerobes. Membership in the human microbiome is evolutionarily determined and is similar across mammals. Comparison studies have shown the human intestinal microbiota is surprisingly similar to that of mice, though similarities stop at the genus (for Firmicutes)35 and species level (for Bacteroidetes)36. However, this membership is not completely static, as exemplified by the changes that occur immediately after humans are born.37 The first inhabitants of the intestine are the facultative anaerobic bacteria, including Escherichia coli and the Firmicutes sp. These bacteria can take advantage of the abundant oxygen in the newborn, but this advantage is gone by the second week of age when the oxidation-reduction potential decreases.38 At this point, obligate anaerobes from both phyla take over (e.g. Clostridia, Bacteroides, and Bifidobacteria). Weaning further favors obligate anaerobes leading to the stabilization of microflora composition.39
In addition to early life events, environmental factors can also influence membership in the individual microbiome.40 An example of this environmental modulation is diet composition. In hosts with a high-fat, high-carbohydrate Western diet, the microbiota is enriched with bacteria from the phylum Firmicutes and underrepresented with Bacteroidetes.41 This is presumably because Firmicutes are more efficient at extracting energy from the given diet.42,43 In a completely different diet, such as one rich in plant polysaccharides, the opposite is true. Actinobacteria and Bacteroidetes are enriched in the gut microbiome of children from Burkina Faso who follow a low-fat, high-fiber diet, whereas the gut bacterial composition is skewed toward more Firmicutes and Proteobacteria in Italian children who have a high-fat, low-fiber diet.8 Though correlative at best, it is important to note the rate of allergies and asthma is almost completely non-existent in the first cohort of children, whereas they continue to rise in the second.44
The mucosal immune environment
The front line of mucosal immunity within the gut is made up of a single layer of columnar epithelial cells continuously renewed from multipotent stem cells located in the crypts of Lieberkühn. These give rise to all lineages of intestinal epithelia, including enterocytes, neuroendocrine cells, M cells, Paneth cells, and Goblet cells. The sum total of these cells is referred to as the gut epithelium. This epithelium is an active component of immunity, with physiologic and anatomical changes when lymphoid areas are present underneath. Some of the key differences between the absorptive and lymphoid-associated epithelium include lower levels of digestive enzymes, a less pronounced brush border, and a large number of M cells. In addition, lymphoid-associated epithelium is distinguishable given that T and B cells, as well as dendritic cells (DCs), infiltrate the diffuse area immediately below the lymphoid-associated epithelium known as the subepithelial dome.45 An illustrative schematic of some of the T-cell-associated processes occurring under the subepithelial dome can be seen in Figure 1. We have chosen to focus on the T-helper 17 (Th17) subset of differentiated CD4 T cells in this illustration, as Th17 cells play crucial roles in mucosal defense and abundantly localize in the lamina propria of the intestine in a commensal microbiota-dependent fashion.46–48
Figure 1.

The interaction between the microbiome and the immune system. Illustration of jejunal enterocyte histology with particular attention to the lamina propria and its resident immune cells (dendritic cells, macrophages, innate lymphoid cells (ILC) and T cells. The enteric immune system provides CD4 T cells with both potential for survival and polarization into different effector subtypes. In terms of survival, naïve CD4 T cells have the capacity to home to the gut and interact with cross-reactive Ag derived from the biomass in the lumen via Ag presentation from a MHC II expressing cell (either traditional APC, specialized intestinal epithelia or due to induced expression of MHC II on enterocytes). In addition, survival factors such as TSLP may be expressed by the enterocytes. Memory CD4 T cells, which can be directly reactive or cross-reactive to the Ag in question, can receive survival cues from similar immune components in the gut environment. Both cell subsets can receive polarizing cues (e.g. IL-6 and IL-21) predisposing the CD4 T cells in question to adopt a Th17 phenotype, which is central in both gut immunity and inflammatory bowel diseases.
The gut epithelial layer is short-lived and regenerates every three to five days. This is perhaps due (in part) to a large amount of functional stress—namely, the constant surveillance of commensal flora and simultaneous impedance of pathogen penetrance into the subluminal environment. Close interaction with luminal bacteria causes increased signaling through pattern recognition receptors (PRRs) in the epithelial barrier, specifically Toll-like receptors (TLRs) and NOD-like receptors (NLRs). The downstream effects of this recognition include polarized transport of Ag for presentation and the secretion of proliferative (e.g. IL-7 and IL-22) or inhibitory (e.g. IL-25 and IL-33) cytokines to directly influence activation or inhibition of specific innate immune cells.49 Collectively, the terminally differentiated layer forms the first line of immune defense in the gut as a fairly impenetrable barrier system. The integrity of this barrier is key to the control of immune surveillance and Ag sampling.
In addition to epithelial barriers, the enteric immune system is composed of highly organized anatomical structures associated with the lamina propria and epithelial cells of the gut, collectively known as gut-associated lymphoid tissue (GALT). GALT can be divided into effector (e.g. infiltrating groups of effector immune cells within the subepithelial dome) and inducer sites (secondary lymphoid tissues responsible for the induction phase of the immune response). Peyer's patches (PPs), in particular, are secondary lymphoid organs of the enteric immune system that facilitate Ag sampling from the lumen. PPs do not have afferent lymph flow, but instead depend on the large number of associated M cells in the gut epithelium.45 M cells specialize in transport of macromolecules and Ag into the subepithelial dome, sampling and presenting such macromolecules locally within the PP to T cells.
