

Mutations in the Caenorhabditis elegans dynein heavy chain enhance LET-23 EGFR signaling during vulva cell-fate specification. LET-23 EGFR accumulates in juxtamembrane foci of dhc-1 mutants, suggesting that dynein functions at an early step of LET-23 EGFR trafficking.
Abstract
Epidermal growth factor receptor (EGFR) signaling is essential for animal development, and increased signaling underlies many human cancers. Identifying the genes and cellular processes that regulate EGFR signaling in vivo will help to elucidate how this pathway can become inappropriately activated. Caenorhabditis elegans vulva development provides an in vivo model to genetically dissect EGFR signaling. Here we identified a mutation in dhc-1, the heavy chain of the cytoplasmic dynein minus end–directed microtubule motor, in a genetic screen for regulators of EGFR signaling. Despite the many cellular functions of dynein, DHC-1 is a strong negative regulator of EGFR signaling during vulva induction. DHC-1 is required in the signal-receiving cell and genetically functions upstream or in parallel to LET-23 EGFR. LET-23 EGFR accumulates in cytoplasmic foci in dhc-1 mutants, consistent with mammalian cell studies in which dynein is shown to regulate late endosome trafficking of EGFR with the Rab7 GTPase. However, we found different distributions of LET-23 EGFR foci in rab-7 versus dhc-1 mutants, suggesting that dynein functions at an earlier step of LET-23 EGFR trafficking to the lysosome than RAB-7. Our results demonstrate an in vivo role for dynein in limiting LET-23 EGFR signaling via endosomal trafficking.
INTRODUCTION
Epidermal growth factor receptor (EGFR) signaling regulates many cellular processes, most notably cell proliferation in mammalian cells (Normanno et al., 2006). Thus it is not surprising that EGFR and family members are overexpressed in many human cancers. Lysosomal degradation is an important mechanism to turnover activated EGFR (Sorkin and Goh, 2009). On activation, EGFR is internalized into early endosomes, where the cytoplasmic tail is still able to engage downstream signaling proteins. Maturation of early endosomes into late endosomes is accompanied by the formation of multivesicular bodies and the internalization of EGFR from the outer membrane into intraluminal vesicles and thus sequestration of EGFR from downstream signaling proteins. Subsequent fusion with lysosomes results in the ultimate destruction of EGFR. Rab GTPases are important mediators of endosome trafficking (Stenmark, 2009). The coordinated functions of the Rab5 and Rab7 GTPases regulate early-to-late endosome maturation and lysosomal fusion. Inhibition of Rab7 in mammalian cells results in accumulation of EGFR in multivesicular bodies and in both Caenorhabditis elegans and mammalian cells leads to increased EGFR signaling (Vanlandingham and Ceresa, 2009; Skorobogata and Rocheleau, 2012; BasuRay et al., 2013). In addition to mediating late endosome fusion with lysosomes along with the HOPS complex, Rab7 couples with the dynein minus-end microtubule motor via the Rab7-interacting proteins RILP and ORP1L, which tether to the dynactin complex, a regulator of dynein (Johansson et al., 2007). Inhibition of dynactin by overexpression of the p50 subunit blocks endosome movement from the cell periphery, inhibits EGFR degradation, and causes sustained Erk1/2 activation (Taub et al., 2007). However, dynein can be recruited by other Rab GTPases to mediate membrane trafficking in the endocytic and secretory pathways (Horgan and McCaffrey, 2011; Hunt and Stephens, 2011). In Drosophila, dynein promotes EGFR signaling during eye and wing development by promoting secretion of the EGFR ligand, Spitz (Iyadurai et al., 2008). Thus dynein has both positive and negative effects on EGFR signaling in fruit fly and mammalian tissue culture cells, respectively.
C. elegans vulva development provides an in vivo model to study EGFR signaling in epithelial cells (Sundaram, 2013; Schmid and Hajnal, 2015). During larval development, six vulval precursor cells (VPCs), P3.p–P8.p, express LET-23 EGFR on both the apical and basolateral membranes. The anchor cell in the overlying gonad secretes an EGF-like ligand, LIN-3, which engages LET-23 EGFR on the basolateral membrane, initiating a conserved Ras/mitogen-activated protein kinase (MAPK) signaling pathway in the P6.p cell and specifying the primary vulval cell fate. Graded LIN-3 signaling and LIN-12 Notch signaling specify the neighboring P5.p and P7.p cells to adopt the secondary vulval fates. P5.p–P7.p cells undergo three rounds of cell division to generate the 22 cells of the mature vulva. The remaining uninduced VPCs (P3.p, P4.p, and P8.p) divide once (P3.p does not divide 50% of the time) and fuse with the surrounding hypodermis. Mutations that decrease LET-23 EGFR signaling result in a vulvaless (Vul) phenotype in which fewer than three VPCs are induced. Mutations that increase LET-23 EGFR signaling result in a multivulva (Muv) phenotype in which more than three VPCs are induced. The LIN-2 Cask/LIN-7 Veli/LIN-10 Mint (LIN-2/7/10) complex binds the C-terminal tail of LET-23 EGFR and is required for the basolateral localization of LET-23 EGFR in the VPCs and vulva induction. Mutations in the lin-2/7/10 complex result in primarily apical localization of LET-23 EGFR and a strong Vul phenotype.
In a suppressor screen of the lin-2 mutant Vul phenotype, we identified two suppressors with embryonic lethal phenotypes, vh4 and vh22 (Skorobogata et al., 2014). We previously reported that vh4 is a mutation in the agef-1 gene that partially restores basolateral membrane localization of LET-23 EGFR. Here we report that vh22 is a temperature-sensitive mutation in dhc-1, the heavy chain of the dynein minus end–directed microtubule motor. We demonstrate that dhc-1 is a strong negative regulator of LET-23 EGFR signaling that functions upstream of or in parallel to LET-23 EGFR. LET-23::GFP accumulates in foci in the P6.p descendants of dhc-1(vh22) animals, consistent with a defect in endocytic trafficking of LET-23 EGFR. Unlike rab-7 mutants, many of the LET-23::GFP foci in dhc-1(vh22) mutants remain adjacent to the plasma membrane, suggesting that dhc-1 regulates an earlier step of LET-23 EGFR trafficking to the lysosome.
