

In Tay–Sachs disease, mutations in HEXA can lead to aberrant α subunits of the HexA enzyme. Two such mutants have folding defects and are cleared by endoplasmic reticulum-associated degradation. Toward the pursuit of therapeutic treatments, it was found that manipulating endoplasmic reticulum quality control can impair mutant α degradation and improve cellular Hex activity.
Abstract
Loss of function of the enzyme β-hexosaminidase A (HexA) causes the lysosomal storage disorder Tay–Sachs disease (TSD). It has been proposed that mutations in the α chain of HexA can impair folding, enzyme assembly, and/or trafficking, yet there is surprisingly little known about the mechanisms of these potential routes of pathogenesis. We therefore investigated the biosynthesis and trafficking of TSD-associated HexA α mutants, seeking to identify relevant cellular quality control mechanisms. The α mutants E482K and G269S are defective in enzymatic activity, unprocessed by lysosomal proteases, and exhibit altered folding pathways compared with wild-type α. E482K is more severely misfolded than G269S, as observed by its aggregation and inability to associate with the HexA β chain. Importantly, both mutants are retrotranslocated from the endoplasmic reticulum (ER) to the cytosol and are degraded by the proteasome, indicating that they are cleared via ER-associated degradation (ERAD). Leveraging these discoveries, we observed that manipulating the cellular folding environment or ERAD pathways can alter the kinetics of mutant α degradation. Additionally, growth of patient fibroblasts at a permissive temperature or with chemical chaperones increases cellular Hex activity by improving mutant α folding. Therefore modulation of the ER quality control systems may be a potential therapeutic route for improving some forms of TSD.
INTRODUCTION
Lysosomal storage disorders (LSDs) are a devastating class of more than 50 pathologies, with a combined frequency in the human population of ∼1:5000 births (Poorthuis et al., 1999; Vellodi, 2005; Platt et al., 2012). Their unifying feature is a defect in the function of the endosome–lysosome system, which in many cases leads to the toxic accumulation of metabolites and cell death. A subset of LSDs, GM2 gangliosidoses, are a series of associated disorders resulting from insufficiency of active β-hexosaminidase A (HexA), the enzyme that processes GM2 ganglioside to GM3 ganglioside in the lysosome (Mahuran, 1999). The loss or inactivation of HexA causes the toxic buildup of GM2, leading to cell death and disease (Svennerholm, 1962; Huang et al., 1997).
The HexA enzyme is produced from the HEXA and HEXB genes, which encode the α and β subunits, respectively. These are 60% identical at the amino acid level and are synthesized at the endoplasmic reticulum (ER), where they are glycosylated (Sonderfeld-Fresko and Proia, 1989; Weitz and Proia, 1992), form intramolecular disulfide linkages (Maier et al., 2003), and dimerize. Because of the structural similarities between the chains, multiple isozymes can be formed via differential association: HexA (αβ), HexS (αα), and HexB (ββ) (Robinson and Stirling, 1968; Hou et al., 1996). Although HexB is the most stable of these complexes, HexA is the only species capable of processing GM2 ganglioside (Lemieux et al., 2006). Therefore it is hypothesized that the production of β is kept low to promote heterodimerization over β homodimerization (Mahuran, 1991). In the Golgi apparatus, specific glycans are modified with mannose-6-phosphate (M6P), allowing for the trafficking of Hex enzymes to lysosomes (Sonderfeld-Fresko and Proia, 1989; Weitz and Proia, 1992). However, this recognition and transport is not entirely efficient, resulting in constitutive secretion both in vitro and in vivo (Hasilik and Neufeld, 1980; Yang et al., 2015). Once in the lysosome, the α and β chains are proteolytically processed to their mature forms (Little et al., 1988; Mahuran et al., 1988; Hubbes et al., 1989; Sagherian et al., 1993). These cleavage events appear to be dispensable for activity—extracellular HexA is full length yet enzymatically active. In the lysosome, presentation of the GM2 ganglioside substrate from the bilayer to the active site of HexA additionally requires the adaptor protein GM2-Activator (Xie et al., 1991).
Loss of function in either subunit of HexA or its adaptor protein can lead to GM2 gangliosidosis. Tay–Sachs disease (TSD) is clinically defined by mutations in the HEXA gene, Sandhoff disease by mutations in the HEXB gene, and AB variant gangliosidosis by inactivating mutations in GM2-Activator (Schepers et al., 1996; Mahuran, 1999). Of these related disorders, TSD is the best characterized, with well over 100 known mutations identified in the HEXA gene (Triggs-Raine et al., 2001; see also an online database at www.hexdb.mcgill.ca). Depending on the mutations inherited, there may be no signs of disease, a late-onset and chronic form of the disease, or severe juvenile-onset disease causing death by the age of 4. Based on phenotypic information, along with activity assays of patients and healthy people, the currently accepted model posits a “critical threshold” of ∼10% activity, below which TSD presents in some manner (Conzelmann and Sandhoff, 1983). From a therapeutic point of view, this may allow for the rescue of certain disease-causing mutants, as attempted by recent clinical trials (Clarke et al., 2011; Osher et al., 2011).
Even with the identification of many disease-associated alleles, there are few options for TSD treatment and no cures. The lack of information about mutant HexA synthesis and trafficking during disease pathology, as opposed to its normal biosynthetic route, is likely a contributing factor to the poor availability of therapeutic avenues. Therefore we chose to study two important variants of HEXA, one that produces G269S α and one that produces E482K α.
G269S is the most commonly identified allele in patients with adult-onset TSD, making it a clear target for future treatments (Navon and Proia, 1989; Brown and Mahuran, 1993). On the other hand, the E482K mutant is a relatively rare variant, but a severe one that exhibits little residual activity and causes infantile forms of TSD (Proia and Neufeld, 1982). Unlike very rare mutations that affect the active site of α, these mutations have been predicted to interfere with folding pathways (Proia and Neufeld, 1982; Nakano et al., 1988; Brown and Mahuran, 1993; Tropak et al., 2004; Ohno et al., 2008). In vitro, G269S appears to lose activity at physiological temperatures (Brown and Mahuran, 1993). Structurally, the sterically larger serine side chain in position 269 may displace local backbone conformation, disallowing proper folding and trafficking of the enzyme (Lemieux et al., 2006).
Computer modeling of the E482K mutation predicted that it has a much more drastic effect on protein conformation than G269S (Ohno et al., 2008), correlating with a previous report that E482K can form insoluble aggregates (Proia and Neufeld, 1982). Additionally, phosphorylated E482K has not been observed, perhaps signifying that it cannot leave the ER (Proia and Neufeld, 1982). The crystal structure of HexA confirms that E482 is a buried residue important for a salt bridge with an arginine residue (Lemieux et al., 2006). The change in charge of this residue likely explains the massive conformational changes predicted to occur (Lemieux et al., 2006; Ohno et al., 2008).
In patients, it is clear that many α mutants such as G269S and E482K are properly synthesized but never reach the lysosome. Though it has been speculated that these mutants (and others) are degraded early in the secretory pathway, there has been little direct evidence to support these hypotheses (Mu et al., 2008; Wang et al., 2011). In fact, mechanisms of protein turnover from the ER were uncovered well after much of the initial characterization of TSD mutations. We now have a deeper understanding of ER-associated degradation (ERAD), the processes by which terminally misfolded proteins in the ER are sensed, escorted to membrane complexes, dislocated to the cytosol, and degraded by the proteasome. Here we asked whether ERAD is indeed involved in the retention and disposal of mutant α in TSD and whether we could interfere with ER homeostasis to promote cellular activity in cell models of the disease.
RESULTS
E482K and G269S α mutants remain unprocessed and have impaired activities
For characterization of folding and trafficking of HexA, the human HEXA and HEXB genes were cloned into expression plasmids, with an N-terminal FLAG tag appended to the HEXA gene immediately after the ER-targeting signal sequence, and a C-terminal HA tag fused to HEXB (Figure 1A). It has been previously shown that small tags do not interfere with Hex activity, indicating that the FLAG and HA peptides would not significantly impact the proteins (Yamada et al., 2013). Site-directed mutagenesis was used to recapitulate the disease-associated mutants of α, G269S and E482K.
FIGURE 1:

Characterization of cloned HEXA and HEXB gene products. (A) Schematic representation of the tagged HEXA and HEXB genes and their predicted cellular processing. SS, signal sequence; FLAG, epitope tag; disulfide bonds are indicated by “S-S”; αp, α propeptide; αm, α mature protein; βp, β propeptide; βa and βb, β mature polypeptides. (B) The indicated FLAG-HEXA constructs were expressed in HEK293T cells and harvested the following day for immunoblot analysis with an anti-α antibody to detect both precursor and mature species. Brefeldin A was added to the indicated cells at a concentration of 0.5 μg/ml for 17 h to inhibit forward trafficking. 14-3-3, cytosolic loading control. (C) The indicated FLAG-HEXA constructs were expressed as in B, but the resulting Western blot was probed with the M1 anti-FLAG antibody. (D) HEXB-HA was expressed in HEK293T cells, with or without brefeldin A treatment (2 μg/ml for 15 h), and processed for immunoblot analysis with the anti-β antibody to observe precursor and mature forms. Endog, endogenous mature β; exog, exogenously expressed mature β. (E) FLAG-HEXA or HEXB-HA was expressed for 24 h and analyzed by Western analysis in both reducing and nonreducing gels; anti-α and anti-β were used for detection, respectively.
