

ABSTRACT
Although the PINK1-PARK2 pathway contributes to the pathogenesis of Parkinson disease, its roles in sepsis (a major challenge for critical care) were previously unknown. Here, we show that pink1−/− and park2−/− mice are more sensitive to polymicrobial sepsis-induced multiple organ failure and death. The decrease in the circulating level of the neurotransmitter dopamine in pink1−/− and park2−/− mice accelerates the release of a late sepsis mediator, HMGB1, via HIF1A-dependent anaerobic glycolysis and subsequent NLRP3-dependent inflammasome activation. Genetic depletion of Nlrp3 or Hif1a in pink1−/− and park2−/− mice confers protection against lethal polymicrobial sepsis. Moreover, pharmacological administration of dopamine agonist (e.g., pramipexole), HMGB1-inhibitor (e.g., neutralizing antibody or glycyrrhizin), or NLRP3-inhibitor (e.g., MCC950) reduces septic death in pink1−/− and park2−/− mice. The mRNA expression of HIF1A and NLRP3 is upregulated, whereas the mRNA expression of PINK1 and PARK2 is downregulated in peripheral blood mononuclear cells of patients with sepsis. Thus, an impaired PINK1-PARK2-mediated neuroimmunology pathway contributes to septic death and may represent a novel therapeutic target in critical care medicine.
KEYWORDS: HMGB1, hypoxia, IL1A, inflammasome, mitophagy, PARK2, PINK1, sepsis
Introduction
Sepsis, including severe sepsis and septic shock, is a clinical condition occurring in patients following infection or injury.1 It remains a major challenge in the intensive care unit despite advances in modern medicine. The pathogenesis of sepsis was partly attributable to dysregulated neuroimmune activation with altered production of neurotransmitters2 and inflammatory mediators.3 Immunometabolism is an emerging field that intersects with key metabolic pathways involved in the immune response.4 Although a patient with sepsis often displays or exhibits symptoms complicated by encephalopathy and hyperlactatemia,5 the molecular mechanism underlying neurotransmitter-mediated regulation of immunometabolism has not yet been established.
Inflammation, the primary response of the innate immune system to damage and invasion, is associated with many neurodegenerative diseases, including Parkinson disease (PD). It has been well established that genetic deletion of the genes encoding PINK1 (PTEN-induced putative kinase 1) and PARK2 (parkin RBR E3 ubiquitin protein ligase) causes progressive mitochondrial damage and development of PD.6 Mitochondria play a central role in energy metabolism and cell death. Although the PINK1-PARK2 pathway plays a major role in the maintenance of mitochondrial quality control by triggering mitophagy (a form of selective autophagy to remove damaged or superfluous mitochondria),7,8 its involvement in sepsis-induced immunometabolism dysregulation remain largely unknown.
In the current study, we show that pink1−/− and park2−/− mice are more sensitive to polymicrobial sepsis than wild-type (WT) mice partly due to impairment of immunometabolism. We demonstrate that the decrease in circulating levels of dopamine (a neurotransmitter released by the brain) in pink1−/− and park2−/− mice mediates HMGB1 (high mobility group box 1; a late mediator of lethal sepsis) release through activation of the NLRP3 (NLR family, pyrin domain containing 3) inflammasome (a platform for processing proinflammatory cytokines). Moreover, we demonstrate that HIF1A (hypoxia inducible factor 1 α subunit; a master transcriptional regulator of hypoxia)-mediated aerobic glycolysis contributes to NLRP3 inflammasome activation and lethal sepsis in pink1−/− and park2−/− mice. Genetic or pharmacological inhibition of Hif1a- and Nlrp3-dependent HMGB1 release confers protection against polymicrobial sepsis in pink1−/− and park2−/− mice. Thus, an impaired PINK1-PARK2 pathway occupies a previously unidentified pathogenic role in critical care medicine and may represent a novel target for patients with sepsis.
Results
pink1−/− and park2−/− mice are more sensitive to polymicrobial sepsis due to impaired dopamine production
Experimental sepsis can be induced in mice by cecal ligation and puncture (CLP) because it closely mimics many clinical features of polymicrobial infection observed in human sepsis.9 To determine the effect of PINK1 and PARK2 on polymicrobial sepsis, pink1−/−, park2−/−, and WT mice were subjected to moderate CLP using single punctures with 22-gauge syringe needles. The survival of pink1−/− and park2−/− mice was significantly lower than that of WT mice within 8 d of CLP (Fig. 1A). Histological analysis revealed more pronounced damage in multiple organs (e.g., liver, lung, kidney, heart, pancreas, brain, and small intestine) of the pink1−/− and park2−/− mice compared with age- and sex-matched control mice (Fig. 1B). Biochemical measurement of tissue enzymes also exhibited a greater elevation of liver (e.g., GPT/ALT [glutamic pyruvic transaminase, soluble]), heart (e.g., TNNI [troponin I]), kidney (e.g., creatinine), and pancreas (e.g., AMY2 [amylase]) enzymes in the pink1−/− and park2−/− mice (Fig. 1C). Similarly, serum MPO (myeloperoxidase) and LDH (lactate dehydrogenase) activities were remarkably higher in pink1−/− and park2−/− mice than in WT mice (Fig. 1C), suggesting that sepsis-induced tissue injury is more significant in the pink1−/− and park2−/− mice.
Figure 1.
