

Abstract
Daphnia species have become models for ecological genomics and exhibit interesting features, such as high phenotypic plasticity and a densely packed genome with many lineage-specific genes. They are also cyclic parthenogenetic, with alternating asexual and sexual cycles and environmental sex determination. Here, we present a de novo transcriptome assembly of over 32,000 D. galeata genes and use it to investigate gene expression in females and spontaneously produced males of two clonal lines derived from lakes in Germany and the Czech Republic. We find that only a low percentage (18%) of genes shows sex-biased expression and that there are many more female-biased gene (FBG) than male-biased gene (MBG). Furthermore, FBGs tend to be more conserved between species than MBGs in both sequence and expression. These patterns may be a consequence of cyclic parthenogenesis leading to a relaxation of purifying selection on MBGs. The two clonal lines show considerable differences in both number and identity of sex-biased genes, suggesting that they may have reproductive strategies differing in their investment in sexual reproduction. Orthologs of key genes in the sex determination and juvenile hormone pathways, which are thought to be important for the transition from asexual to sexual reproduction, are present in D. galeata and highly conserved among Daphnia species.
Keywords: sexual dimorphism, males, environmental sex determination, crustaceans, juvenile hormone pathway, genetic variation
Introduction
Males and females of many animal and plant species differ phenotypically while sharing the same genome. Most of these differences can be attributed to sex-biased genes, i.e., genes expressed differentially between males and females (Ellegren and Parsch 2007). In species with genetic sex determination, an individuals’ sex is determined by its complement of chromosomes. However, this is not the case in species with environmental sex determination, such as water fleas (Daphnia), crocodilians, and Crepidula snails. In these species, males and females are genetically identical and sex chromosomes are absent. Thus, sex is a phenotypically plastic trait; differences between males and females arise through the influence of the environment on gene expression.
Daphnia is a wide-spread genus of freshwater crustaceans that serves as an important food source for many fish species. Because of their dominant position within the phytoplankton feeders, Daphnia are considered keystone species in many freshwater habitats (Carpenter et al. 1987). Daphnia are cyclically parthenogenetic, which means that they can switch between an asexual and a sexual mode of reproduction (Hebert 1978). Under favorable conditions, females reproduce asexually via parthenogenesis (Hebert 1978; Ebert 2005). If environmental conditions change, external biotic and/or abiotic stimuli lead to a switch to sexual reproduction, resulting in the production of male, in addition to female, offspring. The inducing stimuli include high population densities, reduced food availability, shorter days, and lower temperatures (Stross and Hill 1965; Carvalho and Hughes 1983; Kleiven et al. 1992). As sex determination is entirely environmental and no sex chromosomes are present, both daughters and sons are exact genetic clones of their mothers. Females also produce haploid sexual eggs, which form resting eggs once they have been fertilized by males. Pairs of resting eggs are encased in ephippia and can then endure adverse conditions such as the drying out of ponds or the freezing over of a lake (Hebert 1978; Ebert 2005). The ephippia can stay in the sediment for multiple years and can be dispersed over very large distances (Cáceres 1998; Vanschoenwinkel et al. 2008). Once conditions improve, the next generation of sexually produced females can emerge and resume asexual propagation (Ebert 2005). Because parthenogenesis is the predominant mode of reproduction, the sex ratio of natural Daphnia populations is highly skewed towards females.
With the publication of the genome of Daphnia pulex (Colbourne et al. 2011), Daphnia has become a powerful model to study to effect of ecotoxins, phenotypic plasticity, and environmental sex determination. Daphnia pulex has a very compact genome of ∼200 million base pairs (Mb) (Colbourne et al. 2011). Nevertheless, it harbors an extremely high number of protein coding genes (30,810), >36% of which have no described ortholog in the insects, the sister class of the crustaceans, for which many genomes have been sequenced. These orphan genes, as well as duplicated genes, have been shown to play an important role in local adaptation and phenotypic plasticity (Colbourne et al. 2011). However, without more genome sequences from the genus Daphnia and the crustaceans in general, it is not possible to determine how many of these orphans and gene duplications are lineage-specific. In other species, gene duplications are thought to play a role in the resolution of sexual conflict (Connallon and Clark 2011), which may contribute to their abundance in Daphnia.
A prime example of phenotypic plasticity in Daphnia is the switch between sexual and asexual reproduction. The genome must encode everything necessary for both types of reproduction and multiple studies in D. pulex and other Daphnia species have identified key genes that are involved in this switch (Olmstead and LeBlanc 2002; Kato et al. 2011; Chen, Xu et al. 2014; Toyota, Miyakawa, Hiruta, et al. 2015; Toyota, Miyakawa, Yamaguchi, et al. 2015). Genes involved in sex determination in systems with genetic sex determination such as doublesex (dsx) and transformer (tra) appear to have been coopted (Toyota et al. 2013; Chen, Xu et al. 2014) for signaling sexual differentiation in Daphnia. Furthermore, methyl farnesoate (MF), the unepoxylated form of insect juvenile hormone III (JH III) produced in the mandibular gland, has been shown to induce sexual reproduction in Daphnia (Olmstead and LeBlanc 2002). The signaling cascade leading to MF production in response to environmental cues remains to be elucidated. A recent study showed that the NMDA receptor seems to be involved in signaling upstream of MF and may be a sensor of environmental stimuli (LeBlanc and Medlock 2015; Toyota, Miyakawa, Yamaguchi, et al. 2015). In addition, exogenous treatment with MF and other JH III analogs induces male production in a dose-dependent manner (Olmstead and LeBlanc 2002; Tatarazako et al. 2003; Ginjupalli and Baldwin 2013). Some of these JH III analogs are common insecticides that reach freshwater systems with rain washout, where they can disturb the natural cycles of sexual and asexual reproduction in Daphnia. In Daphnia studies, males are often induced with MF. It is not clear, however, how exogenous MF-induced male production differs from natural male production. There is evidence indicating that exogenously induced males are worse at fertilizing resting eggs than naturally produced males and that JH III homologs reduce fertility and life span in general (Tatarazako et al. 2003; Ginjupalli and Baldwin 2013; LeBlanc et al. 2013). Thus, it seems that MF treatment can have an effect that differs from natural sexual reproduction.
Although much is known about the involvement of MF and the juvenile hormone (JH) pathways in triggering male production, there have been very few studies on the evolution of sex-biased genes or gene expression in Daphnia. One early study by Eads et al. (2007) identified sex-biased genes in D. pulex by comparing females with MF-induced males However, this study was conducted before the genome sequence was available; the microarray used therefore contained only ∼1,500 genes (∼5% of all genes). Furthermore, due to the lack of genome sequences from closely related species, the sex-biased genes found in D. pulex were compared with Drosophila melanogaster, which further limited the number of orthologous genes available for comprehensive evolutionary analysis. Together with the D. pulex genome, a much more extensive characterization of sex-biased gene expression in D. pulex was published (Colbourne et al. 2011). These data were generated using a genome tiling microarray, but also using MF-induced males. As the identification and characterization of sex-biased genes was not the main focus of the genome paper, no in-depth analysis on this subset of genes has been carried out.
Since the publication of the D. pulex genome (Colbourne et al. 2011), the amount of genomic and transcriptomic resources for arthropod species has grown substantially, although crustaceans are still poorly represented in comparison to the insects. In this study, we present a high quality de novo transcriptome assembly of D. galeata (Sars 1864), with extensive annotations and an analysis of sex-biased gene expression. We used RNA-sequencing (RNA-seq) to identify differentially expressed genes between females and naturally produced males of two clonal lineages derived from geographically separated populations and compare our findings to the available data from D. pulex. We also describe key genes in the sex determination and the juvenile hormone pathways that are linked to the switch from asexual to sexual reproduction. Our results allow for comparative genomics of sex-biased genes within the Daphnia genus and shed light on the influence of cyclic parthenogenesis on the evolution of sex-biased gene expression.
Materials and Methods
Sampling
A set of D. galeata resting stages (ephippia) was collected from the sediment of four lakes (Jordan Reservoir in the Czech Republic, Müggelsee and Lake Constance in Germany, and Greifensee in Switzerland) and hatched under laboratory conditions. The hatchlings were used to establish clonal lines in a laboratory setting. The lines were maintained for at least eight months and up to two years at 20 °C and a constant 15:9 hour light-dark photoperiod prior to the start of the present study. The semi-artificial media (based on ultrapure and spring water, trace elements and phosphate buffer (Rabus and Laforsch 2011) was regularly changed, and the Daphnia were fed every other day with a suspension of the unicellular algae Acutodesmus obliquus. The species identity was checked by sequencing a fragment of the 12S mitochondrial locus and 10 microsatellite markers, following protocols by Taylor et al. (1996) and Yin et al. (2010), respectively. All clonal lines were found to have a D. galeata mitochondrial background. A clustering approach (NEWHYBRIDS, version 1.1; Anderson and Thompson 2002) with the microsatellite markers showed they belong to the D. galeata cluster. Furthermore, the level of genetic differentiation was estimated by π (Tajima 1989), which was calculated from SNP data using vcftools –window-pi (version 0.1.11; Danecek et al. 2011). The mean value of π for the pair of clones this study focuses on was 0.006, averaged over 53,800 windows. For all the clonal lines used for the assembly, the mean π value was 0.004, averaged over 96,751 windows.