RESULTS
Identification of vh22 as an essential suppressor of the lin-2(-) Vul phenotype
Mutations in the lin-2, lin-7, or lin-10 gene result in a strong Vul phenotype that is easily suppressed by loss of a negative regulator of LET-23 EGFR signaling, such as the SLI-1 Cbl E3 ubiquitin ligase homologue, the GAP-1 Ras GAP, or the LIP-1 MAPK phosphatase (Jongeward et al., 1995; Yoon et al., 1995; Hajnal et al., 1997; Berset et al., 2001). We previously identified the rab-7(ok511) deletion as a strong suppressor of mutations in the lin-2, lin-7, and lin-10 genes, reverting a strong Vul phenotype back to wild-type and Muv phenotypes (Skorobogata and Rocheleau, 2012). The maternal effect embryonic lethality of the rab-7(ok511) mutation would preclude its identification in screens for viable suppressors. Therefore we conducted a clonal forward genetic screen for essential suppressors of the lin-2(e1309) Vul phenotype to identify additional genes that might function with rab-7 to antagonize LET-23 EGFR signaling (Skorobogata et al., 2014). We identified the vh22 mutation as a strong suppressor of the lin-2(e1309) Vul phenotype. Whereas lin-2(e1309) mutants are 100% Vul, the vh22; lin-2(e1309) double mutants are only 15% Vul at 20°C, with 85% of the animals developing a wild-type vulva (Figure 1, A–D, and Table 1). Like mutations in other negative regulators, vh22 single mutants have essentially normal vulva induction (Figure 1C and Table 1). In addition, vh22 animals are ∼80% embryonic lethal at 20°C and have a small-body morphology phenotype (Table 2; unpublished data). Embryos often arrest with undifferentiated multinucleate cells, indicative of an early cell division defect (Figure 1, E and D). However, no cell division defects were observed in the vulval cell lineages. The embryonic lethality and the suppression of the lin-2(e1309) Vul phenotypes were temperature sensitive, as both phenotypes were considerably less severe at 15°C and more severe at 25°C (Tables 1 and 2).
FIGURE 1:

dhc-1(vh22) suppresses the lin-2(e1309) Vul phenotype. (A–D) Representative DIC images of vulvas of wild-type, lin-2(e1309), dhc-1(vh22), and dhc-1(vh22); lin-2(e1309) L4-stage larvae. The lin-2(e1309) larva lacks a vulva, whereas in dhc-1(vh22); lin-2(e1309), the vulva is mostly restored. dhc-1(vh22) single mutants have a wild-type vulva. (E–G) DIC images of early embryos of wild-type, dhc-1(vh22), and dhc-1(or195). Bars, 10 μm.
TABLE 1:
Loss of dhc-1 suppresses the lin-2(e1309) Vul phenotypes.
| Genotype | Temperature(°C) | Muv(%) | Vul (%) | Average number of VPCs induced | Number of animals scored |
|---|---|---|---|---|---|
| Wild type | All | 0 | 0 | 3.0 | Many |
| dhc-1(vh22) | 20 | 0 | 5 | 2.98 | 20 |
| lin-2(e1309) | 20 | 0 | 100 | 0.29 | 21 |
| dhc-1(vh22); lin-2(e1309) | 15 | 5 | 80** | 1.5*** | 20 |
| 20 | 0 | 15*** | 2.85*** | 20 | |
| 25 | 0 | 11*** | 2.98*** | 20 | |
| GFP(RNAi); dhc-1(vh22); lin-2(e1309) | 20 | 12 | 24 | 2.82 | 25 |
| dhc-1(RNAi); dhc-1(vh22); lin-2(e1309) | 20 | 4 | 8 | 2.94 | 25 |
| dhc-1(vh22); lin-2(e1309); orIs17[GFP::DHC-1] | 15 | 0 | 100* | 0.44 | 26 |
| 20 | 12 | 58*** | 1.80**** | 33 | |
| dhc-1(or195); lin-2(e1309) | 15 | 0 | 100 | 0.13 | 15 |
| 20 | 0 | 96 | 0.78** | 23 | |
| 25 | 0 | 89** | 0.9** | 29 | |
| dhc-1(or352); lin-2(e1309) | 15 | 0 | 100 | 0.23 | 15 |
| 20 | 0 | 88** | 0.69** | 18 | |
| 25 | 0 | 76** | 1.53*** | 44 | |
| GFP(RNAi); lin-2(e1309) | 20 | 0 | 100 | 0.32 | 31 |
| dhc-1(RNAi); lin-2(e1309) | 20 | 7 | 85* | 1.59**** | 17 |
| elpc-1(RNAi); lin-2(e1309) | 20 | 0 | 95 | 0.36 | 40 |
| cpd-1(RNAi); lin-2(e1309) | 20 | 0 | 98 | 0.3 | 40 |
Statistical significance was determined using Fisher’s exact test (graphpad.com/quickcalcs). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. dhc-1; lin-2 mutants were compared with lin-2 single mutants. orIs17-carrying strains were compared with dhc-1(vh22); lin-2(e1309) at the same temperatures. dhc-1(RNAi), elpc-1(RNAi), and cpd-1(RNAi) were compared with gfp(RNAi). The penetrance of the lin-2(e1309) phenotype is similar at all temperatures.
TABLE 2:
dhc-1(vh22) is temperature-sensitive embryonic lethal.
| Percentage embryonic lethality (n) | |||
|---|---|---|---|
| Genotype | 15°C | 20°C | 25°C |
| dhc-1(vh22) | 23 (211) | 81 (274) | 96 (126) |
| dhc-1(or195) | 22 (369) | 97 (514) | 99 (304) |
| dhc-1(or352) | 26 (507) | 90 (460) | 97 (934) |
| dhc-1(vh22)/dhc-1(or195) | 96 (1218) | ||
| dhc-1(vh22)/dhc-1(or352) | 95 (1167) | ||
| dhc-1(vh22); orIs17(DHC-1::GFP) | 1.3 (382)**** | 0.5 (583)**** | 2.3 (264)**** |
Statistical analysis was performed as in Table 1. The dhc-1(vh22); orIs17 strain has the lin-2(e1309) mutation in the background and was compared to dhc-1(vh22) single mutants at the same temperature. n, number of embryos scored.