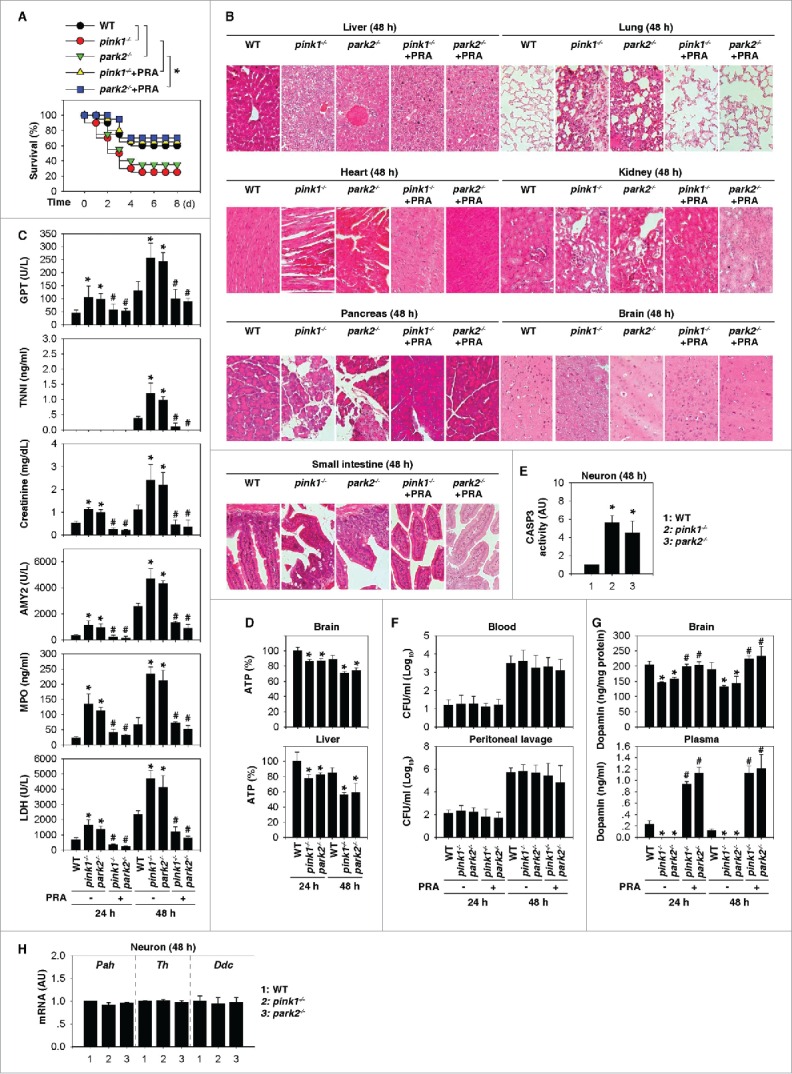
pink1−/− and park2−/− mice are more sensitive to polymicrobial sepsis due to impaired dopamine production. (A) Survival after moderate CLP in wild-type (WT), pink1−/−, and park2−/− mice with or without pramipexole (PRA; 1 mg/kg/i.p. at 2, 12, 24, and 48 h after CLP) treatment (n=20 mice/group; *, P < 0.05). (B to H) In parallel, tissue H&E staining (B), serum enzyme activity (C), ATP levels (D), CASP3 activity in neurons (E), bacterial loads (F), dopamine concentrations (G), and the indicated gene mRNA in neurons (H) were assayed (n = 3 to 5 mice/group; *, P < 0.05 versus WT group; # vs. the group without PRA).
Previous studies have suggested that loss of PINK1 or PARK2 results in a mitophagy deficiency-associated decrease of ATP synthesis and an increase of apoptosis in multiple cells, including neurons.6,10,11 Consistently, the tissue levels of ATP were significantly reduced in brains and livers (Fig. 1D) and CASP3/caspase-3 activity (an apoptosis marker) were increased in brain tissue neurons (Fig. 1E) from pink1−/− and park2−/− mice after CLP. The PINK1-PARK2 pathway also plays a role in xenophagy,12 a selective autophagic pathway that delivers intracellular bacteria for degradation in lysosomes. However, the number of colony-forming units from blood and peritoneal lavage after CLP did not differ between pink1−/−, park2−/−, and WT mice (Fig. 1F), suggesting that the PINK1-PARK2 pathway might not influence bacterial elimination during experimental sepsis.
Given the essential role of PINK1 and PARK2 in dopaminergic neuronal function, we next compared the levels of dopamine between pink1−/−, park2−/−, and WT mice. Compared with WT mice, pink1−/− and park2−/− mice showed a significantly larger reduction in central (brain) and peripheral (serum) dopamine levels after CLP (Fig. 1G). The gene expression of the enzymes (e.g., Pah [phenylalanine hydroxylase], Th [tyrosine hydroxylase], and Ddc [dopa decarboxylase]) in the dopamine biosynthetic pathway did not significantly change in brain tissue neurons from pink1−/− and park2−/− mice after CLP (Fig. 1H). These findings suggest that decreases in neuron survival (but not in impaired dopamine biosynthesis) contribute to loss of dopamine production in pink1−/− and park2−/− mice.
To determine whether the reduced dopamine levels contribute to septic death, we supplemented (intraperitoneally) the septic mice with pramipexole (PRA; a Food and Drug Administration-approved dopamine agonist for treating PD patients13) at +2, +12, +24, and +48 h after CLP. Remarkably, administration of PRA restored both brain and serum dopamine levels in septic pink1−/− and park2−/− mice (Fig. 1G) and cocurrently increased their survival rates (Fig. 1A). Consistently, PRA also conferred protection against tissue injury, as determined by tissue histology (Fig. 1B) and enzyme release (Fig. 1C), although it did not affect tissue bacterial loads in septic pink1−/− and park2−/− mice (Fig. 1F). Thus, these findings suggest that the reduced circulating dopamine levels contribute to the exacerbated septic lethality in animals that are defective in the PINK1 and PARK2 pathways.
HMGB1 is a late mediator of septic death in pink1−/− and park2−/− mice
The pathogenesis of lethal sepsis is partly attributable to a dysregulated inflammatory response, as manifested by the overproduction of proinflammatory mediators. We next compared the circulating levels of inflammatory mediators between pink1−/−, park2−/−, and WT mice. Serum levels of early (IL [interleukin] 1B,14 TNF/TNF-α [tumor necrosis factor],15 and IL616) and late (HMGB117 and CIRBP [cold inducible RNA binding protein]18) mediators in pink1−/−, and park2−/− mice were higher than those in the WT mice over a wide time period (6 to 48 h after CLP, Fig. 2A). However, treatment with PRA only markedly decreased the circulating levels of IL1B and HMGB1 (but not TNF, IL6, and CIRBP) in the septic pink1−/− and park2−/− mice (Fig. 2A), suggesting that dopamine may specifically modulate the release of inflammasome-dependent cytokines during experimental sepsis.
