

Point mutations in Arl13b disrupt cilia morphology, as well as transcriptional and nontranscriptional Shh signaling. Although cilia themselves are not required for nontranscriptional Shh signaling, restricting Arl13b from cilia interferes with its regulation of Shh-dependent chemotaxis.
Abstract
The regulatory GTPase Arl13b localizes to primary cilia, where it regulates Sonic hedgehog (Shh) signaling. Missense mutations in ARL13B can cause the ciliopathy Joubert syndrome (JS), and the mouse null allele is embryonic lethal. We used mouse embryonic fibroblasts as a system to determine the effects of Arl13b mutations on Shh signaling. We tested seven different mutants—three JS-causing variants, two point mutants predicted to alter guanine nucleotide handling, one that disrupts cilia localization, and one that prevents palmitoylation and thus membrane binding—in assays of transcriptional and nontranscriptional Shh signaling. We found that mutations disrupting Arl13b’s palmitoylation site, cilia localization signal, or GTPase handling altered the Shh response in distinct assays of transcriptional or nontranscriptional signaling. In contrast, JS-causing mutations in Arl13b did not affect Shh signaling in these same assays, suggesting that these mutations result in more subtle defects, likely affecting only a subset of signaling outputs. Finally, we show that restricting Arl13b from cilia interferes with its ability to regulate Shh-stimulated chemotaxis, despite previous evidence that cilia themselves are not required for this nontranscriptional Shh response. This points to a more complex relationship between the ciliary and nonciliary roles of this regulatory GTPase than previously envisioned.
INTRODUCTION
The Sonic hedgehog (Shh) signaling pathway regulates many biological processes by controlling the processing and consequently the activity of Gli transcription factors. This transcriptional form of Shh signaling requires the primary cilium; mutations in any of a large number of cilia-associated genes cause serious disruption or total ablation of transcriptional Shh signaling (Huangfu et al., 2003; Mariani and Caspary, 2013). Indeed, most components of the Shh pathway, including the Shh receptor Patched1 (Ptch1), the obligate Shh transducer Smoothened (Smo), and the Gli proteins themselves, are dynamically trafficked in and out of cilia in response to Shh ligand (Corbit et al., 2005; Haycraft et al., 2005; Rohatgi et al., 2007). In addition, components of the Shh signaling pathway can also regulate transcription-independent cellular processes. This nontranscriptional branch of the pathway regulates axon guidance (Charron et al., 2003; Sánchez-Camacho and Bovolenta, 2008; Jin et al., 2015) and fibroblast migration (Bijlsma et al., 2007, 2012). Of note, Shh-dependent chemotaxis in fibroblasts occurs even in cells completely lacking cilia, suggesting that nontranscriptional Shh signaling is cilia-independent (Bijlsma et al., 2012).
Joubert syndrome (JS) can result from mutations in >20 different cilia-associated genes (Parisi and Glass, 1993–2016; Romani et al., 2014; Thomas et al., 2014; Tuz et al., 2014), including the gene on which we focus, ARL13B. JS is diagnosed by a neuroanatomical abnormality of the hindbrain known as the molar tooth sign. JS has been described as a disorder of axon guidance (Engle, 2010) and leads to abnormal development of many white matter tracts (Yachnis and Rorke, 1999; Poretti et al., 2007). Some JS signs and symptoms, such as cerebellar hypoplasia, are reminiscent of transcriptional Shh signaling defects, but the etiology of the axon guidance defects associated with JS remain elusive. Whether cilia-associated genes regulate nontranscriptional Shh signaling is unknown.
The regulatory GTPase Arl13b regulates the transcriptional response to Shh in a number of ways. Loss of Arl13b misregulates ciliary traffic of Ptch1, Smo, Gli2, Gli3, and other Shh-associated proteins (Larkins et al., 2011). In addition to its role in protein traffic, Arl13b regulates Shh transcriptional output downstream of Smo activation. Loss of Arl13b also results in constitutive, ligand-independent production of Gli activator (GliA) but leaves the normal regulation of Gli repressor production intact (Caspary et al., 2007). Arl13b is primarily known as a ciliary protein, and loss of Arl13b leads to abnormalities in cilia structure (Caspary et al., 2007). However, recent studies show that Arl13b is also present in early endosomes and cell surface circular dorsal ruffles (Barral et al., 2012; Casalou et al., 2014), indicating that it also functions outside of the cilium.
ARL13 is ancient, predicted to be present in the last eukaryotic common ancestor. The ARL13 gene appears to have been lost during evolution in organisms that lack cilia and duplicated (to ARL13A and ARL13B) in the urochordates, a common ancestor to all vertebrates. Intriguingly, this duplication coincides with the vertebrate-specific link between cilia and Hedgehog signaling (Logsdon and Kahn, 2003; Li et al., 2004; Kahn et al., 2008; East et al., 2012; Schlacht et al., 2013). In Caenorhabditis elegans, which lacks, Hedgehog signaling, arl-13 mutations disrupt cilia structure and ciliary protein traffic (Cevik et al., 2010; Li et al., 2010; Warburton-Pitt et al., 2014). Although no functional or loss-of-function mutations have been reported for Arl13a orthologues, mutations in Arl13b cause a number of defects in humans and model organisms, such as developmental defects in zebrafish (Sun et al., 2004; Duldulao et al., 2009). Total loss of Arl13b is embryonic lethal in mammals (Caspary et al., 2007). Some point mutations in ARL13B are compatible with life and are linked to the human ciliopathy JS (Cantagrel et al., 2008). Previous studies identified JS patients with homozygous R79Q and Y86C mutations in ARL13B, as well as with an R200C mutation in combination with a truncating mutation (Cantagrel et al., 2008; Thomas et al., 2015).
Arl13b is so named because of its sequence homology to the Arf and Arf-like (Arl) family of small GTPases, although this homology is limited to the N-terminal GTPase domain of Arl13b, which comprises only about half of the protein. Arl13b’s N-terminal Arf domain includes all four of the consensus nucleotide-binding motifs, as well as the canonical switch 1 and switch 2 loops that mediate GTPase interactions with effectors and modulators (Joneson et al., 1996; Kuai and Kahn, 2000). However, Arl13b lacks the highly conserved glutamine in the second nucleotide-binding motif: the amino acid sequence is DVGGQ in Arfs and heterotrimeric G proteins but is DLGGG in Arl13b. This change is predicted to alter Arl13b’s biochemical activities because mutation of this glutamine in other GTPases, including Arfs (Zhang et al., 1994), Arls (Van Valkenburgh et al., 2001; Zhou et al., 2006), and Ras (Cox and Der, 2010), leads to a dominant-activating protein with loss of both intrinsic and GTPase-activating protein (GAP)–stimulated GTP hydrolysis. Another key feature of Arfs is that they have a conserved glycine residue at their N-terminus that is the site of N-myristoylation (Kahn et al., 1988), which facilitates their association with membranes (reviewed in Donaldson and Jackson, 2011). Many Arls, including Arl13b, lack this conserved glycine. Uniquely among Arls, Arl13b contains near its N-terminus a pair of cysteines that become palmitoylated and are essential for its membrane association and cilia localization (Cevik et al., 2013). Also unique within the Arf family, Arl13b contains a cilia localization signal (CiLS) motif that is homologous to the VxP-containing CiLS in rhodopsin, polycystin-1, and polycystin-2 (Deretic et al., 2005; Geng et al., 2006; Ward et al., 2011). This motif in Arl13b is required for its cilia localization (Higginbotham et al., 2012; Cevik et al., 2013).
We investigated the effects of disease-causing and other predicted functional mutations in murine Arl13b on cilia morphology and Shh signaling. To establish a model for Shh signaling and advance the process of determining the specific roles and mechanisms of action of Arl13b in multiple, distinct pathways, we generated a series of point mutations and examined their abilities to rescue transcriptional and nontranscriptional responses to Shh in mouse embryo fibroblasts deleted for the endogenous protein. Previous studies of Arl13b’s function in Shh signaling relied on constitutive or conditional null alleles of Arl13b (Caspary et al., 2007; Horner and Caspary, 2011; Larkins et al., 2011; Su et al., 2012). Furthermore, these studies examined only transcriptional Shh signaling. Our goals include a molecular understanding of Arl13b actions as a regulatory GTPase in Shh signaling in both normal and disease states.