To sequence the complete transcriptome, six clonal lines per lake were raised in high numbers. Mature females were placed at equal densities (40 individuals per liter) in semi-artificial medium for a week, and the juveniles were regularly removed. Gravid females from the equal density beakers were then collected within three days during a time window of a few hours. Twenty to thirty individuals were homogenized in a 1.5-ml centrifuge tube in 1 ml Trizol (Invitrogen, Waltham, MA) immediately after removing the water. The Trizol homogenates were kept at −80 °C until further processing. These 24 genotypes (6 lines×4 lakes) constitute the “assembly” set.
Male and female samples were collected from one clone from Jordan Reservoir (J2 clone) and one clone from the Müggelsee (M10 clone). These clones naturally produced males under standard laboratory conditions and hence, no MF or short-day induction was necessary. Clones were kept at densities of ∼20 − 30 individuals in 250-ml beakers and juveniles were removed every day and put into fresh beakers. Juveniles were sexed and separated at day five after birth and then harvested at day nine. Only females with an empty brood pouch were collected and used for RNA extraction. Because sexually reproducing females usually show signs of developing ephippia by this age, the collected females were assumed to be asexuals. Individuals were homogenized and stored in Trizol at −80 °C until RNA extraction was performed. For the female samples, 10 − 20 individuals were pooled, whereas 20 − 30 individuals were pooled for the smaller males. Two biological replicates per sex and clone were collected, resulting in a total of eight samples for RNA-seq library preparation.
RNA Preparation and Sequencing
Total RNA was extracted following a modified phenol/chloroform protocol. Briefly, chloroform was added to the Trizol homogenate and the mixture was centrifuged in a Phase Lock Gel tube (5 PRIME, Gaithersburg, MD), which allowed for the clean separation of the upper aqueous and lower phenol phases. Absolute ethanol was added to the upper phase containing the RNA and the samples were further processed using the RNeasy kit (Qiagen, Venlo, The Netherlands) for the” assembly” samples or the Isolate II RNA Mini Kit (Bioline, London, UK) for the “sex” samples and following the manufacturers’ instructions. The total RNA was eluted in RNAse free water.
The RNA concentration was checked using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE) and the quality was assessed with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). In case of phenol contamination (skewed A230/260 ratio), the RNA was precipitated over-night with a 3M sodium acetate solution at −20 °C and washed with ethanol.
The 24 “assembly” samples were pooled in equimolar proportions prior to the sequencing library preparation. In total, eight “sex” samples and the “assembly” sample were sent to the company GATC (Konstanz, Germany) for library preparation and sequencing. To create a complete transcriptome (i.e., as many transcripts as possible), the cDNA from the “assembly” sample was normalized prior to sequencing, by first denaturing and re-associating of the cDNA strands, and subsequently by filtering out the single stranded cDNA through hydroxyapatite chromatography.
The “assembly” sample was size-sorted and only fragments with a minimum length of 650 base pairs (bp) were retained. Sequencing was carried out on a complete Illumina (San Diego) MiSeq lane, with 250-bp paired-end (PE) reads. The “sex” samples were sequenced on an Illumina HiSeq 2000, with 50-bp single-end (SE) reads.
Read Quality Control and Trimming
Pre-processing of the sequence reads generated from the D. galeata libraries consisted of several steps. First, TruSeq adapter trimming of paired-end reads was performed using Trimmomatic (version 0.32; Bolger et al. 2014). Some overlap of the paired-end reads was expected; these were merged using FLASH (version 1.2.10; Magoc and Salzberg 2011). A custom Perl script was used to remove all reads with uncalled bases (N’s) in their sequence. Afterwards, quality filtering of the reads was done using the Trimmomatic sliding window function to remove fragments of 40 bases that had an average Phred score <30. Further, reads were trimmed at the 5′ and 3′ ends, removing bases with a Phred score <10. Reads shorter than 150 bp after trimming and quality filtering were discarded. For short single-end reads (the “sex” samples), we used a sliding window function to remove fragments of 10 bases that had average Phred score <35, retaining only those reads longer than 25 bp.
Transcriptome Assembly
We used a combined approach to obtain the best transcript predictions for D. galeata, as described in Nakasugi et al. (2014). Instead of running only one assembly algorithm, we ran several using the PE “assembly” set, and combined the resulting transcripts. The input requirements and parameter settings for each of these assemblers are slightly different and are listed in supplementary table S1, Supplementary Material online. In order to ensure the representation of potentially male-specific transcripts in the final transcript list, short reads generated for the “sex” study were also assembled independently from the PE reads with SOAPdenovo-Trans (Xie et al. 2014) and Oases-Velvet (Schulz et al. 2012).
Four different assembly programs based on de Bruijn graphs were used: Trinity (release r20131110; Grabherr et al. 2011), Trans-ABySS (ABySS version 1.4.8, Trans-ABySS version 1.5.1; Simpson et al. 2009), SOAPdenovo-Trans (version 1.03; Xie et al. 2014) and Oases-Velvet (Oases version 0.2.8, Velvet version 1.2.10; Schulz et al. 2012). MiSeq PE reads merged with FLASH were treated as SE reads and were used for the assembly for Trinity, Trans-ABySS and SOAPdenovo, whereas only PE nonoverlapping reads were used for Oases-Velvet. For Trans-ABySS, k-mer lengths of 22 − 36 with a step-size of 2 and 40 − 68 with a step-size of 4 were used for assembly. The Trinity assembly was performed with k-mer length 25 because it does not support multiple k-mer length assembly. PE assemblies with k-mer lengths of 21 − 63 with a step-size of 6 were generated using Oases-Velvet. The SOAPdenovo-Trans assembly was performed using k-mer lengths of 21 − 61 with a step-size of 10 (for SE short read assembly parameters see supplementary table S2, Supplementary Material online).
EviGene Pipeline
In order to reduce the data redundancy, assemblies were first processed either with built-in utilities of the assemblers or with CD-HIT-EST (version 4.6.1; Fu et al. 2012) using a threshold of 98% identity to remove nearly identical contigs. A slightly lower threshold than that used by Nakasugi et al. (2014) was chosen because several genotypes were mixed in the sequencing library. If this threshold were set too high, many variants would be retained, which would reduce the efficiency of the subsequent analysis steps. The merging utility of Trans-ABySS was applied for Trans-ABySS assembled contigs. Instead of using the Oases-Velvet merging utility, all Oases-Velvet contigs were pooled and clustered using CD-HIT-EST for both SE and PE read assemblies (table 1). For SOAPdenovo-Trans, the scaffolding option was not used, the SE and PE read assemblies were pooled, and the contigs were directly clustered with CD-HIT-EST. No merging or clustering was necessary for Trinity because it only implements one k-mer size.
Table 1.
Number of Transcripts Produced by the Individual Assembly Programs and Their Contribution to the Final Transcriptome
| Assembler | Total contigs | Contigs after CD-HIT-EST | Transcripts in okay-main set | Transcripts in alternative set |
|---|---|---|---|---|
| Trinity | 100,749 | 100,749 | 9,848 | 5,251 |
| Trans-ABySS | 185,991 | 185,991 | 4,962 | 13,729 |
| Oases-Velvet | 442,560 | 126,055 | 9,692 | 19,225 |
| SOAPdenovo | 489,649 | 170,562 | 8,401 | 9,644 |
| Total | 1,218,949 | 583,357 | 32,903 | 47,849 |
The contigs set resulting from pooling all assembler outputs was further processed with the EvidentialGene tr2aacds script (Gilbert 2013). This script from the EvidentialGene package (short EviGene pipeline) selects de novo assembled transcripts from the meta-assembly, based on coding potential. First, coding DNA sequences (CDS) and amino acid sequences are produced for each of the transcripts. Highly similar sequences are identified through a “BLAST on self” (default settings: 98% similarity or higher). The EviGene pipeline then outputs three sets of transcripts: “okay-main” for primary transcripts, “okay-alt” for alternative transcripts (variants may be found here) and “drops” for redundant or uninformative transcripts (i.e., perfect duplicates of okay-main transcripts, perfect fragments of okay-main transcripts, very short amino acid sequences). Unless stated otherwise, the transcript, coding sequences or translated protein sequences of the okay-main set of the EviGene pipeline were used for further quality assessment and downstream analyses.