Figure 2.

HMGB1 is a late mediator of septic death in pink1−/− and park2−/− mice. (A) Time course of serum proinflammatory mediator levels in CLP-induced wild-type (WT), pink1−/−, and park2−/− mice with or without pramipexole (PRA; 1 mg/kg/i.p. at 2, 12, 24, and 48 h after CLP) treatment (n = 3 to 5 mice/group; *, P < 0.05 versus the group without PRA). (B) Survival after moderate CLP in pink1−/− and park2−/− mice with or without IgG (10 mg/kg/i.p. at +12, +24, and +48 h after CLP), HMGB1-neutralizing antibody (HMG1 Ab; 10 mg/kg/i.p. at +12, +24, and +48 h after CLP), and glycyrrhizin (Glz; 10 mg/kg/i.p. at +12, +24, and +48 h after CLP) (n = 10 mice/group; *, P < 0.05). (C and D) In parallel, tissue H&E staining (C) and serum enzyme activity (D) were assayed (n = 3 to 5 mice/group; *, P < 0.05 vs. IgG group). (E) Survival after moderate CLP in pink1−/− and park2−/− mice with or without IgG (10 mg/kg/i.p. at +12, +24, and +48 h after CLP), IL1B-neutralizing antibody (late: 10 mg/kg/i.p. at +12, +24, and +48 h after CLP; early: 10 mg/kg/i.p. at +2, +6, and +12 h after CLP) (n = 10 mice/group; *, P < 0.05).
To confirm whether increased IL1B or HMGB1 contribute to septic death in pink1−/− and park2−/− mice, we treated these mice with IL1B- or HMGB1-neutralizing antibodies in parallel experiments. Delayed administration of HMGB1- (but not IL1B-) specific neutralizing antibodies at +12, +24, and +48 h after CLP rescued pink1−/− and park2−/− mice from lethal sepsis (Fig. 2B), and attenuated tissue injury (Fig. 2C) and the release of tissue enzymes (Fig. 2D). Similarly, treatment with IL1B-neutralizing antibodies at +2, +6, and +12 h after CLP partly conferred protection against CLP-induced death in pink1−/− and park2−/− mice (Fig. 2E). Moreover, delayed administration of glycyrrhizin (a direct HMGB1 inhibitor19) at +12, +24, and +48 h after CLP also promoted protection against CLP-induced animal lethality (Fig. 2B) and tissue injury (Fig. 2C and 2D) in pink1−/− and park2−/− mice. These findings suggest that excessive systemic accumulation of inflammasome-dependent cytokines (e.g., IL1B and HMGB1) contributes to exacerbated septic lethality in pink1−/− and park2−/− mice.
Activation of the NLRP3 inflammasome contributes to septic death in pink1−/− and park2−/− mice
Inflammasomes, the platforms that detect pathogens and sterile stressors, contribute to the release of proinflammatory mediators such as IL1B and HMGB1 by activating CASP1/caspase-1 or CASP4/caspase-4 (formerly murine CASP11).20,21 Please note that murine CASP11 is no longer the primary acronym in the MGI database and both murine and human acronyms are now CASP4; care should also be taken to prevent confusion with SCAF11/SFRS2IP, which is involved in pre-mRNA processing and is sometimes also termed CASP11. Dopamine has recently been identified as a negative regulator of NLRP3 inflammasome activation.22 To assess the contribution of NLRP3 inflammasome activation to the elevated susceptibility of pink1−/− and park2−/− mice to septic insult, we generated double-knockout mice (pink1−/− nlrp3−/− and park2−/− nlrp3−/−). The disruption of Nlrp3 attenuated the oversensitivity of pink1−/− or park2−/− mice to lethal sepsis, as manifested by increased animal survival (Fig. 3A) and decreased tissue injury (Fig. 3B and 3C). Serum concentrations of IL1B and HMGB1 (but not IL6, TNF, and CIRBP) were also significantly reduced in these double-knockout mice compared with respective pink1−/− or park2−/− mice following CLP (Fig. 3D), suggesting that the sepsis-induced excessive accumulation of IL1B and HMGB1 in pink1−/− and park2−/− mice is regulated by NLRP3 inflammasome pathways.
Figure 3.

Activation of the NLRP3 inflammasome contributes to septic death in pink1−/− and park2−/− mice. (A) Survival after moderate CLP in the indicated knockout mice with or without MCC950 (20 mg/kg/i.p. at +2, +12, +24, and +48 h after CLP) or same volume phosphate-buffered saline treatment (n = 10 mice/group; *, P < 0.05). (B to D) In parallel, tissue H&E staining (B), serum enzyme activity (C), and serum proinflammatory mediators (D) were assayed (n = 3 to 5 mice/group; *, P < 0.05 versus pink1−/− or park2−/− group).
To further test this possibility, we next determined whether pharmacological inhibition of NLRP3 inflammasomes by MCC95023 affects the septic outcome in pink1−/− and park2−/− mice. As with knockout of Nlrp3 in mice, administration of MCC950 at +2, +12, +24, and +48 h after CLP also increased animal survival and decreased tissue injury, as confirmed by histology (Fig. 3B) and enzyme assay (Fig. 3C) in pink1−/− and park2−/− mice. Consistently, MCC950 also reduced the serum levels of IL1B and HMGB1 (but not IL6, TNF, and CIRBP) in septic pink1−/− and park2−/− mice (Fig. 3D). Thus, activation of the NLRP3 inflammasome contributes to septic death in pink1−/− and park2−/− mice by regulating IL1B and HMGB1 release.