We first studied the steady-state distribution of α in HEK293T cells. Immunoblotting whole-cell lysates with an anti-α antibody revealed that only wild-type (WT) α was proteolytically cleaved to the predicted mature species (Figure 1B; Hasilik and Neufeld, 1980). This processing was dependent on forward trafficking in the secretory pathway, since it was inhibited by brefeldin A treatment. Mature forms were not observed upon expression of either G269S or E482K, indicating that these variants either do not reach the lysosome or cannot be cleaved by the proper proteases. Additionally, the cloning strategy used for FLAG-HEXA allowed for the specific identification of ER species, using the anti-FLAG M1 antibody that only recognizes free N-terminal FLAG sequences (i.e., after cleavage of the signal sequence). This epitope is clearly lost upon proteolytic processing of WT α in the lysosome (Figure 1C).
Expression of the HEXB-HA construct also yielded both immature and mature β species, and the appearance of the latter was sensitive to brefeldin A (Figure 1D). The heterogeneity in the molecular weights is due to glycosylation, as described previously (Sonderfeld-Fresko and Proia, 1989; unpublished data). Finally, the immature and mature species of both α and β collapsed into single species when lysates were resolved on nonreducing gels (Figure 1E), indicating that disulfide bonds covalently link the mature species, even after proteolysis (Schuette et al., 2001). Therefore both FLAG-HEXA and HEXB-HA are expressed properly and undergo the expected biosynthesis in our cell system.
To measure the activity of each α construct, we monitored the fluorescence from the breakdown of 4-methylumbelliferone N-acetylglucosamine-6-sulfate (MUGS). Because of structural differences in the active sites of α and β, coupled with the sulfate group and negative charge of this substrate, MUGS is specifically cleaved by the α subunit in HexA (αβ) and HexS (αα) but not by HexB (ββ) (Mark et al., 2003; Lemieux et al., 2006). Extracts of the HEK293T cell line tested here have basal activity toward MUGS, because they express endogenous Hex dimers, yet ectopic expression of WT α greatly enhanced this activity (Figure 2A). While expression of WT α increased intracellular activity approximately sevenfold, neither E482K nor G269S were detectably active (Figure 2B). Therefore, as expected, the E482K and G269S mutations inhibit the formation of active HexA and HexS.
FIGURE 2:

Activity and secretion of α mutants. (A, B) The indicated FLAG-HEXA constructs were transiently expressed in HEK293T cells for 48 h, lysed in a pH 4.2 lysis buffer, and subjected to the MUGS activity assay at 37°C, as described in Materials and Methods. Samples in A were tested for fluorescence at the indicated time points (without quenching the reaction); measurements in B represent endpoint readouts after quenching with a high-pH buffer. Activity was normalized to untransfected cells and is indicated under the graph. Values are plotted as averages of three experiments with SDs. (C) Supernatant and whole-cell lysates from HEXA-expressing cells were collected after 24 h of protein expression. α was detected after immunoblot analysis with the anti-FLAG antibody. (D) Conditioned medium from C was subjected to the MUGS assay to test for extracellular HexA activity. Increase in activity was normalized to the activity present in conditioned medium from untransfected cells. Values are plotted as averages of three experiments with SDs.
Because the targeting of lysosomal enzymes via M6P receptors is imperfect, we asked whether each α subunit was secreted. Indeed, WT α was observed in the supernatant of cells after transient expression (Figure 2C), confirming previous reports (Hasilik and Neufeld, 1980). Surprisingly, G269S was also found in the conditioned medium, though to a much lesser extent than WT (∼20%). No E482K was detected, indicating that it was completely retained intracellularly. The presence of G269S in the conditioned medium without detection of the mature species in the cell (Figure 1B) implies that either G269S targeting to the lysosome is ineffective (e.g., poor recognition by GlcNAc-1-phosphotransferase, the first step in M6P tag generation; Tiede et al., 2005) or that some fraction of G269S reaches the lysosome but cannot be properly cleaved. Correlating with the secretion data, activity was enhanced in the medium of cells expressing G269S by approximately fourfold (Figure 2D). This level of activity is lower than would be predicted by the band intensities in Figure 2C, indicating that, even though G269S has extracellular activity, its specific activity may be lower than the WT enzyme at physiological temperature.
Biochemical and cell biological differences among α mutants
Because the lysosomal processing, secretion, and activity of E482K and G269S α were impaired, we pursued the hypothesis that they are misfolded. Molecular modeling had suggested that both mutations affect the structure of the α chain, with the E482K mutation predicted to affect a much larger area of the protein than G269S (Ohno et al., 2008). Experimentally, soluble G269S had been found to become inactivated at physiological temperatures (Brown and Mahuran, 1993; Tropak et al., 2004). Because folding may be dependent on glycan addition, subunit assembly, and other cell-specific factors and modifications, we avoided cell-free expression or assays of purified proteins and instead focused on probing the conformations of α and its mutants in whole-cell extracts.
We first discovered that when pellets from detergent lysis were examined, E482K was detected more readily in the NP-40 insoluble fraction than either WT or G269S (Figure 3A), confirming a previous report that showed E482K may aggregate (Proia and Neufeld, 1982).
FIGURE 3:

Conformational and biochemical differences among α mutants. (A) The indicated FLAG-HEXA constructs were expressed in HEK293T cells for 20 h and harvested in lysis buffer containing 1% NP-40. Insoluble material was resuspended in 3% SDS, heated, and sonicated. Samples of the NP-40–soluble (5% of total) and NP-40–insoluble (10% of total) were subjected to gel electrophoresis and Western blotting with anti-FLAG. Band quantifications were used to determine the overall fraction of insoluble material of each α protein. (B) FLAG-HEXA constructs were transiently expressed for 24 h, and cell lysates were prepared in nonreducing Laemmli sample buffer before SDS–PAGE and immunoblot analysis. Black arrows, slower migrating species only present upon WT α expression; black arrowhead, G269S-specific band migrating at a size corresponding to a dimer; white arrowhead, species resistant to gel migration. (C) HEK293T cells expressing the indicated FLAG-HEXA constructs were harvested and lysed without protease inhibitors. Equal amounts of total protein were incubated with decreasing concentrations of trypsin (1:20 K ratio indicates 1 μg trypsin per 20,000 μg whole-cell lysate) for 1 h at 37°C. Reactions were quenched with SDS sample buffer before SDS–PAGE and Western blotting. (D) Quantification of band intensity from C, normalized to the “no trypsin” sample of each set. Fifty percent of starting material is indicated by dashed line. (E) HEK293T cells were transiently transfected with FLAG-HEXA constructs and were left untreated or treated with brefeldin A (0.5 μg/ml) or bafilomycin A1 (50 nM) for 18.5 h. Cell lysates were then subjected to IP with anti-FLAG agarose to determine the co-IP of endogenous β with FLAG-α for each mutant/condition. *, heavy chain of the anti-FLAG antibody used for IP.
The second piece of evidence that mutant α chains had altered conformations was obtained from nonreducing gels. On immunoblotting of whole-cell lysates, the WT α yielded one major band and two minor, slower mobility species (Figure 3B, black arrows). WT α is known to contain three disulfide bonds, one of which retains the propeptide to the main chain (Hubbes et al., 1989; Mark et al., 2003; Lemieux et al., 2006); it is therefore possible that these species represent folding intermediates with distinct electrophoretic mobilities. Alternatively, these bands may represent phosphorylated species in the Golgi. Neither E482K nor G269S displayed such minor forms under nonreducing conditions. Instead, E482K was significantly more enriched at the top of the gel (Figure 3B, white arrowhead), correlating with its relative insolubility in NP-40 and probable aggregation (Figure 3A). The G269S protein ran as a major monomer species and a band corresponding to the size of a dimer (Figure 3B, black arrowhead). While the precise nature of these unique redox species remains to be identified, the data suggest that the oxidative folding of E482K and G269S differs from that of WT α.
Third, limited trypsin proteolysis of α chains in cell lysates revealed that, while WT and G269S α were equally sensitive to trypsin, E482K was significantly more resistant (Figure 3, C and D). We conclude that the E482K mutation causes a change in protein conformation (e.g., aggregation), resulting in lowered trypsin accessibility.
Fourth, we probed the ability of α to bind to β, which reports on the fold of the α dimerization surface (Maier et al., 2003; Mark et al., 2003). Coimmunoprecipitations (co-IP) confirmed that while WT and G269S were able to associate with endogenous β, E482K was not (Figure 3E). Considering that the E482K mutation is not proximal to the dimerization domain (Ohno et al., 2008), these results strongly suggest that the mutation causes large-scale alterations to the protein, which may underlie its lack of lysosomal targeting, secretion, and activity.
On the basis of these four experimental approaches, we conclude that the E482K mutation causes global misfolding of α and that the G269S mutation has a subtler effect that appears to alter at least its oxidative folding pathway.
