

Abstract
Disruption of sleep/wake activity in Alzheimer’s disease (AD) patients significantly affects their quality of life, and that of their caretakers, and is a major contributing factor for institutionalization. Levels of amyloid-β (Aβ) have been shown to be regulated by neuronal activity and to correlate with the sleep/wake cycle. Whether consolidated sleep can be disrupted by Aβ alone is not well understood. We hypothesized that Aβ42 can increase wakefulness and disrupt consolidated sleep. Here we report that flies expressing the human Aβ42 transgene in neurons have significantly reduced consolidated sleep compared to control flies. Fatty acid binding proteins (Fabp) are small hydrophobic ligand-carriers that have been clinically implicated in Alzheimer’s disease. Aβ42 flies that carry a transgene of either the Drosophila Fabp or the mammalian brain-type Fabp show a significant increase in nighttime sleep and long consolidated sleep bouts, rescuing the Aβ42-induced sleep disruption. These studies suggest that alterations in Fabp levels and/or activity may be associated with sleep disturbances in AD. Future work determining the molecular mechanisms that contribute to Fabp-mediated rescue of Aβ42–induced sleep loss will be important for the development of therapeutics in the treatment of AD.
Keywords: neurodegeneration, fruit fly, beta-amyloid
Graphical Abstract
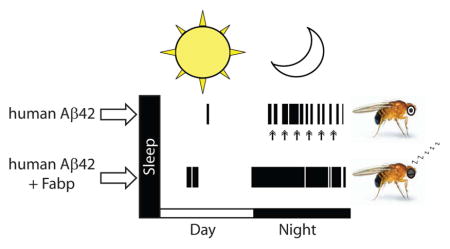
Introduction
Circadian disturbances and nocturnal problems such as sleeplessness are common among Alzheimer’s disease (AD) patients, and are major contributing factors for institutionalization (Pollak and Perlick 1991; Weldemichael and Grossberg 2010). Evidence suggests sleep is disrupted in AD (Prinz et al. 1982), and sleep disorders may increase risk of AD (Osorio et al. 2015; Osorio et al. 2011). Recent studies in humans and animal models suggest that a bidirectional relationship exists between sleep/wake behavior and amyloid-β (Aβ), a small peptide that is associated with plaque formation found in AD patients (Ju et al. 2013; Kang et al. 2009; Liguori et al. 2014; Mander et al. 2015; Roh et al. 2012; Rothman et al. 2013; Spira et al. 2013; Sprecher et al. 2015). Aβ is produced following sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretases (Haass and Selkoe 2007), and begins to aggregate in the human brain years before clinical symptoms, neuronal damage and synaptic loss in AD (Holtzman et al. 2011a; Sperling et al. 2011). Aβ aggregation forms the hallmark of insoluble plaques observed in AD pathology, it is known that clinically asymptomatic people already have reduced steady-state functional connectivity in brain areas containing amyloid deposits (Sheline et al. 2010; Sperling et al. 2009). This suggests that other biomarkers of preclinical AD associated with Aβ deposition may exist, and the identification of such factors are essential to develop treatments to slow or prevent AD pathogenesis (Holtzman et al. 2011b).
Levels of Aβ in cerebrospinal fluid (CSF) cycle based on time of day in humans, with a progressive decline in circadian amplitude occurring with age and amyloid deposition (Huang et al. 2012). Diurnal cycling of Aβ has also been shown to be negatively correlated with Aβ aggregation and increased wakefulness (Roh et al. 2012). Aβ concentrations are elevated following periods of chronic sleep restriction in AD mouse models (Kang et al. 2009; Rothman et al. 2013), supporting a positive feedback loop hypothesis between Aβ levels, clearance, aggregation, and the sleep/wake cycle (Gerstner et al. 2012a; Ju et al. 2013; Lim et al. 2014). Given that Aβ levels are higher during periods of wakefulness (Kang et al. 2009) and the peak in amplitude of cycling Aβ levels in human CSF precedes the peak in total sleep time by 6 hours (Huang et al. 2012), we were interested in testing the hypothesis that neural expression of Aβ increases wakefulness and disrupts consolidated sleep. Sleep disturbance is thought to occur at ages prior to the onset of cognitive deficits in AD, and therefore could represent a preclinical marker (Ju et al. 2014). Due to the link between sleep disturbance and Aβ levels, we also wanted to examine if any Aβ-induced sleep disruption exists at ages prior to the onset of traditionally established AD phenotypic markers.
The fruit fly, Drosophila melanogaster, is an excellent animal model system to test these questions for three reasons. First, the fly AD model recapitulates most of the clinically relevant hallmark traits of the disease, including the formation of amyloid deposits, progressive memory loss, neurodegeneration and premature death (Iijima et al. 2004; Iijima-Ando and Iijima 2010). Second, Drosophila is an established model to study sleep/wake behavior, and unlike wild-type mice which have comparatively more fragmented sleep and are nocturnal, Drosophila has a very consolidated period of sleep during the dark phase of the light-dark cycle (Cirelli and Bushey 2008; Ho and Sehgal 2005; Zimmerman et al. 2008). This becomes more fragmented with aging (Bushey et al. 2010; Koh et al. 2006; Koudounas et al. 2012). Third, the fly model offers the opportunity to rapidly evaluate specific genetic targets that could inform the therapeutic potential for treatments that delay or prevent neurodegenerative disease (Bilen and Bonini 2005; Lu and Vogel 2009).
In our study, we show that flies that express human Aβ have significant sleep fragmentation at 2–3 days post-eclosion, an age prodromal to memory deficits previously reported in adult flies (Iijima et al. 2004). AD flies that contain either the fly Fabp or the murine Fabp7 transgene rescue the Aβ-induced disruption in consolidated sleep. These findings suggest 1) Aβ peptide neuronal expression affects sleep quality, 2) Aβ-induced sleep fragmentation may be an early marker in the course of human AD, and 3) therapeutic approaches targeting Fabp may have clinical implications for reducing sleep-associated disturbances observed in AD.
Materials and Methods
Drosophila genetics and stocks
Male flies (Drosophila melanogaster) carrying a human Aβ42 transgene expressed in the presence of neuronal promoter elav (C155-GAL4) (Finelli et al. 2004; Iijima et al. 2004) were examined for changes in sleep compared with flies overexpressing either dFabp or mFabp7 (Gerstner et al. 2011a; Gerstner et al. 2011b) using a video monitoring assay (Zimmerman et al. 2008). The parental genotypes of flies used in this study are as follows: elavc155-GAL4/Y (elav-GAL4, obtained from J. Williams), GeneSwitch-elav-GAL4 (GS-elav-GAL4; Bloomington), UAS-Aβ42 (obtained from M. Konsolaki), hs-dFabp (101-4), hs-mFabp7 (103-5), and the isogenic w (isoCJ1) background strain (Gerstner et al. 2011b). Fly genotypes are all backcrossed into the w (isoCJ1) background for a minimum of 6 generations, and summarized in Table 1, with descriptions of each genotype and abbreviations. For the GeneSwitch experiments (Vanderheyden et al. 2013), food was prepared by vortexing the RU486 drug with melted food and allowing mixture to solidify. Briefly, 500μM RU486, dissolved in 80% ethanol, was prepared in 1% agar, 5% sucrose minimal media. Flies were first recorded in tubes containing only 1% agar, 5% sucrose minimal media, and after 2 days were switched to minimal media containing drug or vehicle and were replaced under the camera for the duration of the recording. Heat-shock treatment consisted of increasing the ambient room temperature to 29°C for 3 days following baseline recording.
Table 1.