Transcriptome Quality Assessment
The basic metrics used for quality assessment in genome sequencing projects such as N50 and contig length have limited validity for transcriptome quality analysis, because the expected transcript length is unknown for this species. Thus, the quality of the transcriptome assembly was assessed with multiple strategies in order to determine its completeness and reliability.
We first mapped the reads that were used to construct the transcriptome back to the assembled transcripts. This was done using the alignment tool NextGenMap (version 0.4.10; Sedlazeck et al. 2013) with the default parameters except for increased sensitivity (-i 0.8).
The Core Eukaryotic Genes Mapping Approach (CEGMA) (version 2.5; Parra et al. 2007) was used to assess how many of the 248 highly conserved core genes can be identified in the D. galeata de novo transcriptome. Furthermore, we annotated orthologs of genes that are presumably present in single copy in arthropod genomes. For this, we used the Benchmarking set of Universal Single-Copy Orthologs (BUSCO) (version 1.2; Simao et al. 2015) with the Arthropod BUSCO set from OrthoDB (version 7; Waterhouse et al. 2013), which contains all genes found in single copy in >90% of the included 38 species. Daphnia pulex was chosen as the reference species, as it is the closest relative to D. galeata that is contained in OrthoDB (genome version JGI060905). The D. pulex BUSCO set contains 2,421 genes that were then used to search the D. galeata de novo transcriptome, as well as the D. magna genome early release (version 7; Daphnia Genomics Consortium 2015).
In order to assess contamination in our transcriptome, we investigated how many and which transcripts might have their origin in other species. For this purpose, all amino acid sequences were blasted against the NCBI nonredundant (nr) database (downloaded Feb. 2015) with an E value cut-off of 1e-05. Subsequently, the results were loaded into MEGAN (version 5.8.4; Huson et al. 2011) to visualize in which species the sequences have their best hit based on the hierarchy of NCBI taxonomy.
Annotation and Functional Analysis
In order to obtain functional annotation of the D. galeata de novo transcriptome and be able to compare it to other species, OrthoMCL (version 2.0.9; Fischer et al. 2011) was used to cluster amino acid sequences of D. galeata, D. pulex (version JGI060905; Colbourne et al. 2011), D. magna (version 7; Daphnia Genomics Consortium 2015), as well as D. melanogaster (version 5.56; St. Pierre et al. 2014) and Nasonia vitripennis (version 1.2; Werren et al. 2010) into orthologous groups and determine “inparalogs”. The initial OrthoMCL BLAST step was run with an E value cut-off of 1e-05. In addition to the ortholog annotation, protein domains were annotated for all three Daphnia species using PfamScan (version 1.5) to search the Pfam A database (version 27.0; Finn et al. 2014) together with hmmer3 (version 3.1b; Mistry et al. 2013). Gene Ontology (GO; Ashburner et al. 2000) terms were transferred from the annotated domains with the Pfam2GO mapping (version 1.8; Hunter et al. 2009).
Pfam A domain annotations were also used for quality control. Transcripts containing annotated domains not found in any of the other Daphnia species or in any of 15 completely sequenced insect protein sets (supplementary table S3, Supplementary Material online) were manually checked for contamination. These transcripts were compared with those found to be of noncrustacean origin by MEGAN (version 5; Huson et al. 2011).
Proteins were also annotated with InterPro domains via InterProScan (version 5.13-52.0; Jones et al. 2014). We used all databases implemented in InterProScan with the exception of Coils. In addition to default parameters, the -pathways and -iprlookup options were set to annotate genes with InterPro terms and pathway identifiers. This additional domain annotation was done in order to both validate the Pfam annotation and increase the number of annotated proteins. This approach increases the statistical power of the over-representation analysis for the sex-biased genes. For the same reason, the results from the BLAST search against NCBI nr were used as input for BLAST2GO (version 3.0; Conesa et al. 2005). The GO terms transferred from Pfam domains are more reliable as they are based on functional modules that were experimentally validated. However, this approach can be too conservative because only genes that have an annotated Pfam A domain can be annotated with GO terms. Thus, the Pfam2GO annotation was complemented with the less stringent BLAST2GO annotation. As an additional source of functional terms, we annotated K-numbers for KEGG pathways using BLASTKOALA (release 72.0, accessed 1 Oct 2014; Kanehisa et al. 2014).
Differential Gene Expression
Trimmed 50bp single-end reads produced by sequencing the male and female samples were mapped to the D. galeata de novo transcriptome using NextGenMap with increased sensitivity (-i 0.8 - -kmer-skip 0 -s 0.0). Different mapping parameters were initially tested to find the best sensitivity threshold. By default, NextGenMap uses only a subset of the reference seeds to increase mapping speed. However, this can be disadvantageous for genomes with a high proportion of duplicated genes, which is why we increased sensitivity at the expense of computational time. With an identity of 80%, a large percentage of reads (∼90%) still map, whereas at the very conservative 95% identity, much more data is lost (10 − 20% more reads are discarded than at 80% identity). The required identity was increased from the default setting (65% identity) due to the high number of recent gene duplications that can be expected based on the D. pulex genome (Colbourne et al. 2011). For the same reason, all seeds were used to build the lookup table from the transcriptome and the highest sensitivity setting was used.
The statistical analysis of differentially expressed genes was done with DESeq2 (version 1.6.3; Love et al. 2014) as implemented in the Bioconductor package (version 3.0; Gentleman et al. 2004) in R (version 3.2.0; R core Team 2015). A two-factor design was used to account for the factors sex (male vs. female) and clone (J2 vs. M10). All P values were adjusted for multiple testing using the Benjamini–Hochberg correction (Benjamini and Hochberg 1995) as implemented in DESeq2. To reduce multiple testing, DESeq2 only considers genes that have a minimum number of reads. For our study design, this minimum was 12 mapped reads across all libraries. Furthermore, adjusted P values cannot be calculated for genes with high variation among replicates or low median expression across libraries (Love et al. 2014). A gene was considered sex-biased if the comparison for the factor sex yielded an adjusted P value of 0.05 or lower. The degree of sex or clone bias was determined by the fold-change difference between the sexes (or clones) as calculated in DESeq2. Based on this, sex-biased genes were binned into four groups: <2-fold, 2- to 5-fold, 5- to 10-fold, and >10-fold difference in expression. In addition to the two-factor analysis, a one-factor analysis was also used with DESeq2 to study the effect of sex and clone separately. All sex-biased genes in either the two- or the one-factor analysis can be found in supplementary table S9, Supplementary Material online.
Comparisons with D. pulex sex-biased genes were based on the differential gene expression data set from Colbourne et al. (2011). These sex-biased genes were detected on a tiling microarray and the number of genes in each of the fold-change groups mentioned above can be found in supplementary table S4, Supplementary Material online. For comparisons with sex-biased genes in D. melanogaster, we ran DESeq2 on an adult whole fly RNA-seq data set with two biological replicates per sex by Meisel et al. (2012) with the same parameters as for D. galeata (supplementary table S4, Supplementary Material online).
To infer the function of sex-biased genes, the annotations from Pfam, InterPro, and KEGG were used, along with any available GO terms. Over-representation of these annotated features was tested with Fisher’s exact test. To take into account the hierarchy between terms and the classification into the three separate GO categories (molecular function, cellular component, biological process), over-representation analysis for GO terms was done with the Bioconductor package topGO (version 2.18.0; Alexa and Rahnenfuhrer 2010) together with the GO hierarchy from the GO.db package (version 3.0.0, Carlson 2015). Multiple testing corrections (Benjamini and Hochberg 1995) were also performed in R using the “stats” package. To determine orthologs to D. pulex and the overlap of sex-biased genes between species, the clustering into orthologous groups from OrthoMCL was used. To test for the conservation of sex-biased genes, we compared the number of clustering orthologs and BLAST hits between female-biased gene (FBG) and male-biased gene (MBG), and between both sets of sex-biased genes and unbiased genes using Fisher’s exact test. Furthermore, within the MBG and FBG, we tested whether there was a relationship between the degree of sex bias (measured as the log2 fold-change in expression between the sexes) and a gene’s probability of having a BLAST hit or an ortholog. This was done using binomial generalized linear models (GLM) in R.
Evolutionary Rates
We analyzed the correlation of gene expression between the J2 and M10 clones using quantile normalized RPKM values for expressed genes (RPKM > 1). Spearman’s ρ was calculated for male and female libraries separately using the mean of the biological replicates. Furthermore, Spearman rank correlations were calculated for MBG, FBG, and unbiased gene separately based on the one-factor (separately for each clone) differential expression analyses. Genes were required to be sex-biased in at least one of the clones and genes with opposite sex bias were excluded from the analysis. Only genes classified as unbiased in both clones independently were used. Differences in Spearman correlations between groups of genes were identified using the test of the equivalence of two correlation coefficients after Fisher Z-transformation.