Another important type of organized intestinal lymphoid tissue gets its name from the anatomical resemblance to isolated lymphoid follicles (ILFs). ILFs consist of large groups of IgA-secreting B cells, along with other smaller populations of immune cells, such as DCs and several subsets of innate lymphoid cells (ILCs) that produce cytokines to modulate immune responses. ILCs are an interesting group of innate cells that exhibit lymphoid morphology but do not express Ag-specific receptors. Despite this difference with T cells, their cytokine profiles are similar, and as such, they are classified as ILC1 (Tbet expression and a Th1 cytokine profile), ILC2 (GATA3 expression and Th2 cytokine profile), and ILC3 (RORγt expression and Th17 cytokine profile).50 ILCs are believed to be early “orchestrators” of responses by secreting cytokines that prime the Ag-presenting environment towards activation or tolerance.51 This is important because distinct subsets of ILCs can be inhibited or activated by bacterial products or via bacterial signaling on the gut epithelium. Detailed descriptions of each ILC subset and their predominant functions, as they relate specifically to the microbiome, have been reviewed elsewhere.52
Apart from innate cells modulated by non-specific cues, there are cellular components of the adaptive immune system also residing in the gut. For example, CD103+ DCs and CX3CR1+ phagocytes are APCs with a balancing role in the activation and tolerance of Ag-specific T-cell immunity in the enteric lymph.53 CD103+ DCs and CX3CR1+ phagocytes both sample luminal Ag independently using dendritic processes, and migrate to mesenteric lymph nodes (MLNs) to present Ag to B and T cells.54 DCs are major modulators of T-cell responses, because they can serve as potent APCs and at the same time contribute to the local cytokine environment through detection of microbial substances by their PRRs. LP DCs may mediate T-cell induction by commensals in several ways. They may sample commensal-derived Ag, either by directly contacting the bacteria through extension of dendrites into the gut lumen or detecting bacterial products, such as serum-amyloid-A, that gain access to the LP. They may then present these Ag to intestinal T cells to induce commensal-specific T cells. Alternatively, LP DCs may be conditioned by commensals indirectly, e.g. by cytokines produced by other cells in response to the bacteria. Figure 2 depicts a hypothetical setting, where segmented filamentous bacteria (SFB) attachment to the intestinal epithelium induces cytokine production by γδ IECs, which go on to modulate DC function. T-cells activated in these tissues then circulate via the thoracic duct lymph flow and into the bloodstream, but remain gut-tropic in nature and thereafter return to the intestinal lamina propria.55 This return to the gut, in turn, has been hypothesized to limit systemic inflammation.56 Recent work by Geem et al.,57 however, has shown that in lymphotoxin-deficient mice, in which secondary lymphoid organs are underdeveloped, populations of Ag-specific T cells responding to a commensal bacteria (SFB) are still present, but not in MHC II (CIITA) knock-out mice. Therefore, it is likely that the key place of interaction between the microbiome and gut-tropic CD4 T cells is the lamina propria and not the MLNs as previously thought.58 In addition, CD103+ DCs seem to be integral in be presenting Ag to CD4 T cells under this context, although CX3CR1+ phagocytes have been involved in pathogenic interactions between commensal Ag and Ag-specific CD4 T cells.59 Lastly, B-cell activation in the PPs and MLNs may help the host control the microbiota.60 Activated gut B cells will differentiate and produce IgA in a T cell-independent manner, which helps in the control of the bacterial burden by neutralizing bacterial adhesion to luminal epithelium.61 The advantage of IgA produced in the PPs and MLNs is to act locally and limit inflammation systemically.62 A more thorough description of B cell roles in enteric immunity is available elsewhere.63
Figure 2.
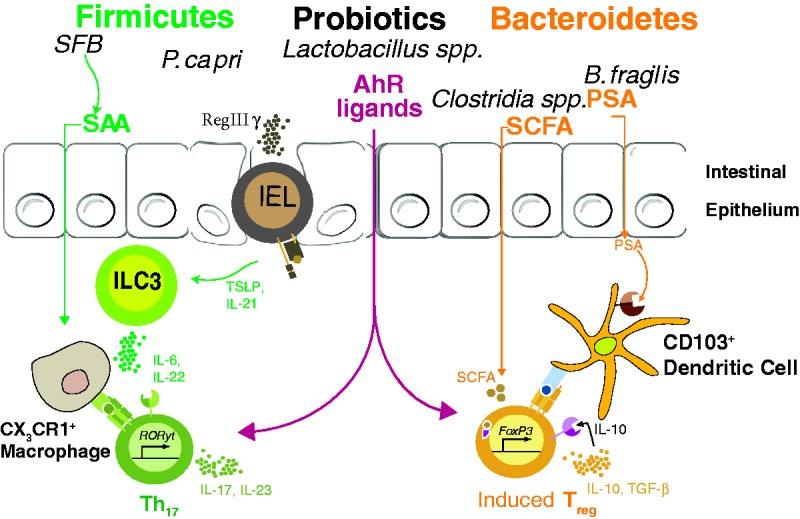
Modulation of commensal-specific CD4 T cells by bacterial metabolites and carefully balanced microbial gut membership. SFB attachment to epithelial surface induces RegIIIγ production by either γδ intrapepithelial lymphocytes or enteric epithelial cells, ultimately influencing the cytokine secretion of type-3 innate lymphoid cells (ILC3s). SFB also promotes Th17 polarization in the intestinal lamina propria via host production of serum amyloid A (SAA) and Ag presentation (currently believed to be through CX3CR1+ phagocytes). In contrast, B. fragilis PSA interacts with TLR2 on DCs to induce colonic Treg. In addition, Clostridial sp., as well as a group of Bacteroidetes en masse—including B. caccae, B. massiliensis, B. thetaiotaomicron, B. vulgatus, and P. distasonis—induce colonic Treg, presumably via bacterial production of SCFAs. Indole metabolism by certain bacterial species (Lactobacilli, in particular) can create Aryl hydrocarbon receptor ligands, thus possibly contributing to both Th17 polarization and enteric Treg induction.
SFB: segmented filamentous bacteria; Th17: T-helper 17; SCFA: short-chain fatty acids; SAA: serum amyloid A.
Although the mechanisms governing host–commensal interactions are still enigmatic, bacterial immunomodulation is an indisputable phenomenon. Mice born in the absence of any microbes (germ-free (GF)) have been essential to demonstrate a clear function for commensalism in the development of the gut immune system. GF mice have smaller PPs, fewer IgA-producing B cells, as well as skewed numbers of innate lymphocyte subsets in the gut.52 Most of these immunologic changes are rapidly reversible by the introduction of normal microbiota, whereas others (particularly changes in specific subsets of ILCs) are reversible only during early infancy. For example, adult restoration of the gut flora in GF mice corrects most immunological defects observed, but the number of iNKT cells in the lamina propria is significantly higher than in specific pathogen-free (SPF) mice. When gut flora is restored in GF mice during the first 24 h of life, iNKT cell numbers are comparable to those of SPF mice.62 The same findings can be seen in mice carrying a microbiome with less Sphingomonas sp. (a genera that produces glycolipids recognized by CD1d in iNKT cells)63 compared with mice carrying a higher burden of Sphingomonas.62 Along the same lines, the number of lymphoid tissue inducer (LTi) cells are lower in GF mice than in wild type mice, and these differences cannot be abrogated in adult GF mice given a normalized microbiota.64 These results suggest there is a teleological imperative for how the residence of innate immune cells, particularly ILCs, are curated—almost as if these cells are instructed by the microbial residents in early life, but are not subject to the plasticity of the microbiota.65 Regardless of their ability to be modulated as adults, those early changes to the composition of ILCs can potentially set the stage for adaptive immune responses with a propensity for inflammation, and the subsequent development of autoimmunity and allergy.
The microbiome and T-cell immunity
CD4 T cells are among the most important peripheral lymphocyte subsets when it comes to the orchestration of successful immune responses. A particularly striking feature of CD4 T cells is their plasticity of function, which is unique for cells with such exquisite Ag specificity. Functional plasticity is key, because it makes each CD4 T cell both an effector of tolerance and an orchestrator of inflammation. This ambivalent change in function, or “polarization”, is reversible, and directed in large part by the local environment in which the CD4 T-cell encounters Ag presented in MHC II molecules. The environmental “set-up” in question includes specific cytokines, small molecular targets of steroid receptors (retinoic acid (RA) derivatives and aryl hydrocarbons, in particular), and yet-to-be-discovered or fully elucidated modulatory pathways.