RESULTS
Model system used in our studies
We engineered mutations in murine Arl13b at residues that are conserved across species and predicted to affect protein function (Figure 1A). We generated a G75Q mutation, which restores the homologous glutamine, to determine the significance of Arl13b’s departure from other Arf/Arl proteins at this residue. We also generated a predicted dominant-negative T35N mutation, which is homologous to the RasS17N and Arf1T31N mutations that disrupt guanine nucleotide binding in those proteins (Feig and Cooper, 1988; Dascher and Balch, 1994). Of note, Arl13bT35N was analyzed in other studies, but its effect on Shh signaling was not measured (Hori et al., 2008; Duldulao et al., 2009; Humbert et al., 2012). To study the functional effects of disrupting Arl13b palmitoylation, we used the C8S,C9S mutation, which was previously found to affect membrane and cilia localization of Arl13b. To test the effects of reduced cilia localization on Arl13b functionality, we generated a V358A mutation, which disrupts the CiLS motif. Finally, we generated the R79Q, Y86C, and R200C point mutations in the mouse Arl13b protein, homologous to the JS-causing mutations in humans, to assess their effects on Shh signaling.
FIGURE 1:

Arl13b-GFP mutants R200C, C8S,C9S, and V358A do not localize in cilia. (A) Schematic of mutations in conserved residues in Arl13b’s Arf (red) and C-terminal domains (blue) at residues (marked by red asterisks below the sequences) that are highly conserved in Arl13b orthologues and/or Arf family GTPases. (B) Representative images showing localization of GFP, Arl13b-GFP, or the specified Arl13b mutants (each shown in green) relative to cilia (acetylated α-tubulin [Ac.Tub], in red) in Arl13bhnn MEFs. Scale bar, 10 μm. (C) Percentage of cells with cilia (based on acetylated α-tubulin staining) in Arl13bhnn MEFs expressing Arl13b mutants. (D) Percentage of GFP-positive cilia in Arl13bhnn MEFs expressing GFP alone, Arl13b-GFP, or each of the mutants. Asterisk, significant difference (p < 0.05) from Arl13bWT-GFP.
To test the effects of Arl13b mutations in the absence of wild-type Arl13b, we used protein null Arl13bhnn mouse embryonic fibroblasts (MEFs; Caspary et al., 2007). We infected the immortalized MEFs with lentiviruses driving the expression of wild-type or mutant Arl13b with C-terminal green fluorescent protein (GFP) fusion proteins or GFP alone as a negative control. We previously showed that fusion of GFP to Arl13b’s C-terminus does not interfere with Arl13b function (Larkins et al., 2011). To enrich for transduced cells, we used fluorescence-activated cell sorting (FACS) to purify GFP-positive cells and performed all assays on the resulting heterogeneous cell populations. By analyzing heterogeneous populations, we effectively increase our sample size within a single experiment, as opposed to analyzing multiple clonal populations separately. Our cell populations are composed of MEFs with different insertion sites for the lentiviral expression vector and variable Arl13b-GFP expression levels. Thus the average level of expression within each population may shift, depending on environmental factors such as growth conditions and passage number. Western blot analysis provides an unbiased estimate of the true mean expression level in the population (Supplemental Figure S1). We addressed concerns over the effects of levels of protein expression by performing Western blots on the cell populations at different times. This enabled us to monitor changes in population-level averages. In fact, we observed consistent results in assays done at a variety of passage numbers, suggesting that even if population average protein expression levels fluctuated between assays, this did not affect the overall results.
Arl13b mutations affecting its localization to cilia
We found no significant differences between the percentage of ciliated cells in MEFs expressing GFP alone, Arl13bwild type(WT)-GFP, or any of the variants (Figure 1C). In primary MEFs, loss of Arl13b results in fewer ciliated cells, but a significant fraction of cells (∼20%) retain their cilia (Larkins et al., 2011). In contrast, our immortalized Arl13bhnn MEFs, expressing Arl13b-GFP or GFP alone, were ciliated at rates ranging from 40 to 55% (Figure 1C). These data raise the possibility that the immortalization process may have altered other aspects that control ciliogenesis—for example, a cell-cycle checkpoint.
When we analyzed the percentage of GFP-positive cilia in each cell population, we observed that most of the fusion proteins localized strongly to cilia, with the exceptions of Arl13bR200C-GFP, Arl13bC8S,C9S-GFP, and Arl13bV358A-GFP (Figure 1, B and D; summarized in Table 1). Previous data suggested that Arl13bC8S,C9S-GFP and Arl13bV358A-GFP would fail to localize to cilia due to the disruption of Arl13b’s palmitoylation site and cilia localization signal, respectively. The behavior of Arl13bR200C-GFP was more surprising, especially given the fact that other JS-causing mutants had no effect on cilia localization, suggesting that different JS-associated ARL13B mutations likely act through distinct mechanisms to cause disease.
TABLE 1:
Summary of experimental results testing the effects of expressing Arl13b mutants in Arl13hnn MEFs.
| Arl13b variant (in hnn MEFs) | Mutation type | GFP in cilia | Cilia number | Smo in cilia | qPCR Shh response | Shh migration |
|---|---|---|---|---|---|---|
| WT-GFP | Yes | Normal | Normal | Normal | Normal | |
| GFP only | Null | No | Normal | Abnormal | Reduced | Impaired |
| R79Q-GFP | JS | Yes | Varied | Normal | Normal | Normal |
| Y86C-GFP | JS | Yes | Normal | Normal | Normal | Normal |
| R200C-GFP | JS | No | Normal | Normal | Normal | Normal |
| T35N-GFP | GTPase | Yes | Normal | Abnormal | Reduced | |
| G75Q-GFP | GTPase | Yes | Normal | Normal | Reduced | |
| V358A-GFP | CiLS | No | Normal | Normal | Normal | Impaired |
| C8S,C9S-GFP | Palm. | No | Normal | Abnormal | Reduced |
Statistically significant differences between Arl13bWT-GFP and other mutants are shown in bold.
Arl13b mutations affect nontranscriptional Shh signaling in MEF migration
To determine whether Arl13b regulates nontranscriptional Shh signaling, we tested the effect of Arl13b mutations on Shh-dependent chemotaxis (Bijlsma et al., 2007, 2012). We found that Arl13bhnn MEFs expressing GFP alone showed significant impairment in migration toward either recombinant Shh ligand (rShhN, Figure 2A) or the Smo agonist purmorphamine (Figure 2D) compared with Arl13bhnn MEFs rescued with Arl13bWT-GFP. This supports the conclusion that Arl13b does indeed play a role in mediating Shh-dependent chemotaxis in MEFs. As a control for general cell motility defects, we demonstrated that Arl13bhnn MEFs expressing GFP alone or Arl13bWT-GFP migrate similarly toward fetal calf serum, a nonspecific chemoattractant (Figure 2D).
FIGURE 2:
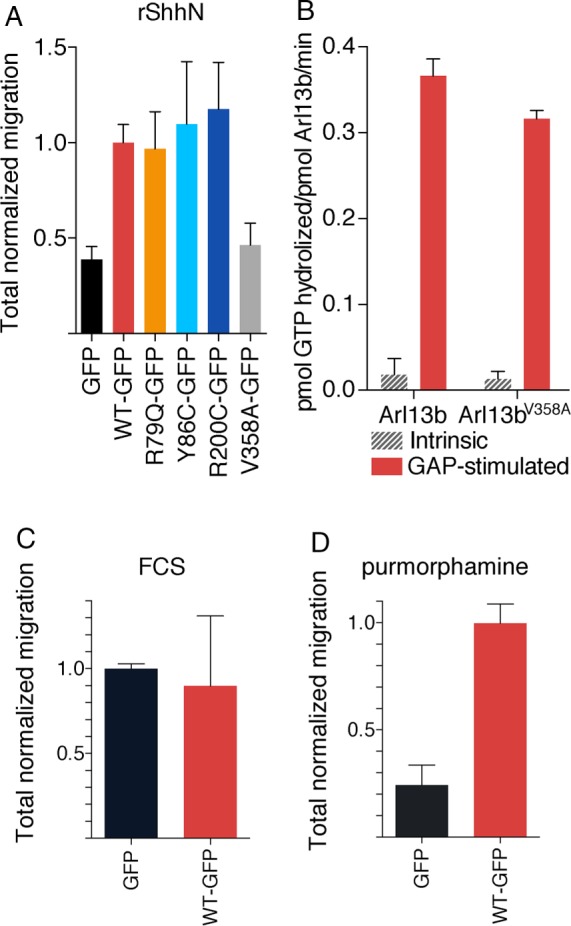
Nonciliary Arl13bV358A mutant is defective in Shh-stimulated MEF migration despite Arl13bV358A being biochemically indistinguishable from wild-type Arl13b protein. (A) Average total migration for Arl13bhnn MEFs expressing Arl13b-GFP mutants, normalized to wild-type Arl13b-GFP, migrating toward rShhN. N = 12. Analysis described in Materials and Methods. (B) Recombinant mouse Arl13b and Arl13bV358A display indistinguishable intrinsic and GAP-stimulated rates of GTP hydrolysis. (C) Arl13bhnn MEFs expressing GFP or Arl13bWT-GFP, normalized to the GFP-only control, migrate similarly toward fetal calf serum (FCS). (D) Arl13bhnn MEFs expressing GFP show impaired migration toward the Smo agonist purmorphamine, which can be rescued by Arl13bWT-GFP.