In addition to within-species comparisons, protein-coding sequences were compared between species. For this, 1-to-1 orthologs based on bidirectional best-hit BLAST against D. pulex proteins at a 1e-10 E value cut-off were determined. These protein sequences were then aligned with MUSCLE (Edgar 2004). Corresponding nucleotide sequences were aligned based on the protein alignment using tranalign (Rice et al. 2000) and Ka/Ks values were calculated using KaKs_Calculator (version 2.0; Wang et al. 2010) with the default parameters. Based on the two-factor differential expression analysis, Ka/Ks ratios of MBG and FBG were compared with each other and those of unbiased genes with the Wilcoxon test. Genes were classified as positively selected if they had Ka/Ks > 1 and P < 0.05 (Fisher’s exact test) as calculated by KaKs_Calculator.
Annotation of the Sex Determination and Juvenile Hormone Pathway
We searched the literature for key genes involved in sex determination and the switch from asexual to sexual reproduction in Daphnia. To annotate these genes in our D. galeata transcriptome, we used the domain annotations, the BLAST results and the OrthoMCL clustering. We compared the number of genes with those found in D. pulex and D. magna and searched the alternative transcript set from the EviGene pipeline for possible isoforms. Furthermore, we checked whether these genes showed differential expression between the sexes or clones and compared the intensity and direction of the bias in Daphnia species.
Sequence Availability
Raw sequence reads used for the de novo assembly (MiSeq and HiSeq) were deposited in the European Nucleotide Archive (ENA) (accessions PRJEB14950 and ERR1551394–ERR1551397). The experimental set up for the analysis of sex-biased genes is available in ArrayExpress (accession E-MTAB-4981). The assembled contigs (okay-main set) are available at the ENA as a Transcriptome Shotgun Assembly Project PRJEB14950. Annotation details (Pfam, nr BLAST, Uniprot), as well as the “okay-alt” set of transcripts are available upon request from the corresponding author. In order to retain the assembler information, the prefix “Dgal” was used for all transcripts names, followed by a character indicating the origin of the contig: “o” for Oases-Velvet, “t” for Trinity, “s” for SOAPdenovo, and “a” for Trans-ABySS.
Results
Transcriptome Sequencing
A total RNA sample from D. galeata from a mixture of 24 clonal lines from four different lakes (“assembly” set) was sequenced using the Illumina MiSeq platform, producing a total of 40.6 million reads. These consisted of roughly 20.3 million PE reads of 250bp length. After trimming and removing reads that were too short and/or contained ambiguous bases, 16.7 million reads remained for the assembly. The majority (12.1 million) of these pairs could be merged together using FLASH.
An additional 328 million SE RNA-seq reads of 50bp in length were generated from males and females of two clonal lines from two different lakes using the Illumina HiSeq platform. Depending on the library, 0.30 − 10.44% of the reads were excluded after trimming and quality control. In total, nearly 317 million reads were available for subsequent differential expression analysis, giving an average of 39.6 million reads per library (supplementary table S2, Supplementary Material online). In addition, 164.4 million reads from male libraries were used in the de novo transcriptome assembly.
De Novo Assembly
For the de novo transcriptome assembly, multiple assemblies with four different programs (Trinity, SOAPdenovo, Oases-Velvet, Trans-ABySS) and different k-mer sizes were combined. The de novo assemblers produced between 100,749 (Trinity) and 489,649 (SOAPdenovo) contigs, with a combined total of 1,218,949 (table 1). Applying CD-HIT-EST where necessary considerably reduced the redundancy of the data set; 583,357 contigs were merged together and further processed with the EviGene pipeline. The EviGene pipeline, used to merge different assemblies, classified 32,903 transcripts into the okay-main set and 47,849 transcripts into the alternative set. No particular assembler stood out as delivering very few or many transcripts, but there were differences among assemblers (table 1). Furthermore, Trinity was better in recovering the longest proteins: 532 of the 1,000 longest proteins were obtained with this assembler. The number of obtained transcripts agrees well with the number of described genes in the related species D. pulex (30,810) (Colbourne et al. 2011). In addition to the 32,903 transcripts, the tr2aacds script from the EviGene pipeline also produced a set of CDS and proteins. According to the output, we assembled 33,555 coding sequences and proteins. These numbers differ because one transcript from the okay-main set can have multiple open reading frames (ORFs) and thus result in multiple protein sequences.
Transcriptome Metrics and Quality
The EviGene pipeline bases contig selection on sequences’ coding potential. Thus, a greater protein length is expected from the EviGene pipeline assemblies compared with the assemblies obtained by individual programs. The average length of the EviGene protein okay-main set was 306 amino acids (median = 191). The average contig length of 1,189bp is slightly longer than for D. pulex (1,063bp). In general, the metrics for the D. galeata transcriptome are very similar to those for D. pulex (supplementary table S5, Supplementary Material online). Using NextGenMap, 86.83% of the PE reads and 86.16% of the SE male reads could be mapped back to the newly assembled transcriptome.
A statistical analysis of the longest 1,000 proteins in a transcriptome can be informative regarding the quality of the assembly. These very long proteins are described for several species, tend to be conserved, and are usually difficult to assemble completely. For D. galeata, the average length of the 1,000 longest proteins was 1,637 amino acids, which is lower than the observed maximum of around 2,000 amino acids for insects and crustaceans, and falls below that obtained for D. magna (2,080bp) (Daphnia Genomics Consortium 2015). Thus, D. magna appears to have longer and more completely assembled longest proteins. However, 85% of the longest proteins for D. galeata were classified as complete according to the EviGene pipeline classification algorithm. Furthermore, >99% of the 1,000 longest proteins had a homolog in D. pulex (BLASTx, E value ≤1e-07), which further supports their high quality.
CEGMA is an analysis tool to find ultra-conserved core eukaryotic genes (CEGs) that should be present in any de novo genome or transcriptome assembly. This analysis was carried out by performing a BLAST search of the CDS (okay-main set) obtained with the EviGene pipeline for 248 CEGs. To assess the completeness of the assembly, these results were compared with the number of CEGs in D. pulex and D. magna. For D. galeata, 80% of the CEGs were identified as complete in the CDS okay-main set, and this percentage increased to 93% when considering also partial hits. In comparison, more CEGs were identified in D. pulex (93% complete and 98% including partial hits) and D. magna (94% and 100%, respectively; table 2). However, considering that there is no genome sequence available for this species, the recovery for D. galeata CEGs was exceptionally high.
Table 2.
Quality Measurements to Assess Transcriptome Completeness
| Daphnia pulex | D. magna | D. galeata | |
|---|---|---|---|
| Complete CEGs | 231 (93%) | 234 (94%) | 197 (80%) |
| Partial CEGs | 12 (5%) | 13 (6%) | 34 (13%) |
| BUSCO (total) | 2,421 | 2,418 | 2,378 |
| 95 − 100% complete | 2,421 | 2,076 | 2,093 |
| 90 − 95% complete | 0 | 161 | 97 |
| 80 − 90% complete | 0 | 121 | 77 |
| 60 − 80% complete | 0 | 42 | 54 |
| 40 − 60% complete | 0 | 12 | 42 |
| <40% complete | 0 | 6 | 15 |
| No hits | 0 | 3 | 43 |
Another way to assess the quality of a genome or transcriptome is to quantify the duplication rate of highly conserved orthologs that are present in only a single copy in related species. This is important because genetic diversity, gene duplications, and/or the presence or multiple isoforms can lead to misassemblies, where one gene is represented by multiple contigs. The BUSCO analysis can be used to assess the frequency of such misassemblies. Applying BUSCO to the CDS okay-main set allowed the recovery of 2,378 D. galeata transcripts corresponding to 2,421 single-copy genes of D. pulex (98%; table 2). However, 59 of the recovered single copy orthologs (2%) were present in multiple copies. This is not unusual, as even the BLAST of D. pulex BUSCO genes against itself produced 2% multi-copy orthologs. Overall, the majority of the single-copy orthologs in D. galeata showed very high recovery, with 88% of the genes being over 95% complete (table 2).
Functional Annotation
We annotated the D. galeata transcriptome using BLAST, orthologous clustering, protein domain annotation, GO, and metabolic pathway annotation. In total, 24,641 of the 33,555 D. galeata proteins (73%) could be annotated with at least one of the methods used in this study. The BLAST search against NCBI nr yielded the largest number of annotations: 24,285 proteins (72%) had a good BLAST hit with E <1e-05. According to the MEGAN analysis, 88% of these (21,210) have their best BLAST hit to D. pulex proteins, 389 have their best hit to a bacterial sequence, and 97 are most similar to a plant protein. The others mainly have similarities with other arthropod sequences (supplementary fig. S1, Supplementary Material online).