HIF1A-mediated aerobic glycolysis contributes to inflammasome activation in pink1−/− and park2−/− mice
Others and we have demonstrated that aerobic glycolysis contributes to sepsis through promoting the release of proinflammatory mediators, including IL1B and HMGB1.24,25 We next analyzed the levels of circulating lactate (the end product of aerobic glycolysis) in pink1−/− and park2−/− mice. Compared with WT mice, the pink1−/− and park2−/− mice were slightly hyperlactatemic at baseline, and this phenotype was significantly enhanced following CLP surgery (Fig. 4A). The administration of a dopamine agonist, PRA, led to a reduction of circulating lactate levels in septic pink1−/− and park2−/− mice (Fig. 4A), suggesting that dopamine may participate in the regulation of aerobic glycolysis and lactate production.
Figure 4.

HIF1A-mediated aerobic glycolysis contributes to inflammasome activation in pink1−/− and park2−/− mice. (A, B) Time course of serum lactate levels (A) and the indicated gene expression at 48 h (B) in CLP-induced wild-type (WT), pink1−/−, and park2−/− mice with or without pramipexole (PRA; 1 mg/kg/i.p. at 2, 12, 24, and 48 h after CLP) treatment (n = 3 to 5 mice/group; *, P < 0.05 vs. the group without PRA). (C) In parallel, brain GSH levels at 48 h after CLP were assayed (n = 3 mice/group; *, P < 0.05 versus the group without PRA). (D) Survival after moderate CLP in the indicated knockout mice (n = 10 mice/group; *, P < 0.05). (E to G) In parallel, tissue H&E staining (E), serum enzyme activity (F), serum lactate (G), IL1B (G), and HMGB1 (G) were assayed (n = 3-5 mice/group; *, P < 0.05 vs. pink1−/− or park2−/− group).
To test this possibility, we examined the impact of dopamine inhibition on the expression of HIF1A, a transcriptional activator responsible for regulating cellular and systemic aerobic glycolysis during inflammation.26 Administration of PRA suppressed the sepsis-induced upregulation of Hif1a mRNA expression, as well as that of its target genes (e.g., Ldha and Pdk1) in the brain, skeletal muscle, and peritoneal macrophages from pink1−/− and park2−/− mice (Fig. 4B). Reactive oxygen species have been implicated as important signaling molecules to enhance Hif1a mRNA expression and activity, 27 as well as inflammasome activation.28 PRA displays antioxidant activity in neuroprotection through increased glutathione (GSH) production.29 Similarly, administration of PRA suppressed the sepsis-induced GSH depletion in the brain from pink1−/− and park2−/− mice (Fig. 4C), suggesting that dopamine inhibits HIF1A signaling through regulation of GSH production.
To confirm whether HIF1A-mediated aerobic glycolysis contributes to lethal sepsis in pink1−/− and park2−/− mice, we generated double-knockout mice (pink1−/− hif1a−/− and park2−/− hif1a−/−). The disruption of Hif1a attenuated the oversensitivity of pink1−/− or park2−/− mice to lethal sepsis, as manifested by increased animal survival (Fig. 4D) and decreased tissue injury judged by histological improvement (Fig. 4E) and reduction of tissue enzyme release (Fig. 4F). Importantly, serum levels of lactate, IL1B, and HMGB1 were significantly decreased in pink1−/− hif1a−/− and park2−/− hif1a−/− mice after CLP as compared to respective pink1−/− and park2−/− mice (Fig. 4G). These data suggest that HIF1A is required for the enhanced glycolytic phonotype and inflammasome activation caused by the loss of pink1 or park2.
Gene changes in the PINK1- and PARK2-dependent neuroimmune pathways in human sepsis
Although the murine CLP paradigm has some features that warrant its recommendation, this model of severe sepsis has some flaws and cannot fully mimic human sepsis.9 We next determined whether the PINK1-PARK2-HIF1A-NLRP3 pathway changes in peripheral blood mononuclear cells (PBMCs) of patients with sepsis. Compared with a healthy control group, the mRNA levels of HIF1A (Fig. 5A) and NLRP3 (Fig. 5B) in PBMCs were increased in the sepsis group. In contrast, the mRNA levels of PINK1 (Fig. 5C) and PARK2 (Fig. 5D) in PBMCs were decreased in the sepsis group compared with the healthy control group (Table S1). These findings indicate a potential pathogenic role of the PINK1-PARK2-HIF1A-NLRP3 pathway in human sepsis.
Figure 5.

Gene changes in the PINK1- and PARK2-dependent neuroimmune pathways in human sepsis. (A to D) Box plots comparing measures of HIF1A (A), NLRP3 (B), PINK1 (C), and PARK2 (D) mRNA levels in PBMC samples of sepsis patients (n = 10) and healthy controls (n = 10). The mRNA levels are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line). *, P < 0.05 versus control group. (E) Schematic depicting PINK1- and PARK2-dependent protective neuroimmune pathways in lethal sepsis.
Discussion
Increasing evidence has supported the important role of the nervous system and neurotransmitters in the regulation of the systemic inflammatory response to infection and injury.30 Here, we demonstrate that disruption of the PINK1-PARK2 pathway exacerbates polymicrobial sepsis partly through impairing the release of the neurotransmitter dopamine. Conversely, dopamine limits the systemic inflammatory response through interfering with the immunometabolism pathway, including HIF1A-dependent anaerobic glycolysis and lactate production, which contributes to NLRP3-dependent inflammasome activation and subsequent HMGB1 release (Fig. 5E). Thus, neuroimmune signaling pathways could represent novel therapeutic targets for the treatment of sepsis and other lethal systemic inflammatory diseases.
As key regulators of the mitochondrial quality control pathway, PINK1 and PARK2 are critically involved in the regulation of mitochondrial dynamics in the brain and other organs. Genetic mutations of the PINK1 and PARK2 genes in humans are associated with early-onset of PD, a neurodegenerative disease characterized by the loss of dopaminergic neurons.6 In mice, pink1 deficiency worsens aging-associated lung fibrosis, possibly through accelerating mitochondrial dysfunction.31 Given the high prevalence of PD and sepsis in elderly patients,32 as well as the currently observed exacerbation of septic lethality in pink1- and park2-deficient mice, it may be important to investigate whether PD patients also exhibit worse sepsis outcomes.