We next tested several Arl13b mutants—the JS-causing Arl13bR79Q-GFP, Arl13bY86C-GFP, and Arl13bR200C-GFP, as well as the CiLS mutant Arl13bV358A-GFP. We hypothesized that JS-causing mutations would disrupt nontranscriptional Shh signaling, consistent with a model of JS in which axon guidance defects arise from abnormal nontranscriptional Shh signaling in the growth cone. Furthermore, because nontranscriptional Shh signaling does not require the cilium, we predicted that the CiLS mutation disrupting Arl13b’s ciliary localization would not affect migration. Although the cilium is the signal transduction hub for transcriptional Shh signaling, we posited that Arl13b should be able to serve its functions regulating nontranscriptional Shh-dependent chemotaxis from outside of the cilium. Surprisingly, we found that MEFs expressing the JS-causing Arl13b mutants migrated toward rShhN indistinguishably from MEFs expressing Arl13bWT-GFP. In contrast, MEFs expressing the ciliary localization mutant Arl13bV358A-GFP behaved similarly to Arl13bhnn MEFs expressing only GFP. We eliminated the possibility that there was an insufficient level of Arl13bV358A-GFP expression by performing Western blot analysis on the cell populations used in the migration assays. We observed that Arl13bV358A-GFP was expressed at a higher level than Arl13bWT-GFP, as were the other mutants that rescued chemotaxis (Supplemental Figure 1B).
One explanation of these results is that JS-causing Arl13b mutations have only subtle effects on Shh-related Arl13b functions, whereas mutating Arl13b’s CiLS domain ablates or severely disrupts overall protein function. However, on the basis of previous work involving Arl13b mutants (Higginbotham et al., 2012; Humbert et al., 2012; Gotthardt et al., 2015) and Arl13b functioning outside of cilia (Barral et al., 2012; Casalou et al., 2014), we believe it most likely that Arl13b outside of the cilium retains its biochemical properties and functions. To test this, we generated purified recombinant preparations of mouse glutathione S-transferase (GST)–Arl13b and GST-Arl13bV358A after expression in human embryonic kidney 293T (HEK293T) cells. The two proteins expressed to comparable levels, remained soluble throughout purification, and yielded similar levels of purified protein. To assess their biochemical properties and abilities to interact with other biologically relevant partners, we determined the intrinsic and GAP-stimulated rates of GTP hydrolysis, using an assay described previously to purify ARL2 GAPs (Bowzard et al., 2005, 2007). We found strong (∼20-fold) GAP-stimulated activity using a detergent-solubilized preparation from bovine brain as the source of Arl13b GAP. The intrinsic rates of GTP hydrolysis were 0.018 ± 0.019 and 0.013 ± 0.009 pmol of GTP hydrolyzed/min/pmol Arl13b for GST-Arl13b and GST-Arl13bV358A, respectively. The GAP-stimulated rates of hydrolysis for these same two preparations were 0.366 ± 0.020 and 0.316 ± 0.010 pmol GTP hydrolyzed/min/pmol Arl13b. Thus there were no differences between the abilities of GST-Arl13b and GST-Arl13bV358A to serve as substrates in this reaction (Figure 2B).
Chlamydomonas Arl13 was recently shown to have exchange factor activity toward Arl3 (Gotthardt et al., 2015), and so we tested for Arl3 guanine nucleotide exchange factor (GEF) activity of murine Arl13b using a comparable assay, as described in Materials and Methods. The rate-limiting step in activation of regulatory GTPases is typically the release of bound GDP, so we measured the ability of purified Arl13b proteins to increase the rate of dissociation of prebound [3H]GDP from purified human Arl3. We used N-terminal truncations, termed Δ19-Arl13b or Δ19-Arl13bV358A, as used previously (Zhang et al., 1994). We found that the rate of dissociation of GDP from Arl3 (1 μM) alone under these conditions was 3.2 × 10−4 s−1, yielding a half-life of the bound species of ∼37 min. Addition of GST-Δ19-Arl13b or GST-Δ19-Arl13bV358A (2 μM) resulted in dramatic increases in this rate to 3.4 × 10−3 and 3.6 × 10−3 s−1, respectively, yielding half-lives of only 3 min for Arl3-GDP in the presence of either Arl13b protein. We separately measured the rate of [35S]GTPγS binding in the same assay because the binding of the activating ligand is limited by the release of bound GDP and found comparable effects of added Arl13b proteins in this assay (unpublished data). Cleavage of the GST tag did not alter these rates, suggesting that the tag did not interfere with any of these activities. We interpret these biochemical data as strong evidence that the V358A mutation does not cause defects in protein folding or the ability to bind to either of these two binding partners, including effectors. Instead, this mutation is expected to interfere with a different protein–protein interaction that is required for cilia localization.
Arl13b mutations affect transcriptional Shh signaling
Gli1 and Ptch1 messages are both up-regulated upon treatment of cells with Shh. Thus, to measure transcriptional Shh signaling in Arl13bhnn MEFs expressing wild-type or mutant Arl13b-GFP, we used quantitative real-time PCR (qPCR) for the Shh target genes Gli1 and Ptch1 (Figure 3). Gli1 is not expressed unless transcriptional Shh signaling is active (Bai et al., 2002). Ptch1 is expressed at baseline levels to serve its functions as a Shh receptor in the presence of ligand and Smo inhibitor when ligand is absent; Ptch1 becomes up-regulated upon Shh stimulation, in a form of negative feedback (Marigo et al., 1996; Goodrich et al., 1997). Therefore we expected the magnitude of change upon Shh treatment to be higher for Gli1 than Ptch1 as a result of the lower baseline of Gli1, which we observed consistently. Several Arl13b mutants caused statistically significant loss of the Gli1 transcriptional response in Arl13bhnn MEFs (Figure 3). In accordance with previous studies using primary Arl13bhnn MEFs (Su et al., 2012), Arl13b-GFP was able to restore a strong (∼14-fold) Shh-induced Gli1 expression in the immortalized Arl13bhnn cells, whereas those expressing GFP alone exhibited a very limited (∼3- to 4-fold) Shh response. Furthermore, Arl13bC8S,C9S-GFP, Arl13bT35N-GFP, and Arl13bG75Q-GFP each showed a significantly decreased response to Shh, similar to that found with GFP alone. These results suggest that membrane association of Arl13b, as well as GTP binding and hydrolysis, is important for Arl13b regulation of the Shh transcriptional response, as predicted. Intriguingly, Arl13bV358A-GFP and Arl13bR200C were each able to rescue the transcriptional response to Shh in Arl13bhnn MEFs despite their apparent absence from cilia (Figure 1B). This suggests that, although cilia serve as a critical hub for transcriptional Shh signaling, nonciliary Arl13b may also be able to regulate the transcriptional Shh signaling cascade.
FIGURE 3:

Arl13b-GFP mutants T35N, G75Q, and C8S,C9S disrupt the transcriptional response to Shh stimulation. Real-time qPCR of Shh targets Gli1 and Ptch1 in Arl13bhnn MEFs expressing Arl13b-GFP mutants. Data are shown as fold change relative to the baseline expression level of each target gene in the Arl13bWT-GFP cell line. Asterisk, significant difference (p < 0.05) from Arl13bWT-GFP.