Drosophila Stocks
| St# | Genotype | Notes | Abbreviation |
|---|---|---|---|
| 1 | w; UAS-Aβ42; GeneSwitch-elav-GAL4 | GeneSwitch-elav-GAL4 with UAS-Aβ42 (for conditional expression of Aβ42 specifically in neurons upon treatment with RU486) | Aβ42; GS-elav |
| 2 | w; UAS-Aβ42; + | UAS-Aβ42 (control) | UAS-Aβ42/+ |
| 3 | w; + ; GeneSwitch-elav-GAL4 | GeneSwitch-elav-GAL4 Driver (control) | GS-elav/+ |
| 4 | w (isoCJ1); +; + | wild type isogenic background strain (also known as 2202U; control) | w (isoCJ1) |
| 5 | w, elav-GAL4; + ; + | elav-GAL4 Driver (control) | elav/Y |
| 6 | w, elav-GAL4; UAS-Aβ42; + | elav-GAL4 with UAS-Aβ42 (for constitutive expression of Aβ42 specifically in neurons) | Aβ42/+ |
| 7 | w; UAS-Aβ42; hs-dFabp | UAS-Aβ42 with hs-dFabp (control) | UAS-Aβ42/+; dFabp/+ |
| 8 | w, elav-GAL4; + ; hs-dFabp | elav-GAL4 with hs-dFabp (control) | dFabp/+ |
| 9 | w, elav-GAL4; UAS-Aβ42; hs-dFabp | elav-GAL4 with UAS-Aβ42 and hs-dFabp (for constitutive expression of Aβ42 specifically in neurons, with dFabp overexpression) | Aβ42/+;dFabp/+ |
| 10 | w, elav-GAL4; UAS-Aβ42; hs-mFabp7 | elav-GAL4 with UAS-Aβ42 and hs-mFabp7 (for constitutive expression of Aβ42 specifically in neurons, with mFabp7 overexpression) | Aβ42/+;mFabp7/+ |
Sleep behavior
Flies were collected following eclosion and placed on standard cornmeal agar media. 2–3 days after eclosion, flies were transferred individually under CO2 anaesthesia into tubes 6 cm long containing food at one end and a yarn plug at the other. Behavioral recordings of fly sleep were collected under 12 hours white light:12 hours infrared light (wavelength 950 nm) at 25°C in Precision Instruments 818 incubators equipped with two fluorescent bulbs and 2 infrared lights mounted horizontally beside cameras (Zimmerman et al. 2008). Cameras took an image every 5 seconds and Matlab CSCN analysis used pixel subtraction to track movement of flies. Analytic software can be obtained by contacting Dr. Pack (allan.pack@uphs.upenn.edu). Sleep was defined as five or more minutes of continuous inactivity, and was the minimum amount of time to qualify as a single sleep bout. The assessment of activity/inactivity is explained more fully below. ZT (zeitgeber time) 0 was defined as the time of lights on, and ZT 12 as the time of lights off. Daytime sleep measures were from lights on to lights off (ZT0-ZT12), nighttime measures were from lights off to lights on (ZT12-ZT24). Average sleep bout duration was defined as the mean of all sleep bouts for the given period for the group. Average maximal sleep bout duration was defined as the mean of the longest sleep bout of all flies within a group. Bout counts were defined as the mean number of transitions between sleep and wake states for the group. Latency is defined as the amount of time to first sleep episode following lights off (ZT12).
Consolidation index
Consolidation index (CI) is a measure of consolidation of sleep for a group. Greater CI values reflect less interrupted sleep (i.e., less sleep fragmentation), while lower CI values indicate more sleep fragmentation. CI was calculated by summing the square of all sleep bout lengths (in minutes) and dividing by the total amount of sleep (Pitman et al. 2006).
Locomotor activity
The digital image analysis method previously described is based on frame subtraction and was used to determine a quiescent fly from a moving fly (Zimmerman et al. 2008), as well as calculate locomotor activity. The amount of movement of a fly was calculated by summing the number of pixels changed between consecutive subtracted images, at the same sampling rate used to determine sleep (5 seconds). Briefly, corresponding pixels from 2 temporally adjacent images are subtracted and a resulting DIFFERENCE image is generated. Each pixel in the DIFFERENCE image has the value GS(XiYj)=[(GS2(XiYj)–GS1(XiYj))/2]+127, where GS(XiYj) is the DIFFERENCE image grayscale value centered around a value of 127 at pixels X position i and Y position j and GS2 and GS1 are the grayscale values at that same pixel for the second and first video frames, respectively, in a pair of temporally adjacent frames. The images were digitized using 8 bits per pixel; therefore, the range in grayscale values for a single pixel was from 0 to 255. Areas of interest corresponding to each monitor tube, ie, each fly, were analyzed separately. Gray pixels, ones in which motion did not occur, have values close to 127. They are not exactly 127 because of noise in image acquisition. Since the degree of noise varied between incubators and cameras, for every experiment, the degree of this noise was determined by analyzing a portion of the image that was outside the monitoring tubes and therefore contained no movement. Movement per minute wake was defined by summing the number of pixels changed between images accounting for this inherent noise, and dividing by the amount of time (minutes) spent in wakefulness, over each one hour bin.
Reverse transcription quantitative polymerase chain reaction
RNA transcript levels were determined using reverse transcription quantitative polymerase chain reaction (qPCR). For qPCR experiments, 50 fly heads were collected at ZT6 for each of six biological replicates. RNA was isolated using the TRIzol reagent (Life Technologies) per the manufacturer’s protocol, with the addition of an overnight incubation in isopropanol at −20°C before a 30 minute centrifugation at 13,000g in 4°C to precipitate the RNA. RNA pellets were resuspended in nuclease-free water and subjected to an RQ1 RNase-Free DNase (Promega) treatment following the manufacturer’s protocol, followed by a second RNA purification as before. RNA was quantified using an ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, Delaware). Complementary DNA was generated using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and 4μg of total RNA. The reaction volume was brought to 20μl total volume with PCR grade water and the cDNA synthesis was completed with the PTC-100 Programmable Thermal Controller (MJ Research, Inc.) per the kit’s instructions. Quantitative PCR was carried out using PowerSYBR Green PCR Master Mix (Applied Biosystems) and 10ng cDNA per reaction, with technical duplicates of each sample. All reactions were run using the 7900HT Fast Real-Time PCR System (Applied Biosystems) and analyzed with the 7900HT software, Science Detection System 2.4. The primers used were as follows: dFabp-AB forward primer, ATCATCACCCTGGATGGCAA, reverse primer, ATGGTGAGGGTGGTGATCAG; mFabp7 forward primer, CCAGCTGGGAGAAGAGTTTG, reverse primer, TTTCTTTGCCATCCCACTTC; hAB42 forward primer, GCAGAATTCCGACATGACTCA, reverse primer, TGCACCTTTGTTTGAACCCA; dGapdh2 forward primer, TTTCTCAGCCATCACAGTCG, reverse primer, CCGATGCGACCAAATCCATT; actin forward primer, ACACACCCATTGCAGACAAA, reverse primer, TTCTGAAGGAGCGGAAGTGT; and RPL32 forward primer, GCTAAGCTGTCGCACAAATGG, reverse primer GGTGGGCAGCATGTGG. The results were normalized against the geometric mean of the mRNA levels of the three housekeeping genes, dGapdh2, actin and RPL32, to determine the VCt (cycle threshold) values. For GeneSwitch studies, 50 fly heads were collected at ZT6 for each of six biological replicates with either 3–4 days of RU486 or vehicle treatment (as described above), and RNA was prepared and quantified as above.
Western blotting
For each genotype, 50 fly heads were homogenized in Tris-HCl buffer with Protease Inhibitor Cocktail (Sigma P2714) for 45 seconds and centrifuged at 14,000 rpm for 1 minute. Lysate samples were separated on 12% Tris-HCl 0.75mm gels and transferred to nitrocellulose membranes (Invitrogen). The membranes were blocked with 5% nonfat dry milk (Nestle) and blotted with anti-dFabp antibody (1:1000), anti-actin antibody (1:1000, Cell Signaling Technology), and anti-rabbit secondary antibody IR 680 (1:5000, LI-COR Biotechnology) according to previously published methods (Gerstner et al. 2011a; Gerstner et al. 2011b). Each timepoint had three biological replicates and consisted of N=50 fly heads pooled per timepoint (N=50 × 3 biological replicates).
Antibody Characterization
Please see Table 2 for a list of all antibodies used.
Table 2.
Table of Primary Antibodies Used
| Antigen | Description of Immunogen | Source, Host Species, Cat.#, Clone or Lot#, RRID | Concentration/Dilution Used |
|---|---|---|---|
| Actin | Monoclonal antibody is produced by immunizing animals with a synthetic peptide corresponding to residues near the amino-terminus of human β-actin protein. | Cell Signaling Technology; β-Actin (13E5) Rabbit mAb #4970 | 1:1000 |
| dFabp | Recombinant Protein corresponding to amino acids 1–130 of Drosophila dFabp-B | Generated at Cocalico Biologicals Inc., Rabbit (#181); (Gerstner et al., 2011b) | 1:1000 |
The beta-Actin antiserum recognized only the expected monomeric (45 kD) band on western blot of Drosophila head lysate.
The dFabp antiserum recognized a single band of 14.5 kD molecular weight on western blots of Drosophila head lysate (Gerstner et al. 2011b).
Statistical analyses
Statistical significance was determined using a two-tailed Student’s t-test, and stated whether equal variance is assumed for each test as indicated; unless otherwise indicated when appropriate, we used an ANOVA (SigmaPlot 11), the type (i.e. one-way or two-way) and post-hoc tests (for example, Holm-Sidak to test multiple comparisons) as indicated in text and figure legends. Values were accepted as significant if P < 0.05. Flies were excluded from sleep analysis if they died during the course of the experiment, and figures where multiple days are present, sample sizes (N) are consistent throughout and indicated in figure legends.
