

Abstract
The human gastric pathogen Helicobacter pylori attaches to antral epithelial cells in vivo. Cultured human antral epithelial cells, AGS and NCI-N87 cell lines, were grown in the absence or presence of H. pylori and compared with respect to gene transcript levels, protein expression, organization of the actin cytoskeleton, and the regulation of cell migration. The Clontech Neurobiology array detected differentially expressed transcripts, while Western blots were used to investigate related changes in protein levels. Infection with H. pylori consistently upregulated annexin II, S100 A7, Rho-GTP, and IQGAP-1, whereas SSTR-1 was downregulated upon H. pylori infection. In the adherens junction, E-cadherin and IQGAP-1 were translocated from the plasma membrane to intracellular vesicles. The primary and NCI-N87 cells were similar with respect to cell-cell and cell-matrix adhesion and cell migratory behavior; in contrast the AGS cells were significantly different from the primary gastric epithelial cell preparations, and thus caution must be used when using this cell line for studies of gastric disease. These studies demonstrate a correlation between H. pylori infection and alterations to epithelial cell adhesion molecules, including increased levels of Rho-GTP and cell migration. These data indicate that destabilizing epithelial cell adherence is one of the factors increasing the risk of H. pylori-infected individuals developing gastric cancer.
Helicobacter pylori is a spiral, gram-negative rod that attaches specifically to the gastric epithelial cells lining the antrum of the stomach (2). H. pylori is able to withstand the hostile environment of the stomach by secretion of urease buffers, which neutralize the pH of its immediate surroundings. Flagella allow these highly motile bacteria to cross the mucous lining of the stomach and to attach to the apical surface of the mucosal epithelial cells. These antral epithelial cells are linked together at the apical surface by a system of interacting proteins that comprise the tight and adherens junctions (12, 31). These junctions effectively seal off the lumen of the stomach, preventing access of gastric acid and pathogens to the interstitial space and, hence, to the general circulation.
Individuals infected by H. pylori have increased gastrin levels and decreased levels of somatostatin (SST) hormones that regulate gastric acid secretion. As a result, infected individuals develop mucosal gastritis, increasing their risk of ulceration and, in the longer term, gastric cancer (10, 11). Gastric adenocarcinomas show characteristic changes in the expression of E-cadherin, a transmembrane protein forming the core of the adherens junction. Loss of the E-cadherin complex at the apical pole is thought to induce loss of cell-cell adhesion (34). Direct evidence of E-cadherin mutations triggering tumorigenesis has come from recent studies linking inactivating germ line mutations of the E-cadherin gene (CDH1) in hereditary diffuse gastric cancer (33). Furthermore, it has been shown that the degree of loss or dysfunction of E-cadherin is proportional to the migratory behavior of the tumor cells and hence metastatic potential (18). Modulation of cell-cell adhesion involves the cadherin-catenin complex. IQGAP-1 induces the dissociation of α-catenin from this complex through signal transduction pathways involving the small Rho GTPases (21). Previous studies investigating the effects of H. pylori infection on the gastric epithelium have used the gastric epithelial cell line AGS. However, the AGS cell line does not form a polarized epithelium and does not express the critical adherens junction protein, E-cadherin (18), resulting in a lack of tight junction formation (1). Previous studies of H. pylori infection using AGS cells have indicated a role for members of the Rho family of GTPases in cell migration. Members of this family act as molecular switches that control a large array of signaling events in a temporal and spatial manner (23); in particular, at the leading edge of migrating cells they regulate the extension of lamellipodia and filopodia and the formation of new adhesions during cell migration (24). While it is established that Rho GTPases have an important role to play in cell migration, it remains unclear what effect these molecules have on the migration of cells with established tight and adherens junctions at the apical pole, such as primary gastric cell preparations and the NCI-N87 cell line.
Using primary epithelial cell culture preparations with fully functional adherens junctions, we confirmed that H. pylori preferentially attaches at cell-cell interfaces (15). Research using the AGS cell line has provided conflicting evidence, with bacterial attachment either directed toward cell-cell interfaces (1, 2) or occurring in a random pattern over the cell surface and not confined to cell junctions (32). For several years we have been using the gastric epithelial cell line NCI-N87 alongside our primary epithelial cell culture preparations to mimic bacterial adhesion to epithelia in vivo. The suitability of NCI-N87 cells as a model system has been previously indicated by comparison of Western blot and reverse transcription-PCR (RT-PCR) data from the AGS, KATO III, Hs746t, and NCI-N87 cell lines (6). Bacterial adhesion to epithelia has been previously shown to involve the S100 proteins forming complexes with members of the annexin family of calcium-binding proteins (14). The focus of the present study was to determine how infection of primary epithelial cell preparations and cultured gastric cells with a virulent form of H. pylori modulated expression levels of genes encoding G protein-coupled receptors (GPCRs), cell adhesion molecules, and intracellular signaling molecules. Experiments were performed to determine whether such changes in gene expression levels affected cellular function and migratory capabilities.
MATERIALS AND METHODS
Cell culture.
Human antrum was obtained from six multiple organ donors (four males and two females, ages 24 to 50 years) in association with the British Columbia Transplant Association, with ethical approval by the University of British Columbia Clinical Screening Committee. To test for a prior infection by H. pylori, before digestion, a small piece of antral mucosa was placed in rapid urea medium (PML Microbiologicals) and incubated at 37°C. After 24 h of incubation, the tube was observed for a color change indicative of urea digestion. None of the organ donors had a preexisting infection by H. pylori. A single-cell suspension of mucosal cells was prepared, separated by centrifugal elutriation, and cultured as described previously (8). The F2 fraction of the antral cells was plated into either 6-well Costar/Corning cell culture plates at a density of 106/ml at 2 ml/well or 24-well plates with 120-mm-diameter 3-aminopropyltriethoxy silane solution (APES)-coated glass coverslips at 1 ml/well in growth medium (50:50 F10 and Dulbecco's modified Eagle medium supplemented with 5% fetal bovine serum, 10 μg of penicillin/ml, 10 μg of streptomycin/ml, 2 mM glutamine, and 8 μg of insulin/ml). The gastric cell lines AGS and NCI-N87 (American Type Culture Collection cell bank) were grown in the same medium. The plates were maintained in 5% CO2 at 37°C for 48 h prior to either collection of mRNA or protein or fixation for immunocytochemical studies.
Bacterial cell culture.
Wild-type H. pylori strain G27 (11) was grown from frozen stocks on horse blood agar plates containing 5% horse blood and the antibiotics trimethoprim (10 μg/ml), cefsulodin (6 μg/ml), and vancomycin (5 μg/ml) and cultured under microaerophilic conditions at 37°C for 72 h. The bacteria were then subcultured in brucella broth (Difco) supplemented with 10% fetal calf serum (FCS; Gibco) and 1% IsoVitaleX (Becton Dickinson, Cockeysville, Md.). The culture was incubated with shaking under microaerophilic conditions in a CampyPak jar (Becton Dickinson) at 37°C for 24 h. NCI-N87 cells were infected with Helicobacter felis to determine if alterations in mRNA expression were a generalized phenomenon in response to a bacterial infection. H. felis (a gift from AstraZeneca, Boston, Mass.) was grown on Mueller-Hinton blood plates (5% horse blood), cultured under microaerophilic conditions at 37°C for 96 h, and subcultured in Brucella broth minus bisulfite medium supplemented with 5% FCS (Gibco). Infection protocols were as described above for H. pylori.
Inoculation of epithelial cells with H. pylori and H. felis.