vh22 is an allele of dhc-1
To identify the gene mutated by vh22, we used the associated small-body morphology and embryonic lethal phenotypes to map vh22. First, we found that vh22 was balanced by the hT2 balancer consisting of a translocation between chromosomes I and III (Zetka and Rose, 1992). Single nucleotide polymorphism (SNP) mapping located vh22 between SNPs W03D8 at −6 map units (mu) and pkP1057 at 1 mu on chromosome I (Figure 2A). The location was further delineated by complementation with chromosomal deficiencies: vh22 complemented qDf3 and mnDf111 but failed to complement sDf4 and hDf6, indicating that vh22 was located between −2.21 and 0.1 mu (Figure 2A). Whole-genome sequencing identified two missense mutations in the dhc-1 gene, which was previously reported to have early cell division defects (Figure 1G; Gonczy et al., 1999; Schmidt et al., 2005), as well as homozygous nonsynonymous mutations in the elpc-1 and cpd-1 genes (see Materials and Methods). We found that RNA interference (RNAi) of dhc-1 but not elpc-1 or cpd-1 significantly suppressed the lin-2(e1309) Vul phenotype (Table 1). dhc-1(RNAi) did not enhance or suppress the vh22; lin-2(e1309) vulval phenotype (Table 1). Consistent with vh22 being a mutation in dhc-1, two conditional alleles of dhc-1—or195 and or352—suppressed the lin-2(e1309) Vul phenotype at nonpermissive temperature (Table 1; Hamill et al., 2002). Furthermore, vh22 fails to complement dhc-1(or195) and dhc-1(or352) for the embryonic lethal phenotype (Table 2). Finally, we can rescue the vh22 embryonic lethality and suppression of lin-2(e1309) Vul phenotype with a green fluorescent protein (GFP)::dynein heavy chain-1 (DHC-1) transgene, orIs17 (Tables 1 and 2; Gassmann et al., 2008). Therefore we conclude that vh22 is a loss-of-function mutation in dhc-1.
FIGURE 2:

dhc-1(vh22) mapping and the lesions. (A) Schematic representation of the middle of chromosome I (LGI). The SNPs used for interval mapping are indicated on top with their chromosomal locations (map units [mu]). The number of recombinant animals positive for the Hawaiian SNP out of the total number of animals tested for each SNP is indicated below. The two chromosomal deficiencies that fail to complement vh22—sDf4 and hDf6—are shown in red, and the complementing deficiencies—qDf3 and mnDf111—are shown in blue. A bracket indicates the 2.1-mu interval between the left end of hDf6 and the right end of sDf4 to which vh22 maps. (B) Homology domains of the DHC-1 protein in common with the dynein heavy chains from other organisms: dimerization and accessory chain interaction domains (DHC_1 and DHC_2) in blue, AAA-type ATPase domains (AAA1–6) in red, and the microtubule-binding stalk (MT binding) in green. The locations of the two missense mutations in vh22 and the or195 and or352 alleles are shown below. Valine 3228 in the microtubule-binding stalk is changed to an aspartic acid. Valine 3228 in C. elegans (Cel) is conserved in D. melanogaster (Dme) NP_001261430.1, Homo sapiens (Hsp) NP_0011367.2, and Strongylocentrotus purpuratus (Spu) XP_797645.3 but not in the fungi Aspergillus nidulans (Ani) XP_657722.1, Schizosaccharomyces pombe (Spo) XP_001713108.1, or Saccharomyces cerevisiae (Sce) GAA24775.1. Aspartic acid 4344 resides in a C-terminal region and is changed to an asparagine. Aspartic acid 4344 and the surrounding region are conserved among the metazoans analyzed but not present in the fungi. Amino acid (aa) identities are highlighted in black and similarities in gray.
DHC-1 is the C. elegans dynein heavy chain subunit of the cytoplasmic dynein complex, a minus end–directed microtubule motor (Lye et al., 1995). DHC-1 comprises the motor and microtubule-binding domain (C-terminal half) and a stem (N-terminal) that is involved in dimerization and recruitment of numerous accessory chains and regulators (Figure 2B; Allan, 2011). The missense mutations in dhc-1 change two amino acid residues that are conserved in metazoans but not fungi. The first mutation changes a valine 3228 to an aspartic acid in the microtubule-binding domain, the second, an aspartic acid 4344 to asparagine in a metazoan-specific C-terminal domain. We do not know which change or whether both changes contribute to the dhc-1(vh22) phenotypes, but vh22 is a stronger suppressor of the lin-2(-) Vul phenotype than the or195 and or352 alleles, which have been deemed strong mutations with respect to embryonic phenotypes (Table 1; Schmidt et al., 2005).
dhc-1 functions upstream or in parallel to let-23 EGFR
Suppression of the lin-2(e1309) Vul phenotype by dhc-1 alleles suggests that DHC-1 is a negative regulator of LET-23 EGFR signaling. Consistent with this hypothesis, we find that dhc-1(vh22) enhances the Muv phenotype of a gain-of-function mutation in let-60 Ras, n1046 (Table 3). The let-23(sy1) allele truncates the last six amino acids shown to be required for binding the LIN-2/7/10 complex and thus has an identical vulval phenotype as mutations in lin-2/7/10 (Aroian et al., 1994; Kaech et al., 1998). We find that dhc-1(vh22) strongly suppresses the Vul phenotype of the let-23(sy1) and lin-7(e1413) mutations and that dhc-1(RNAi) suppresses the Vul phenotype of the lin-10(e1439) mutation (Table 3). The let-23(sy97) mutation is a more severe truncation of LET-23 EGFR, which blocks signaling to LET-60 Ras in multiple tissues (Aroian and Sternberg, 1991). We find that dhc-1(vh22) fails to suppress the Vul phenotype of let-23(sy97), consistent with dhc-1 functioning upstream or in parallel to let-23 EGFR (Table 3). lin-3(e1417) is a strong hypomorphic mutation in the promoter of lin-3 EGF that specifically abrogates lin-3 EGF expression in the Anchor Cell (Hwang and Sternberg, 2004). We find that dhc-1(vh22) significantly suppresses the Vul phenotype of lin-3(e1417), suggesting that dhc-1 functions downstream of or in parallel to lin-3 EGF (Table 3). Taken together, these data suggest that DHC-1 is genetically a negative regulator of LET-23 EGFR signaling and functions at the level of LET-23 EGFR.