Arl13b mutations affect Shh-dependent Smo enrichment in cilia
After observing the effects of Arl13b mutants on transcriptional Shh signaling, we investigated whether any of these effects could be related to abnormal traffic of Smo. Our previous work showed that Arl13bhnn MEFs exhibit abnormal Shh-dependent traffic of Shh pathway components, including Smo itself (Larkins et al., 2011). In wild-type cells, Smo is enriched in the cilium only after Shh stimulation. In Arl13bhnn cells, however, Smo is constitutively enriched in cilia and forms abnormal puncta along the length of the cilium, suggesting that Arl13b plays a critical role in the Shh-dependent enrichment of Smo in cilia. It remains unclear whether Arl13b regulates the entry and/or the exit of Smo. However, Arl13b also acts downstream of Smo to regulate Shh signaling (Caspary et al., 2007), so the effects we observed on transcriptional signaling could be due to a Smo-independent mechanism. Therefore we assayed for Smo enrichment in the cilia of Arl13bhnn MEFs expressing Arl13b-GFP mutants at baseline and under Shh-stimulated conditions. Because a small amount of Smo is present in cilia even in the absence of Shh signaling (Ocbina and Anderson, 2008; Milenkovic et al., 2015) and we can detect low levels of Smo in almost all cilia under control conditions, we refer to “Smo-enriched” cilia rather than “Smo-positive” cilia when describing the accumulation of ciliary Smo that normally reflects active Shh signaling.
Consistent with previous reports, in Arl13bhnn MEFs expressing Arl13bWT-GFP, Smo enrichment in cilia occurred upon Shh stimulation, whereas in MEFs expressing GFP alone, Smo enrichment in cilia was evident regardless of Shh stimulation (Figure 4). We observed elevated baseline Smo enrichment, consistent with a deficiency in Arl13b activity, in MEFs expressing Arl13bT35N-GFP and Arl13bC8S,C9S-GFP. Even in cell populations with abnormally high ciliary enrichment of Smo at baseline, we saw a trend toward further Smo accumulation in cilia upon Shh stimulation, suggesting that Arl13b is not the sole determinant of Smo’s Shh-dependent ciliary traffic. Still, results from these studies suggest that Arl13b plays an important role in regulating Smo enrichment in cilia that is sensitive to Arl13b’s presence on cell membranes and its normal regulation of guanine nucleotide cycling. Of interest, this regulation of Smo does not explicitly require Arl13b to be present in cilia: Arl13bR200C-GFP and Arl13bV358A-GFP, each of which failed to localize to cilia, show normal patterns of Smo ciliary enrichment under control and Shh-stimulated conditions. This suggests that Arl13b functions outside the cilium to regulate ligand-dependent Smo enrichment.
FIGURE 4:
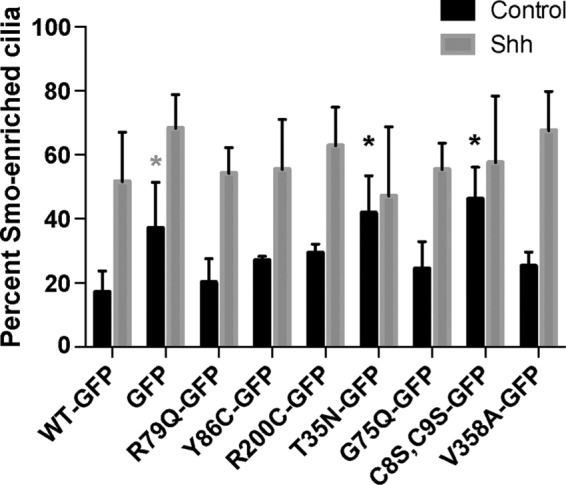
Arl13b’s role in ciliary Smo enrichment is altered by Arl13b-GFP mutants T35N and C8S,C9S. Shh-dependent ciliary enrichment of Smo in response to Shh stimulation in Arl13bhnn MEFs expressing Arl13b-GFP mutants. Smo enrichment was quantified based on amount of Smo staining colocalized with acetylated α-tubulin as described in Materials and Methods. Black asterisks, significantly different from Arl13bWT-GFP with multiple comparison adjusted p < 0.05. Gray asterisk, adjusted p = 0.05.
DISCUSSION
We set out to assign function to particular amino acid residues in the Arl13b protein to see whether we could deconvolve the distinct phenotypes we observe in Arl13bhnn cells and better understand the cellular defects associated with the disease-linked mutations. We examined several classes of mutants, including GTP-handling mutants (T35N and G75Q), JS-causing mutants (R79Q, Y86C, and R200C), a palmitoylation-deficient mutant (C8S,C9S), and a nonciliary mutant (V358A). We found that the mutants affected ciliary localization of Arl13b protein, the transcriptional response to Shh, and Shh-dependent ciliary Smo enrichment in distinct ways. In addition, we extended Arl13b’s functions to include regulation of nontranscriptional, Shh-dependent MEF migration. Because nontranscriptional, Shh-dependent MEF migration occurs in the absence of cilia, we were surprised to find that Arl13bV358A, which was not evident in cilia, was unable to mediate MEF migration (summarized in Table 1). Taken together, our data support Arl13b functioning as a GTPase, albeit an atypical one, and operating both in and out of the cilium in a complex, and potentially interdependent, manner.
Both of the GTPase-handling mutants, T35N and G75Q, localized to cilia, yet neither could rescue the decreased Shh transcriptional response of Arl13bhnn MEFs, consistent with alterations in their biochemical activity. However, the mutations affect Arl13b function distinctly: only the T35N mutant, with its presumptive decrease in binding of guanine nucleotides, failed to rescue ligand-independent ciliary Smo enrichment in Arl13bhnn MEFs. In contrast, restoration of the homologous Q in the G75Q mutant was functional in this assay. The palmitoylation-deficient mutant (C8S,C9S) behaved similarly to the T35N mutant, although the C8S,C9S mutant did not localize to cilia. Association of a variety of regulatory GTPases with membranes is fundamental to their actions, and so the profound effects of removing sites of palmitoylation are readily understood. We interpret these data as evidence that both membrane association and nucleotide binding are critical components in Arl13b cellular actions. Thus, despite Arl13b being an atypical member of the Arf family with regard to the absence of the key glutamine that is directly involved in GTP hydrolysis in other family members, it retains both intrinsic and GAP-stimulated GTPase activities, suggesting that it uses a distinctive mechanism of nucleotide hydrolysis.
With Arl13b shown to function in key aspects like other members of the Arf family, we are increasingly confident that we can draw on analogies with previous results to predict mechanistic consequences of at least two of the three JS mutations. The R79Q and Y86C mutations are each within the guanine nucleotide binding-sensitive switch 2 and, by analogy, should not alter protein folding or nucleotide handling but instead alter one or a subset of specific GTPase-effector interaction(s). Consistent with this, each mutant localizes to cilia and rescued Shh-dependent ciliary Smo enrichment, as well as both transcriptional and nontranscriptional Shh signaling. In contrast, the R200C mutation, in the nonhomologous C-terminal domain, rarely localized to cilia and yet behaved like the wild-type protein in all other regards assayed here. Although this might be unexpected, given that these are pathogenic mutations, the fact that loss of Arl13b is embryonic lethal would be consistent with subtle hypomorphic alleles being compatible with life. Cells in vivo respond to both the amount and duration of Shh signaling (Dessaud et al., 2007, 2008). Because several of the phenotypes in JS patients are consistent with a loss in Shh signaling, it is important to note that our assays were performed at a single time point, and follow-up studies of these mutations in vivo might yet reveal a role for the JS-causing mutations in Shh signaling. Identifying binding partners whose interactions are affected by the switch 2 mutations is also predicted to reveal novel mechanisms of Arl13b actions relevant to JS disease processes.
Regulation of nontranscriptional Shh signaling
Arl13b acts at multiple steps in the transcriptional Shh pathway: it regulates the ligand-dependent ciliary enrichment of Smo and also acts downstream of Smo to regulate GliA (Caspary et al., 2007; Larkins et al., 2011). We found that the Arl13bV358A mutant is deficient in localization to cilia and disrupts Shh-dependent chemotaxis but has no effect on Shh-dependent ciliary enrichment of Smo, suggesting that Arl13b’s role in nontranscriptional Shh signaling lies downstream of Smo ciliary enrichment. Little is known about the downstream mechanisms of Shh signaling in chemotaxis, but Arl13b’s common role in both the transcriptional and nontranscriptional pathways suggests that other components may be shared between the two.
The C-terminal domain of Arl13b is not homologous to any other domain, and we lack any structural information about it. Thus, given our observation that the V358A mutation is defective in cilia localization, as well as in Shh-stimulated cell migration, we tested its integrity in biochemical assays. The facts that this point mutant expressed to the same levels in HEK cells, purified the same, remained soluble throughout purification and storage, and displayed indistinguishable intrinsic and GAP-stimulated GTPase activity with the wild-type protein are all consistent with retention of protein folding and some biologically important protein–protein interactions. Of course, this mutant is predicted to be deficient in at least one protein–protein interaction: that involved in sorting of its traffic to cilia and involving the mutated VxP motif.