Before inoculation the human cell cultures were washed in sterile phosphate-buffered saline (PBS) twice to remove nonadherent cells and antibiotics, and then 1 ml of fresh growth medium (without penicillin and streptomycin) containing the appropriate amount and strain of bacteria was added. Following growth of the bacteria overnight in brucella broth the bacteria were washed in sterile PBS and an optical density reading at 600 nm (OD600) was taken. Ten microliters of the bacteria (5 μl of H. felis) with an OD600 of 0.5 was used to inoculate 1 ml of growth medium. The infected epithelial cells were maintained in 10% CO2 at 37°C for 48 h prior to either collection of mRNA or protein or fixation for immunocytochemical studies. In one series of experiments, cells were infected with heat-killed H. pylori to confirm that viable bacteria were required to induce changes in mRNA expression. Heat-killed H. pylori (G27) cells were prepared by incubating the bacteria in a 56°C water bath for 30 min, chilling them on ice, and then incubating them in an 80°C water bath for a further 10 min. NCI-N87 cells were infected with 10 μl of bacteria at an OD600 of 0.5. A dilution of the bacteria was plated onto a blood plate and maintained in 10% CO2 at 37°C for several days to verify that the bacteria were dead.
Collection of RNA and protein.
All the cells were harvested with Trizol (Gibco/BRL). Total RNA and protein were collected from the Trizol suspensions by the supplied protocols. RNA samples were resuspended in diethyl pyrocarbonate-treated distilled water (dH2O), aliquoted, and stored at −70°C. The integrity of the RNA was verified by ethidium bromide visualization following electrophoresis through an agarose gel. Protein pellets were resuspended in 1% sodium dodecyl sulfate (SDS) in dH2O and stored at −20°C.
Gene array analysis.
The human neurobiology arrays were obtained from Clontech. Five micrograms of total RNA was used to synthesize cDNA. RNA was first treated with DNase, and then the RNA was reverse transcribed into cDNA with Superscript II reverse transcriptase (Gibco/BRL) with the Clontech specific primer mixture and [α-32P]dATP according to the manufacturer's directions. Hybridized filters were placed in a Molecular Dynamics phosphor screen cassette for 3 days prior to development of the image. The intensity of the spots was determined with the Clontech software package.
Analysis of the arrays involved normalizing for interarray variation with the housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. After background subtraction and normalization, all genes below the level of detection of the array (density values < 0) were assigned a value of 20 to facilitate interarray analyses. In all cases, arrays were run in pairs of a control sample (uninfected) with the corresponding infected sample. To confirm the presence of virulent bacteria in the cultures, a prescreen of the mRNA by RT-PCR for urease expression was performed. The expression pattern of six different primary cell preparations (four from males and two from females, ages 24 to 50 years) was analyzed. The expression pattern of the gastric cell lines was determined by analysis of three arrays prepared with mRNA from different cell preparations but the same passage number. The data were expressed as the means of the ratios of infected to control, and statistical significance was assessed by Student's unpaired t test, with a P value of <0.05 considered significant.
Since small changes in mRNA levels can translate into large changes in the resultant protein levels, genes were considered to be significantly increased by H. pylori infection if they fulfilled one of the following criteria: absent in controls and expressed in infected cells or increased >1.3-fold above control levels with a P value of <0.05. A decrease in expression levels was defined as a level <75% of control levels and a P value of <0.05.
cDNA synthesis for RT-PCR.
First-strand cDNA was prepared from 5 μg of DNase-pretreated RNA in first-strand buffer (Gibco Life Technologies, Grand Island, N.Y.) according to manufacturer's instructions. A gene-specific primer (2 pmol) and random hexamers (250 ng; Gibco Life Technologies) were used to prime cDNA synthesis.
PCR analysis of gene expression.
Quantitative PCR was used to evaluate the relative difference in SSTR-1 and S100 A7 expression between infected and uninfected samples (since antibodies against these proteins were either unavailable or did not work well for Western blotting applications). This was done with Ambion's QuantumRNA 18S internal standard system. The PCRs were carried out with 2 μl of cDNA in a 25-μl total volume of PCR buffer (67 mM Tris [pH 9.01], 1.5 mM MgSO4, 166 mM [NH4]2SO4, 10 mM β-mercaptoethanol) containing 3 mM MgCl2, 0.2 mM deoxynucleoside triphosphates (Pharmacia), 50 ng of gene-specific primer, and 2 μl of 18S primer-competimer (competing primer). SSTR-1 primers had the following sequences: forward, 5′GGAGGAGCCGGTTGACTATT3′; reverse, 5′AAGGTAGCCTGAAAGCCTTCC3′. S100 A7 primers had the following sequences: forward, 5′GAAAGCAAAGATGAGCAACACTC3′; reverse, 5′TTGGTGGGGCTGGGTCACTG3′. The PCR products were separated by electrophoresis through a 1.2% agarose gel. The DNA was visualized and photographed with the Eagle Eye II video system (Stratagene). The band intensities were measured by computer-assisted densitometric analysis (Eagle Eye II; Stratagene). Ratios of the band intensity for the gene of interest to that for the internal standard for uninfected samples were compared to corresponding ratios for infected samples.
Northern blot analysis.
For uninfected and H. pylori-infected NCI-N87 samples, 25 ng of sequenced PCR product was loaded per lane on a 1% formaldehyde-agarose gel, electrophoresed for 1.5 h at 80 V, and transferred to a nylon membrane. Probes were generated according to the Strip EZ DNA protocol (Ambion). Probe hybridization and subsequent analysis were carried out as described for the Northern Max protocol (Ambion). Membrane exposure to film was for 2 h. GAPDH was used as the internal standard to ensure equal loading.
Western blot analysis.
Equal amounts of protein (range, 10 to 20 μg of total protein, as determined by bicinchoninic acid colorimetric assay; Pierce) from both control and infected cells were run in tandem on polyacrylamide electrophoresis gels of the appropriate densities and then transferred to nitrocellulose membranes with the Bio-Rad Mini-Protean system. Positive controls for the proteins were run at the same time whenever available. After being blocked in 5% milk protein the membranes were incubated with primary antibodies (in 5% heat-treated horse serum) against annexin II, annexin IV, E-cadherin, IQGAP-1 (BD Transduction Laboratories), and histone 2B (Chemicon). Binding was visualized with horseradish peroxidase-conjugated secondary antibodies (Jackson) against the primary antibodies and ECL (Amersham PharmaciaBiotech). After exposure and development the films (BioMax Light-1; Kodak) were scanned and the bands were quantified with ImageQuant software. Histone protein levels were used to normalize between samples.
Immunoprecipitation experiments.
The cells were lysed with lysis buffer (50 mM Tris [pH 7.5], 1% Triton X-100, 500 μl of 1× complete protease inhibitors [Roche]), and the lysates were incubated for 1 to 2 h at 4°C with or without mouse anti-E-cadherin (BD Transduction Laboratories), followed by a 0.5-h incubation with protein G beads (Amersham Pharmacia). The immunoprecipitates were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis.
Ca2+ switch.
Confluent NCI-N87 monolayers were trypsinized in the presence of EDTA until a single-cell suspension was obtained. Cells (primary and NCI-N87) were seeded at 4 × 105 on APES-coated glass coverslips in 24-well plates and allowed to attach in 50:50 F10-Dulbecco's modified Eagle medium containing 1 mM Ca2+ for 2 h. Cells were washed three times in 0 mM Ca2+ culture medium before fresh 0 mM Ca2+ culture medium was added. Medium was removed, and cells were immediately fixed in 4% paraformaldehyde at 30-min intervals over a 2-h period. Cells were then processed for immunofluorescence.
Immunocytochemistry.