Results
Short-term neuronal human Aβ expression induces wakefulness in Drosophila
To test the hypothesis that neuronal Aβ expression increases wakefulness and disrupts consolidated sleep, we employed the GeneSwitch (GS) GAL4 system of Drosophila, which allows for the spatial and temporal control of transgene expression (Nicholson et al. 2008; Osterwalder et al. 2001; Roman et al. 2001). In the presence of the steroid RU486 (mifepristone), the upstream activating sequence (UAS) effector line UAS-Aβ42 (Iijima et al. 2004) conditionally expresses the human Aβ42 via the pan-neural driver GeneSwitch-elav-GAL4 (Osterwalder et al. 2001). Previous reports using a similar model show that the GeneSwitch-elav-GAL4 system can induce UAS-driven Arctic Aβ42 mRNA levels with 2 days of exposure to RU486 (Rogers et al. 2012), and that the GS-inducible drug does not have any effect on sleep (Vanderheyden et al. 2013). Short-term neuronal expression (3 days) of Aβ42 of adult w; UAS-Aβ42; GeneSwitch-elav-GAL4 (Aβ42; GS-elav) flies yielded a significant increase in Aβ42 transcript levels, but not in either w; UAS-Aβ42; + (UAS-Aβ42/+) or w; + ; GeneSwitch-elav-GAL4 (GS-elav/+) control flies (Fig. 1A). We also observe a decrease in sleep specific to the daytime in Aβ42; GS-elav flies following short-term expression of Aβ42 (Fig. 2A). The average daytime sleep bout length and the average maximum daytime sleep bout length are both reduced upon short-term neural Aβ42 activation (Fig. 2, B and C). The number of daytime bouts was also significantly increased upon Aβ42 neural expression (Fig. 2D), while the consolidation index (CI, a measure of consolidated sleep; see methods for definition) was concomitantly reduced (Fig. 2E). No difference was observed in nighttime consolidation (Fig. 2F). The reduction in daytime sleep was not due to increased activity (defined as pixels moved, see methods), since daytime movements were unchanged between RU486-treated and untreated control flies (Fig. 2G). However, Aβ-induced a reduction in nighttime activity (Fig. 2H). Total nighttime sleep, average night bout length, average maximum night bout length, average number of night bouts, and latency to sleep following lights off were not significantly affected between RU486-treated and untreated flies (data not shown). Analysis of individual days of RU486 treatment revealed a progressive decrease in average daytime sleep bout length and average maximum daytime bout length (Fig. 3, A and B) over the 3 days of observation. This progressive loss in consolidated daytime sleep was not associated with any changes in daytime activity (Fig. 2G, Fig. 3C). However, Aβ-induced nighttime activity was reduced on days 1–3 (Fig. 3D). These data indicate that Aβ expression alone can modulate daytime sleep, and these effects are not explained by changes in movement.
Fig. 1.

RU486 induces Aβ42 and does not alter dFabp mRNA expression in w; UAS-Aβ42; GeneSwitch-elav-GAL4 flies. (A) Induction of Aβ42 mRNA in w; UAS-Aβ42; GeneSwitch-elav-GAL4 (Aβ42; GS-elav) flies compared to w; UAS-Aβ42; + (UAS-Aβ42/+) and w; + ; GeneSwitch-elav-GAL4 (GS-elav/+) control flies treated (+) or untreated (−) with RU486 for 3 days assessed by qPCR. (B) Induction of dFabp mRNA in Aβ42; GS-elav flies compared with UAS-Aβ42/+ and GS-elav/+ control flies treated (+) or untreated (−) with RU486 for 3–4 days assessed by qPCR. (n = 50 per replicate; 6 replicates/genotype; two-tailed Student’s t-test, equal variance). ***P < 0.001. Values represent means +/− SEM.
Fig. 2.

Short-term neuronal Aβ induction fragments daytime sleep. (A to F) Comparison of daytime sleep profile in w; UAS-Aβ42; GeneSwitch-elav-GAL4 (Aβ42; GS-elav) flies treated (+) or untreated (−) with RU486 averaged across 3 days assessed as minutes (min) of Sleep (A to C), Number of Bouts (D), Consolidation (E) during the 12 hour light period, and Consolidation (F) during the 12 hour dark period. (G to H) Comparison of activity profile in Aβ42; GS-elav flies treated (+) or untreated (−) with RU486 averaged across 3 days assessed as pixels moved during the light period (G) dark period (H) (n = 14 per group; A–G, two-tailed Student’s t-test, equal variance). *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means +/− SEM. ZT, zeitgeber time.
Fig. 3.

Conditional neuronal expression of Aβ induces progressive sleep loss. (A to C) Comparison of daytime profile in w; UAS-Aβ42; GeneSwitch-elav-GAL4 (Aβ42; GS-elav) flies treated (+) or untreated (−) with RU486 assessed as minutes (min) of Sleep (A and B), and Pixels Moved (C) during the 12 hour light period across 3 days (d1, d2, and d3). (D) Comparison of activity profile in Aβ42; GS-elav flies treated (+) or untreated (−) with RU486 assessed as pixels moved during the night period across 3 days. (n = 14 per group; p<0.001 two-way ANOVA, factors DAY and RU486 treatment, post-hoc Holm-Sidak). *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means +/− SEM. ZT, zeitgeber time.
Constitutive neuronal expression of human Aβ disrupts both daytime and nighttime sleep in Drosophila
Since we observed that short-term neuronal expression of Aβ can modulate sleep, and that sleep disturbance may occur at ages prior to cognitive impairments in AD, we next wanted to characterize the effect of constitutive Aβ expression on sleep in flies. We used a well-established genetic model that was previously determined to possess the negative effects of Aβ-induction such as reduced cognitive performance, climbing ability and survival, as well as neurodegeneration (Iijima et al. 2004). We specifically determined whether Aβ-induced sleep disruption might exist at ages prior to the onset of traditionally established AD phenotypic markers. To do this, we examined sleep in 2–3 day old male w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies, which express UAS-Aβ42 under control of the constitutive pan-neural driver elav-GAL4, and compared them to controls. Previous work has established that learning impairments not yet observed in 2–3 day old Aβ42/+ flies occur later on, first appearing in males (but not yet in females) by 6–7 days old, with progressive loss in both males and females by 14–15 days old (Iijima et al. 2004). Compared to 2–3 day old male controls, we observed sleep reduction in age-matched Aβ42/+ male flies (Fig. 4, A–C, and G). This Aβ-induced sleep loss in Aβ42/+ flies was accompanied by a reduction in daytime maximal sleep bout length (Fig. 4E), with a decrease in day bout number (Fig. 4F). The average day and night sleep bout length was not affected in Aβ42/+ flies compared to w, elav-GAL4; + ; + (elav/Y) controls (Fig. 4D and H, respectively). Similarly, the average maximal night sleep bout and night bout counts were not affected in Aβ42/+ flies compared to w, elav-GAL4; + ; + (elav/Y) controls (Fig. 4I and J, respectively). Control UAS-Aβ42/+ flies that carry UAS-Aβ42 in the absence of the driver line had more sleep time (24 hour) and more total nighttime sleep, with no differences in night bout number, night bout length, maximum night bout length, or consolidation index compared to w (isoCJ1); +; + (w (isoCJ1)) isogenic background strain controls (Fig. 5, A–F). These data indicate that constitutive neuronal Aβ expression decreases total sleep and increases sleep fragmentation, and that these changes in sleep likely precede other phenotypic markers of AD, including cognitive impairments.
Fig. 4.

Constitutive neuronal Aβ42 disrupts sleep, and is rescued by Fabp overexpression. (A and B) Comparison of diurnal sleep profile in 2–3 day old w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies against control w, elav-GAL4; + ; + (elav/Y) flies, w, elav-GAL4; + ; hs-dFabp (dFabp/+) flies, and w, elav-GAL4; UAS-Aβ42; hs-dFabp (Aβ42/+; dFabp/+) flies assessed as minutes (min) of Sleep during the full 24 hour period. Significance for each point is shown below the x-axis in (A). (C to F) Comparison of daytime sleep profile in 2–3 day old Aβ42/+ flies against control elav/Y, w, dFabp/+ and Aβ42/+; dFabp/+ flies assessed as minutes (min) of Sleep (C to E) and Number of Bouts (F) during the 12 hour light period. (G to J) Comparison of nighttime sleep profile in 2–3 day old Aβ42/+ flies against control elav/Y, w, dFabp/+ and Aβ42/+; dFabp/+ flies assessed as minutes (min) of Sleep (G to I) and Number of Bouts (J) during the 12 hour dark period. (elav/Y and Aβ42/+ n = 40/group; dFabp/+ n = 36; Aβ42/+; dFabp/+ n = 34; A, p<0.001 two-way ANOVA, factors GENOTYPE and TIME, post-hoc Holm-Sidak; B-J, p<0.001 one-way ANOVA, post-hoc Holm-Sidak). *P < 0.05, **P < 0.01, ***P < 0.001, n.s. (not significant). Values represent means +/− SEM. ZT, zeitgeber time. Light bars = light period; dark bars = dark period.