Decoupling Arl13b function from cilia
One of the most surprising results of this study was the fact that Arl13b mutants that are undetectable in cilia retain some functions. Arl13b is a ciliary protein, and JS, like other diseases resulting from cilia-related genetic mutations, is considered a ciliopathy. Transcriptional Shh signaling is tightly linked to cilia, and yet, nonciliary Arl13bV358A-GFP, as well as nonciliary, JS-causing Arl13bR200C-GFP, exhibits normal induction of Shh target genes upon ligand stimulation in Arl13bhnn MEFs. Meanwhile, in assays of nontranscriptional Shh signaling, Arl13bV358A-GFP MEFs show significant impairment in their Shh response, despite the fact that the cilium itself is not necessary for Shh-dependent cell migration. This suggests distinct roles for Arl13b in transcriptional versus nontranscriptional Shh signaling, one of which requires its CiLS motif and one of which does not. In contrast, Arl13bR200C-GFP did rescue the nontranscriptional Shh signaling, which could be due to it being undetectable in cilia for a reason distinct from Arl13bV358A-GFP.
Because the cilium itself is not required for Shh-dependent chemotaxis, it is difficult to understand why Arl13b’s CiLS would be required for this process. One model to explain our results is that Arl13b must interact with a binding partner found only in cilia in order to potentiate nontranscriptional Shh signaling, and Arl13bV358A and this binding partner never encounter each other in ciliated cells. Alternatively, the CiLS in Arl13b may regulate more than just cilia localization. Arf and Arl proteins are major regulators of protein traffic at the Golgi and trans-Golgi network, where VxP motifs target rhodopsin, polycystin1, and polycystin2 to cilia in a process that requires Arf4 and Arl3 (Kahn, 2009; Mazelova et al., 2009; Kim et al., 2014). Recent work shows that Arl13b acts as an Arl3 GEF, and our data argue that Arl13bV358A retains this activity (Gotthardt et al., 2015). Thus mutation of Arl13b’s CiLS may prolong its ability to interact with Arl3 or other effectors at the Golgi/trans-Golgi network, which could affect traffic of proteins to nonciliary regions of the membrane that are important for regulating chemotaxis.
Future studies investigating Arl13b’s role in nontranscriptional Shh signaling are needed to dissect the function of the CiLS motif in regulating Arl13b’s cilia localization and its role in potential cilia-independent functions. Because cells lacking cilia are capable of Shh-mediated chemotaxis, it would be informative to test whether nonciliated cells that also lack Arl13b can migrate toward a Shh source. It may be that Shh-dependent chemotaxis in nonciliated cells depends on interactions between proteins normally found in the cilium that are also capable of interacting when forced into nonciliary compartments. If Arl13b mediates these interactions, one would predict that loss of Arl13b should disrupt Shh-dependent chemotaxis in nonciliated cells, which could be rescued by Arl13bV358A. If the CiLS motif is involved in the formation of protein complexes in nonciliary compartments, however, Arl13bV358A may not be capable of restoring Shh-dependent chemotaxis. Alternatively, because the loss of the cilium results in dysregulation of the many molecules that make up the ciliary proteome, it is possible that Shh-dependent chemotaxis in nonciliated cells occurs via a different mechanism than in ciliated cells. Indeed, previous data suggest that there is some gain of function in Shh-dependent fibroblast migration in nonciliated cells (Bijlsma et al., 2012). Therefore it is possible that Arl13b is involved only in the ciliary Shh signaling cascade and that nonciliated cells would be capable of Shh-dependent chemotaxis even without Arl13b. Our data indicate that the relationship between Arl13b, cilia, and Shh signaling is more nuanced than previously believed and provide a foundation for exploring these questions.
In sum, we found that Arl13b plays multiple roles in the regulation of Shh signaling both within and outside of cilia and separated these functions using point mutations in Arl13b. Our results also showed that JS-causing Arl13b mutants disrupt Arl13b function through multiple mechanisms and yet give rise to the same disease phenotypes. This work emphasizes the importance of investigating the diverse functions of ciliary gene products as we continue to unravel the molecular etiology of JS and other ciliopathies.
MATERIALS AND METHODS
Mice and genotyping
All mouse work was performed at Emory University under approved Institutional Animal Care and Use Committee protocols. Arl13bhnn mice were generated in an N-ethyl, N-nitrosourea screen (Caspary et al., 2007) on a C57/Bl6 background and maintained on a C3H background. Genotyping for the Arl13bhnn allele uses strain-specific linked markers to detect whether mice are Bl6 or C3H both upstream and downstream of the Arl13b locus. Arl13bhnn genotyping primers were hnn147, forward, AAT GCC TCA AGT GCC TCT TT, and reverse, GGG ACT CAT CTT TGG GAA CA; and hnn174, forward, TGT GGG TGG CAT ATG TAG GA, and reverse, GCT AGC TAT TTT CTG TTG CTG GA.
Generating MEFs
Pregnant dams at embryonic day 12.5 (E12.5; determined by the presence of a vaginal plug at E0.5) were killed by cervical dislocation and embryos dissected into cold phosphate-buffered saline (PBS). After removal of the head and visceral organs, embryos were mechanically dissociated by being passed several times through an 18-gauge needle in warm MEF medium (DMEM plus10% fetal bovine serum [FBS] and 1× penicillin/streptomycin). Dissociated MEFs were cultured in gelatin-coated tissue culture dishes (0.1% gelatin in water, autoclaved and filtered, was placed on dishes for at least 1 h before use and then aspirated off before plating MEFs).
Primary MEFs at passage two or three were cultured until 50–75% confluent and then transfected with a SV40 large T antigen construct to induce immortalization. Immortalizing MEFs were cultured until confluent and split 1:10 six times (a final dilution factor of 1:1,000,000 from the original primary culture) before being considered immortalized. Immortalizing MEFs that took >14 d to become confluent after a 1:10 split were discarded.
Generating lentiviruses
Arl13bWT-GFP and Arl13bV358A-GFP lentiviruses were produced by the Emory University Center for Neurodegenerative Disease Viral Vector Core. All other lentiviruses used were generated in the Caspary lab by cotransfecting HEK293FT cells with envelope (pMD2.G), packaging (psPAX2) and lentiviral Arl13b expression (generated from L13-Arl13b-GFP) plasmids and collecting virus-containing supernatant from the cells. At 24 h after transfection, medium was changed to 10 ml of DMEM plus 10% FBS. This medium was left on the virus-producing transfected cells for 24 h and then passed through a 0.45-μm filter and stored at 4°C. This collection procedure was repeated the following day.
FACS purification of lentiviral-infected MEF lines
Immortalized MEFs were plated at low density in 50% MEF medium plus 50% lentiviral supernatant. MEFs were cultured for 48–72 h to allow time for lentiviral infection and protein expression before FACS. On the day of sorting, MEFs were detached using 0.25% trypsin and resuspended in sort buffer (Ca2+/Mg2+-free PBS with 1% FBS, 1 mM EDTA, and 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.0, sterilized through a 0.22-μm filter).
Sorting was performed by the Emory University School of Medicine Core Facility for Flow Cytometry or the Emory·Children’s Pediatric Research Center Flow Cytometry Core using a BD FACS Aria II Cell Sorter. A 100-μm nozzle was used to accommodate the relatively large size of the MEFs. Before sorting lentivirus-infected MEFs for GFP, an uninfected negative control was used to define gates for single cells using forward scatter and side scatter. GFP-positive infected cells were purified through a two-step process: a presort enrichment to isolate GFP-positive MEFs, followed by single-cell sorting of the GFP-enriched population into a 24-well plate. Depending on the number of infected cells in a given sample, from one to four wells were filled with 750–5000 GFP-positive MEFs. This process creates a genetically heterogeneous cell population produced from multiple founder cells representing a variety of genomic insertion sites for the lentiviral construct with a variety of GFP expression levels.
Immunofluorescence staining
Arl13b-GFP MEF lines were labeled with primary antibodies for acetylated α-tubulin (1:2500; T6793; Sigma-Aldrich, St. Louis, MO), GFP (1:1000; ab13970; Abcam, Cambridge, UK), and/or Smo (1:500; a gift from the Kathryn Anderson lab, Memorial Sloan Kettering Cancer Center, New York, NY), fluorescent secondary antibodies (Alexa Fluor 488 or 568 for the appropriate species, 1:500; Life Technologies, Carlsbad, CA), and Hoechst nuclear stain (1:3000).
Image analysis
Fluorescent micrographs were acquired with a 40× objective on a Leica CTR6000 microscope using SimplePCI software. For experiments quantifying the number of cilia, microscopy fields for imaging were chosen based only on Hoechst staining to ensure unbiased sampling of ciliated versus nonciliated cells. For experiments quantifying the number of GFP- or Smo-positive cilia, microscopy fields for imaging were chosen based on the acetylated α-tubulin staining to ensure that an appreciable number of cilia were present in each image.