Cells (primary and cell lines) were plated on APES-coated glass coverslips and incubated for 48 h to facilitate attachment prior to infection with H. pylori. Control and infected cells were fixed 48 h postinfection in 4% paraformaldehyde for 10 min at room temperature. The coverslips were washed in PBS and permeabilized with 0.1% Triton X-100 for 10 min at room temperature. Cells were then washed three times in PBS before incubation with the primary antibody. After 18 h at 4°C the primary antibodies were removed and the coverslips were washed three times in PBS prior to incubation in the relevant secondary antibody (AlexaFluor 488 and 594; Molecular Probes, Eugene, Oreg.) for 1 h at room temperature. After removal of the secondary antibody, the coverslips were washed three times in PBS and then mounted in Vectashield (Vector Laboratories, Burlington, Ontario, Canada) on glass slides and screened with a Zeiss Axiophot microscope at ×100. Representative cells were scanned with the Spot 2 camera, and the digitized image was converted with Adobe Photoshop, version 5.0, or imaged by deconvolution microscopy. Control incubations included incubation in the secondary antibody in the absence of primary antibodies and preabsorption of the antibodies with control antigen when available.
Deconvolution microscopy.
Fixed, labeled cells were examined with a Nikon Diaphot 200 microscope equipped for epifluorescence. The image detector was a thermoelectrically cooled charge-coupled device camera with a SITe SI502AB chip, a peak quantum efficiency of 80%, and a 16-bit dynamic range. Forty to 60 images at 0.25-μm intervals were acquired with a 60/1.4 PlanApo objective, giving pixel dimensions of 100 by 100 by 250 nm. The point spread function of the microscope was determined with 100-nm Fluorospheres (Molecular Probes) of the appropriate wavelength. Image stacks were then deconvolved and aligned, and three-dimensional images were generated.
Affinity precipitation of Rho-GTP.
Cells were grown to confluence on 100-cm-diameter petri dishes and serum starved 24 h prior to infection. Cells were infected with H. pylori (200 μl of H. pylori/ml of cells corrected to OD600) and incubated for 24 h at 37°C. Cells were washed in PBS at room temperature, and lysis buffer was added for 5 min at 4°C. Cells were scraped from the dish and centrifuged at 6,000 × g for 5 min at 4°C to remove nuclei. The supernatant was harvested, added to beads, and agitated for 1 h at 4°C. Supernatant was discarded, and beads were washed with lysis buffer. After being centrifuged at 6,000 × g for 3 min at 4°C the supernatant was removed, and wash buffer was added to beads. The centrifugation step was repeated (as described above), and supernatant was removed. The pellet was resuspended in 15 μl of loading buffer at 37°C for 5 min before loading onto gel.
Cell migration.
Cells were grown to confluence in six-well plates and were serum starved 24 h prior to the experiment (0.3% FCS and no insulin). Cells were infected with H. pylori (200 μl/ml corrected to OD600) and incubated at 37°C for 5 h. A 10-μl pipette tip was used to create a wound across the center of the well. After plates were washed to remove cell debris, bacteria were reapplied (see concentrations above). The plates were returned to the incubator, and the rate of wound closure was measured at 12-h intervals over a 48-h period.
To assess the effect of SST on cell migration, the cells were either untreated or treated with 1 or 10 nM CH275 (kind gift from J. Rivier, Salk Institute), an SSTR-1-specific analog, in the absence of H. pylori infection. The rate of migration was expressed as millimeters per 12-h period, and the effect of SSTR-1 analog treatment was assessed at each time point by the unpaired Student t test. The cell line data are from six different experiments (n = 8 for H. pylori migration data); for the primary cells the data are from three experiments.
RESULTS
Gene arrays.
RNA was collected from three types of cells (primary antral cells and NCI-N87 and AGS cell lines) either infected for 48 h with H. pylori or uninfected (controls). 32P-cDNA was screened in the gene arrays. For details of the gene expression patterns of the three cell preparations refer to http://www.physiology.ubc.ca/Buchanarray.htm.
The primary antral cells showed the greatest number of mRNAs significantly affected by bacterial infection, with 19 genes upregulated (increased >1.3-fold above control levels with a P value of <0.05) and 48 genes downregulated (decreased to <75% of control levels and a P value of <0.05; for details see http://www.physiology.ubc.ca/Buchanarray.htm). For the AGS cell line very few mRNAs were significantly affected, with eight upregulated and two downregulated (http://www.physiology.ubc.ca/Buchanarray.htm). NCI-N87 showed more mRNAs significantly upregulated than downregulated (http://www.physiology.ubc.ca/Buchanarray.htm).
To determine whether live bacteria were required to induce the changes in gene expression profiles, we infected NCI-N87 cells with heat-killed H. pylori. This had no effect on the levels of gene transcripts detected with the array compared to levels for the live, infected cells (data not shown). An additional possibility was that infection of the cell cultures by any bacterial species would result in cellular stress and that this alone could be responsible for the changes in gene expression. We examined this possibility by infecting NCI-N87 cells with the related species H. felis. In these experiments significant changes in gene expression were observed (http://www.physiology.ubc.ca/Buchanarray.htm) although these were distinct from those seen with H. pylori. Infection with H. felis failed to significantly alter the levels of expression of S100 A7 or annexins. However, expression of the adenosine A1 receptor and hexokinase was increased.
Receptors and signaling.
The results of the gene arrays indicated that H. pylori infection alters genes encoding epithelial cell receptors. Of the genes downregulated by H. pylori, the primary and NCI-N87 cells showed more overlap than the AGS cell line. The former two cell preparations showed small yet significant decreases in the SST receptor subtype SSTR-1 and KDEL receptor mRNA levels, which were absent from AGS cells. Due to the important role played by SST in regulating gastrin release, cell proliferation, and migration, we determined whether the change in SSTR-1 mRNA in the primary cells could be confirmed by a different technique. In view of the low levels of expression, we used semiquantitative PCR and confirmed a consistent decrease in SSTR-1 mRNA of 50% (control band density, 1.0; H. pylori-infected cell band density, 0.57; P < 0.05, n = 4; Fig. 1).
FIG. 1.

Semiquantitative RT-PCR, using ribosomal 18S as the internal standard, confirmed that SSTR-1 transcripts were decreased in H. pylori-infected (+H.p) cells (arrow). The PCRs were carried out with 2 μl of cDNA in a 25-μl total volume of PCR buffer. SSTR-1 primers had the following sequences: forward, 5′GGAGGAGCCGGTTGACTATT3′; reverse, 5′AAGGTAGCCTGAAAGCCTTCC3′. The PCR products were electrophoresed in a 1.2% agarose gel. The DNA was visualized and photographed with the Eagle Eye II video system (Stratagene). A ratio of the band intensities between SSTR-1 and the internal standard for the uninfected sample was compared with that for the infected samples. Cnt, control uninfected cells. Results are representative of the results of nine different reactions.
Bacterial adherence.
Two of the genes expressed at high levels in control cells, the annexin II and IV genes, were significantly upregulated by H. pylori infection in the primary antral cells and NCI-N87 cell line but not the AGS cell line. The availability of specific antibodies against members of the annexin family allowed the use of Western blot analysis to determine if increased annexin II transcripts resulted in increased protein levels. We used the housekeeping protein histone 2B to normalize the loading of proteins for quantitation of the Western blots. While the annexin II protein was increased 1.6- ± 0.2-fold (n = 5, P < 0.05) above control, the levels of annexin IV remained unchanged (Fig. 2A).
FIG. 2.