Fig 5.

Control w; UAS-Aβ42; + and w; UAS-Aβ42; hs-dFabp flies do not exhibit any difference in sleep consolidation compared to w (isoCJ1) isogenic background strain. (A to F) Comparison of sleep profile in 2–3 day old isogenic background strain control w (isoCJ1) flies against control w; UAS-Aβ42; + (UAS-Aβ42/+) and w; UAS-Aβ42; hs-dFabp (UAS-Aβ42/+; dFabp/+) flies assessed as minutes (min) of Sleep for the 24 Hour period (A), and as minutes of Sleep (B, C, and D), Number of Bouts (E), and Consolidation Index (F) during the 12 hour dark period. (w (isoCJ1) n = 74/group; UAS-Aβ42/+ n = 74; UAS-Aβ42/+; dFabp/+ n = 49; one-way ANOVA, post-hoc Holm-Sidak). *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means +/− SEM.
Constitutive neuronal Aβ expression effects on dFabp protein and dFabp mRNA levels
In order to determine whether Aβ can influence the expression of Fabp, we measured dFabp mRNA levels using reverse transcription quantitative PCR (qPCR) in Aβ42 transgenic and control flies. First, we evaluated dFabp expression in Aβ42; GS-elav flies with and without RU486. We did not observe any changes in dFabp mRNA in Aβ42; GS-elav flies compared with control flies (Fig. 1B). Second, we measured levels of dFabp mRNA in Aβ42/+ flies and compared with elav/Y and UAS-Aβ42/+ controls, and also did not see differences (Fig. 6A). To determine whether Aβ42 affects dFabp at the protein level, we next measured dFabp at four timepoints across the light-dark cycle in 2–3 day old male Aβ42/+ flies compared to controls using western blotting. Flies that express Aβ42 appeared to have a disruption of dFabp protein levels in the light-to-dark transition compared to controls (Fig. 7A). We then compared dFabp expression in Aβ42/+ flies compared to elav/Y controls (Fig. 7B), and found a trend for reduction of dFabp in Aβ42/+ flies (p=0.054 two-way ANOVA, factors GENOTYPE and TIME). These data indicate that constitutive neuronal Aβ expression may regulate diurnal dFabp protein levels but does not affect dFabp mRNA levels.
Fig. 6.

Expression of dFabp mRNA is normal in w, elav-GAL4; UAS-Aβ42; + flies. (A) qPCR results of dFabp mRNA in 2–3 day old w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) fly heads compared to w, elav-GAL4; + ; + (elav/Y) and w; UAS-Aβ42; + (UAS-Aβ42/+) control flies. Also shown are w, elav-GAL4; UAS-Aβ42; hs-dFabp (Aβ42/+; dFabp/+) and w, elav-GAL4; UAS-Aβ42; hs-mFabp7 (Aβ42/+; mFabp7/+) flies. No significant differences were observed between groups (one-way ANOVA n = 6/group; each group consists of ~50 heads). (B) qPCR results of mFabp7 mRNA in the heads of 2–3 day old Aβ42/+; mFabp7/+ flies compared to elav/Y, UAS-Aβ42/+, Aβ42/+, and Aβ42/+; dFabp/+ flies. Significant differences were observed between groups (one-way ANOVA, P<0.001, Holm-Sidak post-hoc; n=6/group; each group consists of 50 heads). Values represent means +/− SEM.
Fig. 7.

Expression of dFabp protein is reduced in w, elav-GAL4; UAS-Aβ42; + flies. (A to B) Western blots show a trend in reduction of dFabp protein in the heads of 2–3 day old w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies compared to control w, elav-GAL4; + ; + (elav/Y) flies. (A) Diurnal western blot of Actin (44kD) and dFabp (14.5kD) in elav/Y and Aβ42/+ fly heads. (B) Average normalized dFabp expression level shows a trend in reduction in Aβ42/+ flies compared to elav/Y controls (P=0.054 two-way ANOVA, factors for GENOTYPE and TIME). Values represent means +/− SEM. ZT, zeitgeber time. Light bars = light period; dark bars = dark period.
Aβ-induced sleep loss is rescued by fatty acid binding proteins
Previously we have shown that overexpression of either dFabp or the mouse brain fatty acid binding protein (mFabp7) in Drosophila is able to modulate sleep and enhance cognitive performance (Gerstner et al. 2011a; Gerstner et al. 2011b). Therefore, we next wanted to examine if the Aβ42-induced sleep disruption in 2–3 day old flies could be rescued by an extra copy of Fabp. We observed that the reduced sleep in Aβ42/+ flies could be rescued in age-matched w, elav-GAL4; UAS-Aβ42; hs-dFabp (Aβ42/+;dFabp/+) flies over the course of the day (Fig. 4A; p<0.001 two-way ANOVA, factors GENOTYPE and TIME, post-hoc Holm-Sidak). We then determined the amount of total sleep (24 hours) in Aβ42/+ flies, and found a significant reduction in sleep compared to elav/Y and elav-GAL4; + ; hs-dFabp (dFabp/+) controls, which was rescued by dFabp (Fig. 4B; p<0.001 one-way ANOVA, post-hoc Holm-Sidak). Similar effects were observed for total sleep during day (Fig. 4C) and night (Fig. 4G). This dFabp rescue of Aβ-induced sleep loss is accompanied by an increase in daytime maximal bout length (Fig. 4E), with nominal effects on other sleep measures (Fig. 4, D, F, H–J). Control w; UAS-Aβ42; hs-dFabp (UAS-Aβ42/+; dFabp/+) flies (without the elav-GAL4 driver) had less total sleep time (24 hour) compared to UAS-Aβ42/+ control flies, with nominal effects on other sleep measures (Fig. 5). Control UAS-Aβ42/+ and UAS-Aβ42/+; dFabp/+ flies did not exhibit any differences in sleep consolidation compared to the w (isoCJ1) isogenic background strain (Fig. 5I). Previously we have shown heat-shock induction of dFabp in flies further augments sleep and enhances cognitive performance (Gerstner et al. 2011b). Aβ-induced sleep loss that was observed in Aβ42/+ flies was rescued in Aβ42/+; dFabp/+ flies with heat-shock treatment (Fig. 8, p<0.001 two-way ANOVA, factors GENOTYPE and TIME, post-hoc Holm-Sidak). These data suggest that Aβ42-induced sleep loss can be rescued by fatty acid binding proteins.
Fig. 8.

Heat-shock induction of Fabp rescues neuronal Aβ42-induced sleep/wake disruption. Comparison of diurnal sleep profile with heat-shock treatment (5-days at 29°C) in w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies against control w, elav-GAL4; + ; + (elav/Y), w, elav-GAL4; + ; hs-dFabp (dFabp/+), and w, elav-GAL4; UAS-Aβ42; hs-dFabp (Aβ42/+; dFabp/+) flies assessed as minutes (min) of Sleep during the full 24 hour period. Significance for each point is shown below the x-axis in. (elav/Y and Aβ42/+ n = 39/group; dFabp/+ n = 32; Aβ42/+; dFabp/+ n = 34; A, p<0.001 two-way ANOVA, factors GENOTYPE and TIME, post-hoc Holm-Sidak, *P < 0.05. Values represent means +/− SEM. ZT, zeitgeber time. Light bars = light period; dark bars = dark period.
Aβ42-induced sleep fragmentation is rescued by fatty acid binding proteins
Since Aβ induces reductions in total sleep (Fig. 4B) and maximal sleep bout length (Fig. 4E), this suggests that constitutive neuronal Aβ expression disrupts consolidated sleep. Therefore, we were interested in examining aspects of sleep fragmentation in Aβ42/+ flies. Examples of individual diurnal sleep bout histograms for each genotype show that timing and length of sleep bouts are altered in Aβ42/+ flies compared to controls, and that the Aβ42-induced sleep fragmentation is rescued in Aβ42/+; dFabp/+ flies (Fig. 9, A to D). We also measured nighttime sleep consolidation, and observed Aβ42/+ flies have a reduced consolidation index compared to controls, which is rescued in Aβ42/+; dFabp/+ flies (Fig. 9E).
Fig. 9.