Protein domain annotations yielded 16,237 annotated proteins with the Pfam database alone and 19,729 using all databases in InterPro except for Coils. GO terms based on Pfam2GO could be found for 10,431 proteins and K-numbers for the KEGG pathway analysis could be found for 7,192 proteins of the okay-main set. Furthermore, 22,887 proteins from the D. galeata okay-main set (68%) could be clustered in orthologous groups with proteins from D. pulex, D. magna, D. melanogaster, and N. vitripennis (see “Materials and Methods” section). In a total of 21,403 orthologous groups resulting from clustering of these five proteomes, 14,535 contain D. galeata proteins. The clustering shows that many Daphnia-specific groups (∼5,000 per species) contain sequences from at least two Daphnia species, but none from the insect species (fig. 1). The insect-specific groups are much fewer in comparison (∼1,500 depending on how many species were used). Although the inclusion of more insect proteomes results in a slight increase in the number of insect-specific clusters, it does not change the overall pattern (supplementary fig. S2, Supplementary Material online).
Fig. 1.—
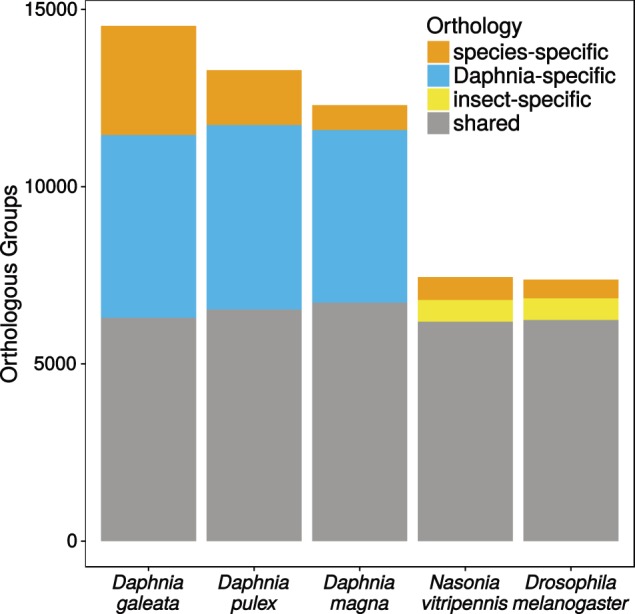
Number of orthologous groups per species that are shared between Daphnia and insects, are shared between multiple species within either the insects or Daphnia or contain only proteins from one species (species-specific).
Shared orthologs between the Daphnia species do not reflect their phylogenetic relationship, with D. pulex showing the greatest similarity to D. magna, rather than to D. galeata. Using the okay-main set of transcripts, 9,461 orthologous groups are shared by all three Daphnia species, 1,065 are shared by D. pulex and D. galeata, 1,080 are shared by D. pulex and D. magna, and 902 are shared by D. galeata and D. magna. However, the annotation qualities differ among the species and many D. magna genes were manually annotated based on the D. pulex annotation. This has not been done for D. galeata because the genome sequence is not available. If putative alternative transcripts for D. galeata are included (the okay-alt set), we find that there are more shared orthologous groups between D. galeata and D. pulex (1,193) than between D. galeata and D. magna (788) or between D. pulex and D. magna (985), correctly reflecting the phylogenetic relationship.
Among the protein domains annotated with Pfam A, we found 330 domain IDs that were present only in Daphnia, but not in insects. Of these 330 domains that are present in at least one of the three Daphnia species, 72 are found only in D. galeata (supplementary fig. S3, Supplementary Material online). These 72 different domain types are present in 83 proteins, of which 72 are mono-domain proteins. Of these proteins, 34 have no BLAST hit against NCBI nr, 10 have their best hit to plants and 23 to bacteria. Plant and bacterial sequences might reflect contaminations, as the animals were not treated with antibiotics before sequencing and had fed on algae the day before they were harvested. Horizontal gene transfer is also possible, but rather unlikely. Only three proteins have a BLAST hit to a protein of an insect species and might thus represent valid proteins. A complete D. galeata genome sequence will make it possible to evaluate the presence or absence of these genes.
Sex- and Clone-Biased Gene Expression
To compare gene expression between the sexes, adapter-trimmed 50bp SE reads from a total of four male and four female RNA-seq libraries were mapped to the D. galeata transcriptome (supplementary table S2, Supplementary Material online). The male and female libraries were prepared from two clonal lineages derived from different lakes: Jordan Reservoir, Czech Republic (clone J2) and Müggelsee, Germany (clone M10). Even though most assemblies that contributed to the transcriptome are based solely on long paired-end reads from females, we observe similar mapping efficiencies for the male and female libraries (91 − 92% and 92 − 94% of reads mapped, respectively), which suggests that our transcriptome has nearly equal representation of male and female transcripts.
Using a two-factor model that accounts for both sex and clone, we find a total of 10,253 differentially expressed genes (adjusted P < 0.05). Of these, 5,842 genes are differentially expressed between males and females, and 5,492 are differentially expressed between the clones. Of the sex-biased genes, 2,518 are MBG and 3,324 are FBGs (table 3). For the clone-biased genes, 2,873 are upregulated in M10 compared with J2, whereas the opposite case holds for 2,619 genes (table 3). There are 1,081 genes (10.5%) that are both sex- and clone-biased (supplementary fig. S4, Supplementary Material online). The majority of the sex-biased genes show only low expression differences between males and females, with almost half of the genes having a fold-change <2 (table 3). The 3,041 genes with more than a 2-fold expression difference between the sexes are depicted in figure 2. Some of these genes show highly sex-biased expression in one clone, but only a low degree of sex bias in the other. In contrast to sex-biased genes, clone-biased genes have a stronger expression difference between clone J2 and M10: the median fold-changes of sex- and clone-biased genes are 2.0 and 3.1, respectively (Wilcoxon test P < 0.001).
Table 3.
Numbers of Sex- and Clone-Biased Genes in Daphnia galeata and Their Degree of Bias as Determined in the Two-Factor Analysis
| All | <2-fold | 2- to 5-fold | 5- to 10-fold | >10-fold | |
|---|---|---|---|---|---|
| Sex-biased | 5,842 | 2,801 | 2,416 | 294 | 331 |
| MBG | 2,518 | 1,264 | 984 | 126 | 144 |
| FBG | 3,324 | 1,537 | 1,432 | 168 | 187 |
| Clone-biased | 5,492 | 1,276 | 2,634 | 884 | 698 |
| J2 | 2,619 | 685 | 1,187 | 419 | 328 |
| M10 | 2,873 | 591 | 1,447 | 465 | 370 |
Fig. 2.—
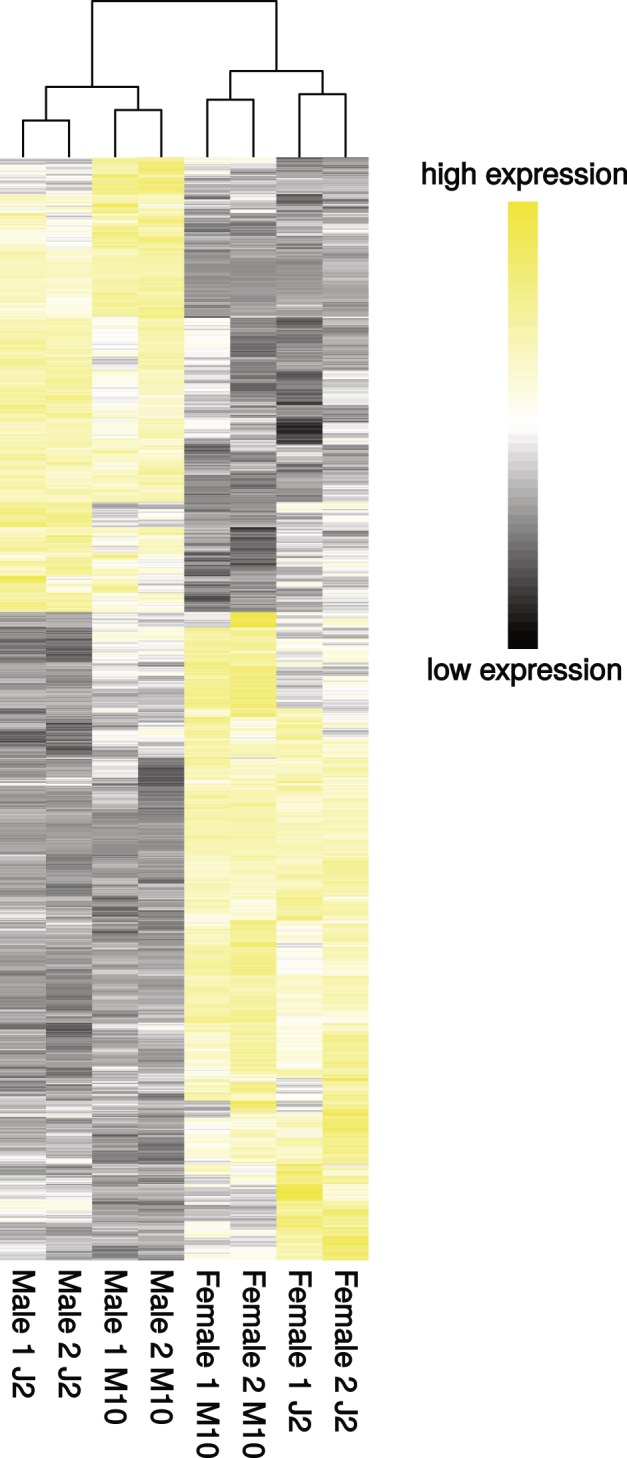
Heatmap showing all sex-biased genes with more than a 2-fold expression difference between males and females in the two-factor (combined) analysis. Clustering is based on Euclidean distances of RPKM values.