α Mutants are bona fide ERAD substrates
The cellular location of the α–β dimerization has been historically unclear, but we found that WT α associated with β, even in cells pretreated with brefeldin A, which disrupts traffic to the Golgi complex, or bafilomycin A1, which disrupts acidic compartments (Figure 3E). Thus α–β dimerization occurs early in the biosynthesis of HexA, and we speculate that dimerization is likely a prerequisite for HexA to leave the ER, as has been observed with many other multisubunit proteins. Assembly in the ER is also consistent with the secretion of WT and G269S, but not E482K (Figure 2C), since only WT and G269S are competent to bind β (Figure 3E).
Because the ER plays an important role in quality control of secretory pathway proteins, and because E482K and G269S are not properly trafficked, we sought to identify ER-resident components that may be important for the regulation of HexA and took a targeted co-IP approach to determine which common ER factors interact with α and its mutants (Supplemental Figure 1). The Hsp70 master chaperone BiP and two of its cofactors, GRP170 and PDIA6, associated with all three forms of α. Notably, WT α associated less well with BiP and its cofactors, implying that either WT α resides in the ER for less time than E482K and G269S or that the mutant α subunits require more rounds of chaperone assistance. The association of α with the BiP machinery was selective, since two of the most abundant lumenal chaperones, GRP94 and PDI, were not found in the complex.
In addition to BiP, two components of the ERAD machinery associated with α—OS-9 and SEL1L (Supplemental Figure 1). OS-9 is an ER-resident lectin that functions in the recognition of terminally misfolded proteins and their subsequent escort (Christianson et al., 2008; Hosokawa et al., 2009) to membrane-bound retrotranslocons, sites at which ERAD substrates are dislocated to the cytosol for proteasomal degradation. Indeed, α mutants also associated with SEL1L, a key scaffold of these retrotranslocon complexes (Mueller et al., 2006, 2008). Similar to its association with BiP, WT α associated less well than E482K and G269S with both OS-9 and SEL1L. This was most clear in the case of SEL1L, where WT α bound poorly, if at all, to the retrotranslocon scaffold, whereas E482K and G269S bound SEL1L as clearly as the well-studied ERAD substrate NHK (Supplemental Figure 1B). That these interactions were detected at all suggested that some conformers of α were recognized by the ERAD machinery.
The evidence for a degradation pathway for mutant α was significant: 1) α mutants showed little/no secretion, lysosomal processing, or activity; 2) E482K, and to a lesser extent G269S, showed evidence for altered conformation; and 3) mutant forms of α were found in complex with the ERAD factors OS-9 and SEL1L. We hypothesized that mutant α chains were triaged via ERAD and therefore used pulse-chase analysis to monitor the ER forms of WT, E482K, and G269S α. In addition to measuring their basal, constitutive turnover (half-lives of ER species: 2.9 h for WT, 2.5 h for E482K, 4.1 h for G269S), we also quantified turnover in cells treated with the proteasome inhibitor MG-132 and in cells expressing a dominant-negative version of Hrd1 (Hrd1C294A), an E3 ubiquitin ligase and key component of the mammalian retrotranslocon (Bernasconi et al., 2010; Chen et al., 2011).
Both treatment with MG-132 and expression of Hrd1C294A stabilized all α proteins over the indicated chase times. This included even the WT protein, although the stabilization was most significant for the E482K mutant, the most severely misfolded (Figure 4, A, C, and E; quantification in Figure 4, B, D, and F). However, the turnover measured in these experiments is a combination of protein degradation and exit from the ER, and because MG-132 can induce ER stress and cause off-target cellular responses, we also examined whether inhibition of proteasomes affected α secretion coordinately. Indeed, the persistence of intracellular WT α was explained by inhibition of its secretion, indicating that the stress of proteasomal inhibition impacts the forward trafficking of WT α (Figure 5, A and B). E482K, on the other hand, was not secreted (Figure 5C), consistent with our observations at steady state (Figure 2C). Therefore the pulse-chase results (Figure 4, C and D) represent the true sensitivity of intracellular E482K to proteasomal degradation. Finally, although a small fraction of G269S is secreted, MG-132 stabilization of G269S is not due to an alteration in the secreted pool (Figure 5, D and E). Taken together, these results indicate ER pools of E482K and G269S are stabilized by proteasomal inhibition, implicating ERAD as their mechanism of degradation. The sensitivity of WT α to MG-132 stems from a defect in secretion, not intracellular turnover.
FIGURE 4:

WT and mutant α are sensitive to impairment of ERAD. (A) WT FLAG-HEXA was expressed in HEK293T cells alone or in combination with the dominant-negative mutant of Hrd1, C294A. At 40 h posttransfection, a set of single-transfection cells were pretreated with MG-132 (25 μM, 2.5 h), whereas the other sets were treated with DMSO. Cells were metabolically labeled with [35S]methionine/cysteine for pulse-chase analysis, maintaining MG-132 or DMSO throughout the chase. At the indicated time points, cells were harvested and lysates were subjected to IP with anti-FLAG agarose to observe the kinetics of intracellular pro-α. (B) Quantification of three experiments as depicted in A. Percent α remaining was calculated relative to t = 0 from within each condition and is plotted with SD. (C) Same as A but with FLAG-E482K α. (D) Quantification of three experiments as depicted in C. Percent α remaining is plotted with SD. (E) Same as A but with G269S. Owing to a slower rate of turnover for the G269S mutant, chase points were taken at 7 h instead of at 5 h. (F) Quantification of three experiments as depicted in E. Percent α remaining is plotted with SD.
FIGURE 5:

Quantification of intracellular and extracellular α. (A) WT FLAG-HEXA was expressed in HEK293T cells. At 40 h posttransfection, cells were pretreated with DMSO or MG-132 (25 μM, 2.5 h). Cells were metabolically labeled for pulse-chase analysis, as in Figure 4. Intracellular and secreted (sec) fractions were isolated as indicated, subjected to IP with anti-FLAG agarose for α isolation. (B) Quantification of bands from two independent experiments as in A. Values were quantified relative to t = 0 within each treatment set. (C) WT and E482K FLAG-HEXA were expressed for pulse-chase analysis as previously described; the α constructs were isolated from the indicated intracellular and secreted fractions via anti-FLAG agarose. Lanes were rearranged digitally at the indicated vertical lines to better display data. (D, E) Same as in A and B but with G269S FLAG-HEXA.
To provide more definitive evidence for α turnover by ERAD, we asked whether the α chains were in fact dislocated to the cytosol. If true, this would be the most compelling argument for ER quality control, since dislocation is one of the most proximal activities of ERAD. To ask whether the Hex proteins were exposed to the cytosol for degradation, we adapted an elegant assay developed by Burrone and colleagues (Petris et al., 2011; Vecchi et al., 2012). In this retrotranslocation assay, an ER protein of interest is first designed with a biotin acceptor peptide (BAP) tag. The construct is then coexpressed with a cytosolic enzyme BirA, which has the ability to conjugate biotin to the BAP tag. Because this can occur only when both proteins are present in the same compartment, an ER-resident/secretory protein must be first retrotranslocated to the cytosol to be biotinylated (scheme in Figure 6A).
FIGURE 6:

α mutants, but not WT α, are retrotranslocated to the cytosol. (A) Schematic of the retrotranslocation assay. An ER-targeted protein, engineered with a BAP tag is coexpressed in cells with cytosolic BirA. Upon treatment of cells with biotin, BirA can attach a biotin molecule to the BAP tag, but only when the protein of interest is retrotranslocated from the ER to the cytosol. This modification is detected using streptavidin to retard gel mobility of biotinylated molecules. Glycans on substrates present in the cytosol can be removed by PNGase, an activity that is inhibited by Z-VAD(OMe)-FMK (Misaghi et al., 2004, 2006). (B) For confirmation of the validity of the assay, V5 and BAP-tagged WT α1-antitrypsin (α1AT, top panel) and NHK α1AT (bottom panel) were coexpressed with BirA in HEK293T cells and treated with 0.1 mM biotin for 8 h. For the last 4 h of treatment, cells were also treated with DMSO, Z-VAD(OMe)-FMK, MG-132, or MG-132 + Z-VAD(OMe)-FMK, as indicated (Z-VAD(OMe)-FMK at 30 μM; MG-132 at 20 μM). Cells were lysed and alkylated in a 6% SDS lysis buffer supplemented with 20 μM N-ethylmaleimide. After sonication, samples were either left untreated or treated with 4 μg streptavidin, resolved by gel electrophoresis, and subjected to Western blot analysis with the anti-V5 antibody. Arrows on the right indicate the size shifts of bands due to streptavidin binding. α1AT:strept, α1AT-biotin-streptavidin complex; NHK:strept, NHK-biotin-streptavidin complex; dgNHK, deglycosylated NHK. Small red arrows indicate where cytosolic, deglycosylated species should run on the gel. (C) Retrotranslocation assays of the indicated HEXA-V5-BAP constructs, performed as in B. dg-α, deglycosylated α; fragment, a fragment observed in the cytosol during proteasomal inhibition.
For confirmation of the fidelity of the assay, V5-tagged WT α1-antitrypsin (α1AT)-BAP or the canonical ERAD substrate NHK-BAP were expressed in cells and examined for retrotranslocation. For detection of biotinylated molecules, streptavidin was incubated with lysate samples before SDS–PAGE; this allowed for the identification of streptavidin-shifted bands by immunoblot analysis (Figure 6B). The well-folded WT α1AT is very efficiently secreted (unpublished data) yet shows some biotinylation, indicating that the assay does have a small background of mistargeted substrate or biotinylation during cell death. However, upon MG-132 treatment, size-shifted bands of the ERAD substrate NHK, but not WT α1AT, were detected below the main species, and these bands were entirely sensitive to streptavidin (Figure 6B). They represent cytosolic, deglycosylated forms of NHK, since treatment with the cytosolic PNGase inhibitor, Z-VAD(OMe)-FMK, inhibits their formation.