Constitutive neuronal Aβ42 fragments sleep, and is rescued by Fabp overexpression. (A to D) Sleep bout histograms showing four representative examples of each group by genotype. Dark bars in histograms are sleep bouts, open space is wake. Fragmented sleep in Aβ42/+ flies is indicated by arrows. (E) Consolidation Index during the ZT12–24 hour dark period in 2–3 day old w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies against w, elav-GAL4; + ; + (elav/Y) flies, w, elav-GAL4; + ; hs-dFabp (dFabp/+) flies, and w, elav-GAL4; UAS-Aβ42; hs-dFabp (Aβ42/+; dFabp/+) flies. (elav/Y n = 31; Aβ42/+ n = 32; dFabp/+ n = 37; Aβ42/+; dFabp/+ n = 26; p<0.001 one-way ANOVA, post-hoc Holm-Sidak). **P < 0.01, ***P < 0.001. Values represent means +/− SEM. ZT, zeitgeber time. Light bars = light period; dark bars = dark period.
We were also interested in examining sleep in Aβ42/+ flies carrying a copy of the mouse Fabp, mFabp7, in order to determine if these Aβ42-induced effects can also be rescued by the mammalian ortholog. Similar to Aβ42/+; dFabp/+ flies, we observed an increase in nighttime sleep in w, elav-GAL4; UAS-Aβ42; hs-mFabp7 (Aβ42/+; mFabp7/+) flies (Fig. 10A). This increase in sleep was also accompanied by an increase in average night sleep bout length (Fig. 10B), and average nighttime maximal bout length (Fig. 10C). No change was observed in night bout count (Fig. 10D). The sleep consolidation index was also augmented in Aβ42/+; mFabp7/+ flies (Fig. 10E). Expression of mFabp7 mRNA was only observed in Aβ42/+; mFabp7/+ overexpressing flies, and not detected in elav/Y, UAS-Aβ42/+, or Aβ42/+; dFabp/+ flies (Fig. 6B). Together, these data indicate that Aβ42 induces sleep fragmentation, and that expressing either fly or mammalian Fabp7 is sufficient to rescue this effect.
Fig. 10.

Murine Fabp7 rescues constitutive neuronal Aβ42-induced sleep loss and fragmentation. (A to E) Comparison of night sleep profile in w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies against w, elav-GAL4; UAS-Aβ42; hs-mFabp7 (Aβ42/+; mFabp7/+) flies assessed as minutes (min) of Sleep (A to C) and Number of Bouts (D) during the 12 hour dark period. (E) Consolidation Index during the 12 hour dark period. (Aβ42/+ n = 31; Aβ42/+;mFabp7/+ n = 25; two-tailed Student’s t-test, equal variance). *P < 0.05, **P < 0.01. Values represent means +/− SEM.
Constitutive neuronal expression of human Aβ increases activity
Since we previously demonstrated that constitutive neuronal Aβ expression induces fragmented sleep, we also tested whether the presence of Aβ is associated with changes in locomotor activity. Therefore, we examined movement in Aβ42/+ flies and found that there was an increase in pixels moved compared to controls (Fig. 11, A and B), which was more pronounced at night (Fig. 11, C and D). Rescue of Aβ-induced activity was observed in Aβ42/+; dFabp/+ flies (Fig. 11 A and B, p<0.001 two-way ANOVA, factors GENOTYPE and TIME, post-hoc Holm-Sidak). Movement per minute of wakefulness, a measure used to indicate differences in hyperactivity, differs from normal activity measures (pixels moved), since it accounts for the amount of activity per unit time spent in wake. Hyperactivity did not differ in Aβ42/+ flies compared to controls during the day or night (Fig. 11, E and F), suggesting that the mechanism for Aβ–induced activity is at least partially distinct from that for Aβ–induced sleep disturbance associated with hyperactivity.
Fig 11.

Neuronal expression of Aβ42 peptide induces activity. (A and B) Comparison of movement profiles in 2–3 day old w, elav-GAL4; UAS-Aβ42; + (Aβ42/+) flies against control w, elav-GAL4; + ; + (elav/Y) flies, w, elav-GAL4; + ; hs-dFabp (dFabp/+) flies, and w, elav-GAL4; UAS-Aβ42; hs-dFabp (Aβ42/+; dFabp/+) flies assessed as pixels moved (A and B). (C to F) Comparison of movement profiles in 2–3 day old Aβ42/+ flies against elav/Y, dFabp/+ and Aβ42/+; dFabp/+ flies assessed as pixels moved (C and D) and pixels moved per minute wake (Movement/min Wake; E and F). (elav/Y and Aβ42/+ n = 40/group; dFabp/+ n = 36; Aβ42/+; dFabp/+ n = 34; (A and B; p<0.001 two-way ANOVA, factors GENOTYPE and TIME, post-hoc Holm-Sidak; C-F one-way ANOVA, post-hoc Holm-Sidak). *P < 0.05, **P < 0.01, ***P < 0.001. Values represent means +/− SEM. ZT, zeitgeber time.
Discussion
In this study, we observed that human Aβ disrupts sleep in a Drosophila model of Alzheimer’s disease. Inducible transgenic neuronal expression of Aβ disrupted daytime consolidated sleep. This data provides evidence to suggest that neuronal Aβ expression alone is able to disrupt sustained periods of sleep, prior to long-term accumulation and subsequent aggregation. Constitutive neuronal Aβ expression disrupted consolidated nighttime sleep as well as reduced total sleep time in flies 2–3 days post-eclosion (or 2–3 day old flies). We were able to rescue this Aβ-induced sleep fragmentation in transgenic flies carrying an additional copy of dFabp. Similar findings were observed in transgenic flies carrying a copy of the mouse brain-type fatty acid binding protein, mFabp7. These findings indicate that Aβ alone can perturb consolidated sleep behavior, and that fatty acid binding protein genes rescue Aβ-induced sleep fragmentation.
The sleep/wake cycle is a behavior that occurs broadly throughout animal phyla, and is an essential property of normal brain function. Disturbances in sleep across many species, including flies, mice and humans, have been shown to cause cognitive impairment and memory deficits (Abel et al. 2013; Ganguly-Fitzgerald et al. 2006; Goel et al. 2013; Graves et al. 2001; McDermott et al. 2003). Aβ accumulation is also believed to cause cognitive deficits across these species (Iijima et al. 2004; Kittelberger et al. 2012; Lesne et al. 2006; Lesne et al. 2013). It has been shown that circadian rhythms are affected by neural expression of Aβ in flies while the molecular clock remains intact (Chen et al. 2014), suggesting Aβ may influence other behaviors such as sleep. Recently, one study observed that increases in Aβ led to reduced and fragmented sleep in Drosophila (Tabuchi et al. 2015), similar to our observations here. Studies on AD mouse models and AD patients also support a role for Aβ-associated sleep/wake perturbations (Jyoti et al. 2010; Vitiello and Borson 2001; Wisor et al. 2005; Wu and Swaab 2007; Zhang et al. 2005). Whether Aβ affects consolidated sleep at ages prior to the onset of amyloid-associated cognitive decline has remained elusive, however sleep disturbance may be a preclinical marker of AD, occurring at ages before cognitive disturbance (Ju et al. 2014; Lim et al. 2014). Here we found that short-term expression of Aβ disrupted consolidated sleep in flies, although these effects were specific to daytime. Short-term Aβ expression reduced daytime sleep consolidation (Fig. 2E) but trended to increase night time sleep consolidation (Fig. 2F). This induced expression occurred on a time-scale within days (Fig. 1), therefore the differences in day and night time sleep consolidation likely are from short term effects of Aβ, as opposed to long-term effects that may include more extracellular accumulation. We also provide evidence that long term expression of Aβ is able to disrupt daytime and night time sleep (Fig. 4). Flies with constitutive neuronal Aβ expression exhibit loss of both daytime and nighttime sleep at an age that precedes learning deficits previously reported using this same model (Iijima et al. 2004). Since short term expression of Aβ already disrupts sleep (albeit daytime sleep; Figure 2 and 3), while long term Aβ expression disrupts both daytime and night time sleep that is rescued by dFabp and mFabp7 (Figures 4, 9, 10), this suggests that 1) chronic Aβ expression generates progressive sleep loss, and 2) this progressive sleep loss can be rescued by Fabp. This also suggests that disrupted sleep may be prodromal to Aβ-associated cognitive decline in Alzheimer’s disease. Previously it was shown that interstitial levels of Aβ increase in the brains of young AD model mice during wakefulness and decrease during sleep (Roh et al. 2012), even in the absence of Aβ plaques, similar to wakefulness-associated levels of Aβ in human cerebrospinal fluid (Bateman et al. 2007; Huang et al. 2012). Our results show that short-term Aβ expression results in decreased sleep in flies, indicating that the Aβ peptide can induce wakefulness, albeit only in the daytime. On the other hand, our results demonstrate that long-term constitutive neuronal expression of Aβ is capable of disrupting both daytime and nighttime sleep, suggesting that Aβ aggregation may be responsible for perturbing consolidated nighttime sleep, similar to recent reports (Tabuchi et al. 2015). Our study did not discriminate between intracellular and extracellular Aβ accumulation over time, however other studies using similar models have indicated a relationship between Aβ accumulation and sleep disruption (Ju et al. 2014; Lim et al. 2014). It has been shown that Aβ aggregation leads to dysregulation of the sleep/wake cycle, and sleep deprivation in turn produces further Aβ aggregation (Roh et al. 2012; Tabuchi et al. 2015). Recently, sleep has been shown to facilitate clearance of Aβ in mammals (Xie et al. 2013) and reduce Aβ accumulation in flies (Tabuchi et al. 2015). Levels of Aβ in human CSF associated with deposition were also linked to sleep disruption in preclinical AD (Ju et al. 2013). Taken together, these data suggest a positive feedback loop between sleep/wake behavior and Aβ levels, metabolism and aggregation (Gerstner et al. 2012a; Lim et al. 2014). An early disruption in consolidated sleep may accelerate Aβ accumulation, which would lead to further wakefulness-associated Aβ release, aggregation, sleep disruption and cognitive loss (Gerstner et al. 2012a; Kang et al. 2009). Future longitudinal studies in animal models and humans will be needed to assess whether such a relationship between AD pathology and disruption in sleep/wake states exists.