The one-factor analysis, in which the two clones were analyzed separately for sex-biased genes (see “Materials and Methods” section), revealed that clone J2 has many more sex-biased genes than clone M10. In the combined two-factor analysis, we find 5,842 sex-biased genes. Using only clone J2 leads to a similar number of 5,488 sex-biased genes (2,648 MBG and 2,840 FBG). However, using only clone M10 results in only 1,573 sex-biased genes (545 MBG and 1,028 FBG; fig. 3 and supplementary table S6, Supplementary Material online). If we exclude the genes that were already identified as sex-biased in the combined two-factor analysis, we find an additional 1,691 differentially expressed genes between males and females in clone J2, but only 106 additional differentially expressed genes in clone M10 (fig. 3). Consistent with the two-factor model, there are more FBG than MBG in the two individual one-factor analyses (supplementary table S6, Supplementary Material online). Interestingly, in clone J2, the majority of genes show low sex bias, similar to the two-factor analysis. In clone M10, however, there are only very few genes in the category with the lowest degree of sex bias (supplementary table S6, Supplementary Material online). The median fold change of sex-biased genes in clone M10 is twice as high as in clone J2 (4.6 and 2.3, respectively, Wilcoxon test, P < 0.001). The one-factor analyses identify 333 MBG and 736 FBGs that are significant in both clones. In addition, this analysis finds seven genes with the opposite sex bias in the two clones. Six of these are male-biased in clone J2, but female-biased in clone M10. The function of these genes is unknown. They either have no annotation and no BLAST hit or are homologous to “hypothetical proteins” of D. pulex. Two of the genes that are female-biased in J2 but male-biased in M10 harbor Pfam A domains. The gene Dgal_o31139t1 contains a homeobox domain and Dgal_o39721t1 contains an RNA binding domain. The latter is also more highly expressed (3.5-fold) in M10 than in J2 in the two-factor analysis. These domains could indicate that the proteins play a role in transcriptional regulation. Overall, the one-factor analyses reveal that the sex bias we observed is mainly driven by clone J2 (fig. 3 and supplementary table S6, Supplementary Material online). We also find that, in the one-factor analyses, there are more clonal differences between males than between females (3,453 and 2,249 clone-biased genes for males and females, respectively; Fisher’s exact test, P < 0.001; fig. 3 and supplementary table S6, Supplementary Material online).
Fig. 3.—

Results of the one-factor analysis showing sex-biased genes in each clone separately. (A) Dot plot showing the ratio of male/female expression for the J2 clone and the M10 clone, highlighting those genes that show differences in sex bias in the one-factor analysis compared with the two-factor (combined) analysis. Genes identified as sex-biased in the two-factor analysis or in the one-factor analysis for both clones are depicted in gray. Genes detected as sex-biased only in the J2 clone are shown in orange, whereas those detected as sex-biased only in the M10 clone are shown in yellow. Green dots indicate genes that are significantly female-biased in one clone, but significantly male-biased in the other. (B) Dot plot showing the ratio of M10/J2 expression for females and males. Genes identified as clone-biased in the two-factor analysis or in the one-factor analysis in both sexes are depicted in gray. Genes detected as clone-biased only in females are shown in orange, whereas those detected as clone-biased only in males are shown in yellow. Green dots indicate genes that are significantly M10-biased in one sex, but significantly J2-biased in the other. (C) Proportional Venn diagram for female-biased genes in the two-factor analysis and in each of the clones in the one-factor analysis. (D) Proportional Venn diagram for male-biased genes in the two-factor analysis and in each of the clones in the one-factor analysis.
GO and protein domain annotations were used to analyze the differentially expressed genes from the two-factor analysis for over-represented functional terms. Among the highly FBG (>10-fold), we found mainly terms related to lipid metabolism (5 genes) and protein-binding (24 genes). For highly MBG, only the term “serine-type endopeptidase inhibitor activity” is over-represented and found in three genes. Additionally, when looking at all sex-biased genes, MBGs show enrichment for multiple terms involved in G-protein coupled receptor signaling. Among the FBG, transcription factors and terms involved in nucleic acid binding, transcription and GTPase binding were found. A complete list of over-represented GO terms and Pfam domains is provided in supplementary tables S7 and S8, Supplementary Material online. The genes that are found in the one-factor analyses in both clones show very similar GO-term enrichment. MBG are also enriched for serine-type endopeptidase inhibitor activity and, additionally, for RNA binding. FBG in the one-factor analyses show the same terms for protein binding and lipid transport, but are also enriched for pseudouridine synthase activity, which is important in post-translational modification of cellular RNAs. As there were only few annotations with K-numbers in the KEGG analysis (21% of proteins have a K-number), no over-represented K-numbers could be found among the sex-biased genes. For the FBG, 1,212 genes (36%) could be annotated with K-numbers, whereas for the MBG only 420 genes (17%) could be annotated.
We find that more FBG than MBG have orthologs in other Daphnia or insect species (64% and 52% for FBG and MBG, respectively; Fisher’s exact test, P < 0.001) and the higher the sex bias, the less likely a gene is to have an ortholog (table 4). However, this is only significant for FBGs, probably due to the relatively low number of strongly MBGs (GLM, P < 0.05 and odds ratio = 0.84 for FBG, P = 0.07 and odds ratio = 0.88 for MBG). Not only are FBG more likely to have orthologs in Daphnia or insect genomes, but they also have more BLAST hits to the NCBI nr database (Fisher’s exact test, P < 0.001), suggesting that they are more conserved than MBG. Furthermore, highly sex-biased genes have fewer hits to the NCBI database, indicating that there is a negative relationship between sex-biased expression and evolutionary conservation (table 4; GLM, P < 0.001 and odds ratio = 0.75 for FBG, P < 0.001 and odds ratio = 0.74 for FBG). Comparing sex-biased genes in D. galeata with those found in D. pulex shows that 1,280 genes are FBG in both species (39% of FBG in D. galeata), whereas 601 are MBG in both species (24% of MBG in D. galeata). The direction of the sex bias is different for 192 D. galeata genes. Ninety-five genes are male-biased in D. galeata but female-biased in D. pulex, whereas 96 show the opposite pattern. The genes that differ in sex bias between Daphnia species are not limited to those with weakly sex-biased expression, as 44% show >2-fold sex bias in D. galeata and 9% show >5-fold sex bias. The genes that are male-biased in D. galeata, but female-biased in D. pulex, do not show an enrichment of any functional terms. However, the genes that are female-biased in D. galeata, but male-biased in D. pulex, are enriched for collagens and genes that play a role in calcium-dependent phospholipid binding.
Table 4.
Conservation of Sex-Biased Genes in Daphnia galeata Based on Homology to Other Arthropod Species
| Orthologous groups |
BLAST hit |
|||
|---|---|---|---|---|
| Degree of bias | FBG | MBG | FBG | MBG |
| All | 64% | 52% | 87% | 65% |
| >10-fold | 49% | 41% | 73% | 40% |
| 5- to 10-fold | 56% | 47% | 85% | 60% |
| 2- to 5-fold | 51% | 46% | 85% | 65% |
| <2-fold | 79% | 58% | 90% | 69% |
Evolutionary Rates
The annotation and orthology mapping indicated differences in conservation between FBG and MBG between species, with MBGs being less conserved. To see if this pattern was present within species, we investigated gene expression divergence between the J2 and M10 clones. Even though the number of sex-biased genes differs greatly between the clones (fig. 3), expression between clone J2 and M10 is highly correlated in both males and females (Spearman’s ρ = 0.9 in both sexes, fig. 4A and B). We also looked at the correlation of gene expression between the clones for the different classes of sex-biased genes. If MBGs evolve faster in their expression level than unbiased genes (UBG), we would expect lower correlations between clones for MBGs. Our data indeed support this. While unbiased genes show the highest degree of expression correlation (test of the equivalence of two correlation coefficients using Fisher Z-transformation, P < 0.001 compared with MBG and FBG), the effect is not large (Spearman’s ρ only 0.02 higher than for FBG and MBG). However, MBG and FBG do not differ significantly (P = 0.72, fig. 4C). Hence, within species, we do observe faster evolution of gene expression in MBGs, as well as in FBGs compared with genes with unbiased expression.