Current medical practice guidelines recommend the use of dopamine as a first-line drug to treat hypotension (low blood pressure), low cardiac output, and reduced perfusion of body organs due to shock, trauma, and sepsis.33 However, dopamine therapy suffers from a relatively short half-life (<2 min in serum) and adverse side effects, particularly for patients with septic shock.34 In contrast, dopamine agonists have much longer half-lives (usually 6 to 12 h in serum),13 but have not yet been tested for efficacy in experimental sepsis. In this study, we demonstrated that PRA, a dopamine agonist widely used to treat PD, confers protection against lethal sepsis in pink1−/− and park2−/− mice. These findings support the potential use of dopamine agonist in the treatment of sepsis and other lethal systemic inflammatory diseases.
Dopamine does not cross the intact blood brain barrier and additional dopaminergic neurons, such as those in the adrenal medulla and mesentery, exist in the peripheral nervous system.35 In addition to its central role in the regulation of motor function, peripheral dopamine may also serve as an endogenous regulator of immune functions. Consistent with a recent report,22 we found that the dopamine agonist PRA inhibited NLRP3 inflammasome activation. Although PRA can activate D2-like receptors with low affinity for the 5-hydroxytryptamine and α2-adrenergic receptors, the functional contribution of dopamine D1-like receptors (DRD1 and DRD5) or D2-like receptors (DRD2, DRD3, and DRD4) to PRA-induced NLRP3 inflammasome inhibition remains unknown. Nevertheless, the essential role of NLRP3 inflammasome activation in worsening the septic lethality of pink1−/− or park2−/− mice has been supported by the results from the current study.
HMGB1 is actively secreted by activated innate immune cells during infection and passively released by necrotic cells during injury.36 We observed that the release of HMGB1 is significantly increased in septic pink1−/− or park2−/− mice compared with septic WT controls. The regulatory role of the neuroimmune network in the regulation of HMGB1 release has been supported by our findings that pharmacological dopamine receptor activation (by PRA), NLRP3 inflammasome inactivation (by MCC950), or genetic Nlrp3 deletion concurrently attenuated lethal sepsis and systemic HMGB1 accumulation in pink1−/− or park2−/− mice. Importantly, delayed administration of HMGB1-specific antibody or inhibitor (e.g., glycyrrhizin19) protects against animal death in CLP-induced pink1−/− or park2−/− mice, confirming HMGB1 as a late mediator of lethal systemic inflammation.37
Elevated lactate production, hyperlactatemia, often occurs in patients with severe sepsis or septic shock partly due to anaerobic glycolysis.38 The anaerobic glycolysis is regulated by a major transcription factor, HIF1A, which is upregulated during sepsis at both the mRNA and protein levels. We found that absence of dopamine in pink1−/− or park2−/− mice resulted in HIF1A-mediated aerobic glycolysis induction, although the mechanisms remain unknown. In contrast, PRA impaired sepsis-induced upregulation of HIF1A and several glycolysis-associated genes in the brain, skeletal muscle, and peritoneal macrophages of pink1−/− or park2−/− mice, suggesting that neurotransmitters participate in the counter-regulation of systemic HIF1A expression during sepsis.
It has been shown that Hif1a deficiency renders mice resistant to lethal endotoxemia39 and knockdown of Hif1a by RNAi inhibits glucose metabolism in fibroblasts from pink1−/− mice.40 Here, we provide evidence that HIF1A-mediated aerobic glycolysis may contribute to sepsis-induced NLRP3 inflammasome activation and HMGB1 release in pink1−/− or park2−/− mice. This observation was consistent with our previous findings that inhibition of PKM2 (pyruvate kinase, muscle), a transcriptional target and transcriptional coactivator of HIF1A, concomitantly reduced aerobic glycolysis, HMGB1 release, and septic lethality.24,41 Given the essential role of the PKM2-HIF1A metabolic pathway in controlling IL1B production by activated macrophages,42 it is possible that the HIF1A-dependent immunometabolic dysfunction might be a hallmark of sepsis.
In summary, understanding the complex pathogenesis of sepsis, a complex systemic response to injury and infection, may be an important first step in improving outcomes. Our study has revealed a novel PINK1- and PARK2-dependent neuroimmune pathway that regulates peripheral inflammation through controlling dopamine release, HIF1A and NLRP3 inflammasome activation, and HMGB1 secretion in a mouse model. This neuroimmune pathway orchestrates metabolic and inflammatory responses, leading to hyperlactatemia, tissue injury, and even organ dysfunction. Human monocytes from patients during sepsis display increased HIF1A and target gene expression, which cause immune suppression.43 Persistent elevation of HMGB1 in patients with severe sepsis and septic shock may prolong inflammation and organ injury.44 In addition, uncoupling protein-2 (the positive regulator of NLRP3 inflammasome activation) is significantly increased in myeloid cells of patients with sepsis.45 We observed that the mRNA expression of HIF1A and NLRP3 is upregulated, whereas the mRNA expression of PINK1 and PARK2 is downregulated in PBMCs of patients with sepsis. Thus, the PINK1-PARK2-HIF1A-NLRP3-HMGB1 pathway might represent a novel therapeutic target for future clinical management of sepsis and other lethal inflammatory diseases.
Materials and methods
Reagents
We purchased PRA (Selleck Chemicals, S2011), glycyrrhizin (Sigma, CDS020796), MCC950 (AdipoGen, AG-CR1-3615), and monoclonal IL1B/IL-1β-neutralizing antibody (Clone B122; BioLegend, 503501) from the indicated manufacturers. Monoclonal HMGB1-neutralizing antibody (Clone 2G7) was generated as reported previously.46
Mice
pink1−/−, park2−/−, nlrp3−/−, and hif1a−/− mice were all in the C57BL/6 background and purchased from Jackson Laboratories. These mice were crossed to generate the indicated double-knockout animals. All mice were housed on a 12-h light-dark cycle with controlled temperature (21°C to 23°C) and provided with standard rodent diet and water ad libitum throughout all experiments. Isolation and culture of adult mouse neurons were performed as previously described.47 We conducted all animal care and experimentation in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org) and with approval from the Institutional Animal Care and Use Committees from the University of Pittsburgh and Third Military Medical University.