H. pylori infection of primary-cell preparations and NCI-N87 cells induces increased levels of annexin II and S100 A7 protein expression. Ten microliters of H. pylori with an OD600 of 0.5 was used to inoculate 1 ml of growth medium. The infected epithelial cells were maintained in 10% CO2 at 37°C for 48 h prior to collection of protein. (A) Whole-cell lysates were prepared from control and infected-primary-cell preparations, and protein was analyzed by SDS-PAGE using 7.5% polyacrylamide gels. Annexin II protein expression was analyzed by blotting with a monoclonal anti-annexin II antibody. Annexin IV protein was analyzed by blotting with a monoclonal anti-annexin IV antibody. The density of the bands was related to that of the housekeeping protein histone 2B. Cnt, control uninfected cells; +H.p, H. pylori-infected cells. (B) Semiquantitative RT-PCR, using ribosomal 18S as the internal standard, confirmed that S100 A7 transcripts were upregulated in H. pylori-infected primary cells. The PCRs were carried out with 2 μl of cDNA in a 25-μl total volume of PCR buffer. S100 A7 primers had the following sequences: forward, 5′GAAAGCAAAGATGAGCAACACTC3′; reverse, 5′TTGGTGGGGCTGGGTCACTG3′. The PCR products were electrophoresed in a 1.2% agarose gel. The DNA was visualized and photographed with the Eagle Eye II video system (Stratagene). The ratio of the band intensity for SSTR-1 to that for the internal standard for the uninfected sample was compared with the corresponding ratio for the infected samples. (C) Total RNA (25 ng) was extracted from NCI-N87 cells. The RNA was then electrophoresed in a denatured 1% formaldehyde agarose gel and transferred to a nylon membrane. The membrane was then hybridized with a 32P-labeled probe against S100 A7 and GAPDH. After 2 h the filter was washed and autoradiographed with Kodak XAR5 film for 16 h at −70°C. GAPDH was used as the internal standard and confirmed that S100 A7 levels were upregulated in H. pylori-infected NCI-N87 cells. Data are representative of six independent experiments.
All three cell preparations showed a consistent pattern of change in the expression of members of the S100 family of calcium-binding proteins, previously reported to play a role in bacterial adherence to epithelial cells. In control cells, S100 C was the only member of the family consistently expressed. After infection with H. pylori, S100 A7 was detected at high levels (primary cells: 14.1 ± 0.39, n = 6, P < 0.05; NCI-N87 cells: 5.6 ± 0.1, n = 6, P < 0.05). The increase in S100 A7 was confirmed by semiquantitative PCR and Northern blot analysis (Fig. 2B and C). Although the gene arrays failed to detect S100 A7 mRNA in control samples, semiquantitative PCR demonstrated a low level of expression.
Cell adhesion proteins.
The array data represent cDNA for a number of adhesion proteins that have been reported to play a pivotal role in carcinogenesis. The mRNAs that showed an increase in more than one cell type following H. pylori infection were IQGAP-1 (primary and NCI-N87) and α-2,8-sialyltransferase (primary, NCI-N87, and AGS cells; http://www.physiology.ubc.ca/Buchanarray.htm). Of these, specific antibodies against IQGAP-1, a protein known to regulate the formation of adherens junctions, were available. To investigate whether the changes in IQGAP-1 mRNA levels were reflected in protein levels and whether E-cadherin protein levels were affected, we performed Western blot analyses of control and infected primary gastric epithelial (n = 3) and NCI-N87 cells. Infection with H. pylori did not significantly increase the levels of IQGAP-1 or E-cadherin protein levels (NCI-N87 and IQGAP-1, 1.39 ± 0.5 [n = 3, P > 0.05]; E-cadherin, 1.12 ± 0.2 [n = 3, P > 0.05]; primary and IQGAP-1, 1.17 ± 0.25 [n = 3, P > 0.05]; E-cadherin, 1.36 ± 0.4 [n = 3, P > 0.05]; Fig. 3).
FIG. 3.
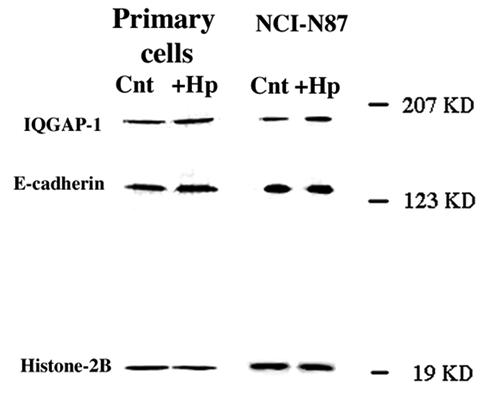
H. pylori infection of primary-cell preparations and NCI-N87 cells has no effect on IQGAP-1 or E-cadherin protein expression. Ten microliters of H. pylori with an OD600 of 0.5 was used to inoculate 1 ml of growth medium. The infected epithelial cells were maintained in 10% CO2 at 37°C for 48 h prior to collection of protein. Whole-cell lysates were prepared from control and infected-cell preparations, and protein was analyzed by SDS-PAGE using 7.5% polyacrylamide gels and then transferred to nitrocellulose membranes. Membranes were incubated with monoclonal anti-IQGAP-1 and monoclonal anti-E-cadherin antibodies. The density of the bands was related to that of the housekeeping protein histone 2B. Data are representative of three to six independent experiments.
In the absence of a change in protein levels, we examined the possibility of a change in protein distribution. These studies demonstrated that H. pylori infection has a significant effect on the distribution of IQGAP-1 and E-cadherin. In control, uninfected primary-cell preparations the majority of the immunoreactivity (IR) for E-cadherin was present on the plasma membrane, while IQGAP-1 was present both at the plasma membrane and in the cytosol. After 48 h of bacterial infection, a significant proportion of the E-cadherin IR was internalized. IQGAP-1 translocated from the cytoplasm to intracellular tubular structures, while E-cadherin IR was present in small vesicles (Fig. 4). This translocation of IQGAP-1 from the cytoplasm to intracellular structures was also observed with H. pylori-infected NCI-N87 cell cultures (Fig. 5). The lack of E-cadherin in the AGS cell line results in a lack of IQGAP-1; therefore, IQGAP-1 could not be detected in the AGS cell line (data not shown).
FIG. 4.

Primary-cell preparations immunostained for IQGAP-1 and E-cadherin. Primary-cell preparations were plated on APES-coated glass coverslips and incubated for 48 h to facilitate attachment prior to infection with H. pylori. Control and infected cells were fixed 48 h postinfection in 4% paraformaldehyde for 10 min at room temperature. (A) Control (Cnt) cells were immunostained with monoclonal anti-IQGAP-1 antibodies for 18 h at 4°C. Note the presence of IR at adherens junctions (arrow). (B) Control cells were immunostained with monoclonal anti-E-cadherin antibodies for 18 h at 4°C. (C) Cells were infected with H. pylori and immunostained for IQGAP-1. Note the dramatic increase in internalized protein in tubulovesicles (arrows). (D) Cells were infected with H. pylori and immunostained for E-cadherin. Note the presence of internalized protein in small vesicles. Scale bar = 5 μm. Images are representative of three independent experiments.
FIG. 5.

H. pylori infection of NCI-N87 cells causes internalization of IQGAP-1. NCI-N87 cells were plated on APES-coated glass coverslips and incubated for 48 h to facilitate attachment prior to infection with H. pylori. Control (Cnt) and infected (+H.p) cells were fixed 48 h postinfection in 4% paraformaldehyde for 10 min at room temperature. (A) Control cells were immunostained with monoclonal anti-IQGAP-1 antibodies for 18 h at 4°C. Note the presence of IR at adherens junctions (arrows). (B) Cells were infected with H. pylori and immunostained for IQGAP-1. Note the dramatic increase in internalized protein in tubulovesicles (arrows). Scale bar = 5 μm. Images are representative of three independent experiments.
Cell matrix proteins.
Array analysis of the two gastric cell lines detected expression of mRNA encoded by 222 (AGS) or 171 (NCI-N87) genes, with the majority being at either medium or low abundance. There were significant differences in the gene expression patterns between the primary antral cells and the two gastric cell lines (for details see http://www.physiology.ubc.ca/Buchanarray.htm). A comparison of the small number of highly expressed genes in the three cell preparations demonstrated that members of the annexin and syntaxin families, RACK1, and dystroglycan were invariably in this group.