Before analysis, images were coded using blindanalysis (Salter, 2016), a script that replaces the original file names with a random alphanumeric string and creates a .csv keyfile pairing original and coded file names to be used for later decoding. All image analysis was done while blinded to Arl13b mutant and (where applicable) control versus Shh treatment conditions.
Images were analyzed using the Fiji distribution of ImageJ software (Schindelin et al., 2012). For experiments testing the ciliogenesis rate of different MEF lines, nuclei were counted based on Hoechst staining, and cilia were counted based on acetylated α-tubulin staining. All other measurements analyzed only ciliated cells. For experiments measuring the presence or enrichment of GFP or Smo in cilia, cilia were defined using acetylated α-tubulin staining in one color channel, and then the color channel representing GFP or Smo was examined to see whether staining colocalized with acetylated α-tubulin (Figure 1B).
Migration assays
MEF migration assays were performed using the FluoroBlok Transwell system (BD Falcon, Corning, NY) as in Bijlsma et al. (2012). Before beginning the migration assay, MEFs were washed with PBS and incubated with 10 μM CellTracker Green in serum-free DMEM for 1 h (cells were equilibrated in serum-free DMEM for 1 h before use). Cells were detached using 5 mM ethylenediaminetetraacetic acid (EDTA) in PBS, resuspended and washed in serum-free DMEM, and transferred to a Transwell insert (8-μm pore size) in 100 μl of medium at a concentration of 50,000 cells/well. The lower portion of the Transwell plate contained 600 μl of control or attractant (0.1 μg/ml rShhN or 2 μM purmorphamine) medium. During migration, the plate reader measured fluorescence in the lower portion of the Transwell plate once every 2 min for ∼3 h. For each cell population, 12 different wells were analyzed using this method.
To analyze migration data, several corrections were applied. Background fluorescence was controlled by subtracting the average value of plate reads from a blank well (containing medium but no cells) over the course of the entire experiment from each individual plate read for the cell-containing wells. Nonspecific migration of each cell line was controlled by subtracting the average fluorescence of cells migrating toward control medium from each individual plate read for the corresponding cells in the attractant condition. Finally, migration curves for each cell line were made more directly comparable by setting the starting point of each normalized migration curve to zero. This was done by subtracting a normalizing value from every point on the migration curve such that the value of the first point on the curve was equal to zero. Pooling of data was done using these normalized curves expressed relative to the Arl13bWT-GFP control from each experiment (Figure 2A) or relative to the GFP-only control (Figure 2C).
Protein expression and purification
Human embryonic kidney 293T (HEK; American Type Culture Collection) cells were grown in 10-cm plates in DMEM (Invitrogen) supplemented with 10% FBS (Atlanta Biologicals) at 37°C in a humidified environment gassed with 5% CO2. The medium was switched to DMEM with 2% FBS, and cells were transfected with 1 μg of DNA/ml of medium at a cell density of ∼90% using 3 μg of polyethyleneimine (PEI Max; 24765-2; Polysciences)/ml of medium. All Arl13b protein expression was achieved using the pLEXm-GST parent plasmid, a gift from James Hurley (National Institutes of Health, Bethesda, MD), which uses a chicken β-actin promoter to drive expression and includes a TEV protease cleavage site after the GST and immediately upstream of the inserted open reading frame (Ivanova et al., 2014). Testing found that 2 d was optimal for protein expression. Thus cells were harvested 48 h after transfection, pelleted by centrifugation at 3000 × g, frozen in liquid nitrogen, and stored at −80°C until used for protein purification. Cells were thawed on ice and lysed in five volumes of lysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate [CHAPS], protease inhibitor mixture [S8830; Sigma-Aldrich], and 10 μg/ml deoxyribonuclease I [D4263; Sigma-Aldrich]). The cells were maintained on ice for 30 min before cell debris was removed by centrifugation at 14,000 × g for 10 min at 4°C. The proteins were purified with GST-affinity chromatography. Specifically, 500 μl of glutathione-Sepharose 4B (17-0756-01; GE Healthcare, Little Chalfont, UK) beads was added to 120 mg of protein lysate and incubated at 4°C for 2 h. The beads were then loaded into a disposable column and washed three times with five column volumes of lysis buffer. The proteins were eluted from the beads with three column volumes of elution buffer (25 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM glutathione) and stored at −80°C.
Arl13b GAP assay
The intrinsic and GAP-stimulated GTPase activity of purified recombinant murine Arl13b proteins was determined using the GAP assay described previously for ARL2 (Bowzard et al., 2005, 2007), with minor modifications. The source of Arl13b GAP was bovine brain, which was homogenized before preparing a detergent (1% CHAPS) extract and clarifying by centrifugation at 14,000 × g. Briefly, GST-Arl13b or GST-Arl13bV358A was preloaded with [γ-32P]GTP at 30°C for 1–3 h in loading buffer (25 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 5 mM ATP, 0.1% cholate/3 mM dimyristoyl phosphatidylcholine). The GTPase reaction, performed in a total volume of 50 μl in the assay buffer (25 mM HEPES, pH 7.4, 2.5 mM MgCl2, 1 mM dithiothreitol [DTT], 2.5 mM GTP), was initiated by the addition of 5 μl of preloaded Arl13b. The reactions were incubated for 4 min at 30°C before being stopped by the addition of 750 μl of ice-cold charcoal suspension (5% Norita charcoal in 50 mM NaH2PO4) with mixing. Intrinsic GTPase activity was determined after subtracting the amount of 32Pi released in parallel incubations lacking proteins. GAP-stimulated activities were those in which the bovine brain GAP was added to the assay; intrinsic GTP hydrolysis rates were deducted to determine the GAP-stimulated rates. Any hydrolysis of GTP contributed by the brain detergent extract that was independent of Arl13b was also subtracted, although this was always very small due to the vast excess of cold GTP added to the assay. Charcoal, with bound nucleotides, was pelleted by centrifugation at 3000 × g for 10 min at 4°C. The amount of 32Pi released during GTP hydrolysis was measured by liquid scintillation counting. The experiments were repeated at least twice with at least two different preparations of each protein performed in triplicate.
Arl3 GEF assay
The ability of purified recombinant murine GST-Δ19-Arl13b or GST-Δ19-Arl13bV358A to serve as a GEF for Arl3 was determined using a modification of the assay described in Gotthardt et al. (2015). Human Arl3 was expressed in and purified from bacteria, as previously described (Van Valkenburgh et al., 2001). Briefly, purified recombinant human Arl3 (final concentration 1 μM) was incubated at 30°C in 25 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, and 1 μM [3H]GDP (specific activity 3000 cpm/pmol; PerkinElmer Life Sciences, Waltham, MA), along with the indicated concentration of purified Arl13b preparations. Radioligand binding/release was stopped by dilution of 10 μl of reaction cocktail into 2 ml of ice-cold buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT), and the amount of [3H]GDP remaining bound was determined by filtration through BA85 nitrocellulose filters (0.45 μm, 25 mm [Whatman, Maidstone, UK]), as described previously (Cavenagh et al., 1994). Binding was quantified by using a liquid scintillation counter. The experiments were repeated at least twice with at least two different preparations of each protein performed in duplicate. The 4-min time point was chosen after previously determining that the rate of GDP dissociation was linear with time for at least 10 min under these conditions. In addition, the GEF activity of Arl13b toward Arl3 was found to increase linearly with Arl13b concentration in the range of 0.1–5 μM Arl13b, approaching saturation at >10 μM, with a half-maximal rate seen at 1–2 μM Arl13b.
Quantitative real-time PCR
MEFs were plated in six-well plates at densities of 3 × 105 cells/well and were treated with 0.5% serum control medium or Shh-conditioned medium (Taipale et al., 2000) 24 h after plating. After 24 h, MEFs were detached using 0.25% trypsin, spun down, and flash frozen on dry ice. Cell pellets were kept at –80°C until RNA extraction.