CLP procedure
Sepsis was induced in male or female C57BL/6 mice (8- to 10-wk-old, 22 to 26 g weight) by CLP as previously described.48 Briefly, anesthesia was induced with 5% halothane and maintained with 2% halothane. A small midline abdominal incision was made and the cecum was exteriorized and ligated with 4–0 silk immediately distal to the ileocecal valve without causing intestinal obstruction. The cecum was then punctured once with a 22-gauge needle. The abdomen was closed in 2 layers and the mice were injected subcutaneously with 1 ml Ringer solution including analgesia (0.05 mg/kg buprenorphine). After CLP, mice did not receive antibiotics. The mortality of WT mice typically was approximately 40% by d 7 post-CLP.
ELISA analysis
The GPT/ALT (Bioo Scientific Corporation, 3460-01), TNNI/troponin-I (Life Diagnostics, CTNI-1-US), creatinine (Bioo Scientific Corporation, 5606-01), AMY2/amylase (Abcam, ab102523), MPO (Abcam, ab155458), ATP (PerkinElmer, 6016736), LDH (Abcam, ab102526), dopamine (NOVUS, KA1887), IL1B/IL-1β (BioLegend, 433404), TNF/TNFα (BioLegend, 430907), IL6/IL-6 (BioLegend, 431307), HMGB1 (Shino-Test Corporation, 326054329), CIRBP/CIRP (CUSABIO, P60824), and lactate (Abcam, ab65331) concentrations in serum and/or supernatant fractions from the indicated tissue homogenate were measured using ELISA according to the manufacturer's protocol.
CASP3 activity assay
The activity of CASP3 in cell lysates was assayed by the CASP3 Activity Assay Kit (Cell Signaling Technology, 5723) according to the manufacturer's protocol. It contained a fluorogenic substrate (N-Acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin or Ac-DEVDAMC) for CASP3. During the assay, activated CASP3 cleaved this substrate between DEVD and AMC, generating highly fluorescent AMC that can be detected using a fluorescence reader with excitation at 380 nm and emission between 420-460 nm.
GSH assay
The relative GSH concentration in tissue lysates was assessed using a Glutathione Assay Kit (Sigma, CS0260) according to the manufacturer's instructions.49,50 The measurement of GSH used a kinetic assay in which catalytic amounts (nmoles) of GSH caused a continuous reduction of 5,5′-dithiobis (2-nitrobenzoic acid) to 5-thio-2-nitrobenzoic acid and the GSSG formed was recycled by glutathione reductase and NADPH. The reaction rate was proportional to the concentration of glutathione up to 2 mM. The yellow product (5-thio-2-nitrobenzoic acid) was measured spectrophotometrically at 412 nm.
Measurement of bacterial counts
The peritoneal cavity was washed with 1 ml phosphate-buffered saline (Sigma, P3813) and the peritoneal lavage was collected under sterile conditions. Samples of blood and peritoneal lavage fluid were serially 10-fold diluted in sterile saline and cultured on tryptic soy agar pour plates (BD, BA-256665.02). Plates were incubated (37°C) for 24 to 48 h and colony counts were performed as described previously.51
Quantitative real-time polymerase chain reaction
Total RNA was extracted using TRI reagent (Sigma, T9424) according to the manufacturer's instructions. First-strand cDNA was synthesized from 1 µg of RNA using the iScript cDNA Synthesis kit (Bio-Rad, 1708890). Briefly, 20 μl reactions were prepared by combining 4 μl iScript Select reaction mix, 2 μl gene-specific enhancer solution, 1 μl reverse transcriptase, 1 μl gene-specific assay pool (20×, 2 μM), 12 μl RNA diluted in RNase-free water. cDNA from various cell samples were then amplified by real-time quantitative PCR with specific primers (mouse Hif1α: 5′-ACCTTCATCGGAAACTCCAAAG-3′ and 5′-CTGTTAGGCTGGGAAAAGTTAGG-3′; mouse Ldha: 5′-GCTCCCCAGAACAAGATTACAG-3′ and 5′-TCGCCCTTGAGTTTGTCTTC-3′; mouse Pdk1: 5′-CCACTGAGGAAGATCGACAGAC-3′ and 5′-AGAGGCGTGATATGGGCAATCC-3′; mouse Pah: 5′-GCTGGACAGATTCGCCAATCAG-3′ and 5′-CAGCAAACTGCTTTCGTCTCGC-3′; mouse Th: 5′-TGCACACAGTACATCCGTCATGC-3′ and 5′- GCAAATGTGCGGTCAGCCAACA-3′; mouse Ddc: 5′-GGAGCCAGAAACATACGAGGAC-3′ and 5′- GCATGTCTGCAAGCATAGCTGG-3′; human HIF1A: 5′-TATGAGCCAGAAGAACTTTTAGGC-3′ and 5′-CACCTCTTTTGGCAAGCATCCTG-3′; human NLRP3: 5′- GGACTGAAGCACCTGTTGTGCA-3′ and 5′- TCCTGAGTCTCCCAAGGCATTC-3′; human PINK1: 5′- GTGGACCATCTGGTTCAACAGG-3′ and 5′- GCAGCCAAAATCTGCGATCACC-3′; human PARK2: 5′- CCAGAGGAAAGTCACCTGCGAA-3′ and 5′- CTGAGGCTTCAAATACGGCACTG-3′) and the data were normalized to mouse Rn18s rRNA (5′-CTTAGAGGGACAAGTGGCG-3′ and 5′-ACGCTGAGCCAGTCAGTGTA-3′) or human GADPH RNA (5′-GTCTCCTCTGACTTCAACAGCG-3′ and 5′-ACCACCCTGTTGCTGTAGCCAA-3′) using an CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, 1855195).