The primary cells expressed significantly higher levels of dystroglycan (laminin receptor) and laminin and integrin subunits (http://www.physiology.ubc.ca/Buchanarray.htm). The dystroglycan gene encodes two major products, α-dystroglycan, a highly glycosylated soluble protein, and β-dystroglycan, a transmembrane protein that binds α-dystroglycan to the extracellular surfaces of cells. Immunocytochemical studies with an antibody against α-dystroglycan confirmed that primary cells produced the protein concentrated to the endoplasmic reticulum and Golgi complex; a similar localization was observed in the NCI-N87 cells (Fig. 6A and B). While the AGS cells produced α-dystroglycan, the intracellular distribution was consistent with incomplete synthesis and transfer to the degradative pathway (Fig. 6C). Parallel Western blot analysis confirmed the presence of high levels of protein in the primary cells with the following rank order for the cell lines: NCI-N87 > AGS (Fig. 6G). Taken together, the immunocytochemistry and Western blot data provided a direct comparison of two available gastric epithelial cell lines with the primary antral epithelial cell preparation and indicated that both dystroglycan and paxillin expression and distribution were more closely aligned between the primary cells and the NCI-N87 cell line than between the primary cells and the AGS cell line.
FIG. 6.

Primary-cell preparations, NCI-N87 cells, and AGS cells immunostained for α-dystroglycan and paxillin. (A to F) Cells were grown on APES-coated coverslips for 48 h prior to fixation with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. All cells were immunostained separately with monoclonal anti-α-dystroglycan and monoclonal antipaxillin antibodies for 18 h at 4°C. (G) α-Dystroglycan protein expression levels within primary-cell preparations, NCI-N87 cells, and AGS cells were analyzed by SDS-PAGE. (H) Paxillin protein expression levels within primary-cell preparations, NCI-N87 cells, and AGS cells were analyzed by SDS-PAGE. The housekeeping protein histone 2B was used to normalize for protein loading. The primary (A) and NCI-N87 (B) cells showed a strong immunostaining of the ER and Golgi complex (arrows), whereas the AGS cells (C) showed staining in intracellular vesicles (arrows). The levels of α-dystroglycan protein expression are highest in primary cells, followed (in order) by NCI-N87 and AGS cells (G). The primary cells (D) showed both cytosolic and focal contact staining, while in the NCI-N87 (E) and AGS (F) cells paxillin IR was predominantly at the focal contacts. The largest amount of paxillin protein was present in the primary cells, followed (in order) by AGS cells and NCI-N87 cells (H). Scale bar = 10 μm. Images are representative of three independent experiments.
While the Clontech array did not measure transcript levels of paxillin, an integral focal complex protein, we determined the distribution and level of protein expression in the primary cells and cell lines. Interestingly the amount of protein contained in the cell lysates did not correlate with the presence or development of the focal contacts. The primary cells showed a combination of cytosolic and focal contact-associated paxillin IR (Fig. 6D and H), whereas the NCI-N87 (Fig. 6E and H) and AGS (Fig. 6F and H) cells contained smaller amounts of protein but showed strong paxillin IR focal contacts.
In view of differences in the expression of actin bundling proteins, integrins, and laminins detected with the microarray (see http://www.physiology.ubc.ca/Buchanarray.htm) we examined the organization of the actin cytoskeleton. Image stacks at 0.3 μm in the Z plane through the cells were collected, and the apical and basolateral regions were differentiated. In primary and NCI-N87 cells, stress fibers were confined to a 1-μm region at the basolateral membrane, with the remainder of the plasma membrane (8 to 12 μm in Z) being defined by E-cadherin and cortical actin (Fig. 7). In AGS cells, stress fiber arrays at the basolateral membrane were poorly developed, with a small number of fine fibers (Fig. 7). Also the AGS cells showed high levels of intracellular actin with a low level of cortical actin (Fig. 7). This distribution did not vary with the region of the cell and did not overlap with the distribution of E-cadherin (data not shown). The E-cadherin/catenin complex at the core of the adherens junction is thought to regulate the formation of cortical actin bundles; however, AGS cells lacking E-cadherin at the plasma membrane showed cortical actin. To determine whether cortical actin assembly was independent of E-cadherin in gastric epithelial cells, we completed calcium switch experiments. In primary and NCI-N87 cells, after 30 min in calcium-free medium, E-cadherin was absent from the plasma membrane and cortical actin was significantly reduced (Fig. 8). The absence of extracellular calcium for up to 120 min failed to alter the distribution of E-cadherin or actin in the AGS cells (data not shown).
FIG. 7.

Immunocytochemistry shows that the primary and NCI-N87 cells show similar patterns of E-cadherin and actin staining. All three cell preparations were grown on APES-coated coverslips for 48 h prior to fixation with 4% paraformaldehyde and permeabilization with 0.1% Triton X-100. All cells were immunostained with a monoclonal anti-E-cadherin antibody for 18 h at 4°C before being incubated with phalloidin-AlexaFluor 594 for 1 h at room temperature. At the basolateral membrane E-cadherin (A, primary; E, NCI-N87) is reduced and actin stress fibers predominate (B, primary; F, NCI-N87). From 1 μm from the substrate to the top of the cells both showed strong E-cadherin (C, primary; G, NCI-N87) and cortical actin (D, primary; H, NCI-N87) staining at the plasma membrane. AGS cells show a punctate pattern of E-cadherin staining that does not localize to adherens junctions (I). AGS cells have a uniform pattern of filamentous actin staining with a small number of stress fibers (arrows) and cortical actin (arrowheads) (J). Scale bar = 10 μm. Images are representative of three individual experiments.
FIG. 8.

Removal of extracellular calcium resulted in the internalization of E-cadherin in primary-cell preparations and NCI-N87 cells. Cells were seeded on APES-coated glass coverslips in 24-well plates and allowed to attach in medium containing 1 mM Ca2+ for 2 h. Cells were washed in 0 mM Ca2+ culture medium before fresh 0 mM Ca2+ culture medium was added. Medium was removed, and cells were immediately fixed in 4% paraformaldehyde at 30-min intervals over a 2-h period. Cells were then processed for immunofluorescence. In the primary cells the loss of peripheral E-cadherin (A) correlated with a loss of cortical actin but not stress fibers (B), while in NCI-N87 cells loss of peripheral E-cadherin (C) correlated with a reduction in the number of stress fibers in addition to a significant reduction in cortical actin (D). Scale bar = 10 μm. Images are representative of at least four independent experiments.
Affinity precipitation of Rho-GTP.
Rho pull-down experiments were performed on NCI-N87 cells in the presence of low serum (0.3% FCS), high serum (5% FCS), and 100 nM SST. Rho pull-down experiments measure the level of active Rho within a cell by isolating the active form of Rho (Rho-GTP). Briefly, beads coated with the binding domain of a specific downstream effector of active Rho, Rhotekin-RBD, are incubated with NCI-N87 cell lysates. The beads bind the active form of Rho only; therefore, any increase observed is an increase in active Rho (Rho-GTP), and the amount of active Rho pulled down is detected via Western blotting using an antibody directed against Rho. In the presence of low serum Rho-GTP levels are low, as expected; increasing serum levels increases the levels of GTP-bound Rho. Addition of SST for 20 min decreases Rho-GTP levels by approximately threefold (data not shown). Infecting NCI-N87 cells for 24 h with H. pylori revealed increased levels of GTP-bound Rho compared to those for control cells (Fig. 9A) (H. pylori-infected cells, 1.25 ± 0.13; P < 0.05, n = 3).
FIG. 9.