Recent studies suggest commensal bacteria are actively involved in the development of adaptive immune responses by the programming of CD4 T-cell differentiation above and beyond just the cognate recognition of Ag and regulation of bacterial communities.66 The effect of gut microbiota on innate immunity to systemic infection with viruses and bacteria has been well documented,67–69 but it has been difficult to understand how the microbiota affects non-mucosal T-cell responses without cognate interaction between peripheral T cells and commensal Ag.70 Since more than 80% of the body's lymphocytes reside in the gut,71 it is not far-fetched to posit that the microbiome indirectly controls the diversity and responsiveness of the peripheral T-cell repertoire. These factors may, in turn, be heavily controlled by bacterial membership in the gut. This is supported by the fact that in GF mice, T-cell responses are aberrant, and their total numbers are systemically lower than in SPF mice.
Several groups have attempted to isolate the effect of a single commensal microbial species on CD4 T cells, reporting interesting data that posit the balance between Firmicutes and Bacteroidetes as an important part of adept immunity in the gut. This is reminiscent of how differences in dietary intake skew the microbiome towards either of these two phyla, and there are epidemiological correlations between these changes and the incidence of autoimmunity and allergy. One leading theory among mucosal immunologists is that certain bacterial species (perhaps from certain phyla) induce pro-inflammatory (RORγt-expressing “Th17” CD4 T cells) or regulatory populations of T cells (FoxP3-expressing CD4 regulatory T cells). These changes are Ag-specific (e.g. Ag from the microbe is specifically recognized by a subset of T cells), but modulated by bacterial metabolites and the cytokine environment in which T cells are first presented the Ag. The first example of this phenomenon came from a specific genus of Firmicutes collectively termed SFB. SFB induce proinflammatory states in the gut by direct priming of CD4 T cells, with a resultant group of SFB-specific T cells polarized toward a Th17 phenotype leading to a greater propensity for the development of autoimmunity in mouse models.72,73 Research into enterocyte function shows that SFB attach to the luminal side of enterocytes, causing a shift in the cytokines produced by the epithelial or M cells present.35 While it is not exactly clear how or what these changes are, the polarization of T cells by SFB is dependent on bacterial secretion of serum amyloid A (SAA) and the delivery of this protein to T cells via enterocyte transcytosis or APC uptake. In addition, another member of the Firmicutes family, Prevotella copri, has also been implicated in autoimmune arthritic responses.74,75
In the other group of the dominant gut microbes (Bacteroidetes), a well-known member promotes immunological tolerance. Mazmanian et al.76 demonstrated Bacteroides fragilis induces tolerance in T-cell subsets.76 Further studies have demonstrated B. fragilis-produced polysaccharide A (PSA), like SFB-produced SAA, is key for this induction of tolerance. PSA directs DCs to prime tolerogenic T-cell responses via TLR2 signaling, with specific effects being attributed to innate and adaptive responses generated by plasmacytoid DCs.77,78 However, tolerance and inflammation do not neatly split depending on the phyla, and it is more likely that only particular strains of certain species are regulators of immunity. In support of this idea, Clostridia sp. (which are Firmicutes) induce tolerance in mouse models of colitis.79–81 Likewise, the human pathogen enterotoxigenic B. fragilis (ETBF) will promote inflammatory diarrhea and the creation of pathogenic Th17 responses.82–84
Commensal organism byproducts also have potent roles in the polarization of CD4 T-cell responses, adding further credence to the idea that the contextual “study” of intestinal ecology by naïve T cells is very important for the correct polarization of immunocompetent responses. For example, CD4 T cells have aryl hydrocarbon receptors (AhRs) that modulate gene expression, but respond to different ligands with different effects, such as tryptophan metabolites. Commensal lactobacilli with tryptophanase activity are a source of indoles that function as AhR ligands and promote regulatory T-cell development.85 Similarly, RA has been implicated as an important balancing metabolite of regulatory T cell and Th17 polarization, and changes in RA and RA-like metabolites produced by commensals likely influence the environmental cues T cells receive while Ag is being presented.86–88
A discussion of changes induced by bacterial metabolites in T cells would not be complete without mentioning short-chain fatty acids (SCFAs). SCFAs are 1–6 carbons in length produced by bacterial fermentation in the colon. Most abundant types of SCFAs are butyrate, propionate, and acetate.89 Apart from being a substantial source of energy for colonic epithelial cells,90 SCFAs have divergent functions in the regulation of the immune system, including recruitment of neutrophils via activation of GPR43,91 inhibition of histone deacetylases,92 and the resultant downregulation of T-cell proliferation, DC Ag-presentation, or secretion of pro-inflammatory cytokines. This is an exciting area of ongoing research with many unanswered questions remaining, but it is clear that changes in microbial composition will change the SCFAs produced in the gut, as bacterial species differ greatly in their ability to produce these fatty acids.
The technological advancements, and new insights into mucosal immunity, hint at a beneficial co-evolution of the immune system and the resident bacterial organisms in our body.23 Although plenty of “unknown unknowns” limit our understanding of enteric immunity, it is unquestionable that the microbiota plays an important role in inducing systemic tolerance in the immune system of the host. Moreover, successful enteric immunity is dependent not just on the bacterial tenants, but on the physical barriers compartmentalizing host and microbe, as well as the residing immune cells along the said barriers. The maintenance of homeostasis with commensal residents, in turn, improves host protection against invading pathogens.35 In addition, the cytokine milieu created by the direct and indirect interactions between the microbes and the enterocyte barrier seems to play a big role in the polarization of T cells, and whether T cell-mediated immune responses to intestinal pathogens veer towards a tolerogenic or an inflammatory Ag-specific response. Indeed, the explosion of microbiome studies within the last decade has cemented the idea that a trifecta of epithelial barrier integrity, microbial flora, and adequate innate immune responses is uniquely important to the development of healthy effector T-cell responses.52
The microbiome and sepsis
Advances in our understanding of gut-initiated or gut-propagated critical illness have led to novel investigations into how microbial membership and gut function may be targeted for therapeutic gain in the ICU.93 This is highly significant, given sepsis (currently defined as a “systemic inflammatory response in the presence of a disseminated infection”)14 is the cause of ∼300,000 deaths every year in the United States, and its treatment (which is essentially supportive) constitutes about 40% of all total hospital costs.94,95 Several changes occur to gut physiology in septic patients, either due to extrinsic factors (antibiotics and parenteral nutrition) or intrinsic factors (systemic inflammation and gut leakage). These changes, in turn, influence the composition of the enteric flora.96 For example, there is evidence of rapid alterations in microbial membership composition during the acute phase of critical illness. When stools of patients with severe sepsis are examined, there is a massive loss of microbe diversity, particularly anaerobic diversity.32 Likewise, parenteral nutrition, which is common in critically ill septic patients, causes thinning of the protective mucus layer and decreasing barrier integrity and the availability of SCFAs in the gut.97,98 Most of these patients are also given opioids, which reduce gut motility and (as a result) promote bacterial overgrowth. Another key metabolite, phosphate (Pi), is crucial for bacterial quorum sensing, and Pi availability to the commensals is abrogated during critical illness.32 Reduction in Pi within the gut microenvironment can change the phenotype and virulence of microbes residing in the gut. For example, the PA-I lectin/adhesin, a potent intestinal epithelial barrier disrupting protein, is expressed by Pseudomonas aeruginosa when Pi is limiting.99 P. aeruginosa is then better able to bind to and breach the intestinal epithelium. Reduced amounts of Pi can shift the phenotype of intestinal Candida albicans to a lethal one by inducing hyphae formation.100,101 Moreover, opioid responsiveness by bacteria (and accordingly, virulence) is enhanced in situations of low Pi.102,103 When Pi is abundant, conversely, incoming cues (such as those from opioids) do not transduce virulence.