For testing whether α mutants were also retrotranslocated, BAP-tagged forms of WT and mutant HEXA constructs were created and coexpressed with BirA in HEK293T cells. Deglycosylated forms of both E482K and G269S were clearly formed upon MG-132 treatment and were sensitive to the presence of Z-VAD(OMe)-FMK (Figure 6C). However, these species were not observed upon WT α expression. Taken together, these results indicate that both the E482K and G269S α mutants give rise to cytosolic, glycosylated species (i.e., ER-derived), providing significant evidence for ERAD.
One potential pitfall of these studies is that overexpression of FLAG-tagged α may affect the stoichiometry of the αβ complex formation, leading to enhanced degradation of the excess subunit. Theoretically, this is not a significant concern, since αα homodimers can form readily and do not absolutely require β for trafficking. Additionally, we showed that E482K α does not associate with β, removing the concern of stoichiometric disparity for this mutant (Figure 3E). However, we asked whether coexpression of G269S α with β might improve lysosomal processing, a readout for proper HexA trafficking. No processed G269S was detected, even with increased coexpression of β (Supplemental Figure 2A). Nor did β expression significantly dampen G269S retrotranslocation or sensitivity to MG-132 (Supplemental Figure 2B). Therefore we conclude that while G269S can associate with β, enhanced levels of β do not rescue its misfolding and degradation.
Manipulations of cellular quality control prevent α-chain disposal and increase Hex activity
We next asked whether we could manipulate ER quality control to interfere with the recognition and degradation of α mutants. Ideally, the modulation of ER chaperones or degradation factors would either enhance folding or inhibit turnover of mutant α. If so, valuable insights would be gained into targets that could potentially rescue HexA activity in patients.
Glycosylated ERAD substrates are known to undergo trimming of their N-linked glycans by slow-acting mannosidases in the ER lumen, thought to be a “timer” mechanism by which terminally malfolded proteins are initially recognized (Tokunaga et al., 2000). Because α has three N-linked glycans (Weitz and Proia, 1992; Lemieux et al., 2006), we asked whether the mannosidase inhibitor kifunensine impaired turnover. Kifunensine was clearly active, as observed by the loss of mannose-trimmed molecules of the ERAD substrate NHK (Figure 7A). By pulse-chase analysis, incubation with kifunensine impaired the degradation of both E482K and G269S α (Figure 7B). Therefore blocking ERAD before retrotranslocation and proteasomal degradation also delays mutant α clearance.
FIGURE 7:

Pharmacological inhibition of ERAD stabilizes α mutants but does not restore enzymatic activity. (A) HEK293T cells expressing an HA-tagged ERAD substrate NHK were treated for 20 h with or without 5 μg/ml kifunensine. Whole-cell lysates were analyzed by Western blot to observe the inhibition of mannose trimming. (B) Cells expressing either FLAG-E482K α or FLAG-G269S α were either left untreated or pretreated with 5 μg/ml kifunensine (kif) for 1.5 h. Cells were then metabolically labeled for pulse-chase analysis in the continued presence of kifunensine. At the indicated time points, each α mutant was isolated with anti-FLAG agarose. Quantification of the bands is depicted under each lane, with the pulse sample (t = 0) set to 100%. (C) Conditioned medium and whole-cell lysates were harvested from untransfected cells and cells expressing the indicated FLAG-HEXA constructs, each left untreated or treated with 5 μg/ml kifunensine for 40 h. Western blotting was performed on each fraction to observe secretion. (D) Untransfected cells or cells transfected with the indicated plasmids were left untreated or treated with 5 μg/ml kifunensine for 40 h, at which time samples of each medium were analyzed via the MUGS activity assay (diluted to avoid enzyme saturation) at room temperature. The specific activity of each is plotted (n = 4 replicates; representative of three independent experiments). (E) Cells expressing the indicated FLAG-HEXA constructs were left untreated or treated with 5 μg/ml kifunensine for 20 h, and lysates were then subjected to anti-FLAG agarose followed by immunoblotting to assess co-IP of endogenous β with the indicated α.
We asked whether a kifunensine-mediated impairment in ERAD might produce more properly folded molecules of the α mutants by affording them a greater amount of time to fold. However, treatment with kifunensine did not significantly alter the fraction of secreted molecules; E482K was still undetectable in the medium, and G269S was not significantly elevated (Figure 7C). Coordinately, although kifunensine increased Hex activity secreted from untransfected cells, it provided no further significant increase in activity for any of the HEXA constructs tested (Figure 7, C and D). On the other hand, treatment of E482K-expressing cells with kifunensine did increase the E482K/β interaction, likely due to its longer ER residency (Figure 7E) but potentially because of a better-folded dimerization domain.
Because our data suggested that E482K α is nearly completely degraded via ERAD, we asked whether overexpression of a relevant molecular chaperone might prevent this outcome. Calreticulin (CRT) is a major ER-resident chaperone for glycoproteins, associating and disassociating with substrates in the “calreticulin/calnexin cycle” (Caramelo and Parodi, 2008). Co-IP confirmed the interaction between E482K and CRT (Figure 8A), while pulse-chase analysis demonstrated a drastic stabilization effect on E482K when it was coexpressed with the chaperone (Figure 8, B and C). This was a property of functional CRT, since an inactivating mutation, H153A (Guo et al., 2003), rendered it unable to stabilize the mutant α (Figure 8, B and C). We speculate that a greater number of CRT molecules leads to a larger number of folding attempts, increasing the residency time of E482K in the ER lumen and preventing recognition by ERAD factors.
FIGURE 8:
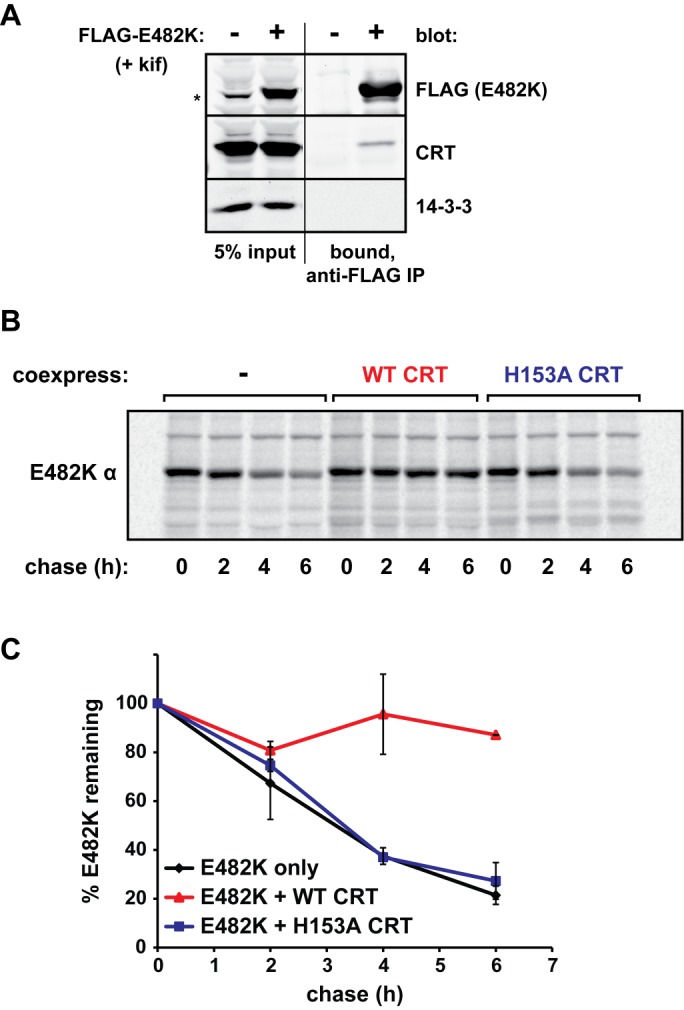
Calreticulin prevents E482K α release to ERAD. (A) HEK293T cells were transiently transfected with FLAG-E482K α or left untransfected, treated with 5 μg/ml kifunensine, and lysates were subjected to anti-FLAG agarose followed by immunoblotting to assess co-IP of CRT with the mutant α. 14-3-3, cytosolic control to ensure stringency of IP. (B) FLAG-E482K α was expressed alone or in combination with WT CRT or the inactive CRT, H153A. Cells were pulse chased 40 h posttransfection; at each of the indicated time points, α was precipitated with anti-FLAG agarose. (C) Quantification of two independent experiments as in B, with SDs.
To study and manipulate mutant α in a more physiological setting, we obtained fibroblastic cell lines from TSD patients via the Coriell Institute cell depository. We used several genetic backgrounds to examine endogenous Hex activity: 1) an apparently healthy person (“WT,” AG02101); 2) a compound heterozygote with one G269S allele and a second allele with a frameshift mutation that disallows proper α production (“G269S,” GM01108); 3) a patient homozygous for the E482K mutation (“E482K,” GM01110); and 4) a patient with a frameshift mutation allele and a splice junction mutation allele, disallowing any α production (complete loss of function, designated “α KO” for α protein knockout, GM02968). As predicted, the G269S, E482K, and α KO cell lines produced < 10% MUGS activity when compared with the WT fibroblasts, in agreement with the critical threshold hypothesis (Figure 9A).