TABLE 3:
DHC-1 negatively regulates LET-23 EGFR signaling downstream or in parallel to LIN-3 EGF.
| Genotype | Muv (%) | Vul (%) | Average number of VPCs induced | Number of animals scored |
|---|---|---|---|---|
| let-60(n1046)a | 65 | 0 | 3.55 | 20 |
| dhc-1(vh22); let-60(n1046) | 92 | 0 | 4.18** | 24 |
| lin-7(e1413) | 0 | 92 | 0.28 | 36 |
| dhc-1(vh22); lin-7(e1413) | 5 | 38**** | 2.5**** | 42 |
| GFP(RNAi); lin-10(e1439) | 0 | 95 | 0.31 | 85 |
| dhc-1(RNAi); lin-10(e1439) | 0 | 91 | 0.69*** | 90 |
| let-23(sy1) | 0 | 100 | 0.06 | 25 |
| dhc-1(vh22); let-23(sy1) | 3 | 57**** | 1.79**** | 35 |
| let-23(sy97)a | 0 | 93 | 0.4 | 29 |
| dhc-1(vh22); let-23(sy97) | 0 | 100 | 0.24 | 23 |
| lin-3(e1417) | 0 | 92 | 0.94 | 36 |
| dhc-1(vh22); lin-3(e1417) | 0 | 82 | 1.56*** | 50 |
Statistical analysis was done as in Table 1. Experiments were performed at 20°C.
aControl data were previously published, as they were collected together with both agef-1(vh4) (Skorobogata et al., 2014) and dhc-1(vh22) double-mutant strains.
DHC-1 functions in the VPCs to negatively regulate LET-23 EGFR signaling
If DHC-1 is regulating LET-23 EGFR, then we anticipated that DHC-1 would function in the VPCs, the signal-receiving cells. To determine whether DHC-1 negatively regulates signaling in the signal-receiving cells, we performed dhc-1(RNAi) specifically in the VPCs. We took advantage of the tissue-specific rescue of rde-1(ne219) mutant animals, which are deficient for RDE-1, an Argonaute required for exogenous RNAi (Tabara et al., 1999; Qadota et al., 2007). Expression of wild-type rde-1(+) under the VPC-specific promoter lin-31 (mfIs70) in the rde-1(ne219) mutant confers sensitivity to RNAi specifically in the VPCs (Tan et al., 1998; Barkoulas et al., 2013). rde-1(ne219); lin-2(e1309) double mutants expressing rde-1(+) in the VPCs were fed RNAi for GFP, dhc-1, and rab-7. Consistent with our previous findings in the whole-animal RNAi experiments, depletion of either dhc-1 or rab-7 by RNAi specifically in the VPCs partially suppressed the lin-2 Vul phenotype, whereas RNAi targeting GFP did not (Table 4). These data are consistent with both DHC-1 and RAB-7 functioning in the VPCs to negatively regulate EGFR signaling.
TABLE 4:
DHC-1 functions in the VPCs.
| RNAi | Muv (%) | Vul (%) | Average number of VPCs induced | Number of animals scored |
|---|---|---|---|---|
| GFP | 0 | 100 | 0.09 | 48 |
| dhc-1 | 0 | 98 | 0.44*** | 50 |
| rab-7 | 2 | 98 | 0.33** | 46 |
The strain QR405: rde-1(ne219); mfIs70[Plin-31::RDE-1]; lin-2(e1309) was used for VPC-specific RNAi feeding. Statistical analysis was done as in Table 1. Experiments were performed at 20°C.
DHC-1 and RAB-7 regulate LET-23 EGFR trafficking
We previously found that RAB-7 regulates the localization of LET-23 EGFR in the VPCs (Skorobogata and Rocheleau, 2012). Specifically, the rab-7(ok511) deletion results in the accumulation of cytoplasmic LET-23::GFP foci in the VPCs of wild type and lin-2(e1309) mutants expressing the gaIs27 LET-23::GFP transgene. However, this transgene did not show the expected localization of LET-23::GFP at the basolateral membrane, which kept us from determining whether RAB-7 regulates apical versus basolateral localization. We reassessed the role of RAB-7 in the regulation of LET-23 EGFR trafficking in the VPCs of live animals using a new LET-23::GFP transgene, zhIs038, which expresses at levels comparable to endogenous LET-23 EGFR and shows clear localization at the basolateral membrane in wild-type VPCs but not lin-2(e1309) mutants (Figure 3, A and B; Haag et al., 2014). The rab-7(ok511) mutation had a small effect on LET-23::GFP localization in the P6.p cell (Supplemental Figure S1). However, the daughter cells, P6.pa and P6.pp, showed a robust accumulation of LET-23::GFP foci in rab-7(ok511) and rab-7(ok511); lin-2(e1309) mutants (Figure 3, C, D, I, and J). This confirms our previous data with the gaIs27 transgene and is consistent with RAB-7 being required for LET-23 EGFR trafficking to lysosomes (Skorobogata and Rocheleau, 2012).
FIGURE 3:

DHC-1 and RAB-7 differentially regulate LET-23 EGFR trafficking. (A–H) Representative confocal images of LET-23::GFP localization in the P6.p daughter cells—P6.pa and P6.pp—of zhIs038 transgene-carrying animals. The receptor is localized to both basolateral and apical membranes of wild-type animals (A) but is restricted to the apical membrane in lin-2(e1309) animals (B). rab-7(ok511) and dhc-1(vh22) mutants accumulate LET-23::GFP in the punctate structures within the vulval cells (C, D) and adjacent to the plasma membrane (E, F), respectively, in both wild-type (C, E) and lin-2(e1309) backgrounds. (G) dhc-1(or195) mutants also accumulate LET-23::GFP foci at 25°C. (H) dhc-1(RNAi) does not significantly change the dhc-1(vh22); lin-2(e1309) phenotype. (I, J) Box-and-whiskers plots of the number of LET-23::GFP foci in the P6.p descendants for the indicated genotypes. The number of animals scored is indicated above. (I) Number of cytoplasmic (pink), juxtamembrane (green), and combined total (yellow) LET-23::GFP foci. For statistical analysis, the average number of foci in the rab-7(ok511), dhc-1(vh22), and dhc-1(or195) mutants was compared with wild type. The dhc-1(vh22); rab-7(ok511) double was compared with the dhc-1(vh22) mutant and was not significantly different from rab-7(ok511). (J) Total number of LET-23::GFP foci. All double mutants were compared with lin-2(e1309). dhc-1(vh22+RNAi); lin-2(-) was not statistically different from dhc-1(vh22); lin-2(-). A two-tailed t test was used to calculate statistical significance (*p < 0.05, **p < 0.01, ****p < 0.0001). Basal and apical membranes are facing in the same direction in all of the images and are indicated in A. Bar, 5 μm.