Patient samples
PBMCs from patients with sepsis and healthy controls were collected from Xiangya Hospital, Central South University. Collection of the samples was approved by Xiangya Hospital's Institutional Review Board. Sepsis was identified according to the 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference guidelines.52
Statistical analysis
Data are expressed as means ± SEM of 3 independent experiments. All data were analyzed using SigmaPlot11.0 software. One-way ANOVA was used for comparison among the different groups. When ANOVA was significant, post hoc testing of differences between groups was performed using a least significant difference test. The Kaplan-Meier method was used to compare the differences in mortality rates between groups. A P value < 0.05 was considered statistically significant.
Supplementary Material
Abbreviations
- AMY2
amylase
- CIRBP
cold inducible RNA binding protein
- CLP
cecal ligation and puncture
- Ddc
dopa decarboxylase
- DRD
dopamine receptor D1
- GPT/ALT
glutamic pyruvic transaminase, soluble
- GSH
glutathione
- HIF1A
hypoxia inducible factor 1, α subunit
- HMGB1
high mobility group box 1
- IL
interleukin; LDH, lactate dehydrogenase
- MPO
myeloperoxidase
- NLRP3
NLR family, pyrin domain containing 3
- Pah
phenylalanine hydroxylase
- PARK2
parkin RBR E3 ubiquitin protein ligase
- PBMC
peripheral blood mononuclear cell
- PD
Parkinson disease
- PINK1
PTEN induced putative kinase 1
- PKM2
pyruvate kinase, muscle
- PRA
pramipexole
- Th
tyrosine hydroxylase
- TNF/TNF-α
tumor necrosis factor
- TNNI
troponin I
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. We thank Dr. Kate Fitzgerald for providing reagents for this study.
Funding
This work was supported by the National Institutes of Health of USA (R01GM115366 and R01CA160417 to D.T; R01AT005076 and R01GM063075 to H.W; R01GM44100 to T.B; and R01GM053396 to D.J.K.), the Natural Science Foundation of Guangdong Province (2016A030308011 to D.T.), and a grant from The National Key Technology R&D Program in China (2012BAI11B01 to J.J.). This project partly utilized University of Pittsburgh Cancer Institute shared resources supported by award P30CA047904.
ORCID
Daniel J. Klionsky http://orcid.org/0000-0002-7828-8118
References
- [1].Angus DC, van der Poll T. Severe sepsis and septic shock. New Engl J Med 2013; 369:840-51; PMID:23984731; http://dx.doi.org/ 10.1056/NEJMra1208623 [DOI] [PubMed] [Google Scholar]
- [2].Freund HR, Muggiasullam M, Peiser J, Melamed E. Brain neurotransmitter profile is deranged during sepsis and septic encephalopathy in the rat. J Surg Res 1985; 38:267-71; PMID:2858604; http://dx.doi.org/ 10.1016/0022-4804(85)90037-X [DOI] [PubMed] [Google Scholar]
- [3].Deutschman CS, Tracey KJ. Sepsis: Current dogma and new perspectives. Immunity 2014; 40:464-76; http://dx.doi.org/ 10.1016/j.immuni.2014.04.001 [DOI] [PubMed] [Google Scholar]
- [4].Mathis D, Shoelson SE. Foreword immunometabolism: an emerging frontier. Nat Rev Immunol 2011; 11:81-3; PMID:21469396; http://dx.doi.org/ 10.1038/nri2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS, Angus DC, Rubenfeld GD, Singer M. Developing a new definition and assessing new clinical criteria for septic shock: For the third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama 2016; 315:775-87; PMID:26903336; http://dx.doi.org/ 10.1001/jama.2016.0289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson disease. Neuron 2015; 85:257-73; PMID:25611507; http://dx.doi.org/ 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/ 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- [8].Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/ 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov 2005; 4:854-65; PMID:16224456; http://dx.doi.org/ 10.1038/nrd1854 [DOI] [PubMed] [Google Scholar]
- [10].Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 2014; 206:655-70; PMID:25154397; http://dx.doi.org/ 10.1083/jcb.201401070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al.. PINK1-associated Parkinson disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 2009; 33:627-38; PMID:19285945; http://dx.doi.org/ 10.1016/j.molcel.2009.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013; 501:512-6; PMID:24005326; http://dx.doi.org/ 10.1038/nature12566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Antonini A, Barone P, Ceravolo R, Fabbrini G, Tinazzi M, Abbruzzese G. Role of pramipexole in the management of Parkinson disease. CNS Drugs 2010; 24:829-41; PMID:20839895; http://dx.doi.org/ 10.2165/11585090-000000000-00000 [DOI] [PubMed] [Google Scholar]
- [14].Dinarello CA, Thompson RC. Blocking IL-1: interleukin 1 receptor antagonist in vivo and in vitro. Immunol Today 1991; 12:404-10; PMID:1838480; http://dx.doi.org/ 10.1016/0167-5699(91)90142-G [DOI] [PubMed] [Google Scholar]
- [15].Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 1987; 330:662-4; PMID:3317066; http://dx.doi.org/ 10.1038/330662a0 [DOI] [PubMed] [Google Scholar]
- [16].Wang P, Ba ZF, Chaudry IH. Mechanism of hepatocellular dysfunction during early sepsis. Key role of increased gene expression and release of proinflammatory cytokines tumor necrosis factor and interleukin-6. Arch Surg 1997; 132:364-9; discussion 9-70; PMID:9108756; http://dx.doi.org/ 10.1001/archsurg.1997.01430280038005 [DOI] [PubMed] [Google Scholar]
- [17].ReferencesWang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al.. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999; 285:248-51; PMID:10398600; http://dx.doi.org/ 10.1126/science.285.5425.248 [DOI] [PubMed] [Google Scholar]
- [18].Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, et al.. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med 2013; 19:1489-95; PMID:24097189; http://dx.doi.org/ 10.1038/nm.3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, Agresti A, Trisciuoglio L, Musco G, Bianchi ME. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol 2007; 14:431-41; PMID:17462578; http://dx.doi.org/ 10.1016/j.chembiol.2007.03.007 [DOI] [PubMed] [Google Scholar]
- [20].Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014; 157:1013-22; PMID:24855941; http://dx.doi.org/ 10.1016/j.cell.2014.04.007 [DOI] [PubMed] [Google Scholar]