Infection of NCI-N87 cells with H. pylori increases Rho-GTP expression levels. Affinity precipitation of Rho-GTP used beads coated with the binding domain of the glutathione S-transferase fusion protein Rhotekin-RBD. (A) Serum-starved NCI-N87 cells were infected for 24 h with H. pylori (+H.p; 200 μl/ml corrected to OD600) and harvested alongside control cells. Cells were centrifuged to remove nuclei, and supernatant was added to beads for 1 h at 4°C. Beads were washed with lysis buffer, and the pellet was resuspended prior to electrophoresis and Western blotting. Membranes were probed with an anti-Rho antibody. Only active Rho is precipitated in these experiments due to the specificity of Rhotekin-RBD for Rho-GTP; therefore the observed increase in Rho protein observed via Western blotting in the presence of H. pylori is an increase in the active form of Rho (Rho-GTP). Cnt, control. (B) Infection of NCI-N87 cells with H. pylori alters basal stress fiber density and formation. Cells were grown on APES-coated coverslips for 48 h prior to experimentation. Control and infected cells were fixed 24 h postinfection in 4% paraformaldehyde prior to permeabilization with 0.1% Triton X-100. Cells were incubated with phalloidin-AlexaFluor 488 for 1 h at room temperature prior to being analyzed by deconvolution microscopy. Control cells show dense compact stress fibers on the basal surface, whereas H. pylori-infected cells show elongated cables of stress fibers. Scale bar = 5 μm. Images are representative of at least four independent experiments.
Cell migration.
Having established that the cell adhesion proteins E-cadherin and IQGAP-1 and the cell matrix proteins dystroglycan and paxillin are present in appropriate locations in the NCI-N87 cell line, migration experiments using this cell line were performed. Of the receptors on the gene array, SSTR-1 was consistently expressed by the cell preparations, and SSTR-1 activation has previously been shown to inhibit CCL39 fibroblast migration (5). Wound healing experiments were undertaken to determine if the changes in cell-cell and cell-matrix adhesion affected migration of control and SST-treated cells. The cell lines were grown to confluence prior to wounding, and the primary cells were plated at high density in the absence of significant cell division. In control experiments the primary and NCI-N87 cells migrated as integrated sheets of cells to cover the surface of the wound, moving at an average rate of 1.2 mm/12 h. The time required for closure of a 5-mm wound was 36 to 48 h (Fig. 10). Activation of SSTR-1 with both concentrations of CH275 significantly inhibited cell migration in both cell preparations (P < 0.05) (Fig. 10B and D). H. pylori infection yields a decrease in SSTR-1 expression that would be consistent with increased migration in the presence of H. pylori. To test this, we determined the effects of H. pylori infection on rates of migration. Wound healing experiments with H. pylori-infected NCI-N87 cells revealed an increase in the rate of epithelial cell migration compared to control, uninfected cells (control, 17.4% wound closure; H. pylori-infected cells, 68.3% wound closure; n = 8; P < 0.05) (Fig. 11).
FIG. 10.

Gastric cell migration is significantly inhibited by the addition of CH275 SSTR-1 analog at 1 and 10 nM. NCI-N87 cells were grown to confluence in six-well plates and were serum starved for 24 h prior to the experiment. Primary cells were plated at high density in the absence of significant cell division. A 10-μl pipette tip was used to create a wound across the center of the well. After a washing to remove cell debris, cells were either untreated or treated with 1 or 10 nM CH275, an SSTR-1-specific analog. The rate of wound closure was measured at 12-h intervals over a 48-h period. The rate of migration is expressed as millimeters per 12-h period, and the effect of SSTR-1 analog treatment was assessed at each time point by the unpaired Student t test. Under control conditions in the presence of 5% serum the primary (A) and NCI-N87 (C) cells migrated as an integrated sheet. Cell migration was significantly inhibited by the addition of CH275 SSTR-1 analog at 1 and 10 nM. (B) Primary cells; (D) NCI-N87 cells.*, P = <0.05. The cell line data are from six individual experiments; the primary-cell preparation data are from three individual experiments.
FIG. 11.

Infection of the NCI-N87 cell line with H. pylori increases gastric epithelial cell migration. NCI-N87 cells were grown to confluence in six-well plates and were serum starved 24 h prior to the experiment. Cells were infected with H. pylori (200 μl/ml corrected to OD600) and incubated at 37°C for 5 h. A 10-μl pipette tip was used to create a wound across the center of the well. After a washing to remove cell debris, bacteria were reapplied (at the concentration above). The rate of wound closure was measured at 12-h intervals over a 48-h period. The rate of migration is expressed as millimeters per 12-h period. All migration experiments were performed in the presence of 5% serum. (A) Both control, uninfected cells (Cnt) and H. pylori-infected cells (+H.p) migrated as an integrated sheet. (B) Over a 24-h period cell migration was significantly faster in the presence of H. pylori. The data report the means ± standard errors for eight wound healing experiments (*, P < 0.05).
DISCUSSION
The majority of studies investigating H. pylori infections to date have used the gastric epithelial cell line AGS. Ideally, studies modeling the effects of H. pylori infection of the human stomach would use human primary gastric epithelial cell preparations. However, access to normal tissue is limited and severely restricts the number and types of experiments that can be performed. Lack of adherens and tight junctions makes the AGS cell line a less-than-optimal model of the gastric epithelium (2). Prior investigations into appropriate gastric cancer cell lines as models to study human diseases have indicated that a more appropriate cell line is NCI-N87 (6). The results of our experiments confirmed that the NCI-N87 gastric epithelial cell line replicated many characteristics of human primary antral cell cultures based on analysis of adherens junction proteins, the actin cytoskeleton, cell matrix protein expression, and cell migration. Our findings were consistent with adherens junctions being specific targets of H. pylori (1, 15)
Our gene array analysis data revealed that H. pylori infection increased the expression of two families of calcium-binding protein genes: the annexin and S100 genes; the resultant proteins are known to regulate bacterial adhesion to epithelia (3, 36). Analysis of paired cell preparations infected with H. pylori revealed that annexin II and S100 A7 transcripts were consistently upregulated. The S100 proteins have been shown to form functional heterotetramers with members of the annexin family of calcium-binding proteins (14, 36). Annexins II and IV were increased after bacterial infection in the primary and NCI-N87 cells. The increase in annexin II transcripts consistently translated into higher protein levels. Although annexin I forms heterotetramers with S100C (28) and annexin II forms heterotetramers with S100 A10 (36), whether either annexin forms complexes with S100 A5 or A7 is unknown. Since the consequence of heterotetramer formation differs depending on the individual partners, it is difficult to predict how the modulation of these genes will affect gastric epithelial cell function. However, it has been previously reported that annexin II was overexpressed in advanced gastric carcinomas and was thought to contribute to gastric carcinoma progression (13). In view of indications that annexin/S100 heterotetramer formation affects membrane stability and actin bundling, the parallel upregulation of S100 and annexin gene transcription by H. pylori may be of significance in adhesion of this bacterium to gastric epithelia and subsequent alterations to the structure of the epithelial cell cytoskeleton.
In 60% of diffuse gastric cancers, E-cadherin-mediated adherens junctions are decreased, the extent of the decrease being proportional to tumor growth and invasiveness (27). While bacterial infection did not alter transcript levels of E-cadherin, levels of IQGAP-1 mRNA were consistently increased. Increased IQGAP-1 results in a loss of E-cadherin at the plasma membrane and increased migration in transfected cells (21, 22). Our studies showed no increase in IQGAP-1 or E-cadherin protein; however, these proteins were translocated to intracellular tubulovesicles (IQGAP-1) or vesicles (E-cadherin), supporting an effect of H. pylori on adherens junction structure and function. The increase in IQGAP-1 mRNA could be due to a compensatory mechanism in response to the shift in IQGAP-1 from the cytoplasm to a sequestered location in tubulovesicles. This recycling phenomenon of IQGAP-1, whereby internalization of the protein causes increased gene expression, resulting in increased protein synthesis, would result in stable IQGAP-1 protein levels even though mRNA concentrations increased. This was consistent with visualization of IQGAP-1 protein redistribution to intracellular structures postinfection. Although significant amounts of both IQGAP-1 and E-cadherin were present in intracellular structures, immunoprecipitation experiments and immunocytochemistry demonstrated the continued presence of a reduced level of E-cadherin at the plasma membrane, mediating a reduced level of cell-cell adherence.