Cell pellets were lysed with RLT lysis buffer and QIAshredder (79656) homogenizer columns (Qiagen, Hilden, Germany). RNA was extracted using the Qiagen RNeasy kit (74104), including the optional on-column DNase digest to minimize contamination with genomic DNA. cDNA was synthesized with iScript Reverse Transcription Supermix (1708840; Bio-Rad, Hercules, CA) using 200 ng of RNA per reaction. Stock primers were made at 50 μM in Tris buffer plus EDTA (TE) and diluted 1:100 with water before use. Primers used were, for Ptch1, TGC TGT GCC TGT GGT CAT CCT GAT T and CAG AGC GAG CAT AGC CCT GTG GTT C; for Gli1, CTT CAC CCT GCC ATG AAA CT and TCC AGC TGA GTG TTG TCC AG; and for Pold3, ACG CTT GAC AGG AGG GGG CT and AGG AGA AAA GCA GGG GCA AGC G.
Samples were run in technical triplicate. Each reaction contained 2 μl of diluted cDNA, 10 μl of Bio-Rad SsoAdvanced Universal SYBR Supermix (1725270), 3 μl of 1:100 forward primer, 3 μl of 1:100 reverse primer, and 2 μl of water. A standard curve of a 1:5 dilution series of cDNA from Shh-treated SmoA1-GFP MEFs (chosen because these cells express very high levels of Shh target genes and thus can be diluted to capture a large dynamic range of transcript levels) was run on each plate.
Each plate was run on a Bio-Rad CFX96 Touch Real-Time PCR Detection System; data were collected and analyzed using Bio-Rad CFX Manager 3.1. The program conditions were 95°C for 5 min; 45 cycles of 95°C for 15 s, 57°C for 30 s, plate read; and generation of a melt curve beginning at 65°C and ending at 95°C. Although primers were optimized for specificity, melt curve data were checked after each experiment to ensure that amplification of only one product had occurred.
Starting quantities (in arbitrary units) were determined from comparison to the standard curve. Each triplicate set of technical replicates was averaged to give a value for a single biological replicate. Gli1 and Ptch1 expression levels were then normalized to the corresponding Pold3 levels for each replicate of a given sample.
Western blots
Cell lysis was performed using modified RIPA buffer plus SIGMAFAST protease inhibitors (S8820). Modified RIPA buffer is 50 mM sodium-Tris, pH 7.4, 150 mM NaCl, 2% (vol/vol) NP-40, 0.5% (wt/vol) deoxycholate, 0.1% (wt/vol) SDS, and 1 mM DTT. Lysates were clarified by centrifugation at 20,000 × g at 4°C for 45 min. A 20-μg amount of protein was separated on 10% Bio-Rad Mini-PROTEAN TGX Stain-Free Precast Gels (4568034). Gels were imaged after activation using the Bio-Rad ChemiDoc Touch Imaging System and transferred to a 0.2-μm nitrocellulose membrane using the Bio-Rad Trans-Blot Turbo Transfer System with the Bio-Rad defined High Molecular Weight setting. Blots were imaged again to ensure proper transfer before being blocked for 30 min with Pierce Superblock T20 (PBS; 37516). Primary antibodies were diluted in T20, and blots were incubated in primary antibody overnight at 4°C.
Blots were rinsed three times for 5 min each in Tris-buffered saline plus Triton X-100 (TBST) and incubated with secondary antibodies diluted in 5% (wt/vol) milk in TBST for 1 h at room temperature. Blots were rinsed three times for 10 min each in TBST before a 5-min incubation in Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare; RPN2232V1). Blots were then imaged for chemiluminescence using the ChemiDoc Touch Imaging System. All analysis of Western blots was done using Bio-Rad ImageLab software. Bands were normalized to an unprobed control protein as measured on the stain-free gel as a loading control.
Statistical analysis
Data were analyzed for statistical significance using Prism 6 (GraphPad Software, San Diego, CA). One-way or two-way analysis of variance (factors: Arl13b genotype; control/Shh treatment) was performed using Dunnett’s multiple comparison test for simple effects of Arl13b genotype, with the mean of each Arl13b mutant compared with a control mean and an overall α level of 0.05.
Supplementary Material
Acknowledgments
We thank Kate Cameron for technical assistance with FACS sorting at the Academic Medical Center, Sarah Bay for comments on the manuscript and heroic figure revisions, and Cheryl Timms Strauss for editing. This work was supported by National Institutes of Health Grants NS056380, GM110663, and NS090029 (T.C.), KWF Project Grant UVA 2012–5607, and continuous support from the AMC Foundation (M.F.B.). L.E.M. was supported by National Institutes of Health training grants (GM08605 and EY007092) and an American Heart Association predoctoral fellowship (11PRE7200011). S.S. received support from National Institutes of Health Grant T32 GM008490. Essential services were provided by the Emory Custom Cloning Core Facility, the Emory·Children’s Pediatric Research Center Flow Cytometry Core, the Emory University School of Medicine Core Facility for Flow Cytometry, and the Emory Viral Vector Core (National Institute of Neurological Disorders and Stroke Core Facilities Grant P30NS055077).
Abbreviations used:
- Arl13bhnn
a protein null allele of Arl13b
- CiLS
cilia localization signal
- FACS
fluorescence-assisted cell sorting
- GliA
Gli activator
- JS
Joubert syndrome
- MEF
mouse embryonic fibroblast
- PBS
phosphate-buffered saline
- Shh
Sonic hedgehog
- Smo
Smoothened
- TBST
Tris-buffered saline plus Triton X-100
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0189) on September 28, 2016.
REFERENCES
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Barral DC, Garg S, Casalou C, Watts GFM, Sandoval JL, Ramalho JS, Hsu VW, Brenner MB. Arl13b regulates endocytic recycling traffic. Proc Natl Acad Sci USA. 2012;109:21354–21359. doi: 10.1073/pnas.1218272110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijlsma MF, Borensztajn KS, Roelink H, Peppelenbosch MP, Spek CA. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007;19:2596–2604. doi: 10.1016/j.cellsig.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Bijlsma MF, Damhofer H, Roelink H. Hedgehog-stimulated chemotaxis is mediated by smoothened located outside the primary cilium. Sci Signal. 2012;5:ra60. doi: 10.1126/scisignal.2002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowzard JB, Cheng D, Peng J, Kahn RA. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J Biol Chem. 2007;282:17568–17580. doi: 10.1074/jbc.M701347200. [DOI] [PubMed] [Google Scholar]
- Bowzard JB, Sharer JD, Kahn RA. Assays used in the analysis of Arl2 and its binding partners. Methods Enzymol. 2005;404:453–467. doi: 10.1016/S0076-6879(05)04040-1. [DOI] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalou C, Seixas C, Portelinha A, Pintado P, Barros M, Ramalho JS, Lopes SS, Barral DC. Arl13b and the non-muscle myosin heavy chain IIA are required for circular dorsal ruffle formation and cell migration. J Cell Sci. 2014;127:2709–2722. doi: 10.1242/jcs.143446. [DOI] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Cavenagh MM, Breiner M, Schurmann A, Rosenwald AG, Terui T, Zhang C, Randazzo PA, Adams M, Joost HG, Kahn RA. ADP-ribosylation factor (ARF)-like 3, a new member of the ARF family of GTP-binding proteins cloned from human and rat tissues. J Biol Chem. 1994;269:18937–18942. [PubMed] [Google Scholar]
- Cevik S, Hori Y, Kaplan OI, Kida K, Toivenon T, Foley-Fisher C, Cottell D, Katada T, Kontani K, Blacque OE. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J Cell Biol. 2010;188:953–969. doi: 10.1083/jcb.200908133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevik S, Sanders AA, Van Wijk E, Boldt K, Clarke L, van Reeuwijk J, Hori Y, Horn N, Hetterschijt L, Wdowicz A, et al. Active transport and diffusion barriers restrict Joubert syndrome-associated ARL13B/ARL-13 to an Inv-like ciliary membrane subdomain. PLoS Genet. 2013;9:e1003977. doi: 10.1371/journal.pgen.1003977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron F, Stein E, Jeong J, McMahon AP, Tessier-Lavigne M. The morphogen Sonic Hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell. 2003;113:11–23. doi: 10.1016/s0092-8674(03)00199-5. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem. 1994;269:1437–1448. [PubMed] [Google Scholar]
- Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Natl Acad Sci USA. 2005;102:3301–3306. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–2503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, Briscoe J. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450:717–720. doi: 10.1038/nature06347. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duldulao NA, Lee S, Sun Z. Cilia localization is essential for in vivo functions of the Joubert syndrome protein Arl13b/Scorpion. Development. 2009;136:4033–4042. doi: 10.1242/dev.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East MP, Bowzard JB, Dacks JB, Kahn RA. ELMO domains, evolutionary and functional characterization of a novel GTPase-activating protein (GAP) domain for Arf protein family GTPases. J Biol Chem. 2012;287:39538–39553. doi: 10.1074/jbc.M112.417477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle EC. Human genetic disorders of axon guidance. Cold Spring Harb Perspect Biol. 2010;2:a001784. doi: 10.1101/cshperspect.a001784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Okuhara D, Yu Z, Tian X, Cai Y, Shibazaki S, Somlo S. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci. 2006;119:1383–1395. doi: 10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse Patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gotthardt K, Lokaj M, Koerner C, Falk N, Gießl A, Wittinghofer A. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. Elife. 2015;4:e11859. doi: 10.7554/eLife.11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Eom T-Y, Mariani LE, Bachleda A, Hirt J, Gukassyan V, Cusack CL, Lai C, Caspary T, Anton ES. Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev Cell. 2012;23:925–938. doi: 10.1016/j.devcel.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y, Kobayashi T, Kikko Y, Kontani K, Katada T. Domain architecture of the atypical Arf-family GTPase Arl13b involved in cilia formation. Biochem. Biophys Res Commun. 2008;373:119–124. doi: 10.1016/j.bbrc.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Horner VL, Caspary T. Disrupted dorsal neural tube BMP signaling in the cilia mutant Arl13b hnn stems from abnormal Shh signaling. Dev Biol. 2011;355:43–54. doi: 10.1016/j.ydbio.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, Seo S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci USA. 2012;109:19691–19696. doi: 10.1073/pnas.1210916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova AA, East MP, Yi SL, Kahn RA. Characterization of recombinant ELMOD (cell engulfment and motility domain) proteins as GTPase-activating proteins (GAPs) for ARF family GTPases. J Biol Chem. 2014;289:11111–11121. doi: 10.1074/jbc.M114.548529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Martinelli DC, Zheng X, Tessier-Lavigne M, Fan C-M. Gas1 is a receptor for sonic hedgehog to repel enteric axons. Proc Natl Acad Sci USA. 2015;112:E73–E80. doi: 10.1073/pnas.1418629112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- Kahn RA. Toward a model for Arf GTPases as regulators of traffic at the Golgi. FEBS Lett. 2009;583:3872–3879. doi: 10.1016/j.febslet.2009.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA, Bruford E, Inoue H, Logsdon JM, Nie Z, Premont RT, Randazzo PA, Satake M, Theibert AB, Zapp ML, et al. Consensus nomenclature for the human ArfGAP domain-containing proteins. J Cell Biol. 2008;182:1039–1044. doi: 10.1083/jcb.200806041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn RA, Goddard C, Newkirk M. Chemical and immunological characterization of the 21-kDa ADP-ribosylation factor of adenylate cyclase. J Biol Chem. 1988;263:8282–8287. [PubMed] [Google Scholar]
- Kim H, Xu H, Yao Q, Li W, Huang Q, Outeda P, Cebotaru V, Chiaravalli M, Boletta A, Piontek K, et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat Commun. 2014;5:5482. [Google Scholar]
- Kuai J, Kahn RA. Residues forming a hydrophobic pocket in ARF3 are determinants of GDP dissociation and effector interactions. FEBS Lett. 2000;487:252–256. doi: 10.1016/s0014-5793(00)02325-5. [DOI] [PubMed] [Google Scholar]
- Larkins CE, Aviles GD, East MP, Kahn RA, Caspary T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell. 2011;22:4694–4703. doi: 10.1091/mbc.E10-12-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kelly WG, Logsdon JM, Schurko AM, Harfe BD, Hill-Harfe KL, Kahn RA. Functional genomic analysis of the ADP-ribosylation factor family of GTPases: phylogeny among diverse eukaryotes and function in C. elegans. FASEB J. 2004;18:1834–1850. doi: 10.1096/fj.04-2273com. [DOI] [PubMed] [Google Scholar]
- Li Y, Wei Q, Zhang Y, Ling K, Hu J. The small GTPases ARL-13 and ARL-3 coordinate intraflagellar transport and ciliogenesis. J Cell Biol. 2010:1–13. doi: 10.1083/jcb.200912001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon JM, Kahn RA. The Arf family tree. In: Kahn RA, editor. ARF Family GTPases. Dordrecht, Netherlands: Kluwer; 2003. pp. 1–21. [Google Scholar]
- Mariani LE, Caspary T. Primary cilia, Sonic Hedgehog signaling, and spinal cord development. In: Tucker KL, Caspary T, editors. Cilia and Nervous System Development and Function. Dordrecht, Netherlands: Springer; 2013. pp. 55–82. [Google Scholar]
- Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28:183–192. doi: 10.1038/emboj.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Weiss LE, Yoon J, Roth TL, Su YS, Sahl SJ, Scott MP, Moerner WE. Single-molecule imaging of Hedgehog pathway protein Smoothened in primary cilia reveals binding events regulated by Patched1. Proc Natl Acad Sci USA. 2015;112:8320–8325. doi: 10.1073/pnas.1510094112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina PJR, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn. 2008;237:2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Glass I. GeneReviews [Internet] Seattle: University of Washington Press; 1993–2016. Joubert syndrome and related disorders. https://www.ncbi.nlm.nih.gov/books/NBK1325/ [Google Scholar]
- Poretti A, Boltshauser E, Loenneker T, Valente EM, Brancati F, Il’yasov K, Huisman TAGM. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol. 2007;28:1929–1933. doi: 10.3174/ajnr.A0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, Sztriha L, Ruta R, Mancini F, Mazza T, Castellana S, et al. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis. 2014;9:72. doi: 10.1186/1750-1172-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter J. 2016. blindanalysis: v1.0 [Data set]. Zenodo. http://doi.org/10.5281/zenodo.44678.
- Sánchez-Camacho C, Bovolenta P. Autonomous and non-autonomous Shh signalling mediate the in vivo growth and guidance of mouse retinal ganglion cell axons. Development. 2008;135:3531–3541. doi: 10.1242/dev.023663. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacht A, Mowbrey K, Elias M, Kahn RA, Dacks JB. Ancient complexity, opisthokont plasticity, and discovery of the 11th subfamily of Arf GAP proteins. Traffic. 2013;14:636–649. doi: 10.1111/tra.12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C-Y, Bay SN, Mariani LE, Hillman MJ, Caspary T. Temporal deletion of Arl13b reveals that a mispatterned neural tube corrects cell fate over time. Development. 2012;139:4062–4071. doi: 10.1242/dev.082321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Thomas S, Cantagrel V, Mariani L, Serre V, Lee J-E, Elkhartoufi N, de Lonlay P, Desguerre I, Munnich A, Boddaert N, et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur J Hum Genet. 2015;23:621–627. doi: 10.1038/ejhg.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, Saada J, Perrault I, Amiel J, Litzler J, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014;35:137–146. doi: 10.1002/humu.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuz K, Bachmann-Gagescu R, O’Day DR, Hua K, Isabella CR, Phelps IG, Stolarski AE, O’Roak BJ, Dempsey JC, Lourenco C, et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2014;94:62–72. doi: 10.1016/j.ajhg.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Valkenburgh H, Shern JF, Sharer JD, Zhu X, Kahn RA. ADP-ribosylation factors (ARFs) and ARF-like 1 (ARL1) have both specific and shared effectors: characterizing ARL1-binding proteins. J Biol Chem. 2001;276:22826–22837. doi: 10.1074/jbc.M102359200. [DOI] [PubMed] [Google Scholar]
- Warburton-Pitt SRF, Silva M, Nguyen KCQ, Hall DH, Barr MM. The nphp-2 and arl-13 genetic modules interact to regulate ciliogenesis and ciliary microtubule patterning in C. elegans. PLoS Genet. 2014;10:e1004866. doi: 10.1371/journal.pgen.1004866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward HH, Brown-Glaberman U, Wang J, Morita Y, Alper SL, Bedrick EJ, Gattone VH, Deretic D, Wandinger-Ness A. A conserved signal and GTPase complex are required for the ciliary transport of polycystin-1. Mol Biol Cell. 2011;22:3289–3305. doi: 10.1091/mbc.E11-01-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachnis AT, Rorke LB. Neuropathology of Joubert Syndrome. J Child Neurol. 1999;14:655–659. doi: 10.1177/088307389901401006. [DOI] [PubMed] [Google Scholar]
- Zhang CJ, Rosenwald AG, Willingham MC, Skuntz S, Clark J, Kahn RA. Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J Cell Biol. 1994;124:289–300. doi: 10.1083/jcb.124.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Cunningham L, Marcus AI, Li Y, Kahn RA. Arl2 and Arl3 regulate different microtubule-dependent processes. Mol Biol Cell. 2006;17:2476–2487. doi: 10.1091/mbc.E05-10-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.