- [21].Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21:677-87; PMID:26121197; http://dx.doi.org/ 10.1038/nm.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015; 160:62-73; PMID:25594175; http://dx.doi.org/ 10.1016/j.cell.2014.11.047 [DOI] [PubMed] [Google Scholar]
- [23].Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, et al.. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21:248-55; PMID:25686105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, Kang R, Lotze MT, Billiar TR, Wang H, et al.. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun 2014; 5:4436; PMID:25019241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al.. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013; 496:238-42; PMID:23535595; http://dx.doi.org/ 10.1038/nature11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al.. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003; 112:645-57; PMID:12628185; http://dx.doi.org/ 10.1016/S0092-8674(03)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arteriosclerosis Thrombosis Vascular Biol 2007; 27:755-61; PMID:17272744; http://dx.doi.org/ 10.1161/01.ATV.0000258979.92828.bc [DOI] [PubMed] [Google Scholar]
- [28].Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221-5; PMID:21124315; http://dx.doi.org/ 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- [29].Le WD, Jankovic J, Xie W, Appel SH. Antioxidant property of pramipexole independent of dopamine receptor activation in neuroprotection. J Neural Transm (Vienna) 2000; 107:1165-73; PMID:11129106; http://dx.doi.org/ 10.1007/s007020070030 [DOI] [PubMed] [Google Scholar]
- [30].Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med 2012; 209:1057-68; PMID:22665702; http://dx.doi.org/ 10.1084/jem.20120571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, et al.. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 2015; 125:521-38; PMID:25562319; http://dx.doi.org/ 10.1172/JCI74942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Powe NR, Jaar B, Furth SL, Hermann J, Briggs W. Septicemia in dialysis patients: incidence, risk factors, and prognosis. Kidney Int 1999; 55:1081-90; PMID:10027947; http://dx.doi.org/ 10.1046/j.1523-1755.1999.0550031081.x [DOI] [PubMed] [Google Scholar]
- [33].Herget-Rosenthal S, Saner F, Chawla LS. Approach to hemodynamic shock and vasopressors. Clin J Am Soc Nephrol: CJASN 2008; 3:546-53; PMID:18256381; http://dx.doi.org/ 10.2215/CJN.01820407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis*. Critical Care Med 2012; 40:725-30; PMID:22036860; http://dx.doi.org/ 10.1097/CCM.0b013e31823778ee [DOI] [PubMed] [Google Scholar]
- [35].Tarazi FI. Neuropharmacology of dopamine receptors:: Implications in neuropsychiatric diseases. J Scientific Res Medical Sci / Sultan Qaboos University 2001; 3:93-104 [PMC free article] [PubMed] [Google Scholar]
- [36].Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, et al.. HMGB1 in health and disease. Mol Aspects Med 2014; 40:1-116; PMID:25010388; http://dx.doi.org/ 10.1016/j.mam.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Critical Care Med 2001; 164:1768-73; PMID:11734424; http://dx.doi.org/ 10.1164/ajrccm.164.10.2106117 [DOI] [PubMed] [Google Scholar]
- [38].Garcia-Alvarez M, Marik P, Bellomo R. Sepsis-associated hyperlactatemia. Crit Care 2014; 18:503; PMID:25394679; http://dx.doi.org/ 10.1186/s13054-014-0503-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 2007; 178:7516-9; PMID:17548584; http://dx.doi.org/ 10.4049/jimmunol.178.12.7516 [DOI] [PubMed] [Google Scholar]
- [40].Requejo-Aguilar R, Lopez-Fabuel I, Fernandez E, Martins LM, Almeida A, Bolanos JP. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nature Commun 2014; 5:4514; PMID:25058378; http://dx.doi.org/ 10.1038/ncomms5514 [DOI] [PubMed] [Google Scholar]
- [41].Zhang Z, Deng W, Kang R, Xie M, Billiar T, Wang H, Cao L, Tang D. Plumbagin protects mice from lethal sepsis by modulating immunometabolism upstream of PKM2. Mol Med 2016; 22:162-172; PMID:26982513; http://dx.dox.org/25565206 10.2119/molmed.2015.00250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, et al.. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 2015; 21:65-80; PMID:25565206; http://dx.doi.org/ 10.1016/j.cmet.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernandez-Jimenez E, Toledano V, Cubillos-Zapata C, Rapisarda A, Chen J, et al.. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity 2015; 42:484-98; PMID:25746953; http://dx.doi.org/ 10.1016/j.immuni.2015.02.001 [DOI] [PubMed] [Google Scholar]
- [44].Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey KJ, Lee ML, Andersson J, Tokics L, Treutiger CJ. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Critical Care Med 2005; 33:564-73; PMID:15753748; http://dx.doi.org/ 10.1097/01.CCM.0000155991.88802.4D [DOI] [PubMed] [Google Scholar]
- [45].Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, Yun M, Kim CK, Howrylak J, Ryter SW, et al.. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 2015; 125:665-80; PMID:25574840; http://dx.doi.org/ 10.1172/JCI78253 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [46].Qin SX, Wang HC, Yuan RQ, Li H, Ochani M, Ochani K, Rosas-Ballina M, Czura CJ, Huston JM, Miller E, et al.. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med 2006; 203:1637-42; PMID:16818669; http://dx.doi.org/ 10.1084/jem.20052203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protocols 2007; 2:1490-8; PMID:17545985; http://dx.doi.org/ 10.1038/nprot.2007.207 [DOI] [PubMed] [Google Scholar]
- [48].Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 2009; 4:31-6; PMID:19131954; http://dx.doi.org/ 10.1038/nprot.2008.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016; 64:488-500; PMID:27015352; http://dx.doi.org/ 10.1002/hep.28574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016; 63:173-84; PMID:26403645; http://dx.doi.org/ 10.1002/hep.28251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Deng MH, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, Hackam D, Billiar TR. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during Sepsis. J Immunol 2013; 190:5152-60; http://dx.doi.org/ 10.4049/jimmunol.1300496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Critical Care Med 2003; 31:1250-6; PMID:12682500; http://dx.doi.org/ 10.1097/01.CCM.0000050454.01978.3B [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.