FIGURE 9:

Endogenous G269S activity can be increased by improving folding conditions. (A) An equivalent number of fibroblasts from Tay–Sachs disease patients of the indicated genetic backgrounds were lysed in a pH 4.2 lysis buffer and subjected to the MUGS activity assay at 37°C as described in Materials and Methods. MUGS activity was normalized to the apparently healthy fibroblast, labeled WT, with SDs (n = 3). HEK293T cells were also measured to determine relative Hex activity. (B) G269S patient fibroblasts were grown at 32 or 37°C in 5% CO2 atmosphere for 2 d; this was followed by lysis and quantification of MUGS activity at room temperature. Specific activity was calculated by normalizing raw fluorescence units to protein content; activity is plotted with SDs (n = 4). Compared with 37°C, *** indicates p ≤ 0.001 in a two-tailed, unequal variance Student’s t test. (C) G269S patient fibroblasts were left untreated or grown in the presence of the following chemical chaperones: 20 mM trehalose, 100 mM proline, 150 mM glutamic acid, or 200 mM TMAO. Cells were lysed after 66 h in a pH 4.2 lysis buffer and subjected to the MUGS activity assay at 30°C. Specific activity was calculated by normalizing data to protein content of each sample (n = 3), and data were normalized to the Hex activity of untreated cells. Compared with untreated cells, * indicates p ≤ 0.05 and ** indicates p ≤ 0.01 in a two-tailed, unequal variance Student’s t test.
To test activity-rescuing manipulations, we chose to further study the G269S mutant, since at least some active G269S can escape ER quality control (Figure 2, C and D). Because in vitro data suggested that the G269S mutation interferes with protein stability at physiological temperatures (Brown and Mahuran, 1993; Tropak et al., 2004), we compared G269S fibroblasts grown at 32 and 37°C. Fibroblasts grown at the lower temperature produced more active G269S, as observed by the specific activity of MUGS cleavage (Figure 9B, roughly increasing G269S from ∼7 to ∼10% of WT activity). Thus we confirmed the temperature sensitivity of G269S and also appear to have improved its folding in cell culture.
Another method to enhance productive folding of cellular proteins is with chemical chaperones, small molecules that can help stabilize the folding of metastable proteins (reviewed in Rajan et al., 2011). Thus far, the only chemical chaperones tested on TSD cells/models have been specific inhibitors of HexA (Tropak et al., 2004; Maegawa et al., 2007), which can act to stabilize intermediates but have their own disadvantages. Instead of inhibitors, we examined enhancement of folding by culturing G269S patient fibroblasts with general chemical chaperones, measuring the resulting intracellular Hex activity after 72 h. While trehalose and proline treatment did not alter enzymatic activity significantly, trimethylamine N-oxide (TMAO) treatment and, even more so, glutamic acid increased endogenous Hex activity in the patient cells (Figure 9C). The fact that up-regulation of G269S does not itself increase cellular activity (Figure 2B) supports the conclusion that these chemical chaperones do not simply alter expression levels of α but may instead promote the folding and activity of G269S.
DISCUSSION
Many common mutations in the HEXA gene are thought to impair folding of the α chain of HexA and thereby decrease the activity of the enzyme to pathological levels. However, many past studies did not distinguish between folding defects per se and defects in the cellular handling of HexA, a distinction with important implications for potential therapies. In the work described here, we analyzed two HEXA mutations yielding the infantile-onset E482K and the adult-onset G269S α-chain mutants and showed that they are defective in folding in the cell and are subjected to disposal by the ERAD quality control system.
Our biochemical and cell biological evidence suggests that the E482K α chain is severely misfolded, forming inactive, trypsin-resistant aggregates in cells, thus confirming a previous report that observed detergent-dependent solubilization (Proia and Neufeld, 1982). E482K is unable to associate with the β subunit and is not observed to be secreted from cells. The G269S mutant, on the other hand, is more similar in conformation to the WT α chain, although it differs in at least its oxidative folding, resulting in a loss of activity. G269S is competent to heterodimerize with β and ∼10–20% is secreted. These findings suggest that clinical severity is proportional to the degree of misfolding, as would be predicted.
We and others have speculated that misfolded TSD-associated mutants would be turned over by ERAD, since this is a dominant pathway in the quality control of proteins in the secretory pathway. However, because much work on the biosynthesis, assembly, and trafficking of HexA was conducted before the characterization of ERAD, there is little direct evidence in the literature to support this hypothesis. Slight gains in activity of G269S fibroblasts had been observed upon treatment with the p97 inhibitor eeyarestatin I, though this treatment broadly affected the expression of ER chaperones and the activation of the unfolded protein response, making the interpretation difficult (Wang et al., 2011). Mu et al. (2008) showed that MG-132 had a moderately beneficial effect on the Hex activity of G269S patient fibroblasts, but this was measured only after several days of treatment during which cells can and do adapt to the chronic stress.
Armed with better tools and a more developed understanding of the ERAD pathways, we examined whether E482K and G269S were bona fide ERAD substrates. Indeed, E482K α is nearly completely stabilized by proteasome inhibition or expression of the dominant negative C294A mutant of the ERAD E3 ligase Hrd1. Because E482K is not secreted or proteolytically cleaved, this confirms that it is nearly completely triaged through ERAD. On the other hand, a small fraction of G269S α does get secreted; however, ∼20% of synthesized G269S is cleared via ERAD.
We further demonstrated ERAD as the mode of α degradation by monitoring ER-to-cytosol retrotranslocation, a more proximal readout than proteasomal degradation. Relying on cytosolic BirA-mediated biotinylation, we found that glycosylated (i.e., ER-derived) forms of both E482K and G269S gained access to the cytosol and furthermore were deglycosylated by cytosolic PNGase. This active process was inhibited by the presence of Z-VAD(OMe)-FMK, an inhibitor of PNGase, providing strong evidence of retrotranslocation.
Because ERAD is the pathway for destruction of disease-associated α mutants, could interference in the recognition of these mutants be a useful strategy in preventing or delaying disease onset? Similar reasoning showed promise in other LSD models, such as Gaucher disease (Sawkar et al., 2006; Wang and Segatori, 2013; Wang et al., 2011). In this work, we provide several proofs of principle that manipulation of folding and quality control may also be useful in TSD. Notably, we show that 1) coexpression of CRT delays E482K α from being identified and escorted to ERAD and that this depends on the activity of CRT; 2) an inhibitor of mannosidase I, kifunensine, delays clearance of both E482K and G269S by impairing early glycan trimming; and 3) patient fibroblasts expressing endogenous levels of G269S can show improved specific activity upon treatments with chemical chaperones or by growth at a permissive temperature. Though the effects of each treatment are modest, optimization and combination of the treatments, as was done for Gaucher disease (Sawkar et al., 2006), may well prove synergistic.
As diagrammed in Figure 10, we interpret the experimental data to show that α is cotranslationally N-glycosylated as it enters the ER, utilizing the calreticulin/calnexin cycle for rounds of attempted folding. Association with β also occurs in the ER and relies on an appropriately folded dimerization interface. Well-folded dimers (either αβ or αα) are competent to exit the ER and traffic to the Golgi for M6P modification and lysosomal sorting or secretion. On the other hand, mutations in α can impact oxidative-folding pathways and overall conformation. These malfolded molecules are recognized by ERAD sensors such as OS-9, a recognition that is likely enhanced by mannose trimming of N-linked glycans. Retrotranslocation of the α mutants occurs via the Hrd1/SEL1L machinery, and cytosolic PNGase removes the cytosol-exposed glycans before proteasomal degradation.
FIGURE 10:

Model for cell pathology in Tay–Sachs disease. Our data support a model whereby α is translocated into the ER, cotranslationally glycosylated, and folded in the calreticulin/calnexin cycle. If properly folded, α and β dimerize in the ER lumen and are exported to the Golgi, where they receive M6P signals to reach lysosomes. If ER folding of α is stalled, for example, in the disease-related mutations G269S and E482K, glycan trimming (perhaps in combination with OS-9 association) can initiate disposal. Molecules of α are brought to the retrotranslocon, where they use at least SEL1L and Hrd1 to access the cytosol. During this process, glycans are removed by cytosolic PNGase. The 26S proteasome is the site of protein destruction.
Because none of the antibodies we tested was able to recognize immature α in patient fibroblasts, we could not assess ERAD of endogenous α. While it is possible that the imbalance between α and β in our overexpression system may impact the propensity to drive α to ERAD, we show 1) E482K cannot associate with β and therefore is not subject to stoichiometric concerns; and 2) G269S targeting, processing, and retrotranslocation are insensitive to coexpression of β. Additionally, since it is thought that the production of β chains is rate limiting to enforce αβ dimers over ββ production, having an excess of α subunits may actually be the biological norm. Ultimately, definitive answers will require examining the turnover of endogenous protein in TSD patient cells and healthy controls. New antibodies or the use of the CRISPR/Cas9 system will enable these studies.