One of the newest (and now widely accepted) theories in sepsis is that T-cell responses are suppressed, or paralyzed, in sepsis survivors. One of the more obvious clues as to why T-cell immunity is particularly impacted during sepsis lies in the organ damage that occurs. The effect of sepsis on T-cell immunity is particularly heinous, and several documented sequelae of sepsis have led us to investigate T-cell function in sepsis-induced immunosuppression. Though simplistic, one can think of sepsis immunosuppression of T cells in terms of a dysregulation in Ag recognition (and successful immunological memory) because of the context, as well as environment in which Ag is encountered.
Context
The gut and spleen are the only solid organs where apoptosis is reliably found during sepsis, and epithelial cells are the only type of cell consistently found to undergo apoptosis in critically ill patients.104 Human autopsy studies and mouse models of sepsis have demonstrated the phenomenon of lymphocyte apoptosis15,16,104–110 is only rivaled by epithelial cell death in the gut lumen of septic hosts.111–115An even more surprising connection is the notion that CD4 T cells might be directly affecting the fate of epithelial integrity in sepsis.
Environment
Dysbiosis, mucosal damage, and overgrowth of pathogenic microbes in the host may overwhelm or confuse the immune system (especially when occurring simultaneously), creating inadequate cytokine environments leading to exacerbated immune responses, like those seen in autoimmunity. It may also create mixed environments; aberrant responses with proliferative and suppressive cues that ultimately cause apoptotic death of T cells.116 Similarly, loss of epithelial barrier integrity in sepsis might lead to mixed cytokine environments that overwhelm correct T-cell function and promote apoptosis aided by other pro-apoptotic factors, including extracellular histone release and oxidative stress.117,118 Supportive of this theory is the fact that a mixture of regulatory and inflammatory T cells are consistently found systemically in septic patients, which might also be the reason why lung injury is common in septic hosts without initial pulmonary septic foci driving the inflammatory response.73,119–122 Finally, studies evaluating the impact of sepsis on DCs have noted the numerical reduction of these cells in both clinical and experimental settings.123–126 There is also less IL-12 and more IL-10 produced by post-septic DCs after stimulation with TLR agonists compared with DCs from sham-treated mice.125–127 The reported deficiencies in DC number and function add an additional layer contributing to a T cell-extrinsic defect, but additional investigation probing the effect of sepsis on DCs is necessary.
Microbiome reactivity and T-cell survival after sepsis
The polymicrobial-driven peritonitis that occurs in the cecal ligation and puncture (CLP) model lends itself well to investigate the direct effect of the gut microbiome on the T-cell response during a septic event. Surprisingly, there has been a paucity of data published with regard to this aspect of sepsis research. Insight into the relationship between the immune system and the microbiome as explored in recent years, aided by improved methods of defining and manipulating the constituents of the gut microbiome, the increased use of GF mice and the identification of endogenous Ag-specific T cells reactive to Ag expressed by gut microbes. As part of the body of literature describing the effect of the gut microbiome on the function of the immune system, recent work from our laboratory examined the number and function of endogenous CD4 T cells specific for an immunodominant epitope within SFB naturally found in commercially available mice128 after CLP-induced sepsis using mice that either contained or lacked SFB.129 Evaluation of the total CD4 T-cell compartment in CLP-treated SFB + and SFB− showed the typical acute reduction in CD4 T-cell numbers followed by a gradual recovery; however, evaluation of endogenous SFB-specific CD4 T cells in these same mice revealed strikingly different outcomes. The number of SFB-specific CD4 T cells decreased in SFB− mice two days post-CLP, and recovered by day 30, similar to the pattern for total CD4 T cells. In contrast, there was a significant increase in number of SFB-specific CD4 T cells in SFB + mice two days after CLP that remained elevated on day 30. The SFB released from the gut after CLP primed an Ag-specific CD4 T-cell response, which in a weird twist of fortune, actually helped the host survive a lethal secondary infection with an experimental pathogen engineered to express a common SFB Ag, whereas secondary lethal infection with the same pathogen lacking the expression of the SFB Ag lead to host death. Thus, the availability of mice containing a specific gut-resident microbe, along with a defined CD4 T-cell population with T-cell receptor (TCR) specificity to Ag expressed by that gut-resident microbe permitted a first investigation into the direct effect of gut microbiome leakage during sepsis on CD4 T cells.
The data showing the influence of a gut-resident microbe on the quantity and function capacity of commensal bacterial Ag-specific CD4 T-cell population introduced the concept that not all T-cell responses are suppressed in sepsis survivors. A key component to this revelation was the interrogation of an endogenous Ag-specific CD4 T-cell population, instead of examining the sepsis-induced effects on T cells at the “total” polyclonal population level. It is likely, however, that other mechanisms can drive CD4 T-cell responses to gut microbiota released during sepsis. The TCR repertoire expressed by the entire T-cell compartment is considered to be of sufficient diversity to protect the host from virtually all foreign pathogens. Recent studies have estimated the theoretical diversity of the T-cell repertoire to consist of 1015–1020 unique αβ TCR,130–132 but the actual number of mature T cells that have successfully navigated the thymic selection process and are in the periphery is considerably less.133–135 Consequently, the peripheral TCR repertoire is significantly less diverse (by an order of several magnitudes) than the number of MHC-expressed peptides.136 To compensate for these numerical differences, TCR cross-reactivity (i.e. recognition of distinct peptide sequences from a variety of proteins by a single TCR) has been considered to be an essential feature of the mature T-cell repertoire.137 Formal proof for T-cell cross-reactivity has come in recent years, coinciding with advancements in the ability to identify small numbers of endogenous Ag-specific T cells in humans and mice. Using peptide:MHC tetramers, there is now convincing data showing that influenza virus-specific and HIV-specific memory CD4 T cells recognize epitopes from unrelated microbes present in the gut (among other locations), and H5N1-specific memory CD4 T cells have been detected in healthy individuals not exposed to H5N1 infection.138–141 These data suggest there is a significant frequency of memory CD4 T cells in the periphery generated as a result of TCR cross-reactivity to commensal Ag and not because of specificity for a commensal pathogen.142 Moreover, certain responses to viral infection change by the presence of specific commensal microbes, and antibiotic-treated mice (where the microbiome community is disrupted) have significantly fewer virus-specific effector CD4 and CD8 T cells when compared with untreated controls during influenza infection.67,70
Our analysis of different endogenous Ag-specific CD4 T-cell populations in CLP-treated mice showed the incomplete numerical recovery of some, but not all, of the populations.143 Among the Ag-specific CD4 T cells examined previously, we were intrigued to see that the influenza A virus (IAV) NP311-specific CD4 T-cell population was the only one (of those we examined) to significantly increase in number in CLP- versus sham-treated mice.143 In addition, these NP311-specific CD4 T cells demonstrated increased frequency of CD44hi cells in CLP-treated mice compared with sham-treated mice—something that was not observed for other pathogen-specific CD4 T-cell populations. Upon subsequent investigation, our unpublished data suggest that the NP311-specific CD4 T cells are indeed responding to some gut-resident microbe released during CLP-induced sepsis. Collectively, these data suggest CD4 T cells are indeed reacting to some commensal bacteria Ag released into the peritoneum during CLP-induced polymicrobial sepsis. Therefore, it is possible that the recovery of some Ag-specific T-cell populations with diminished repertoire diversity is modulated by cross-reactivity with or direct recognition of microbial Ag found in the gut (Figure 3). This is in contrast to the Ag-independent homeostatic proliferation that can occur for other pathogen-specific T-cell populations. The abnormal recovery of TCR repertoire diversity within Ag-specific populations could potentially lead to aberrant responses such as decreased affinity for immune-dominant antigen peptides. This effect of microbes in the recovery of CD4 T cells after sepsis might account for the susceptibility to opportunistic infections, and diminished lymphocyte function seen in sepsis.