The formation of the actin cytoskeleton in epithelial cells is dependent on the presence of adherens junctions and cell- matrix interactions. In polarized epithelial cells the distribution of filamentous actin differs between the apical and basolateral poles. The basolateral membrane is anchored to the substrate through numerous focal contacts that serve as organizational centers for cables of actin stress fibers (26, 29), whereas at the apical regions adherens junctions regulate the formation of cortical actin bundles (20, 25).
To preserve the integrity of the gastric epithelial barrier, apical junctions must be maintained while the base of the cell moves over the basement membrane. Understanding how this process is regulated is vital to determining how the gastric epithelium responds to injury, including the ability to heal ulcers, and how H. pylori infection may affect the reepithelialization process. Clearly the regulation of cell-matrix and cell-cell adhesion complexes must be coordinated to achieve integrated cell migration, with dystroglycan and integrin receptors maintaining adhesion to the basement membrane, while adherens and the tight junction complexes join the apical poles into a continuous sheet (9, 19, 30, 35).
The distribution of the cell matrix protein dystroglycan in the primary-cell preparations was similar to that for the NCI-N87 cell line. In contrast, the AGS cell line exhibited an abnormal distribution, which creates potential problems in the interpretation of migration data using this cell line. In addition to dystroglycan distribution, a requirement for cellular migration is the formation and disassembly of focal contacts at the cell periphery (4, 29). Paxillin is an adaptor protein located at focal contact sites on the basal membrane and involved in adhesion organization and cell migration. The distribution of paxillin in the AGS cell line is abnormal compared to that in the primary-cell preparation and NCI-N87 cell line. The similarity between the primary-cell preparations and the NCI-N87 cell line with regard to expression and distribution of both dystroglycan and paxillin meant that migration studies were performed using these cells only, due to the potential for ambiguous results with the AGS cell line.
H. pylori infection of NCI-N87 cells induced a fourfold increase in the rate of migration compared to that of controls over a 24-h incubatory period. The gene array data revealed that, of genes encoding GPCRs, only members of the SSTR family were significantly regulated by bacterial infection in primary cells and cell lines. Transcripts for SSTR-1 were decreased following the infection of primary and NCI-N87 cells. We have previously reported that SST, acting at the GPCR subtype SSTR-1, inhibits the GTP binding of Rho, attenuates the assembly of actin stress fibers and focal adhesions, and inhibits cell migration in CCL39 cells (5, 7, 16). However, CCL39 cells are not polarized or linked by adherens junctions and do not normally express SST receptors. Our data show for the first time that activation of endogenously expressed SSTR-1 by CH275 inhibited migration in both primary-cell cultures and the NCI-N87 cell line. Infection of NCI-N87 with H. pylori increases migration and Rho-GTP levels, indicating that bacterial infection alone would not promote ulcer formation. The observation that H. pylori increases rates of migration in addition to increasing total Rho-GTP levels may appear to contradict the predicted function of activated Rho; however, epithelial cell migration involves the extension of a leading edge, protrusion, or lamellipodium; the establishment of new adhesion sites at the front; cell body contraction; and detachment of adhesions at the cell rear. All these steps involve the assembly, the disassembly, or the reorganization of the actin cytoskeleton, and each must be coordinated both in space and time to generate productive net forward movement. Rho activity in migrating cells is associated with focal adhesion assembly and cell contractility and is responsible for cell body contraction and rear end retraction. Hence increased levels of Rho-GTP at the rear of a cell and increased Rac at the leading edge induce forward movement. While our Rho-GTP precipitation experiments indicate that H. pylori-induces an increase in total cell Rho-GTP levels, they do not indicate the location of the GTP-bound Rho protein. It is highly probably that the increased Rho-GTP levels observed are compartmentalized at the rear of the cell, thus aiding forward movement. In vivo, the effect of H. pylori would be modulated by activation of the immune system and increased acid loading in the antrum; therefore our data suggest that immunosuppression and antacids in the presence of H. pylori would speed healing. The present studies identified, for the first time, that the expression of genes encoding the S100 and annexin families of calcium-binding proteins, IQGAP-1, and SSTR-1 were modulated by H. pylori infection.
In conclusion, our data indicate that adhesion of H. pylori to gastric epithelial cells initiated rearrangement of the underlying actin cytoskeleton, possibly via S100 and annexin protein interactions. Epithelial cells infected by H. pylori revealed increased IQGAP-1 mRNA and a redistribution of E-cadherin and IQGAP-1 to intracellular locations and a reduction in adherens junctions. Decreased adherens junctions would increase epithelial permeability, allowing nutrient delivery and increased H. pylori survival. The increase in Rho-GTP and epithelial cell migration in H. pylori-infected cultures would indicate that ulceration in vivo is a secondary process triggered by the inflammatory response and increased acid loading.
Acknowledgments
This work was funded by grants from the Canadian Institutes of Health Research (A.M.J.B. and B.B.F.), the Canadian Bacterial Diseases Network of Excellence (B.B.F.), and Genome Canada (B.B.F.).