Clearly, each of the more than 100 HEXA mutations will alter the folding of the α chain in a unique manner. For example, compared with G269S, E482K has a much more dramatic effect on the conformation and trafficking of α. Because E482K is very likely to be globally misfolded, and even aggregation prone, it is unlikely that patients with such a mutation will be helped by buttressing folding pathways or impairing ERAD. Data from the G269S patient fibroblasts, however, suggest that improvement to folding may assist in reaching the ∼10% activity threshold to prevent disease. Interestingly, our pulse-chase data indicate that ∼40% of G269S molecules are unaccounted for, even in the presence of MG-132. Further confirmation will be required, but we speculate that a fraction of G269S in fact reaches the lysosome but is degraded there because it is less stable than WT α. Therefore ERAD is not the only source of cellular clearance and likely depends on the type of mutation and conformational outcomes.
Thus far, TSD therapies have not yet provided patients with useful treatment options. Substrate reduction therapy—the attenuation of GM2 ganglioside production—was unable to prevent neurodegeneration in patients with TSD (Bembi et al., 2006) and may even impact cells undesirably by broadly affecting cell metabolism (Ledeen and Wu, 2010). Gene therapies to restore WT enzymes to patients with LSDs is predicted to essentially cure these diseases, but clearly the types of vectors, timing, and kinetics of enzyme production will require extensive optimization (Kyrkanides et al., 2007). Finally, the use of small molecular inhibitors of HexA has been previously shown to stabilize folding intermediates and increase proper cellular trafficking of α mutants. However, because of the duality of their functions (stabilization of folding vs. inhibition of activity), dosing will be critical to avoid unwanted side effects (Clarke et al., 2011). The data presented in this paper support the idea that manipulating ERAD pathways and improving protein folding may be useful additions to the tools needed to treat some types of TSD.
MATERIALS AND METHODS
Chemicals and antibodies
The following reagents were used: kifunensine and 4-methylumbelliferyl-6-sulfo-N-acetyl-β-d-glucosaminide (Calbiochem, Billerica, MA); bafilomycin A1, MG-132, dimethyl sulfoxide (DMSO), biotin, streptavidin, brefeldin A, proline, glutamic acid, TMAO, and trehalose (Sigma, St. Louis, MO); Z-VAD(OMe)-FMK (Abcam, Cambridge, England). For Western blot analyses, the following antibodies were used: anti-FLAG M1 (Sigma); anti–14-3-3 and anti-SEL1L (Santa Cruz, Dallas, TX); anti–OS-9, anti-PDIA6, anti-calreticulin, anti-HEXA (α), anti-HEXB (β), and anti-PDIA1 (Abcam); anti-GRP94 9G10 (Enzo, Farmingdale, NY); anti-BiP (BD Biosciences, San Jose, CA); anti-GRP170 (kind gift from Linda Hendershot, St. Jude Children’s Research Hospital, Memphis, TN); anti-HA (Cell Signaling Technologies, Danvers, MA); and anti-V5 (Invitrogen, Carlsbad, CA). Secondary antibodies were obtained from Li-Cor, Lincoln, NE (IRDye 680 and 800 nm, for use with the Li-Cor Odyssey system).
Plasmids and cloning
We thank the following colleagues for the gifts of plasmids: Oscar Burrone (ICGEB, Trieste, Italy) for BirA, α1-antitrypsin-V5-BAP, and NHK-V5-BAP; Ron Kopito (Stanford University, Stanford, CA) for NHK-HA; Marek Michalak (University of Alberta, Canada) for WT and H153A calreticulin; and John Christianson (University of Oxford, UK) for Hrd1 C294A.
The FLAG-HEXA plasmid was created by appending a BamHI site to the 5′ end (5′TATGGATCCCTCTGGCCCTGGCCTCAGAACTTCC3′) and a NotI site to the 3′ end (5′ATAGCGGCCGCTCAGGTCTGTTCAAACTCCTGCTC3′) of human pro-HEXA during amplification of the gene from HeLa cDNA. This PCR product was ligated into a BamHI/NotI-digested FLAG-GRP94 pcDNA3.1(+) vector (base plasmid from Invitrogen), allowing for the retention of a murine anionic-trypsin II signal sequence, FLAG tag, and “SGS” linker (BamHI site) upstream of the mature HEXA coding sequence. This signal sequence was used due to its near-complete translocation efficiency—inefficient translocation would be predicted to affect downstream applications such as the retrotranslocation assay.
HEXB-HA was cloned by first adding a KpnI site to the 5′ end (5′ATGGTACCATGGAGCTGTGCGGGCTGGGGC3′) and an AgeI site to the 3′ end (5′ATACCGGTCATGTTCTCGTGGTTACAATATCCAGCATAAAGAGG3′) of the full-length human HEXB during amplification of the gene from 293T cDNA. The PCR product, as well as a NHK-HA vector (Ron Kopito, Stanford), were digested with KpnI and AgeI, purified, and subjected to ligation to produce the final construct of full-length HEXB including its natural signal sequence, a “TG” linker from the AgeI site, followed by an HA tag and stop codon.
For constructing the HEXA-V5-BAP construct, a HindIII site was added to the 5′ end of the signal sequence in the aforementioned FLAG-HEXA construct (5′TATAAGCTTATGTCTGCACTTCTGATCCTAGC3′) and an NheI site introduced at the 3′ end, minus the stop codon (5′AATGCTAGCGGTCTGTTCAAACTCCTGCTCAC3′) during PCR amplification. This product was inserted into a HindIII/NheI-digested NHK-V5-BAP vector (Oscar Burrone, ICGEB), allowing for the production of the final construct: anionic-trypsin II signal sequence–FLAG tag–“SGS” linker–mature HEXA coding sequence–“AS” linker–V5 tag–linker–BAP tag–stop.
Quikchange (Agilent, Santa Clara, CA) was used to design primers for all site-directed mutagenesis, and oligos were purchased from IDT, Coralville, IA. Mutagenesis was conducted by first mixing the following reagents: 5 μl 10× Pfu buffer, 1.25 μl 10 mM dNTP mix, 100 ng plasmid template, 125 ng of each forward and reverse primer, 1 μl Pfu Turbo (Agilent), to 50 μl with water. PCR was run with the following protocol: 95°C for 1:30, [95°C for 0:30, Tm ∼4°C for 1:00, 72°C for 1:00 per 1 kb plasmid] repeated for 18 cycles, followed by 72°C for 10:00 and a 4°C hold. Following transformation, colonies were screened for mutations via restriction analysis (New England Biolabs [Ipswich, MA] enzymes) or sequenced at the Children’s Hospital of Philadelphia Napcore facility.
Cell culture, lysis, and immunoblotting
HEK293T cells and the Tay–Sachs disease patient fibroblasts (Coriell Institute AG02101, GM01108, GM01110, GM02968) were maintained in DMEM (Life Technologies [Carlsbad, CA], Lonza [Basel, Switzerland]) supplemented with PSG (100 U/ml penicillin, 100 μg/ml streptomycin, 2.92 mg/ml l-glutamine; Life Technologies) and 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA) at 37°C in a 5% CO2 incubator. Tay–Sachs disease patient fibroblasts were grown as directed by the Coriell institute, with “WT”, E482K, and HEXA−/− cells grown with non–heat-inactivated serum with supplemental glutamine.
Transient transfection experiments were conducted when cells reached 70–90% confluency, using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were generally harvested for Western analysis 20 h posttransfection for immunoblot analyses. Cells were washed in phosphate-buffered saline (PBS) and lysed in lysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 5 mM KCl, 5 mM MgCl2, 1% NP-40) supplemented with protease inhibitors (Roche, Basel, Switzerland) and 20 mM iodoacetamide (Sigma). After 20 min on ice, samples were spun at 13,000 × g, and postnuclear supernatant fractions were further analyzed. Protein concentration was determined using the Pierce BCA assay (Thermo, Waltham, MA) and quantified on a Synergy HT plate reader (BioTek, Winooski, VT). Lysates were used for immunoprecipitation or diluted in reducing or nonreducing Laemmli buffer directly.
For determination of the NP-40 solubility of α mutants, cells were harvested in the standard 1% NP-40 lysis buffer described above; the insoluble material was resuspended in 3% SDS Tris (pH 6.8) at 56°C for 1.5 h. To shear genomic DNA, we used sonication, using 15× 1-s pulses at intensity 8 on a Microson Ultrasonic Cell Disruptor XL (Misonix, Farmingdale, NY).
Polyacrylamide gels were cast for use in the Owl Separation System (Thermo) using 37.5:1 acrylamide/bis-acrylamide (Bio-Rad, Hercules, CA), generally at 10%. Separated proteins were transferred to nitrocellulose membranes (Bio-Rad) and blocked in 5% nonfat milk in Tris-buffered saline (TBS). Primary antibodies were diluted in 1% nonfat milk in TBS + 0.1% Tween (TBST) and incubated with membranes overnight. Secondary antibodies were diluted 1:10,000 in 5% nonfat milk in TBST. Membranes were scanned directly using the Odyssey imager (Li-Cor).