Figure 3.

The recovery of CD4 T cells after sepsis involves antigen-dependent and independent pathways. The “coloring” of T cells depicted in the top of the illustration represents different antigen-specific populations, amongst a representation of the peripheral CD4 T-cell mass. Sepsis causes a stochastic loss of CD4 T cells by apoptosis, and the end result for a significant group of patients is a state of lymphopenia that is most pronounced for certain cell populations (one such lymphocyte being CD4 T cells). After sepsis, however, a bulk number of peripheral CD4 T cells returns to baseline. Despite the numerical recovery of peripheral CD4 T cells, the diversity and size of Ag-specific CD4 T cell populations change after sepsis. The bottom square “zooms in” in some of the changes occurring to specific CD4 T-cell populations during sepsis that make it possible for antigenic diversity to change. These include both Ag-dependent and independent pathways of recovery. Ag-independent changes in T-cell recovery is mediated mostly by homeostatic proliferation, whereas Ag-dependent changes in T-cell repertoire distribution are influenced by (mostly) cross-reactivity to Ag sampled from the host microbiome.
Concluding remarks
References to the gastrointestinal tract as the “undrained abscess” of multiple organ failure have been predominant in the sepsis literature for decades,144 but our understanding of sepsis has since evolved considerably.145 However, many aspects of sepsis remain undefined, and the interplay between microbiome composition and the immune system during sepsis remains relatively unexplored. The role of T cells in sepsis, particularly, is much more complex and multifactorial in nature than we previously thought. Critical illness and the side-effects stemming from the treatments currently comprising the standard of care may in fact contribute to poor immunological recovery following sepsis. An even more surprising connection is the notion that CD4 T cells might be directly affecting the fate of epithelial integrity in sepsis. Rolf Zinkernagel146 wrote a review targeted to a generation of immunologists who had focused their efforts on dissecting immune responses to haptens and model antigens. In this thought-provoking review, Zinkernagel146 argued that deeper immunological insight could be gained from the vantage point of viruses, echoing Janeway's147 prior assertion that advancements in the field would necessitate “the rediscovery of microbiology by immunologists.” Insights into the interaction between microbiome and mucosal immunity will lead to advances in poorly understood syndromes, particularly those with compromised T-cell function. This represents a hopeful outlook for advances in the treatment of sepsis, a deadly condition that has been called “the graveyard of pharmaceuticals.”148 If understanding pathogenic microbes was the leitmotif of 20th-century medicine, enteric immunity appears poised to take on that role in the 21st century.
Acknowledgements
This work was supported by a U.S. Department of Veterans Affairs Merit Review Award (T.S.G.) and National Institutes of Health Grants T32AI007313 (J.C.-P.), AI119160, AI114543, and GM113961 (V.P.B.), and GM115462 (T.S.G.).
Authors' contributions
J.C.-P. and T.S.G. wrote the manuscript and created figures. T.S.G. and V.P.B. edited the manuscript for structure, scope and content.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol 2007; 7: 379–90. [DOI] [PubMed] [Google Scholar]
- 2.O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep 2006; 7: 688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science 2005; 307: 1915–20. [DOI] [PubMed] [Google Scholar]
- 4.Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 1996; 4: 430–5. [DOI] [PubMed] [Google Scholar]
- 5.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 2016; 14: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, Juncker AS, Manichanh C, Chen B, Zhang W, Levenez F, Wang J, Xu X, Xiao L, Liang S, Zhang D, Zhang Z, Chen W, Zhao H, Al-Aama JY, Edris S, Yang H, Wang J, Hansen T, Nielsen HB, Brunak S, Kristiansen K, Guarner F, Pedersen O, Dore J, Ehrlich SD, Meta HITC, Bork P, Wang J, Meta HITC. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 2014; 32: 834–41. [DOI] [PubMed] [Google Scholar]
- 7.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science 2009; 326: 1694–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Filippo C, Lionetti P. Impact of diet on gut microbiota in the globalized world. Funct Food Rev 2013; 5: 13–22. [Google Scholar]
- 9.Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, Patterson PH, Mazmanian SK. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155: 1451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khosravi A, Mazmanian SK. Disruption of the gut microbiome as a risk factor for microbial infections. Curr Opin Microbiol 2013; 16: 221–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, Mazmanian SK. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014; 15: 374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noval Rivas M, Burton OT, Wise P, Zhang YQ, Hobson SA, Garcia Lloret M, Chehoud C, Kuczynski J, DeSantis T, Warrington J, Hyde ER, Petrosino JF, Gerber GK, Bry L, Oettgen HC, Mazmanian SK, Chatila TA. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J Allergy Clin Immunol 2013; 131: 201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009; 9: 313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions: time for change. Lancet 2013; 381: 774–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, Hotchkiss RS. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011; 306: 2594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr, Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, Karl IE. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 2001; 166: 6952–63. [DOI] [PubMed] [Google Scholar]
- 17.Cabrera-Perez J, Condotta SA, Badovinac VP, Griffith TS. Impact of sepsis on CD4 T cell immunity. J Leukoc Biol 2014; 96: 767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med 2014; 20: 214–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yurist-Doutsch S, Arrieta MC, Vogt SL, Finlay BB. Gastrointestinal microbiota-mediated control of enteric pathogens. Annu Rev Genet 2014; 48: 361–82. [DOI] [PubMed] [Google Scholar]
- 20.Luan YY, Dong N, Xie M, Xiao XZ, Yao YM. The significance and regulatory mechanisms of innate immune cells in the development of sepsis. J Interferon Cytokine Res 2014; 34: 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bermejo-Martin JF, Andaluz-Ojeda D, Almansa R, Gandia F, Gomez-Herreras JI, Gomez-Sanchez E, Heredia-Rodriguez M, Eiros JM, Kelvin DJ, Tamayo E. Defining immunological dysfunction in sepsis: a requisite tool for precision medicine. J Infect 2016; 72: 525–36. [DOI] [PubMed] [Google Scholar]