In mammalian cell culture, Rab7 can recruit dynein to EGFR-containing late endosomes to traffic them along microtubules toward lysosomes in the perinuclear region (Johansson et al., 2007). Therefore we tested whether the VPCs of dhc-1(vh22) mutants accumulated LET-23::GFP foci like rab-7(ok511) mutants. Similar to rab-7(ok511) animals, dhc-1(vh22) and dhc-1(vh22); lin-2(e1309) mutants accumulate LET-23::GFP foci in the P6.p daughter cells, P6.pa and P6.pp (Figure 3, E, F, I, and J). The dhc-1(vh22) mutants accumulated less LET-23::GFP foci, which were primarily adjacent to, or appeared contiguous with, the plasma membrane compared with rab-7(ok511) mutants. The number of LET-23::GFP foci in dhc-1(vh22); lin-2(e1309) animals was not significantly enhanced by dhc-1(RNAi) (Figure 3, H and J), and juxtamembrane LET-23::GFP foci were also seen in dhc-1(or195) mutants at 25°C (Figure 3, G and I). Together these results suggest that DHC-1 might function at an earlier step of LET-23 trafficking than RAB-7. To test whether dhc-1(vh22) is epistatic to rab-7(ok511), we constructed a dhc-1(vh22); rab-7(ok511) double mutant expressing LET-23::GFP. We found that dhc-1(vh22) did not suppress or enhance the rab-7(ok511) cytoplasmic vesicle phenotype (Figure 3I). Although this does not preclude them from functioning in the same pathway, it suggests that dhc-1 is not required for the rab-7(ok511) phenotype. We conclude that the suppression of the lin-2(e1309) Vul phenotype by rab-7(ok511) and dhc-1(vh22) mutations is likely due to a failure to efficiently down-regulate LET-23 EGFR via endosome/lysosome trafficking (Figure 4).
FIGURE 4:
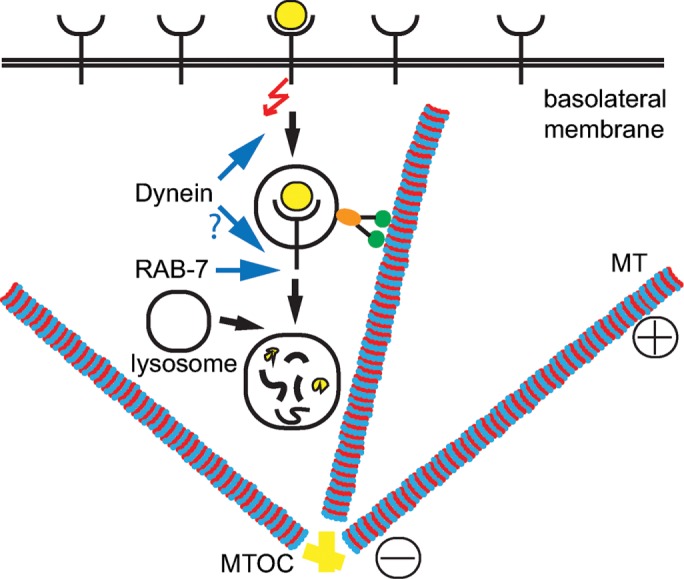
Model of LET-23 EGFR regulation by DHC-1 and RAB-7. DHC-1 (dynein) and RAB-7 promote endocytic trafficking of LET-23 EGFR to the lysosome for degradation. Our data suggest that dynein functions at an early step of receptor trafficking, moving endosomes along microtubules (MT) from the plasma membrane and toward the microtubule-organizing center (MTOC). Then, RAB-7 facilitates lysosomal degradation of LET-23 EGFR, which could also involve dynein. Disruption of either dynein or RAB-7 would result in accumulation of active LET-23 EGFR on endosomes, from which it can continue signaling.
DISCUSSION
Endosome trafficking and lysosome degradation of EGFR is an important mechanism for signal down-regulation. The late endosomal Rab7 GTPase couples with the dynein minus end–directed microtubule motor to traffic late endosomes to lysosomes and facilitate EGFR degradation (Johansson et al., 2007). We previously identified RAB-7 as a potent negative regulator of LET-23 EGFR signaling during C. elegans vulva induction (Skorobogata and Rocheleau, 2012). Here we identified mutations in dhc-1, the dynein heavy chain, in a forward genetic screen for regulators of LET-23 EGFR signaling and intracellular trafficking. DHC-1 is a strong negative regulator of LET-23 EGFR signaling during C. elegans vulva induction. DHC-1 functions in the VPCs, the signal-receiving cells, and genetically functions at the level of LET-23 EGFR. Consistent with a role in EGFR trafficking, RAB-7 and DHC-1 are required for the proper localization of LET-23 EGFR in the VPCs. However, LET-23::GFP foci primarily accumulate adjacent to the plasma membrane in dhc-1 mutant animals, whereas in rab-7 mutants, LET-23::GFP foci are more numerous and distributed throughout the cytoplasm. Together these data suggest that DHC-1 is required for an early step in LET-23 EGFR trafficking to the lysosome in the VPCs and thus plays an important role in signal attenuation.
Cytoplasmic dynein is a heteromeric multiprotein minus end–directed microtubule motor, which is involved in a variety of cellular processes, ranging from movement of a multitude of cargo toward the nucleus to mitotic spindle assembly during mitosis (Roberts et al., 2013). At the core of dynein complex is the DHC dimer, which contains a hexameric AAA+ (ATPase associated with various cellular activities) domain involved in ATP hydrolysis. Among the C-terminally located AAA+ domains lie the stalk and microtubule-binding domain. The N-terminal coiled-coil domains mediate dimerization, interactions with various intermediate, light, and light-intermediate chains, and interactions with LIS1 and the dynactin complex, which regulate cargo binding and motor activity (Allan, 2011). The dhc-1(vh22) allele has two missense mutations. One resides in the very C-terminus, a region specific to metazoan DHCs and of unknown function. The other missense mutation is in the microtubule-binding stalk and thus might alter the conformation of the stalk and potentially its interaction with microtubules. The previously identified conditional alleles of dhc-1—or195 and or352—have been deemed strong alleles of dhc-1 with respect to several embryonic defects (Hamill et al., 2002; Schmidt et al., 2005). The or195 allele also causes a defect in the microtubule-binding stalk, and the or352 allele alters the second AAA+ domain (O’Rourke et al., 2007). Surprisingly, these dhc-1 alleles were less effective suppressors of lin-2(e1309). The lin-2(e1309) allele has often been used to facilitate the identification of embryonic lethal mutations (Kemphues et al., 1988) and was in the background for genetic screens that identified the or195 and or352 alleles, and thus there might have been a negative selection for alleles of dhc-1 that suppress the lin-2 Vul phenotype (Hamill et al., 2002).