Pulse chase and immunoprecipitation
Coprecipitation studies and pulse-chase analyses were conducted using the following purification reagents: anti-FLAG M1 agarose and anti-HA agarose (Sigma). For coprecipitation experiments, cell extracts were incubated with the respective agarose and buffer TNNB (50 mM Tris, pH 7.2, 250 mM NaCl, 0.5% NP-40, 0.1% BSA) for 3–5 h. Beads were spun for 30 s at 1200 rpm, washed in buffer TNN (TNNB without BSA) four times, and finally resuspended in reducing or nonreducing Laemmli buffer. Samples were separated by SDS–PAGE, and Western blot analysis was performed as described above.
For pulse-chase experiments, 60 mm plates of cells were transiently transfected at 70–90% confluency with the plasmid(s) of interest. At 20 h posttransfection, cells were split to poly-d-lysine–coated plates. One day later, cells were washed with Hank’s balanced saline solution (Life Technologies) and starved in labeling medium (DMEM without l-glutamine/l-methionine/l-cystine [Corning, Corning, NY] supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2.92 mg/ml l-glutamine [Life Technologies], 10 mM HEPES, 1 mM sodium pyruvate [Mediatech, Corning, NY], and 3% dialyzed Hyclone serum) for 20 min. Cells were labeled for 20 min with [35S]methionine/cysteine diluted into labeling medium at ∼88 μCi/ml (University of Pennsylvania Office of Environmental Health and Radiation Safety). After labeling, cells were washed in ice-cold PBS supplemented with 5 mM each of methionine and cysteine (Calbiochem) followed by chase medium (growth medium with 5 mM methionine/cysteine) and finally incubated with fresh chase medium for the indicated times. At each time point, cells were harvested using the lysis procedure described above. Labeled proteins of interest were isolated from lysates using the agarose described above for 20 h, washed twice in TNNB, and resuspended in Laemmli buffer. Samples were separated by SDS–PAGE with autoradiography of the dried gels carried out on a Typhoon 9200 (Amersham Biosciences, Little Chalfont, United Kingdom). Quantification was performed using the ImageQuant software (GE Healthcare, Little Chalfont, United Kingdom).
Limited proteolysis
Limited trypsin proteolysis of α mutants was conducted by preparing cell lysates in lysis buffer without protease inhibitors. After protein concentration determination via BCA assay, equal amounts of lysate were aliquoted and subjected to increasing concentrations of dilute trypsin (Promega, Madison, WI). After a 1 h incubation at 37°C, samples were quenched with reducing Laemmli buffer and separated via SDS–PAGE for Western blot analysis.
Retrotranslocation assay
Adapted from Burrone and colleagues (Petris et al., 2011; Vecchi et al., 2012), BAP-tagged ER proteins were coexpressed with cytosolic BirA in HEK293T cells. The following day, the medium was replaced with fresh growth medium supplemented with 0.1 mM biotin for 8 h. MG-132 and Z-VAD(OMe)-FMK were included during the last 4 h before collection. Cells were harvested, washed in PBS, and lysed immediately in SDS lysis buffer (100 mM Tris, pH 6.8, with 6% SDS supplemented with 20 μM N-ethylmaleimide). To shear genomic DNA, we used sonication as described above. Samples of this whole-cell extract were diluted in Laemmli buffer, boiled, split in half, and either left untreated or incubated at room temperature with 4 μg streptavidin for 1 h. Each sample was subjected to SDS–PAGE to observe retardation of biotinylated molecules.
MUGS assay for HexA/S activity
For analysis of conditioned medium of growing cells, aliquots of the medium (typically 20–30 μl) were buffered in equal volume of citrate/phosphate buffer (pH 4.2) in triplicate in a black 96-well plate. Thirty microliters of a 3.2 mM MUGS solution in the citrate/phosphate buffer (pH 4.2) was added to each well, and plates were incubated at the indicated temperatures for 2–4 h. Reactions were quenched by adding 150 μl 0.1 M 2-amino-2-methyl-1-propanol at pH 10.5, and plates were read on the Synergy HT plate reader at 365 nm excitation and 450 nm emission.
Cell lysates were processed in a similar manner, with cells first washed with PBS and lysed with a citrate/phosphate buffer at pH 4.2 containing 1% Triton-X for 20 min at 4°C. Aliquots of lysate were added in triplicate to the 3.2 mM MUGS solution and incubated for 2–4 h before quenching and reading. Control wells such as MUGS only, lysate without MUGS, and buffer only were routinely included to ensure specificity.
Supplementary Material
Acknowledgments
We thank Oscar Burrone (ICGEB, Trieste) for advice on the retrotranslocation assay and Davide Eletto for advice with molecular biology techniques. The following colleagues generously supplied plasmids and antibodies: Oscar Burrone (ICGEB, Trieste), Richard Proia (National Institutes of Health [NIH]), Elizabeth Neufeld (University of California, Los Angels), Ron Kopito (Stanford), Marek Michalak (University of Alberta, Canada), Linda Hendershot (St. Jude Children’s Hospital), and John Christianson (University of Oxford, United Kingdom). This work was supported by training grant F31-NS084666-01 (to D.D.) and NIH grants GM-077480 and AG-18001 (to Y.A.).
Abbreviations used:
- BAP
biotin acceptor peptide
- co-IP
coimmunoprecipitation
- CRT
calreticulin
- DMSO
dimethyl sulfoxide
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- HexA
hexosaminidase A
- LSD
lysosomal storage disorder
- M6P
mannose-6-phosphate
- MUGS
4-methylumbelliferone N-acetylglucosamine-6-sulfate; PBS, phosphate-buffered saline
- TMAO
trimethylamine N-oxide
- TSD
Tay–Sachs disease
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0012) on September 28, 2016.
REFERENCES
- Bembi B, Marchetti F, Guerci VV, Ciana G, Addobbati R, Grasso D, Barone R, Cariati R, Fernandez-Guillen L, Butters T, Pittis MM. Substrate reduction therapy in the infantile form of Tay-Sachs disease. Neurology. 2006;66:278–280. doi: 10.1212/01.wnl.0000194225.78917.de. [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188:223–235. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CC, Mahuran DD. Beta-hexosaminidase isozymes from cells cotransfected with alpha and beta cDNA constructs: analysis of the alpha-subunit missense mutation associated with the adult form of Tay-Sachs disease. Am J Hum Genet. 1993;53:497–508. [PMC free article] [PubMed] [Google Scholar]
- Caramelo JJ, Parodi AA. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. 2008;283:10221–10225. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tukachinsky H, Huang CC, Jao C, Chu YY, Tang HH, Mueller B, Schulman S, Rapoport TT, Salic A. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192:825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JJ, Shaler TT, Tyler RR, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JJ, Mahuran DD, Sathe S, Kolodny EE, Rigat BB, Raiman JJ, Tropak MM. An open-label phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants) Mol Genet Metab. 2011;102:6–12. doi: 10.1016/j.ymgme.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann E, Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6:58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- Guo L, Groenendyk J, Papp S, Dabrowska M, Knoblach B, Kay C, Parker JJ, Opas M, Michalak M. Identification of an N-domain histidine essential for chaperone function in calreticulin. J Biol Chem. 2003;278:50645–50653. doi: 10.1074/jbc.M309497200. [DOI] [PubMed] [Google Scholar]
- Hasilik A, Neufeld EE. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem. 1980;255:4937–4945. [PubMed] [Google Scholar]
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284:17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Tse R, Mahuran DD. Direct determination of the substrate specificity of the alpha-active site in heterodimeric beta-hexosaminidase A. Biochemistry. 1996;35:3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- Huang JJ, Trasler JJ, Igdoura S, Michaud J, Hanal N, Gravel RR. Apoptotic cell death in mouse models of GM2 gangliosidosis and observations on human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1997;6:1879–1885. doi: 10.1093/hmg/6.11.1879. [DOI] [PubMed] [Google Scholar]
- Hubbes M, Callahan J, Gravel R, Mahuran D. The amino-terminal sequences in the pro-alpha and -beta polypeptides of human lysosomal beta-hexosaminidase A and B are retained in the mature isozymes. FEBS Lett. 1989;249:316–320. doi: 10.1016/0014-5793(89)80649-0. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, Miller JJ, Tallents RR, Brouxhon SS, Centola GG, Olschowka JJ. Intraperitoneal inoculation of Sandhoff mouse neonates with an HIV-1 based lentiviral vector exacerbates the attendant neuroinflammation and disease phenotype. J Neuroimmunol. 2007;188:39–47. doi: 10.1016/j.jneuroim.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Ledeen RR, Wu G. In search of a solution to the sphinx-like riddle of GM1. Neurochem Res. 2010;35:1867–1874. doi: 10.1007/s11064-010-0286-0. [DOI] [PubMed] [Google Scholar]