- 22.Condotta SA, Cabrera-Perez J, Badovinac VP, Griffith TS. T-cell-mediated immunity and the role of TRAIL in sepsis-induced immunosuppression. Crit Rev Immunol 2013; 33: 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010; 330: 1768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bantock GG. The modern doctrine of bacteriology, or the germ theory of disease. Br Med J 1899; 1: 846–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morowitz MJ, Babrowski T, Carlisle EM, Olivas A, Romanowski KS, Seal JB, Liu DC, Alverdy JC. The human microbiome and surgical disease. Ann Surg 2011; 253: 1094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wangensteen OH, Wangensteen SD, Klinger CF. Surgical cleanliness, hospital salubrity, and surgical statistics, historically considered. Surgery 1972; 71: 477–93. [PubMed] [Google Scholar]
- 27.Alexander JW. The contributions of infection control to a century of surgical progress. Ann Surg 1985; 201: 423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burns H. Germ theory: invisible killers revealed. BMJ 2007; 334(Suppl 1): s11. [DOI] [PubMed] [Google Scholar]
- 29.Kendall AI. Some observations on the study of the intestinal bacteria. J Biol Chem 1909; 6: 499–507. [Google Scholar]
- 30.Metchnikoff E. Sur la lutte des cellules de l‘organisme contre l’invasion des microbes. Ann Inst Pasteur 1887; 1: 321–36. [Google Scholar]
- 31.Metchnikoff E. The utility of lactic microbes with explanation of the author's views on longevity. Century Magazine 1909; 79: 53–8. [Google Scholar]
- 32.Defazio J, Fleming ID, Shakhsheer B, Zaborina O, Alverdy JC. The opposing forces of the intestinal microbiome and the emerging pathobiome. Surg Clin North Am 2014; 94: 1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, Ugarte E, Munoz-Tamayo R, Paslier DL, Nalin R, Dore J, Leclerc M. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 2009; 11: 2574–84. [DOI] [PubMed] [Google Scholar]
- 34.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005; 308: 1635–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol 2012; 30: 759–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan XC, Segata N, Huttenhower C. Biodiversity and functional genomics in the human microbiome. Trends Genet 2013; 29: 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chierici R, Fanaro S, Saccomandi D, Vigi V. Advances in the modulation of the microbial ecology of the gut in early infancy. Acta Paediatr Suppl 2003; 91: 56–63. [DOI] [PubMed] [Google Scholar]
- 38.Fanaro S, Chierici R, Guerrini P, Vigi V. Intestinal microflora in early infancy: composition and development. Acta Paediatr Suppl 2003; 91: 48–55. [DOI] [PubMed] [Google Scholar]
- 39.Park HK, Shim SS, Kim SY, Park JH, Park SE, Kim HJ, Kang BC, Kim CM. Molecular analysis of colonized bacteria in a human newborn infant gut. J Microbiol 2005; 43: 345–53. [PubMed] [Google Scholar]
- 40.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006; 444: 1027–31. [DOI] [PubMed] [Google Scholar]
- 41.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008; 3: 213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 2014; 20: 159–66. [DOI] [PubMed] [Google Scholar]
- 43.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011; 334: 105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol 2011; 12: 5–9. [DOI] [PubMed] [Google Scholar]
- 45.Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 2003; 3: 331–41. [DOI] [PubMed] [Google Scholar]
- 46.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature 2008; 453: 1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140: 845–58. [DOI] [PubMed] [Google Scholar]
- 48.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008; 28: 454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saenz SA, Siracusa MC, Monticelli LA, Ziegler CG, Kim BS, Brestoff JR, Peterson LW, Wherry EJ, Goldrath AW, Bhandoola A, Artis D. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med 2013; 210: 1823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol 2013; 13: 145–9. [DOI] [PubMed] [Google Scholar]
- 51.Reynders A, Yessaad N, Vu Manh TP, Dalod M, Fenis A, Aubry C, Nikitas G, Escaliere B, Renauld JC, Dussurget O, Cossart P, Lecuit M, Vivier E, Tomasello E. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat-lymphoid cells. EMBO J 2011; 30: 2934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen VL, Kasper DL. Interactions between the intestinal microbiota and innate lymphoid cells. Gut Microbes 2014; 5: 129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott CL, Aumeunier AM, Mowat AM. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol 2011; 32: 412–9. [DOI] [PubMed] [Google Scholar]
- 54.Niess JH. What are CX3CR1+ mononuclear cells in the intestinal mucosa? Gut Microbes 2010; 1: 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenstad H, Ericsson A, Johansson-Lindbom B, Svensson M, Marsal J, Mack M, Picarella D, Soler D, Marquez G, Briskin M, Agace WW. Gut-associated lymphoid tissue-primed CD4+ T cells display CCR9-dependent and -independent homing to the small intestine. Blood 2006; 107: 3447–54. [DOI] [PubMed] [Google Scholar]
- 56.Agace WW. Tissue-tropic effector T cells: generation and targeting opportunities. Nat Rev Immunol 2006; 6: 682–92. [DOI] [PubMed] [Google Scholar]
- 57.Geem D, Medina-Contreras O, McBride M, Newberry RD, Koni PA, Denning TL. Specific microbiota-induced intestinal Th17 differentiation requires MHC class II but not GALT and mesenteric lymph nodes. J Immunol 2014; 193: 431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cerutti A, Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity 2008; 28: 740–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 2000; 288: 2222–6. [DOI] [PubMed] [Google Scholar]
- 60.Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, Geuking MB, Curtiss R, 3rd, McCoy KD, Macpherson AJ. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 2010; 328: 1705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol 2010; 10: 159–69. [DOI] [PubMed] [Google Scholar]
- 62.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 2012; 336: 489–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol 2012; 12: 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen VL, Surana NK, Duan J, Kasper DL. Role of murine intestinal interleukin-1 receptor 1-expressing lymphoid tissue inducer-like cells in Salmonella infection. PLoS One 2013; 8: e65405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kamada N, Nunez G. Regulation of the immune system by the resident intestinal bacteria. Gastroenterology 2014; 146: 1477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mazmanian SK, Lee YK. Interplay between intestinal microbiota and host immune system. J Bacteriol Virol 2014; 44: 1–9. [Google Scholar]
- 67.Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, Wherry EJ, Artis D. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012; 37: 158–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helgeland L, Dissen E, Dai KZ, Midtvedt T, Brandtzaeg P, Vaage JT. Microbial colonization induces oligoclonal expansions of intraepithelial CD8 T cells in the gut. Eur J Immunol 2004; 34: 3389–400. [DOI] [PubMed] [Google Scholar]