Fig. 4.—
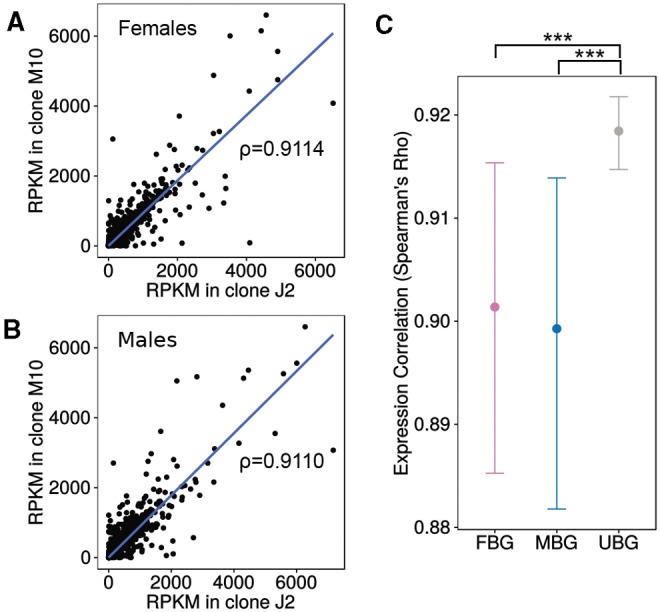
Divergence between clones J2 and M10. (A) Expression correlation between females of clone J2 and clone M10 including Spearman’s ρ. (B) Expression correlation between males of clone J2 and clone M10 including Spearman’s ρ. (C) Expression correlation between clones for male-biased, female-biased, and unbiased genes as a measure of gene expression divergence. Error bars indicate 95% confidence intervals for 1000 bootstrap replicates. Significance was determined by a test of the equivalence of two correlation coefficients after Fisher Z-transformation. *P< 0.05, **P < 0.01, ***P< 0.001.
In order to compare rates of protein evolution of sex-biased genes between species, we determined 1-to-1 orthologs between D. galeata and D. pulex and calculated the ratio of the nonsynonymous and synonymous divergence rates (Ka/Ks). Ka/Ks ratios could be calculated for 7,442 unbiased, 1,409 FBGs, and 815 MBGs (with sex bias determined in the combined two-factor analysis). First, we compared the different categories of sex-biased genes to one another and to unbiased genes. MBGs show significantly higher Ka/Ks than unbiased genes (Wilcoxon test, P < 0.05), whereas FBGs show significantly lower Ka/Ks than unbiased genes (Wilcoxon test, P < 0.001; fig. 5A). There were six genes with Ka/Ks values significantly >1 (supplementary table S9, Supplementary Material online). Interestingly, three of these genes are strongly female-biased (all with a fold change > 20), whereas the other three are unbiased. The first two of these female-biased genes are also female-biased in D. pulex, but the third gene was not present on the tiling array used for sex-biased gene expression and thus, we have no information about its expression in D. pulex. One of the unbiased genes in D. galeata with a Ka/Ks value >1 is female-biased in D. pulex, whereas the other two are also unbiased in D. pulex. None of these genes has a known function, but four of them have a BLAST hit to a hypothetical D. pulex protein. One of the unbiased genes has a Pfam A domain and a corresponding GO term indicating that it may be involved in protein binding and chromatin mediated gene regulation.
Fig. 5.—

Divergence between Daphnia galeata and D. pulex measured as Ka/Ks ratios between 1:1 orthologs. (A) Sequence divergence for male-biased, female-biased, and unbiased genes. (B) Sequence divergence for the different categories of sex-biased genes according to the degree of sex bias. Outlier genes with values > 0.8 are not shown on the plot, but were included in the statistical analysis. Significance was determined by a Wilcoxon test. *P< 0.05, **P < 0.01, ***P< 0.001.
Second, we tested for the relationship between the degree of sex bias and Ka/Ks within the MBG and FBG, respectively. We find a significant, albeit weak, positive correlation between the log2 expression fold change and Ka/Ks for both FBG and MBG (Spearman rank correlation, P < 0.001 for both MBG and FBG; supplementary fig. S5, Supplementary Material online). Hence, the stronger the sex bias, the faster the gene evolves at the protein level (fig. 5B). This is in agreement with our identification of orthologs in other species, where we found that highly sex-biased genes have fewer orthologs than lowly sex-biased genes.
Genes Involved in Sex Determination and the Juvenile Hormone Pathway
We annotated genes that are putatively involved in the sex determination pathway and the juvenile hormone (JH) pathway in D. galeata. Orthology and annotation information were used to identify these genes and we searched the EviGene alternative set for possible isoforms. Furthermore, we tested all genes for differential expression between the sexes or clones, as especially the sex determination genes are known for their sex-specific splicing and expression. We were able to find orthologs of all key genes in these pathways (table 5), which supports the completeness of the transcriptome.
Table 5.
Daphnia galeata Orthologs of Genes Involved in the Sex Determination and Juvenile Hormone Pathways Including Their Isoforms and Differential Expression Information
| Gene name | D. galeata gene ID | Differential expression | D. pulex gene ID |
|---|---|---|---|
| sex-lethal | Dgal_a24_b_768893 | – | JGI_V11_299722 |
| Transformer | Dgal_a30_f_668354a | FBG (<2-fold) | JGI_V11_324323 |
| doublesex1 | Dgal_s258843 | MBG (>10-fold) | JGI_V11_225264 |
| doublesex2 | Dgal_s190135 | MBG (>10-fold) | JGI_V11_299653 |
| Fruitless | Dgal_a80_b_445118b | FBG in J2 (2- to 5-fold) | JGI_V11_290551 |
| fruitless-like | Dgal_s441825 | MBG in J2 (<2-fold) | |
| farnesoic acid O-methyltransferase | Dgal_o10886t1 | – | JGI_V11_208220 |
| juvenile hormone acid O-methyltransferase | Dgal_o22519t1 | – | JGI_V11_300180 |
aIsoform: Dgal_s424561.
bIsoforms: Dgal_ a34_b_717420, Dgal_o5567t3, Dgal_t24935c0t6.
The JH pathway is an essential signaling pathway involved in many functions in arthropods. Male sex-determination in daphnids is initiated by environmental cues leading to synthesis of MF, an analog of JH III produced in the mandibular gland. MF signals are perceived by a methyl-farnesonate receptor (MfR) (LeBlanc and Medlock 2015; Toyota, Miyakawa, Yamaguchi, et al. 2015) leading to a downstream cascade that ultimately produces the male phenotype. We thus searched the D. galeata transcriptome for genes from the JH pathway that have been shown to contribute to MF synthesis and signaling.
Farnesoic acid O-methyltransferase (FAMT) is the key regulator in MF biosynthesis in insects and crustaceans. A FAMT gene has been identified in D. pulex (Colbourne et al. 2011). A putative D. galeata FAMT gene has been identified based on OrthoMCL clustering and BLAST results. It shows no differential expression or alternative transcripts in the D. galeata transcriptome. An alternative path for JH III biosynthesis involves the conversion of farnesoic acid (FA) to JH III acid instead of MF and then further conversion to JH III. This second step is catalyzed by the protein juvenile hormone acid O-methyltransferase (JHAMT) (Shinoda and Itoyama 2003). A candidate JHAMT gene in D. galeata clusters with the D. melanogaster, D. pulex, and D. magna genes. This gene does not show any sex bias and no alternative transcripts were found.
A putative sex-lethal (sxl) homolog has been identified in D. pulex (Kato et al. 2010) and also in the D. galeata transcriptome. We found no alternative transcripts and sxl is the only gene from the sex determination pathway that does not show any sex-biased expression in either the two-factor or the one-factor analyses. In D. magna and D. pulex, only one tra gene, the target of sxl, has been identified and, in contrast to other species, only one isoform is expressed in both sexes (Kato et al. 2010; Chen, Xu et al. 2014). In the D. galeata transcriptome, we also identified only one tra homolog. Although we found a putative tra splice variant in the alternative set, this transcript is shorter than the main transcript due to missing amino acids at the C-terminus and could be an assembly artifact. For the D. galeata tra homolog, we observe a slight female bias in expression. In the one-factor analysis, this was only significant for clone J2, but M10 has a similar fold change. We find two dsx orthologs in the D. galeata transcriptome, both showing extremely male-biased expression with 11- and 34-fold upregulation in males compared with females, respectively (consistent in the two-factor and one-factor analyses). We found two putative fru homologs in the D. galeata transcriptome. One is annotated as fru and clusters with the D. pulex and D. magna fru and the other one is annotated as “sex determination protein fruitless like” (fru-like) and also clusters with a homolog in D. pulex. None of the genes are differentially expressed between the clones in the two-factor analysis. However, the one-factor analysis reveals that fru is female-biased and fru-like is male-biased in clone J2. The alternative set contains three putative splice variants of fru. These may be true variants, as alternative splicing of fru has also been reported in D. melanogaster (Hoshijima et al. 1991; Heinrichs et al. 1998).