Dynein and the dynactin complex promote EGFR trafficking to the lysosome in mammalian cell culture (Taub et al., 2007). There are few data, however, for dynein regulating EGFR signaling. In HeLa cells, indirect inhibition of dynein via overexpression of the p50 dynamitin subunit of the dynactin complex inhibited EGFR degradation and caused sustained Erk1/2 activation (Taub et al., 2007). In Drosophila melanogaster, mutations in dhc and p150 Glued enhanced mutations in EGFR and Star, a regulator of Spitz ligand secretion, suggesting a positive role for dynein and the dynactin complex in Spitz-EGFR signaling (Iyadurai et al., 2008). Our findings may be the first to demonstrate an in vivo role for dynein negatively regulating EGFR signaling and are congruent with the p50 dynamitin overexpression studies in HeLa cells. The differences between the Drosophila study and ours likely reflects the fact that dynein can regulate multiple trafficking pathways, and its effects on cell signaling are likely to be context dependent.
Rab7 recruits the dynein–dynactin complex to late endosomes to drive microtubule-based movement toward lysosomes (Johansson et al., 2007). Because RAB-7 and DHC-1 both negatively regulate LET-23 EGFR signaling in the VPCs, we hypothesized that, as in HeLa cells, RAB-7 and DHC-1 function together to regulate LET-23 EGFR trafficking to the lysosome. We found that LET-23::GFP accumulated in foci adjacent to the plasma membrane of dhc-1 mutants, which is again in line with EGFR localization in HeLa cells overexpressing p50 dynamitin. In rab-7 mutants, LET-23::GFP accumulated in foci that are distributed throughout the cytoplasm. Both phenotypes are consistent with defective endosome-to-lysosome trafficking of LET-23 EGFR in rab-7 and dhc-1 mutants but are inconsistent with DHC-1 functioning exclusively with RAB-7. Instead, we propose that DHC-1/dynein is required at an earlier step of LET-23 EGFR trafficking, promoting movement of endosomes from the plasma membrane deeper into the cytoplasm, whereas RAB-7 regulates later steps of endosome trafficking (Figure 4). The fact that dhc-1 is not required for the rab-7(ok511) phenotype does not preclude it from functioning in the same pathway but might reflect that inhibition of dhc-1 is incomplete or that additional motors or mechanisms might function in parallel with dynein to regulate early stages of LET-23 EGFR trafficking.
The LET-23::GFP localization phenotypes of rab-7 and dhc-1 mutants are more prominent in the P6.p daughters than in P6.p itself (Figure 3 and Supplemental Figure S1). We speculate that this could be cumulative and thus more apparent in the P6.p daughters. Alternatively, there could be increased LET-23 EGFR turnover in the P6.p daughters. LET-23 EGFR signaling in P6.p before the first cell division is sufficient for vulva induction (Kimble, 1981; Ambros, 1999). However, when inductive signaling is compromised, such as in a hypomorphic lin-3 EGF mutant, it is common to see hybrid fates in which only one of the two VPC daughters is induced, consistent with the P6.pa and P6.pp cells still being competent for induction (Wang and Sternberg, 1999). Therefore loss of rab-7 or dhc-1 could potentially increase signaling in P6.p and/or its daughters.
dhc-1(vh22) was identified with agef-1(vh4) as suppressors of the lin-2(e1309) Vul phenotype (Skorobogata et al., 2014). We showed that agef-1(vh4) could partially restore LET-23::GFP localization to the basolateral membrane of lin-2(e1309) mutants, suggesting that AGEF-1 regulates apical versus basolateral distribution of LET-23 EGFR. Neither dhc-1(vh22) nor rab-7(ok511) visibly restored LET-23::GFP to the basolateral membrane of lin-2(e1309) mutants (Figure 3, D and F), although we previously noted that the levels of LET-23::GFP required for induction are below detection (Skorobogata et al., 2014). We suspect that mutations in dhc-1 and rab-7 may reduce turnover of active LET-23 EGFR that is present at low levels on the basolateral membrane of lin-2/7/10 mutants and thus increase signaling above a threshold necessary for vulva induction.
In summary, we identify DHC-1, the core component of the dynein minus end–directed microtubule motor, as a potent negative regulator of LET-23 EGFR signaling in C. elegans. DHC-1 functions in the VPCs to promote an early step in the endocytic trafficking of LET-23 EGFR to the lysosome. This study highlights how alterations to basic cellular machinery such as vesicular trafficking and cytoskeletal transport can strongly affect signal transduction in vivo. In human cells, dynein could have a tumor-suppressive function via its role in endosome trafficking and attenuating signaling by EGFR or other receptors that regulate growth control.
MATERIALS AND METHODS
Strains and alleles
General methods for the handling and culturing of C. elegans were as previously described (Brenner, 1974). C. elegans Bristol strain N2 is the wild-type parent for all of the strains used in this study. Escherichia coli strain HB101 was used as a food source. The Hawaiian strain CB4856 was used for SNP mapping. Unless otherwise noted, experiments were performed at 20°C. Information on the genes and alleles used in this work can be found on WormBase (wormbase.org) and are available through Caenorhabditis Genetics Center (cbs.umn.edu/cgc) unless otherwise noted in the strain list (Supplemental Table S1).