- 69.Pollard M, Sharon N. Responses of the Peyer's patches in germ-free mice to antigenic stimulation. Infect Immun 1970; 2: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spasova DS, Surh CD. Blowing on embers: commensal microbiota and our immune system. Front Immunol 2014; 5: 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, Farber DL. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 2014; 159: 814–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stepankova R, Powrie F, Kofronova O, Kozakova H, Hudcovic T, Hrncir T, Uhlig H, Read S, Rehakova Z, Benada O, Heczko P, Strus M, Bland P, Tlaskalova-Hogenova H. Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis 2007; 13: 1202–11. [DOI] [PubMed] [Google Scholar]
- 73.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010; 32: 815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brusca SB, Abramson SB, Scher JU. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol 2014; 26: 101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, Huttenhower C, Littman DR. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2: e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008; 453: 620–5. [DOI] [PubMed] [Google Scholar]
- 77.Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe 2014; 15: 413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Q, McLoughlin RM, Cobb BA, Charrel-Dennis M, Zaleski KJ, Golenbock D, Tzianabos AO, Kasper DL. A bacterial carbohydrate links innate and adaptive responses through Toll-like receptor 2. J Exp Med 2006; 203: 2853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013; 500: 232–6. [DOI] [PubMed] [Google Scholar]
- 80.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011; 331: 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci Transl Med 2014; 6: 220ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, Rabizadeh S, Golub JE, Mathews LE, Shin J, Sartor RB, Golenbock D, Hamad AR, Gan CM, Housseau F, Sears CL. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun 2009; 77: 1708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sears CL, Islam S, Saha A, Arjumand M, Alam NH, Faruque AS, Salam MA, Shin J, Hecht D, Weintraub A, Sack RB, Qadri F. Association of enterotoxigenic Bacteroides fragilis infection with inflammatory diarrhea. Clin Infect Dis 2008; 47: 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15: 1016–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D'Angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013; 39: 372–85. [DOI] [PubMed] [Google Scholar]
- 86.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007; 317: 256–60. [DOI] [PubMed] [Google Scholar]
- 87.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 2007; 204: 1775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 2008; 181: 2277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 2006; 40: 235–43. [DOI] [PubMed] [Google Scholar]
- 90.Mortensen PB, Clausen MR. Short-chain fatty acids in the human colon: Relation to gastrointestinal health and disease. Scand J Gastroenterol 2009; 31: 132–48. [DOI] [PubMed] [Google Scholar]
- 91.Nilsson NE, Kotarsky K, Owman C, Olde B. Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun 2003; 303: 1047–52. [DOI] [PubMed] [Google Scholar]
- 92.Waldecker M, Kautenburger T, Daumann H, Veeriah S, Will F, Dietrich H, Pool-Zobel BL, Schrenk D. Histone-deacetylase inhibition and butyrate formation: fecal slurry incubations with apple pectin and apple juice extracts. Nutrition 2008; 24: 366–74. [DOI] [PubMed] [Google Scholar]
- 93.Schuijt TJ, van der Poll T, Wiersinga WJ. Gut microbiome and host defense interactions during critical illness. In: Vincent JL (ed) Annual update in intensive care and emergency medicine 2012, Berlin, Heidelberg: Springer, 2012, pp. 29–40. . [Google Scholar]
- 94.Coopersmith CM, Wunsch H, Fink MP, Linde-Zwirble WT, Olsen KM, Sommers MS, Anand KJ, Tchorz KM, Angus DC, Deutschman CS. A comparison of critical care research funding and the financial burden of critical illness in the United States. Crit Care Med 2012; 40: 1072–9. [DOI] [PubMed] [Google Scholar]
- 95.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 2013; 41: 1167–74. [DOI] [PubMed] [Google Scholar]
- 96.Shimizu K, Ogura H, Hamasaki T, Goto M, Tasaki O, Asahara T, Nomoto K, Morotomi M, Matsushima A, Kuwagata Y, Sugimoto H. Altered gut flora are associated with septic complications and death in critically ill patients with systemic inflammatory response syndrome. Dig Dis Sci 2011; 56: 1171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feng Y, Ralls MW, Xiao W, Miyasaka E, Herman RS, Teitelbaum DH. Loss of enteral nutrition in a mouse model results in intestinal epithelial barrier dysfunction. Ann N Y Acad Sci 2012; 1258: 71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Osuka A, Shimizu K, Ogura H, Tasaki O, Hamasaki T, Asahara T, Nomoto K, Morotomi M, Kuwagata Y, Shimazu T. Prognostic impact of fecal pH in critically ill patients. Crit Care 2012; 16: R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu L, Holbrook C, Zaborina O, Ploplys E, Rocha F, Pelham D, Chang E, Musch M, Alverdy J. Pseudomonas aeruginosa expresses a lethal virulence determinant, the PA-I lectin/adhesin, in the intestinal tract of a stressed host: the role of epithelia cell contact and molecules of the Quorum Sensing Signaling System. Ann Surg 2003; 238: 754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romanowski K, Zaborin A, Valuckaite V, Rolfes RJ, Babrowski T, Bethel C, Olivas A, Zaborina O, Alverdy JC. Candida albicans isolates from the gut of critically ill patients respond to phosphate limitation by expressing filaments and a lethal phenotype. PLoS One 2012; 7: e30119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zaborina O, Zaborin A, Romanowski K, Babrowski T, Alverdy J. Host stress and virulence expression in intestinal pathogens: development of therapeutic strategies using mice and C. elegans. Curr Pharm Des 2011; 17: 1254–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DeFazio JR, Zaborin A, Kaiser B, Kim SM, Fleming I, Camp D, Jabri B, Zaborina O, Alverdy JC. Intestinal application of Pi-PEG meinatins the health-promoting microbiota, suppresses the effect of gut pathobiota on immune signaling, and prevents mortality in mice. J Surg Res 2014; 186: 591, . [Google Scholar]
- 103.Romanowski K, Zaborin A, Fernandez H, Poroyko V, Valuckaite V, Gerdes S, Liu DC, Zaborina OY, Alverdy JC. Prevention of siderophore-mediated gut-derived sepsis due to P. aeruginosa can be achieved without iron provision by maintaining local phosphate abundance: role of pH. BMC Microbiol 2011; 11: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol 2006; 6: 813–22. [DOI] [PubMed] [Google Scholar]
- 105.Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood 1996; 87: 4261–75. [PubMed] [Google Scholar]
- 106.Ayala A, Xin Xu Y, Ayala CA, Sonefeld DE, Karr SM, Evans TA, Chaudry IH. Increased mucosal B-lymphocyte apoptosis during polymicrobial sepsis is a Fas ligand but not an endotoxin-mediated process. Blood 1998; 91: 1362–72. [PubMed] [Google Scholar]
- 107.Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol 2005; 174: 5110–18. [DOI] [PubMed] [Google Scholar]
- 108.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med 1997; 25: 1298–307. [DOI] [PubMed] [Google Scholar]