Previously, we and others have shown changes in the time-of-day expression of dFabp mRNA in normal flies (Ceriani et al. 2002; Gerstner et al. 2011b) and Fabp7 mRNA in mice (Gerstner et al. 2008; Gerstner et al. 2006; Gerstner et al. 2012b; Panda et al. 2002). Additionally, we have found that flies that overexpress dFabp or mFabp7 have improved cognitive performance and increased sleep (Gerstner et al. 2011b). We also reported that the relative nuclear to cytoplasmic ratio of Fabp correlates with specific forms of cognitive performance in flies (Gerstner et al. 2011a). Recently it has been shown that increasing dFabp expression rescues memory deficits in learning and memory in mutant flies, and that sleep reverses memory defects in a fly Alzheimer’s model (Dissel et al. 2015). Our studies suggest that the Aβ-induced nighttime sleep fragmentation can be rescued genetically with additional copies of either dFabp or mFabp7. It remains possible that increasing Fabp expression may enhance sleep through compensatory mechanisms rather than restoring sleep because it mitigates the impact of Aβ on sleep. Future studies determining functional relationships between Fabp expression and Aβ aggregation will be important for understanding the relationship between Aβ, Fabp, and sleep. While we used the mFabp7 transgenic flies given our previous studies (Gerstner et al. 2011a; Gerstner et al. 2011b), future work using hFabp7 transgenic flies will be important for determining whether the human Fabp7 ortholog can ameliorate the negative effects of Aβ on sleep. Other future studies determining whether reducing Fabp expression leads to sleep loss, and negatively effects Aβ aggregation will be also be important to elucidate mechanisms relating Aβ, Fabp, and sleep. Because we cannot rule out possible developmental effects in our model, future studies directed at identifying whether Fabp can also rescue cognitive deficits, Aβ42 aggregation and reduced longevity in the inducible Aβ42; GS-elav fly model will be important for our understanding of Fabp pathways in AD. Previous studies have shown progressive learning impairments first appearing in males (but not females) by 6–7 days old, with further learning deficits in both genders by 14–15 days old (Iijima et al. 2004). Future studies examining whether female flies have a similar delay in the progression of sleep deficits compared to males, and whether gender differences in Fabp expression exist, will be important to consider how generalizable the effects of Fabp expression are on Aβ-associated sleep disruption. It is also important to note that the model used in our study does not address the other aspects of AD pathogenesis, such as tau pathology. For example, tau may influence sleep in ways that are independent of Aβ (Di Meco et al. 2014; Menkes-Caspi et al. 2015). Therefore, while our data suggest a pathway involved in Aβ derived sleep fragmentation, it may not fully capture the complex relationship between sleep and AD pathology.
Abnormal Fabp levels in CSF and blood plasma have recently been shown to be associated with AD (Chiasserini et al. 2010; Desikan et al. 2013; Guo et al. 2013; O’Bryant et al. 2013; Ohrfelt et al. 2011; Olsson et al. 2013; Teunissen et al. 2011). While levels of Fabp in the CSF were shown to be elevated in AD patients compared to cognitively normal subjects (Hu et al. 2010), levels of Fabp3 within the brains of AD patients were actually lower (Cheon et al. 2003), in agreement with our results in Drosophila. These data suggest an inverse relationship in relative Fabp levels between brain tissue and CSF in Alzheimer’s disease, in opposition to what is observed for Aβ levels in AD. Studies in human AD support an inverse correlation between brain and CSF amyloid load (Fagan et al. 2006; Forsberg et al. 2008; Grimmer et al. 2009). Alterations in normal Fabp levels observed in AD may contribute to AD-associated sleep disturbances. While the precise mechanisms responsible for Aβ42-induced sleep disruption are not known, our results show that having an extra copy of Fabp can ameliorate these sleep deficits. Fabp7 binds the omega-3 fatty acid DHA with high affinity (Xu et al. 1996) and DHA-mediated nuclear localization of Fabp7 is thought to regulate PPAR transactivity (Mita et al. 2010). Given that polyunsaturated fatty acids such as DHA can rescue cognitive decline in AD animal models and have been suggested to reduce the risk of AD in humans (Boudrault et al. 2009; Calon and Cole 2007), DHA may be operating through Fabp7 nuclear translocation and targeting of PPAR-mediated transcription. PPAR activation has been shown to facilitate the clearance of Aβ in AD animal models (Mandrekar-Colucci et al. 2012; Yamanaka et al. 2012), and since DHA triggers nuclear localization of Fabp7 to regulate PPAR-mediate transcription (Mita et al. 2010), this pathway may represent one mechanism relating Fabp7 with reducing Aβ accumulation and consolidated sleep. DHA is also thought to reduce oxidative stress and inflammation, both of which are associated with AD (Dyall 2010). One way Aβ may contribute to neurodegeneration in AD is by inducing oxidative stress through lipid peroxidation in brain cell membranes and increased production of 4-hydroxynonenal (Butterfield and Lauderback 2002). Fabps have been shown to be modified by 4-hydroxynonenal (Bennaars-Eiden et al. 2002; Hellberg et al. 2010), and therefore may represent one scavenging mechanism to operate as an antioxidant protein in response to lipid peroxidation generated by Aβ. Fabp overexpression in flies has been shown to increase longevity and resistance to oxidative stress (Lee et al. 2012), supporting a role for this concept in the normal aging process. Future work determining whether Fabp expression influences Aβ42 clearance mechanisms or oxidative load, and whether Aβ42 affects Fabp levels or nuclear localization across sleep/wake states will be important for evaluating mechanisms associated with sleep disturbance in AD.
Sleep amount and quality are known to decline with age. This effect is even more pronounced in Alzheimer’s disease, and is a major contributing factor for institutionalization. Here, we observed that sleep quality is significantly disrupted in a Drosophila AD model at an age that precedes other previously established phenotypic markers. Furthermore, we can rescue the sleep fragmentation genetically with Fabp overexpression. These studies implicate Fabp signaling pathways in mediating Aβ-induced sleep/wake fragmentation, and provide further support for Drosophila as a model organism with which to study the relationship between Aβ, sleep/wake behavior, and Alzheimer’s disease.
SIGNIFICANCE STATEMENT.
Sleep problems are common among Alzheimer’s disease (AD) patients, and are a major contributing factor for institutionalization. Clinical studies have suggested a bidirectional relationship between sleep and β-amyloid (Aβ), a small peptide associated with plaque formation in AD. Recent biomarker studies show associations between AD and fatty acid binding proteins. Fabps are reduced in brains of AD patients, but elevated in the cerebrospinal fluid and sera, compared to healthy individuals. Knowledge of the relationships between Aβ, Fabp expression and underlying sleep disturbance would aid in understanding AD pathogenesis, and may guide therapeutic strategies to delay AD disease progression.
Acknowledgments
Funding: This work was supported by National Institutes of Health Grant T32 HL07713 (J.R.G.), T32 HL110952 (W.M.V.), and P01AG017628 (A.I.P.).
Special thanks to M. Konsolaki for sharing the flies, C. Davis, K. Iijima and lab for consultation and M. Lim and K. Singletary for comments on the manuscript.
Footnotes
Conflict of interest statement: J.R.G. is a co-inventor on the U.S. patent application “Methods and compositions for improving sleep and memory” US 13/751,213.
Role of authors: J.R.G. and A.I.P. designed the study. J.R.G., O.L., M.T.C. and C.P. performed the experiments. J.R.G., W.M.V., and C.P. analyzed the data. J.R.G. wrote the manuscript with critical evaluation from co-authors.