Genetic mapping and cloning of dhc-1(vh22)
SNP mapping was used to place vh22 to the middle region of chromosome I (Davis et al., 2005). Chromosome mapping showed linkage of vh22 to SNPs at −6 (W03D8), −1(D1007), 5 (B0205), 13 (F58D5), 14 (T06G6), and 26 (Y105E8B) mu. Interval mapping using two sets of recombinants, 127 animals in total, was conducted using the following SNPs, which result in a restriction fragment length polymorphism: Y71G12A at −17 mu (DraI), W03D8 at −6 mu (DraI), D1007 at −1 mu (DraI), pkP1055 at 0 mu (AvaI), pkP1057 at 1 mu (NaeI), pkP1059 at 3 mu (DraI), B0205 at 5 mu (DraI), F58D5 at 13 mu (DraI), T06G6 at 14 mu (DraI), and Y105E8B at 25 mu (DraI). Genomic DNA from QR180 agef-1(vh4) (Skorobogata et al., 2014) and QR160 dhc-1(vh22) was isolated and submitted to Genome Quebec (Montreal, Canada) for Illumina sequencing. Within the defined map region, dhc-1 (V3228D, D4344N), elpc-1 (A629T), and cpd-1 (T493I) carried homozygous nonsynonymous mutations unique to QR160 dhc-1(vh22) strain.
RNA interference
RNAi feeding was performed essentially as previously described (Kamath et al., 2001) using the dhc-1, elpc-1, cpd-1, and rab-7 clones from the Ahringer RNAi library (Geneservice, Cambridge, United Kingdom). Clones were verified by DNA sequencing. To avoid embryonic and larval lethal phenotypes, synchronized L1 larvae were placed on RNAi plates and scored for vulva induction when the animals reached L4 stage 36–48 h later.
Microscopy and phenotype analysis
General methods for live-animal imaging using Nomarski differential interference contrast (DIC) microscopy were as previously described (Sulston and Horvitz, 1977). Animals were analyzed on an Axio Zeiss A1 Imager compound microscope (Zeiss, Oberkochen, Germany), and images were captured using an Axio Cam MRm camera and AxioVision software (Zeiss). Muv and Vul phenotypes were scored by counting the vulval and nonvulval descendants of P3.p–P8.p in L4-stage larvae as described previously (Skorobogata and Rocheleau, 2012). Fisher’s exact test (graphpad.com/quickcalcs) was used for statistical analysis of the vulval phenotypes. Confocal analysis was performed using Zeiss LSM510 and LSM780 laser scanning microscopes with 63× oil immersion lens (Zeiss) in a single-track mode using 488-nm excitation for GFP. Images were captured using ZEN 2009 and ZEN 2010 Image software (Zeiss). zhIs038 transgene-carrying animals were visualized at the L3 larval stage. The number and location of LET-23::GFP-positive foci in the P6.p descendants were scored blindly.
The percentage embryonic lethality was determined by plating adult hermaphrodites, one per plate, and allowing them to lay eggs for several hours, after which, hermaphrodites were removed and the laid eggs were counted. Approximately 24 h later, the unhatched eggs were counted. To determine the percentage embryonic lethality for each strain, the number of unhatched eggs was divided by the total number of laid eggs.
Supplementary Material
Acknowledgments
We thank Jung Hwa Seo for technical assistance, Richard Roy for sharing reagents, and Stéphane Laporte for access to the LSM510 confocal microscope. We thank Juan Escobar-Restrepo and Alex Hajnal (University of Zürich, Zürich, Switzerland), Karen Oegema and Arshad Desai (University of California, San Diego, La Jolla, CA), and Marie-Anne Felix and Michalis Barkoulas (IBENS, Paris, France) for strains. Many of the strains used in this study were provided by the Caenorhabditis Genetics Center (University of Minnesota, St. Paul, MN), which is supported by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). This work was supported by an operating grant from the Cancer Research Society, a Canadian Institutes of Health Research operating grant (MOP-114935), and a Natural Sciences and Engineering Research Council of Canada discovery grant (RGPIN-341579-13) to C.E.R. C.E.R. was supported by a Fonds de la recherche en santé du Québec Junior II award, O.S. was supported by a Fonds de la recherche en santé du Québec Doctoral training award, J.M. was supported by a Natural Sciences and Engineering Research Council of Canada undergraduate research award, and K.G. was supported by a Natural Sciences and Engineering Research Council of Canada doctoral award. The Research Institute of the McGill University Health Centre is funded in part by the Fonds de la recherche en santé du Québec, and its imaging facility is supported by the Canadian Foundation for Innovation.
Abbreviations used:
- DHC
dynein heavy chain
- EGFR
epidermal growth factor receptor
- MAPK
mitogen-activated protein kinase
- Muv
multivulva
- VPC
vulval precursor cell
- Vul
vulvaless.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-11-0757) on September 21, 2016.
REFERENCES
- Allan VJ. Cytoplasmic dynein. Biochem Soc Trans. 2011;39:1169–1178. doi: 10.1042/BST0391169. [DOI] [PubMed] [Google Scholar]
- Ambros V. Cell cycle-dependent sequencing of cell fate decisions in Caenorhabditis elegans vulva precursor cells. Development. 1999;126:1947–1956. doi: 10.1242/dev.126.9.1947. [DOI] [PubMed] [Google Scholar]
- Aroian RV, Lesa GM, Sternberg PW. Mutations in the Caenorhabditis elegans let-23 EGFR-like gene define elements important for cell-type specificity and function. EMBO J. 1994;13:360–366. doi: 10.1002/j.1460-2075.1994.tb06269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian RV, Sternberg PW. Multiple functions of let-23, a Caenorhabditis elegans receptor tyrosine kinase gene required for vulval induction. Genetics. 1991;128:251–267. doi: 10.1093/genetics/128.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkoulas M, van Zon JS, Milloz J, van Oudenaarden A, Felix MA. Robustness and epistasis in the C. elegans vulval signaling network revealed by pathway dosage modulation. Dev Cell. 2013;24:64–75. doi: 10.1016/j.devcel.2012.12.001. [DOI] [PubMed] [Google Scholar]