- 109.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol 2010; 184: 6766–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wesche-Soldato DE, Lomas-Neira JL, Perl M, Jones L, Chung CS, Ayala A. The role and regulation of apoptosis in sepsis. J Endotoxin Res 2005; 11: 375–82. [DOI] [PubMed] [Google Scholar]
- 111.Coopersmith CM, Stromberg PE, Dunne WM, Davis CG, Amiot DM, 2nd, Buchman TG, Karl IE, Hotchkiss RS. Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. JAMA 2002; 287: 1716–21. [DOI] [PubMed] [Google Scholar]
- 112.Fox AC, McConnell KW, Yoseph BP, Breed E, Liang Z, Clark AT, O'Donnell D, Zee-Cheng B, Jung E, Dominguez JA, Dunne WM, Burd EM, Coopersmith CM. The endogenous bacteria alter gut epithelial apoptosis and decrease mortality following Pseudomonas aeruginosa pneumonia. Shock 2012; 38: 508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hiramatsu M, Hotchkiss RS, Karl IE, Buchman TG. Cecal ligation and puncture (CLP) induces apoptosis in thymus, spleen, lung, and gut by an endotoxin and TNF-independent pathway. Shock 1997; 7: 247–53. [DOI] [PubMed] [Google Scholar]
- 114.Seeley EJ, Matthay MA, Wolters PJ. Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol 2012; 303: L355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turnbull IR, Buchman TG, Javadi P, Woolsey CA, Hotchkiss RS, Karl IE, Coopersmith CM. Age disproportionately increases sepsis-induced apoptosis in the spleen and gut epithelium. Shock 2004; 22: 364–8. [DOI] [PubMed] [Google Scholar]
- 116.Rimoldi M, Chieppa M, Larghi P, Vulcano M, Allavena P, Rescigno M. Monocyte-derived dendritic cells activated by bacteria or by bacteria-stimulated epithelial cells are functionally different. Blood 2005; 106: 2818–26. [DOI] [PubMed] [Google Scholar]
- 117.Grailer JJ, Fattahi F, Dick RS, Zetoune FS, Ward PA. Cutting edge: critical role for C5aRs in the development of septic lymphopenia in mice. J Immunol 2015; 194: 868–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernandez-Jimenez E, Toledano V, Cubillos-Zapata C, Rapisarda A, Chen J, Duan K, Yang H, Poidinger M, Melillo G, Nizet V, Arnalich F, Lopez-Collazo E, Biswas SK. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity 2015; 42: 484–98. [DOI] [PubMed] [Google Scholar]
- 119.Kessel A, Bamberger E, Masalha M, Toubi E. The role of T regulatory cells in human sepsis. J Autoimmun 2009; 32: 211–5. [DOI] [PubMed] [Google Scholar]
- 120.Li J, Li M, Su L, Wang H, Xiao K, Deng J, Jia Y, Han G, Xie L. Alterations of T helper lymphocyte subpopulations in sepsis, severe sepsis, and septic shock: a prospective observational study. Inflammation 2015; 38: 995–1002. [DOI] [PubMed] [Google Scholar]
- 121.Wisnoski N, Chung CS, Chen Y, Huang X, Ayala A. The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock 2007; 27: 251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Leng FY, Liu JL, Liu ZJ, Yin JY, Qu HP. Increased proportion of CD4(+)CD25(+)Foxp3(+) regulatory T cells during early-stage sepsis in ICU patients. J Microbiol Immunol Infect 2013; 46: 338–44. [DOI] [PubMed] [Google Scholar]
- 123.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, Cobb JP, Coopersmith C, Karl IE. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol 2002; 168: 2493–500. [DOI] [PubMed] [Google Scholar]
- 124.Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, Hotchkiss RS. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol 2003; 171: 909–14. [DOI] [PubMed] [Google Scholar]
- 125.Flohe SB, Agrawal H, Schmitz D, Gertz M, Flohe S, Schade FU. Dendritic cells during polymicrobial sepsis rapidly mature but fail to initiate a protective Th1-type immune response. J Leukoc Biol 2006; 79: 473–81. [DOI] [PubMed] [Google Scholar]
- 126.Pastille E, Didovic S, Brauckmann D, Rani M, Agrawal H, Schade FU, Zhang Y, Flohe SB. Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J Immunol 2011; 186: 977–86. [DOI] [PubMed] [Google Scholar]
- 127.Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood 2008; 111: 1797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139: 485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cabrera-Perez J, Babcock JC, Dileepan T, Murphy KA, Kucaba TA, Badovinac VP, Griffith TS. Gut microbial membership modulates CD4 T cell reconstitution and function after sepsis. J Immunol 2016; 197: 1692–8. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature 1988; 334: 395–402. [DOI] [PubMed] [Google Scholar]
- 131.Murugan A, Mora T, Walczak AM, Callan CG., Jr Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc Natl Acad Sci USA 2012; 109: 16161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR, Carlson CS, Warren EH. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med 2010; 2: 47ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999; 286: 958–61. [DOI] [PubMed] [Google Scholar]
- 134.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009; 114: 4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, Webb JR, Holt RA. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 2011; 21: 790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol 2013; 4: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today 1998; 19: 395–404. [DOI] [PubMed] [Google Scholar]
- 138.Lee LY, Ha do LA, Simmons C, de Jong MD, Chau NV, Schumacher R, Peng YC, McMichael AJ, Farrar JJ, Smith GL, Townsend AR, Askonas BA, Rowland-Jones S, Dong T. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J Clin Invest 2008; 118: 3478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Roti M, Yang J, Berger D, Huston L, James EA, Kwok WW. Healthy human subjects have CD4+ T cells directed against H5N1 influenza virus. J Immunol 2008; 180: 1758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Su LF, Kidd BA, Han A, Kotzin JJ, Davis MM. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 2013; 38: 373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang J, James E, Roti M, Huston L, Gebe JA, Kwok WW. Searching immunodominant epitopes prior to epidemic: HLA class II-restricted SARS-CoV spike protein epitopes in unexposed individuals. Int Immunol 2009; 21: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol 2014; 14: 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cabrera-Perez J, Condotta SA, James BR, Kashem SW, Brincks EL, Rai D, Kucaba TA, Badovinac VP, Griffith TS. Alterations in antigen-specific naive CD4 T cell precursors after sepsis impairs their responsiveness to pathogen challenge. J Immunol 2015; 194: 1609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ward PA, Bosmann M. A historical perspective on sepsis. Am J Pathol 2012; 181: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C, Remick DG. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev 2013; 93: 1247–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zinkernagel RM. Immunology taught by viruses. Science 1996; 271: 173–8. [DOI] [PubMed] [Google Scholar]
- 147.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989. 54 (Pt 1):1–13. [PubMed] [Google Scholar]
- 148.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov 2005; 4: 854–65. [DOI] [PubMed] [Google Scholar]