- Lemieux MM, Mark BB, Cherney MM, Withers SS, Mahuran DD, James MM. Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. J Mol Biol. 2006;359:913–929. doi: 10.1016/j.jmb.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little LL, Lau MM, Quon DD, Fowler AA, Neufeld EE. Proteolytic processing of the alpha-chain of the lysosomal enzyme, beta-hexosaminidase, in normal human fibroblasts. J Biol Chem. 1988;263:4288–4292. [PubMed] [Google Scholar]
- Maegawa GG, Tropak M, Buttner J, Stockley T, Kok F, Clarke JJ, Mahuran DD. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem. 2007;282:9150–9161. doi: 10.1074/jbc.M609304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahuran DD. The biochemistry of HEXA and HEXB gene mutations causing GM2 gangliosidosis. Biochim Biophys Acta. 1991;1096:87–94. doi: 10.1016/0925-4439(91)90044-a. [DOI] [PubMed] [Google Scholar]
- Mahuran DD. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- Mahuran DD, Neote K, Klavins MM, Leung A, Gravel RR. Proteolytic processing of pro-alpha and pro-beta precursors from human beta-hexosaminidase. Generation of the mature alpha and beta a beta b subunits. J Biol Chem. 1988;263:4612–4618. [PubMed] [Google Scholar]
- Maier T, Strater N, Schuette CC, Klingenstein R, Sandhoff K, Saenger W. The X-ray crystal structure of human beta-hexosaminidase B provides new insights into Sandhoff disease. J Mol Biol. 2003;328:669–681. doi: 10.1016/s0022-2836(03)00311-5. [DOI] [PubMed] [Google Scholar]
- Mark BB, Mahuran DD, Cherney MM, Zhao D, Knapp S, James MNG. Crystal structure of human β-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay–Sachs disease. J Mol Biol. 2003;327:1093–1109. doi: 10.1016/s0022-2836(03)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaghi S, Korbel GG, Kessler B, Spooner E, Ploegh HH. z-VAD-fmk inhibits peptide:N-glycanase and may result in ER stress. Cell Death Differ. 2006;13:163–165. doi: 10.1038/sj.cdd.4401716. [DOI] [PubMed] [Google Scholar]
- Misaghi S, Pacold MM, Blom D, Ploegh HH, Korbel GG. Using a small molecule inhibitor of peptide: N-glycanase to probe its role in glycoprotein turnover. Chem Biol. 2004;11:1677–1687. doi: 10.1016/j.chembiol.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Mu TT, Ong DD, Wang YY, Balch WW, Yates JJ, III, Segatori L, Kelly JJ. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Klemm EE, Spooner E, Claessen JJ, Ploegh HH. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci USA. 2008;105:12325–12330. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Lilley BB, Ploegh HH. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol. 2006;175:261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Muscillo M, Ohno K, Hoffman AA, Suzuki K. A point mutation in the coding sequence of the beta-hexosaminidase alpha gene results in defective processing of the enzyme protein in an unusual GM2-gangliosidosis variant. J Neurochem. 1988;51:984–987. doi: 10.1111/j.1471-4159.1988.tb01836.x. [DOI] [PubMed] [Google Scholar]
- Navon R, Proia RR. The mutations in Ashkenazi Jews with adult GM2 gangliosidosis, the adult form of Tay-Sachs disease. Science. 1989;243:1471–1474. doi: 10.1126/science.2522679. [DOI] [PubMed] [Google Scholar]
- Ohno K, Saito S, Sugawara K, Sakuraba H. Structural consequences of amino acid substitutions causing Tay-Sachs disease. Mol Genet Metab. 2008;94:462–468. doi: 10.1016/j.ymgme.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Osher E, Fattal-Valevski A, Sagie L, Urshanski N, Amir-Levi Y, Katzburg S, Peleg L, Lerman-Sagie T, Zimran A, Elstein D, et al. Pyrimethamine increases beta-hexosaminidase A activity in patients with late onset Tay–Sachs. Mol Genet Metab. 2011;102:356–363. doi: 10.1016/j.ymgme.2010.11.163. [DOI] [PubMed] [Google Scholar]
- Petris G, Vecchi L, Bestagno M, Burrone OO. Efficient detection of proteins retro-translocated from the ER to the cytosol by in vivo biotinylation. PloS One. 2011;6:e23712. doi: 10.1371/journal.pone.0023712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FF, Boland B, van der Spoel AA. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis BB, Wevers RR, Kleijer WW, Groener JJ, de Jong JJ, van Weely S, Niezen-Koning KK, van Diggelen OO. The frequency of lysosomal storage diseases in the Netherlands. Hum Genet. 1999;105:151–156. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- Proia RR, Neufeld EE. Synthesis of beta-hexosaminidase in cell-free translation and in intact fibroblasts: an insoluble precursor alpha chain in a rare form of Tay-Sachs disease. Proc Natl Acad Sci USA. 1982;79:6360–6364. doi: 10.1073/pnas.79.20.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan RR, Tsumoto K, Tokunaga M, Tokunaga H, Kita Y, Arakawa T. Chemical and pharmacological chaperones: application for recombinant protein production and protein folding diseases. Curr Med Chem. 2011;18:1–15. doi: 10.2174/092986711793979698. [DOI] [PubMed] [Google Scholar]
- Robinson D, Stirling JJ. N-acetyl-beta-glucosaminidases in human spleen. Biochem J. 1968;107:321–327. doi: 10.1042/bj1070321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagherian C, Poroszlay S, Vavougios G, Mahuran D. Proteolytic processing of the pro beta chain of beta-hexosaminidase occurs at basic residues contained within an exposed disulfide loop structure. Biochem Cell Biol. 1993;71:340–347. doi: 10.1139/o93-051. [DOI] [PubMed] [Google Scholar]
- Sawkar AA, Schmitz M, Zimmer KK, Reczek D, Edmunds T, Balch WW, Kelly JJ. Chemical chaperones and permissive temperatures alter localization of Gaucher disease associated glucocerebrosidase variants. ACS Chem Biol. 2006;1:235–251. doi: 10.1021/cb600187q. [DOI] [PubMed] [Google Scholar]
- Schepers U, Glombitza G, Lemm T, Hoffmann A, Chabas A, Ozand P, Sandhoff K. Molecular analysis of a GM2-activator deficiency in two patients with GM2-gangliosidosis AB variant. Am J Hum Genet. 1996;59:1048–1056. [PMC free article] [PubMed] [Google Scholar]
- Schuette CC, Weisgerber J, Sandhoff K. Complete analysis of the glycosylation and disulfide bond pattern of human beta-hexosaminidase B by MALDI-MS. Glycobiology. 2001;11:549–556. doi: 10.1093/glycob/11.7.549. [DOI] [PubMed] [Google Scholar]
- Sonderfeld-Fresko S, Proia RR. Analysis of the glycosylation and phosphorylation of the lysosomal enzyme, beta-hexosaminidase B, by site-directed mutagenesis. J Biol Chem. 1989;264:7692–7697. [PubMed] [Google Scholar]
- Svennerholm L. The chemical structure of normal human brain and Tay-Sachs gangliosides. Biochem Biophys Res Commun. 1962;9:436–441. doi: 10.1016/0006-291x(62)90030-x. [DOI] [PubMed] [Google Scholar]
- Tiede S, Storch S, Lubke T, Henrissat B, Bargal R, Raas-Rothschild A, Braulke T. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat Med. 2005;11:1109–1112. doi: 10.1038/nm1305. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Brostrom C, Koide T, Arvan P. Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J Biol Chem. 2000;275:40757–40764. doi: 10.1074/jbc.M001073200. [DOI] [PubMed] [Google Scholar]
- Triggs-Raine B, Mahuran DD, Gravel RR. Naturally occurring mutations in GM2 gangliosidosis: a compendium. Adv Genet. 2001;44:199–224. doi: 10.1016/s0065-2660(01)44081-8. [DOI] [PubMed] [Google Scholar]
- Tropak MM, Reid SS, Guiral M, Withers SS, Mahuran D. Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff patients. J Biol Chem. 2004;279:13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchi L, Petris G, Bestagno M, Burrone OO. Selective targeting of proteins within secretory pathway for endoplasmic reticulum-associated degradation. J Biol Chem. 2012;287:20007–20015. doi: 10.1074/jbc.M112.355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005;128:413–431. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- Wang F, Segatori L. Remodeling the proteostasis network to rescue glucocerebrosidase variants by inhibiting ER-associated degradation and enhancing ER folding. PLoS One. 2013;8:e61418. doi: 10.1371/journal.pone.0061418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Song W, Brancati G, Segatori L. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J Biol Chem. 2011;286:43454–43464. doi: 10.1074/jbc.M111.274332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz G, Proia RR. Analysis of the glycosylation and phosphorylation of the alpha-subunit of the lysosomal enzyme, beta-hexosaminidase A, by site-directed mutagenesis. J Biol Chem. 1992;267:10039–10044. [PubMed] [Google Scholar]
- Xie B, McInnes B, Neote K, Lamhonwah AA, Mahuran D. Isolation and expression of a full-length cDNA encoding the human GM2 activator protein. Biochem Biophys Res Commun. 1991;177:1217–1223. doi: 10.1016/0006-291x(91)90671-s. [DOI] [PubMed] [Google Scholar]
- Yamada K, Takado Y, Kato YY, Yamada Y, Ishiguro H, Wakamatsu N. Characterization of the mutant beta-subunit of beta-hexosaminidase for dimer formation responsible for the adult form of Sandhoff disease with the motor neuron disease phenotype. J Biochem. 2013;153:111–119. doi: 10.1093/jb/mvs131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WW, Aziz PP, Heithoff DD, Mahan MM, Smith JJ, Marth JJ. An intrinsic mechanism of secreted protein aging and turnover. Proc Natl Acad Sci USA. 2015;112:13657–13662. doi: 10.1073/pnas.1515464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.