- BasuRay S, Mukherjee S, Romero EG, Seaman MN, Wandinger-Ness A. Rab7 mutants associated with Charcot-Marie-Tooth disease cause delayed growth factor receptor transport and altered endosomal and nuclear signaling. J Biol Chem. 2013;288:1135–1149. doi: 10.1074/jbc.M112.417766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berset T, Hoier EF, Battu G, Canevascini S, Hajnal A. Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science. 2001;291:1055–1058. doi: 10.1126/science.1055642. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM. Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics. 2005;6:118. doi: 10.1186/1471-2164-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann R, Essex A, Hu JS, Maddox PS, Motegi F, Sugimoto A, O’Rourke SM, Bowerman B, McLeod I, Yates JR, 3rd, et al. A new mechanism controlling kinetochore-microtubule interactions revealed by comparison of two dynein-targeting components: SPDL-1 and the Rod/Zwilch/Zw10 complex. Genes Dev. 2008;22:2385–2399. doi: 10.1101/gad.1687508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P, Pichler S, Kirkham M, Hyman AA. Cytoplasmic dynein is required for distinct aspects of MTOC positioning, including centrosome separation, in the one cell stage Caenorhabditis elegans embryo. J Cell Biol. 1999;147:135–150. doi: 10.1083/jcb.147.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag A, Gutierrez P, Buhler A, Walser M, Yang Q, Langouet M, Kradolfer D, Frohli E, Herrmann CJ, Hajnal A, et al. An in vivo EGF receptor localization screen in C. elegans identifies the ezrin homolog ERM-1 as a temporal regulator of signaling. PLoS Genet. 2014;10:e1004341. doi: 10.1371/journal.pgen.1004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnal A, Whitfield CW, Kim SK. Inhibition of Caenorhabditis elegans vulval induction by gap-1 and by let-23 receptor tyrosine kinase. Genes Dev. 1997;11:2715–2728. doi: 10.1101/gad.11.20.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill DR, Severson AF, Carter JC, Bowerman B. Centrosome maturation and mitotic spindle assembly in C. elegans require SPD-5, a protein with multiple coiled-coil domains. Dev Cell. 2002;3:673–684. doi: 10.1016/s1534-5807(02)00327-1. [DOI] [PubMed] [Google Scholar]
- Horgan CP, McCaffrey MW. Rab GTPases and microtubule motors. Biochem Soc Trans. 2011;39:1202–1206. doi: 10.1042/BST0391202. [DOI] [PubMed] [Google Scholar]
- Hunt SD, Stephens DJ. The role of motor proteins in endosomal sorting. Biochem Soc Trans. 2011;39:1179–1184. doi: 10.1042/BST0391179. [DOI] [PubMed] [Google Scholar]
- Hwang BJ, Sternberg PW. A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development. 2004;131:143–151. doi: 10.1242/dev.00924. [DOI] [PubMed] [Google Scholar]
- Iyadurai SJ, Robinson JT, Ma L, He Y, Mische S, Li MG, Brown W, Guichard A, Bier E, Hays TS. Dynein and Star interact in EGFR signaling and ligand trafficking. J Cell Sci. 2008;121:2643–2651. doi: 10.1242/jcs.027144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, Olkkonen VM, Neefjes J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol. 2007;176:459–471. doi: 10.1083/jcb.200606077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongeward GD, Clandinin TR, Sternberg PW. sli-1, a negative regulator of let-23-mediated signaling in C. elegans. Genetics. 1995;139:1553–1566. doi: 10.1093/genetics/139.4.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Whitfield CW, Kim SK. The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell. 1998;94:761–771. doi: 10.1016/s0092-8674(00)81735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:RESEARCH0002. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311–320. doi: 10.1016/s0092-8674(88)80024-2. [DOI] [PubMed] [Google Scholar]
- Kimble J. Alterations in cell lineage following laser ablation of cells in the somatic gonad of Caenorhabditis elegans. Dev Biol. 1981;87:286–300. doi: 10.1016/0012-1606(81)90152-4. [DOI] [PubMed] [Google Scholar]
- Lye RJ, Wilson RK, Waterston RH. Genomic structure of a cytoplasmic dynein heavy chain gene from the nematode Caenorhabditis elegans. Cell Motility Cytoskeleton. 1995;32:26–36. doi: 10.1002/cm.970320104. [DOI] [PubMed] [Google Scholar]
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- O’Rourke SM, Dorfman MD, Carter JC, Bowerman B. Dynein modifiers in C. elegans: light chains suppress conditional heavy chain mutants. PLoS Genet. 2007;3:e128. doi: 10.1371/journal.pgen.0030128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadota H, Inoue M, Hikita T, Koppen M, Hardin JD, Amano M, Moerman DG, Kaibuchi K. Establishment of a tissue-specific RNAi system in C. elegans. Gene. 2007;400:166–173. doi: 10.1016/j.gene.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol. 2013;14:713–726. doi: 10.1038/nrm3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid T, Hajnal A. Signal transduction during C. elegans vulval development: a NeverEnding story. Curr Opin Genet Dev. 2015;32:1–9. doi: 10.1016/j.gde.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Schmidt DJ, Rose DJ, Saxton WM, Strome S. Functional analysis of cytoplasmic dynein heavy chain in Caenorhabditis elegans with fast-acting temperature-sensitive mutations. Mol Biol Cell. 2005;16:1200–1212. doi: 10.1091/mbc.E04-06-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorobogata O, Escobar-Restrepo JM, Rocheleau CE. An AGEF-1/Arf GTPase/AP-1 ensemble antagonizes LET-23 EGFR basolateral localization and signaling during C. elegans vulva induction. PLoS Genet. 2014;10:e1004728. doi: 10.1371/journal.pgen.1004728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorobogata O, Rocheleau CE. RAB-7 antagonizes LET-23 EGFR signaling during vulva development in Caenorhabditis elegans. PLoS One. 2012;7:e36489. doi: 10.1371/journal.pone.0036489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sundaram MV. Canonical RTK-Ras-ERK signaling and related alternative pathways. WormBook 2013(Jul 11) 2013:1–38. doi: 10.1895/wormbook.1.80.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Tan PB, Lackner MR, Kim SK. MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell. 1998;93:569–580. doi: 10.1016/s0092-8674(00)81186-1. [DOI] [PubMed] [Google Scholar]
- Taub N, Teis D, Ebner HL, Hess MW, Huber LA. Late endosomal traffic of the epidermal growth factor receptor ensures spatial and temporal fidelity of mitogen-activated protein kinase signaling. Mol Biol Cell. 2007;18:4698–4710. doi: 10.1091/mbc.E07-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009;284:12110–12124. doi: 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Sternberg PW. Competence and commitment of Caenorhabditis elegans vulval precursor cells. Dev Biol. 1999;212:12–24. doi: 10.1006/dbio.1999.9357. [DOI] [PubMed] [Google Scholar]
- Yoon CH, Lee J, Jongeward GD, Sternberg PW. Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-cbl. Science. 1995;269:1102–1105. doi: 10.1126/science.7652556. [DOI] [PubMed] [Google Scholar]
- Zetka MC, Rose AM. The meiotic behavior of an inversion in Caenorhabditis elegans. Genetics. 1992;131:321–332. doi: 10.1093/genetics/131.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.