References
- Abel T, Havekes R, Saletin JM, Walker MP. Sleep, plasticity and memory from molecules to whole-brain networks. Curr Biol. 2013;23(17):R774–788. doi: 10.1016/j.cub.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid-beta levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007;68(9):666–669. doi: 10.1212/01.wnl.0000256043.50901.e3. [DOI] [PubMed] [Google Scholar]
- Bennaars-Eiden A, Higgins L, Hertzel AV, Kapphahn RJ, Ferrington DA, Bernlohr DA. Covalent modification of epithelial fatty acid-binding protein by 4-hydroxynonenal in vitro and in vivo. Evidence for a role in antioxidant biology. J Biol Chem. 2002;277(52):50693–50702. doi: 10.1074/jbc.M209493200. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- Boudrault C, Bazinet RP, Ma DW. Experimental models and mechanisms underlying the protective effects of n-3 polyunsaturated fatty acids in Alzheimer’s disease. J Nutr Biochem. 2009;20(1):1–10. doi: 10.1016/j.jnutbio.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Bushey D, Hughes KA, Tononi G, Cirelli C. Sleep, aging, and lifespan in Drosophila. BMC Neurosci. 2010;11:56. doi: 10.1186/1471-2202-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32(11):1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;77(5–6):287–293. doi: 10.1016/j.plefa.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Ceriani MF, Hogenesch JB, Yanovsky M, Panda S, Straume M, Kay SA. Genome-wide expression analysis in Drosophila reveals genes controlling circadian behavior. J Neurosci. 2002;22(21):9305–9319. doi: 10.1523/JNEUROSCI.22-21-09305.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KF, Possidente B, Lomas DA, Crowther DC. The central molecular clock is robust in the face of behavioural arrhythmia in a Drosophila model of Alzheimer’s disease. Dis Model Mech. 2014;7(4):445–458. doi: 10.1242/dmm.014134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon MS, Kim SH, Fountoulakis M, Lubec G. Heart type fatty acid binding protein (HFABP) is decreased in brains of patients with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl. 2003;(67):225–234. doi: 10.1007/978-3-7091-6721-2_20. [DOI] [PubMed] [Google Scholar]
- Chiasserini D, Parnetti L, Andreasson U, Zetterberg H, Giannandrea D, Calabresi P, Blennow K. CSF levels of heart fatty acid binding protein are altered during early phases of Alzheimer’s disease. J Alzheimers Dis. 2010;22(4):1281–1288. doi: 10.3233/JAD-2010-101293. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Bushey D. Sleep and wakefulness in Drosophila melanogaster. Ann N Y Acad Sci. 2008;1129:323–329. doi: 10.1196/annals.1417.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Thompson WK, Holland D, Hess CP, Brewer JB, Zetterberg H, Blennow K, Andreassen OA, McEvoy LK, Hyman BT, Dale AM. Heart fatty acid binding protein and Abeta-associated Alzheimer’s neurodegeneration. Mol Neurodegener. 2013;8:39. doi: 10.1186/1750-1326-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meco A, Joshi YB, Pratico D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging. 2014;35(8):1813–1820. doi: 10.1016/j.neurobiolaging.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Dissel S, Angadi V, Kirszenblat L, Suzuki Y, Donlea J, Klose M, Koch Z, English D, Winsky-Sommerer R, van Swinderen B, Shaw PJ. Sleep restores behavioral plasticity to Drosophila mutants. Curr Biol. 2015;25(10):1270–1281. doi: 10.1016/j.cub.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyall SC. Amyloid-Beta Peptide, Oxidative Stress and Inflammation in Alzheimer’s Disease: Potential Neuroprotective Effects of Omega-3 Polyunsaturated Fatty Acids. International Journal of Alzheimer’s Disease. 2010;2010:10. [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer’s Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26(3):365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29(10):1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Ganguly-Fitzgerald I, Donlea J, Shaw PJ. Waking experience affects sleep need in Drosophila. Science. 2006;313(5794):1775–1781. doi: 10.1126/science.1130408. [DOI] [PubMed] [Google Scholar]
- Gerstner JR, Bremer QZ, Vander Heyden WM, Lavaute TM, Yin JC, Landry CF. Brain fatty acid binding protein (Fabp7) is diurnally regulated in astrocytes and hippocampal granule cell precursors in adult rodent brain. PLoS One. 2008;3(2):e1631. doi: 10.1371/journal.pone.0001631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Perron IJ, Pack AI. The nexus of Abeta, aging, and sleep. Sci Transl Med. 2012a;4(150):150fs134. doi: 10.1126/scitranslmed.3004815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Vander Heyden WM, Lavaute TM, Landry CF. Profiles of novel diurnally regulated genes in mouse hypothalamus: expression analysis of the cysteine and histidine-rich domain-containing, zinc-binding protein 1, the fatty acid-binding protein 7 and the GTPase, ras-like family member 11b. Neuroscience. 2006;139(4):1435–1448. doi: 10.1016/j.neuroscience.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Vanderheyden WM, LaVaute T, Westmark CJ, Rouhana L, Pack AI, Wickens M, Landry CF. Time of day regulates subcellular trafficking, tripartite synaptic localization, and polyadenylation of the astrocytic Fabp7 mRNA. J Neurosci. 2012b;32(4):1383–1394. doi: 10.1523/JNEUROSCI.3228-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Vanderheyden WM, Shaw PJ, Landry CF, Yin JC. Cytoplasmic to nuclear localization of fatty-acid binding protein correlates with specific forms of long-term memory in Drosophila. Commun Integr Biol. 2011a;4(5):623–626. doi: 10.4161/cib.4.5.16927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Vanderheyden WM, Shaw PJ, Landry CF, Yin JC. Fatty-acid binding proteins modulate sleep and enhance long-term memory consolidation in Drosophila. PLoS One. 2011b;6(1):e15890. doi: 10.1371/journal.pone.0015890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel N, Basner M, Rao H, Dinges DF. Circadian rhythms, sleep deprivation, and human performance. Prog Mol Biol Transl Sci. 2013;119:155–190. doi: 10.1016/B978-0-12-396971-2.00007-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves L, Pack A, Abel T. Sleep and memory: a molecular perspective. Trends Neurosci. 2001;24(4):237–243. doi: 10.1016/s0166-2236(00)01744-6. [DOI] [PubMed] [Google Scholar]
- Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, Shiga T, Wester HJ, Kurz A, Drzezga A. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry. 2009;65(11):927–934. doi: 10.1016/j.biopsych.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LH, Alexopoulos P, Perneczky R. Heart-type fatty acid binding protein and vascular endothelial growth factor: cerebrospinal fluid biomarker candidates for Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 2013;263(7):553–560. doi: 10.1007/s00406-013-0405-4. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hellberg K, Grimsrud PA, Kruse AC, Banaszak LJ, Ohlendorf DH, Bernlohr DA. X-ray crystallographic analysis of adipocyte fatty acid binding protein (aP2) modified with 4-hydroxy-2-nonenal. Protein Sci. 2010;19(8):1480–1489. doi: 10.1002/pro.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KS, Sehgal A. Drosophila melanogaster: an insect model for fundamental studies of sleep. Methods Enzymol. 2005;393:772–793. doi: 10.1016/S0076-6879(05)93041-3. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Goate A, Kelly J, Sperling R. Mapping the road forward in Alzheimer’s disease. Sci Transl Med United States. 2011a:114ps148. doi: 10.1126/scitranslmed.3003529. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011b;3(77):77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, Pickering E, Kuhn M, Chen Y, McCluskey L, Elman L, Karlawish J, Hurtig HI, Siderowf A, Lee VM, Soares H, Trojanowski JQ. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2010;119(6):669–678. doi: 10.1007/s00401-010-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju YE, Kasten T, Morris JC, Mintun M, Duntley S, Bateman RJ. Effects of age and amyloid deposition on Abeta dynamics in the human central nervous system. Arch Neurol. 2012;69(1):51–58. doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101(17):6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Iijima K. Transgenic Drosophila models of Alzheimer’s disease and tauopathies. Brain Struct Funct. 2010;214(2–3):245–262. doi: 10.1007/s00429-009-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat Rev Neurol. 2014;10(2):115–119. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, Morris JC, Holtzman DM. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70(5):587–593. doi: 10.1001/jamaneurol.2013.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyoti A, Plano A, Riedel G, Platt B. EEG, activity, and sleep architecture in a transgenic AbetaPPswe/PSEN1A246E Alzheimer’s disease mouse. J Alzheimers Dis. 2010;22(3):873–887. doi: 10.3233/JAD-2010-100879. [DOI] [PubMed] [Google Scholar]
- Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelberger KA, Piazza F, Tesco G, Reijmers LG. Natural amyloid-beta oligomers acutely impair the formation of a contextual fear memory in mice. PLoS One. 2012;7(1):e29940. doi: 10.1371/journal.pone.0029940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Evans JM, Hendricks JC, Sehgal A. A Drosophila model for age-associated changes in sleep:wake cycles. Proc Natl Acad Sci U S A. 2006;103(37):13843–13847. doi: 10.1073/pnas.0605903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koudounas S, Green EW, Clancy D. Reliability and variability of sleep and activity as biomarkers of ageing in Drosophila. Biogerontology. 2012;13(5):489–499. doi: 10.1007/s10522-012-9393-4. [DOI] [PubMed] [Google Scholar]
- Lee SH, Lee SK, Paik D, Min KJ. Overexpression of fatty-acid-beta-oxidation-related genes extends the lifespan of Drosophila melanogaster. Oxid Med Cell Longev. 2012;2012:854502. doi: 10.1155/2012/854502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain. 2013;136(Pt 5):1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, Albanese M, Mercuri NB, Izzi F, Bernardini S, Nitti A, Sancesario GM, Sica F, Marciani MG, Placidi F. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71(12):1498–1505. doi: 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]
- Lim MM, Gerstner JR, Holtzman DM. The sleep-wake cycle and Alzheimer’s disease: what do we know? Neurodegener Dis Manag. 2014;4(5):351–362. doi: 10.2217/nmt.14.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol. 2009;4:315–342. doi: 10.1146/annurev.pathol.3.121806.151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18(7):1051–1057. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci. 2012;32(30):10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott CM, LaHoste GJ, Chen C, Musto A, Bazan NG, Magee JC. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci. 2003;23(29):9687–9695. doi: 10.1523/JNEUROSCI.23-29-09687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA. Pathological tau disrupts ongoing network activity. Neuron. 2015;85(5):959–966. doi: 10.1016/j.neuron.2015.01.025. [DOI] [PubMed] [Google Scholar]
- Mita R, Beaulieu MJ, Field C, Godbout R. Brain fatty acid-binding protein and omega-3/omega-6 fatty acids: mechanistic insight into malignant glioma cell migration. J Biol Chem. 2010;285(47):37005–37015. doi: 10.1074/jbc.M110.170076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson L, Singh GK, Osterwalder T, Roman GW, Davis RL, Keshishian H. Spatial and temporal control of gene expression in Drosophila using the inducible GeneSwitch GAL4 system. I. Screen for larval nervous system drivers. Genetics. 2008;178(1):215–234. doi: 10.1534/genetics.107.081968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Bryant SE, Xiao G, Edwards M, Devous M, Gupta VB, Martins R, Zhang F, Barber R. Biomarkers of Alzheimer’s disease among Mexican Americans. J Alzheimers Dis. 2013;34(4):841–849. doi: 10.3233/JAD-122074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrfelt A, Andreasson U, Simon A, Zetterberg H, Edman A, Potter W, Holder D, Devanarayan V, Seeburger J, Smith AD, Blennow K, Wallin A. Screening for new biomarkers for subcortical vascular dementia and Alzheimer’s disease. Dement Geriatr Cogn Dis Extra. 2011;1(1):31–42. doi: 10.1159/000323417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson B, Hertze J, Ohlsson M, Nagga K, Hoglund K, Basun H, Annas P, Lannfelt L, Andreasen N, Minthon L, Zetterberg H, Blennow K, Hansson O. Cerebrospinal fluid levels of heart fatty acid binding protein are elevated prodromally in Alzheimer’s disease and vascular dementia. J Alzheimers Dis. 2013;34(3):673–679. doi: 10.3233/JAD-121384. [DOI] [PubMed] [Google Scholar]
- Osorio RS, Gumb T, Pirraglia E, Varga AW, Lu SE, Lim J, Wohlleber ME, Ducca EL, Koushyk V, Glodzik L, Mosconi L, Ayappa I, Rapoport DM, de Leon MJ. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology. 2015;84(19):1964–1971. doi: 10.1212/WNL.0000000000001566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio RS, Pirraglia E, Aguera-Ortiz LF, During EH, Sacks H, Ayappa I, Walsleben J, Mooney A, Hussain A, Glodzik L, Frangione B, Martinez-Martin P, de Leon MJ. Greater risk of Alzheimer’s disease in older adults with insomnia. J Am Geriatr Soc. 2011;59(3):559–562. doi: 10.1111/j.1532-5415.2010.03288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A. 2001;98(22):12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Pitman JL, McGill JJ, Keegan KP, Allada R. A dynamic role for the mushroom bodies in promoting sleep in Drosophila. Nature. 2006;441(7094):753–756. doi: 10.1038/nature04739. [DOI] [PubMed] [Google Scholar]
- Pollak CP, Perlick D. Sleep problems and institutionalization of the elderly. J Geriatr Psychiatry Neurol. 1991;4(4):204–210. doi: 10.1177/089198879100400405. [DOI] [PubMed] [Google Scholar]
- Prinz PN, Vitaliano PP, Vitiello MV, Bokan J, Raskind M, Peskind E, Gerber C. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol Aging. 1982;3(4):361–370. doi: 10.1016/0197-4580(82)90024-0. [DOI] [PubMed] [Google Scholar]
- Rogers I, Kerr F, Martinez P, Hardy J, Lovestone S, Partridge L. Ageing increases vulnerability to aβ42 toxicity in Drosophila. PLoS One. 2012;7(7):e40569. doi: 10.1371/journal.pone.0040569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med. 2012;4(150):150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman G, Endo K, Zong L, Davis RL. P[Switch], a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2001;98(22):12602–12607. doi: 10.1073/pnas.221303998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Herdener N, Frankola KA, Mughal MR, Mattson MP. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Abeta and pTau in a mouse model of Alzheimer’s disease. Brain Res. 2013;1529:200–208. doi: 10.1016/j.brainres.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Raichle ME, Snyder AZ, Morris JC, Head D, Wang S, Mintun MA. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 2010;67(6):584–587. doi: 10.1016/j.biopsych.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63(2):178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM. Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013;70(12):1537–1543. doi: 10.1001/jamaneurol.2013.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher KE, Bendlin BB, Racine AM, Okonkwo OC, Christian BT, Koscik RL, Sager MA, Asthana S, Johnson SC, Benca RM. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol Aging. 2015;36(9):2568–2576. doi: 10.1016/j.neurobiolaging.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi M, Lone SR, Liu S, Liu Q, Zhang J, Spira AP, Wu MN. Sleep interacts with abeta to modulate intrinsic neuronal excitability. Curr Biol. 2015;25(6):702–712. doi: 10.1016/j.cub.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen CE, Veerhuis R, De Vente J, Verhey FR, Vreeling F, van Boxtel MP, Glatz JF, Pelsers MA. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur J Neurol. 2011;18(6):865–871. doi: 10.1111/j.1468-1331.2010.03273.x. [DOI] [PubMed] [Google Scholar]
- Vanderheyden WM, Gerstner JR, Tanenhaus A, Yin JC, Shaw PJ. ERK phosphorylation regulates sleep and plasticity in Drosophila. PLoS One. 2013;8(11):e81554. doi: 10.1371/journal.pone.0081554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello MV, Borson S. Sleep disturbances in patients with Alzheimer’s disease: epidemiology, pathophysiology and treatment. CNS Drugs. 2001;15(10):777–796. doi: 10.2165/00023210-200115100-00004. [DOI] [PubMed] [Google Scholar]
- Weldemichael DA, Grossberg GT. Circadian rhythm disturbances in patients with Alzheimer’s disease: a review. Int J Alzheimers Dis. 2010;2010 doi: 10.4061/2010/716453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisor JP, Edgar DM, Yesavage J, Ryan HS, McCormick CM, Lapustea N, Murphy GM., Jr Sleep and circadian abnormalities in a transgenic mouse model of Alzheimer’s disease: a role for cholinergic transmission. Neuroscience. 2005;131(2):375–385. doi: 10.1016/j.neuroscience.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Wu YH, Swaab DF. Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer’s disease. Sleep Med. 2007;8(6):623–636. doi: 10.1016/j.sleep.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LZ, Sanchez R, Sali A, Heintz N. Ligand specificity of brain lipid-binding protein. J Biol Chem. 1996;271(40):24711–24719. doi: 10.1074/jbc.271.40.24711. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32(48):17321–17331. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Veasey SC, Wood MA, Leng LZ, Kaminski C, Leight S, Abel T, Lee VM, Trojanowski JQ. Impaired rapid eye movement sleep in the Tg2576 APP murine model of Alzheimer’s disease with injury to pedunculopontine cholinergic neurons. Am J Pathol. 2005;167(5):1361–1369. doi: 10.1016/S0002-9440(10)61223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JE, Raizen DM, Maycock MH, Maislin G, Pack AI. A video method to study Drosophila sleep. Sleep. 2008;31(11):1587–1598. doi: 10.1093/sleep/31.11.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]