Editor: J. T. Barbieri
REFERENCES
- 1.Amieva, M. R., R. Vogelmann, A. Covacci, L. S. Tompkins, W. J. Nelson, and S. Falkow. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300:1430-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Athmann, C., N. Zeng, T. Kang, E. A. Marcus, D. R. Scott, M. Rektorschek, A. Buhmann, K. Melchers, and G. Sachs. 2000. Local pH elevation mediated by the intrabacterial urease of Helicobacter pylori cocultured with gastric cells. J. Clin. Investig. 106:339-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babiychuk, E. B., K. Monastyrskaya, F. C. Burkhard, S. Wray, and A. Draeger. 2002. Modulating signaling events in smooth muscle: cleavage of annexin 2 abolishes its binding to lipid rafts. FASEB J. 16:1177-1184. [DOI] [PubMed] [Google Scholar]
- 4.Ballestrem, C., B. Hinz, B. A. Imhof, and B. Wehrle-Haller. 2001. Marching at the front and dragging behind: differential αVβ3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155:1319-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber, D. L., A. M. J. Buchan, C.-Y. Lin, and J. Choi J. 2000. Somatostatin acting at SSTR1 subtype inhibits activation of Rho and actin stress fibre formation. Mol. Biol. Cell 11:100a. (Abstract.) [Google Scholar]
- 6.Basque, J.-R., M. Chenard, P. Chailler, and D. Menard. 2001. Gastric cancer cell lines as models to study human digestive functions. J. Cell. Biochem. 81:241-251. [PubMed] [Google Scholar]
- 7.Buchan, A. M., C.-Y. Lin, J. Choi, and D. L. Barber. 2002. Somatostatin, acting at receptor subtype 1, inhibits rho activity, the assembly of actin stress fibers, and cell migration. J. Biol. Chem. 277:28431-28438. [DOI] [PubMed] [Google Scholar]
- 8.Buchan, A. M. J., R. M. Meloche, Y. N. Kwok, and H. Kofod. 1993. Effect of cholecystokinin and secretin on somatostatin release from cultured antral cells. Gastroenterology 104:1414-1419. [DOI] [PubMed] [Google Scholar]
- 9.Bulitta, C. J., J. V. Fleming, R. Raychowdhury, D. Taupin, I. Rosenberg, and T. C. Wang. 2002. Autoinduction of the trefoil factor 2 (TFF2) promoter requires an upstream cis-acting element. Biochem. Biophys. Res. Commun. 293:366-374. [DOI] [PubMed] [Google Scholar]
- 10.Casella, G., C. A. Buda, R. Maisano, M. Schiavo, D. Perego, and V. Baldini. 2001. Complete regression of primary gastric MALT-lymphoma after double eradication Helicobacter pylori therapy: role and importance of endoscopic ultrasonography. Anticancer Res. 21:1499-1502. [PubMed] [Google Scholar]
- 11.Censini, S., C. Lange, Z. Xiang, J. E. Crabtree, P. Ghiara, M. Borodovsky, R. Rappuoli, and A. Covacci. 1996. Cag, a pathogenicity island of Helicobacer pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 93:14648-14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cereijido, M., L. Shoshani, and R. G. Contreras. 2000. Molecular physiology and pathophysiology of tight junctions. I. Biogenesis of tight junctions and epithelial polarity. Am. J. Physiol. 279:G477-G482. [DOI] [PubMed] [Google Scholar]
- 13.Emoto K, H. Sawada, Y. Yamada, H. Fujimoto, Y. Takahama, M. Ueno, T. Takayama, H. Uchida, K. Kamada, A. Naito, S. Hirao, and Y. Nakajima. 2001. Annexin II overexpression is correlated with poor prognosis in human gastric carcinoma. Anticancer Res. 21:1339-1345. [PubMed] [Google Scholar]
- 14.Garbuglia, M., M. Verzini, A. Hofmann, R. Huber, and R. Donato. 2000. S100A1 and S100B interactions with annexins. Biochim. Biophys. Acta 1498:192-206. [DOI] [PubMed] [Google Scholar]
- 15.Heczko, U., V. C. Smith, R. M. Meloche, A. M. Buchan, and B. B. Finlay. 2000. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes Infect. 2:1669-1676. [DOI] [PubMed] [Google Scholar]
- 16.Hotchin, N. A., T. L. Cover, and N. Akhtar. 2000. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J. Biol. Chem. 275:14009-14012. [DOI] [PubMed] [Google Scholar]
- 17.Hou, C., R. L. Gilbert, and D. L. Barber. 1994. Subtype-specific signaling mechanisms of somatostatin receptors SSTR1 and SSTR2. J. Biol. Chem. 269:10357-10362. [PubMed] [Google Scholar]
- 18.Jawhari, A. U., M. Noda, M. J. Farthing, and M. Pignatelli. 1999. Abnormal expression and function of the E-cadherin-catenin complex in gastric carcinoma cell lines. Br. J. Cancer 80:322-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassis, J., A. Maeda, N. Teramoto, K. Takada, C. Wu, G. Klein, and A. Wells. 2002. EBV-expressing AGS gastric carcinoma cell sublines present increased motility and invasiveness. Int. J. Cancer 99:644-651. [DOI] [PubMed] [Google Scholar]
- 20.Kovacs, E. M., M. Goodwin, R. G. Ali, A. D. Paterson, and A. S. Yap. 2002. Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr. Biol. 12:379-382. [DOI] [PubMed] [Google Scholar]
- 21.Kuroda, S., M. Fukata, M. Nakagawa, K. Fujii, T. Nakamura, T. Ookubo, I. Izawa, T. Nagase, N. Nomura, H. Tani, I. Shoji, Y. Matsuura, S. Yonehara, and K. Kaibuchi. 1998. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell-cell adhesion. Science 281:832-835. [DOI] [PubMed] [Google Scholar]
- 22.Li, Z., S. H. Kim, J. M. G. Higgins, M. B. Brenner, and D. B. Sacks. 1999. IQGAP1 and calmodulin modulate E-cadherin function. J. Biol. Chem. 274:37885-37892. [DOI] [PubMed] [Google Scholar]
- 23.Mackay, D. J., and A. Hall. 1998. Rho GTPases. J. Biol. Chem. 273:20685-20688. [DOI] [PubMed] [Google Scholar]
- 24.Nobes, C. D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53-62. [DOI] [PubMed] [Google Scholar]
- 25.Quinlan, M. P., and J. L. Hyatt. 1999. Establishment of the circumferential actin filament network is a prerequisite for localization of the cadherin-catenin complex in epithelial cells. Cell Growth Differ. 10:839-854. [PubMed] [Google Scholar]
- 26.Rajfur, Z., P. Roy, C. Otey, L. Romer, and K. Jacobson. 2002. Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteins. Nat. Cell Biol. 4:286-293. [DOI] [PubMed] [Google Scholar]
- 27.Ramesh, S., J. Nash, and P. G. McCulloch. 1999. Reduction in membranous expression of β-catenin and increased cytoplasmic E-cadherin expression predict poor survival in gastric cancer. Br. J. Cancer 81:1392-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rety, S., D. Osterloh, J. P. Arie, S. Tabaries, J. Seeman, F. Russo-Marie, V. Gerke, and A. Lewit-Bentley. 2000. Structural basis of the Ca2+-dependent association between S100C (S100A11) and its target, the N-terminal part of annexin I. Struct. Folding Design 8:175-184. [DOI] [PubMed] [Google Scholar]
- 29.Ridley, A. J., W. E. Allen, M. Peppelenbosch, and G. E. Jones. 1999. Rho family proteins and cell migration. Biochem. Soc. Symp. 65:111-123. [PubMed] [Google Scholar]
- 30.Scanga, C. A., J. Aliberti, D. Jankovic, F. Tilloy, S. Bennouna, E. Y. Denkers, R. Medzhitov, and A. Sher. 2002. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168:5997-6001. [DOI] [PubMed] [Google Scholar]
- 31.Sears, C. L. 2000. Molecular physiology and pathophysiology of tight junctions. V. Assault of the tight junction by enteric pathogens. Am. J. Physiol. 279:G1129-G1134. [DOI] [PubMed] [Google Scholar]
- 32.Segal, E. D., S. Falkow, and L. S. Tompkins. 1996. Helicobacter pylori attachment to gastric cells induces cytoskeletal rearrangements and tyrosine phosphorylation of host cell proteins. Proc. Natl. Acad. Sci. USA 93:1259-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suriano, G., C. Oliveira, P. Ferreira, J. C. Machado, M. C. Bordin, O. De Wever, E. A. Bruyneel, N. Moguilevsky, N. Grehan, T. R. Porter, F. M. Richards, R. H. Hruban, F. Roviello, D. Huntsman, M. Mareel, F. Carneiro, C. Caldas, and R. Seruca. 2003. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum. Mol. Genet. 12:575-582. [DOI] [PubMed] [Google Scholar]
- 34.Suriano, G., D. Mulholland, O. de Wever, P. Ferreira, A. R. Mateus, E. Bruyneel, C. C. Nelson, M. M. Mareel, J. Yokota, D. Huntsman, and R. Seruca.. 2003. The intracellular E-cadherin germline mutation V832 M lacks the ability to mediate cell-cell adhesion and to suppress invasion. Oncogene 22:5716-5719. [DOI] [PubMed] [Google Scholar]
- 35.Varro, A., P. J. Noble, L. E. Wroblewski, L. Bishop, and G. J. Dockray. 2002. Gastrin-cholecystokinin (B) receptor expression in AGS cells is associated with direct inhibition and indirect stimulation of cell proliferation via paracrine activation of the epidermal growth factor receptor. Gut 50:827-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zobiack, N., U. Rescher, S. Laarmann, S. Michgehl, M. A. Schmidt, and V. Gerke. 2002. Cell-surface attachment of pedestal-forming enteropathogenic E. coli induces a clustering of raft components and a recruitment of annexin 2. J. Cell Sci. 115:91-98. [DOI] [PubMed] [Google Scholar]