Discussion
We present here the first comprehensive RNA-seq study of sex-biased gene expression in Daphnia from females and males without MF exposure. While MF frequently is used to induce male production in the laboratory, it is not clear how induced and naturally produced males differ. Nevertheless, some differences between MF-induced and naturally produced males have been described. For example, MF-induced males show delayed development and decreased fecundity (Olmstead and LeBlanc 2002; Tatarazako et al. 2003; Ginjupalli and Baldwin 2013). We could also observe this in a preliminary study on our own D. galeata populations. A few clones were kept in control conditions or in medium with MF at a concentration of 400nM. Under MF, D. galeata had fewer offspring (if any) and the mortality rate was much higher. Whereas most previous studies in Daphnia were conducted using microarrays designed from female-derived mRNA, the reference transcriptome for our RNA-seq study was assembled from both female and male mRNA sequences. This increased the number of male RNA-seq reads that could be mapped to the transcriptome: ∼5% more reads from each male library could be mapped to this transcriptome than to a female-derived transcriptome. Thus, we had similar mapping efficiencies in both sexes, allowing identification of strongly male-biased and male-limited genes.
We also present a high quality transcriptome with detailed annotations for the freshwater crustacean D. galeata. This represents a valuable contribution to the available Daphnia genomic resources for at least two reasons. First, its sister species, D. pulex, has become a model organism for studies of phenotypic plasticity and ecotoxicology, as well as for genetics (e.g., to study gene duplication in adaption to new environments) (Colbourne et al. 2011). However, to assess which features of this genome are lineage-specific and which are shared with other species of crustaceans, it is important to extend genomic studies to other Daphnids. Furthermore, additional genomes and transcriptomes make comparative genomic studies possible. Second, all three species for which genome/transcriptome sequences are available (including D. galeata) are dominant phytoplankton grazers in very different habitats: D. pulex is found in large water bodies in Northern America, D. magna occupies all kinds of small ponds and temporarily freshwater bodies in Europe, and D. galeata belongs to a species complex found in permanent freshwater bodies of all sizes in Europe (for an overview see Flössner 1972). Our study thus extends the ecological breadth that is covered and allows for more comparative studies within the Daphnia genus, whereas providing new resources for molecular ecology studies in Europe.
Transcriptome Quality
De novo transcriptome assembly without an available genome presents challenges, including resolving the different transcript isoforms and closely related paralogs. For species with a high gene duplication rate, like those from the genus Daphnia, this task is far from trivial (Colbourne et al. 2011; Nakasugi et al. 2014). We used the tr2aacds script from the EviGene transcriptome assembly pipeline to create a nonredundant, coding-potential optimized set of 32,903 transcripts. This pipeline has been applied for transcriptome assembly in other species, such as Nicothiana benthamiana (Nakasugi et al. 2014), Eleusine indica (Chen, McElroy et al. 2014), and alfalfa cultivars infected with root-knot nematodes (Postnikova et al. 2015), providing biologically useful sets of transcripts for comparative studies.
Transcriptomes are evaluated based on the percentage of reads that map back to the obtained assembly and/or the recovery of conserved genes (Smith-Unna et al. 2015). Recently, Honaas et al. (2016) assessed which quality measurements are the most relevant for high quality transcriptomes based on a comparative study using 90 transcriptomes assembled with six different programs. They concluded that the combination of a high proportion of reads that map back to the assembly, a high proportion of identifiable conserved genes, N50 values close to expected median gene length and obtaining more contigs than the estimated number of genes are the most informative quality features that distinguish high quality assemblies. We used a similar strategy to evaluate our transcriptome.
When performing a quality control analysis, the unusual properties of Daphnia genomes need to be taken into account, as they could cause errors in the assembly (Colbourne et al. 2011). Most importantly, the D. pulex genome has been shown to contain many recent gene duplications with low divergence between the paralogous copies. It is reasonable to expect a similar situation in D. galeata. The assembly of closely related paralogs is a difficult computational problem (Gilbert 2013) and could complicate downstream analysis (Asselman et al. 2016). In addition, our assembly was performed using a pool of individuals from different populations. Thus, we expect some level of polymorphism within individual genes. This can contribute to reduced map-based scores because higher levels of mismatches are expected than when using more homogeneous samples. To reduce the impact of divergence on mapping quality scores, we used NextGenMap for mapping, which has been shown to perform well even when levels of divergence are high (Neme and Tautz 2016). With this strategy, a high percentage (∼86%) of the RNA-seq reads could be mapped back to our de novo transcriptome indicating that it includes a large proportion of the expressed genes in D. galeata.
The high recovery rates of BUSCO genes in our assembly and the CEGMA analysis further indicate that the transcriptome is nearly complete. In these analyses, the numbers are slightly lower than for D. pulex and D. magna (table 2), which is not surprising given that genome sequences are available for these two species. The D. galeata transcriptome shows a relatively low rate of fragmented transcripts as the total number of transcripts (32,903) is similar to the number of protein-coding genes in D. pulex (30,810) and D. magna (29,127), and is 16 times smaller than before the removal of redundant transcripts. BUSCO results indicate that the redundancy in our transcriptome is also similar to D. pulex and D. magna. In addition to the recovery of conserved orthologs in the BUSCO and CEGMA data sets, many genes that are usually highly conserved in arthropods or were identified in Daphnia in previous studies are also present in the D. galeata transcriptome. These include sex-determining genes, genes involved in the response to juvenile hormone, and methyl-transferases.
We were able to annotate the majority of genes (73%). The MEGAN analysis showed that the proportion of proteins that have their best BLAST hit outside the arthropods, and thus may represent putative contamination, is low (7%). However, this might underestimate non-D. galeata transcripts, because 28% of all proteins have no BLAST hit against NCBI nr and may also contain sequences from other species. The Pfam analysis also indicated that the majority of putatively gained domains might be the result of contamination. However, this phylogenetic incongruence only concerns 36 proteins and an alternative explanation for their occurrence in D. galeata could be horizontal gene transfer. More detailed studies of these proteins, as well as the sequencing of the D. galeata genome, will help elucidate the origin of these sequences.
Sex-Biased Gene Expression
We find that only a relatively small proportion of genes in the D. galeata transcriptome shows sex-biased gene expression (18%). If we consider the clones individually, as was done in the one-factor analysis (fig. 3), the percentage is even lower for M10 (5% of all genes are sex-biased). If a more conservative cut-off is applied to the differentially expressed genes from the two-factor analysis and only genes with an expression difference of 2-fold or more between the sexes are considered, then only 9% of the genes are classified as sex-biased. These results are similar to those observed in D. pulex, where 25% of the genes are classified as sex-biased with a P value cut-off of 0.05, and 10% are classified as sex-biased with a fold-change cut-off of 2 (supplementary table S4, Supplementary Material online). The proportion of sex-biased genes is much less than that reported for Drosophila species where as many as two-thirds of all genes can show sex-biased expression when whole animals are assayed (Gnad and Parsch 2006; Jiang and Machado 2009; Meisel et al. 2012). However, in D. melanogaster, the proportion of genes showing sex-biased expression can vary greatly among tissues (Huylmans and Parsch 2015). A possible explanation for the low proportion of sex-biased genes in D. galeata is that there is very little sexual dimorphism outside of the reproductive organs and that the gonads are smaller relative to body size than they are in Drosophila. However, ripe ovaries (just before oviposition) also occupy a large section of the ventral cavity in Daphnia. Another possible explanation is that cyclic parthenogenetic reproduction leads to a loss of sex-biased gene expression, especially among the highly sex-biased genes. This has been observed in aphids, as well as in hermaphrodite species of Caenorhabditis, where either the lower frequency of sexual reproduction or self-fertilization has led to a feminization and overall desexualization of gene expression (Thomas et al. 2012; Jaquiéry et al. 2013). Since the Daphnia genome experiences selection mainly in a female background and only rarely in a male background, one might also expect the loss of strongly MBGs, especially if their expression is detrimental to females. Thus, the low number of sex-biased genes, their low degree of sex bias, and the excess of FBG relative to MBG could be a consequence of cyclic parthenogenesis, which leads to a relaxation of purifying selection on MBGs. However, more studies are needed to test this hypothesis, especially in the light of the high variation in sex-biased expression that is observed between clones.
We find that D. galeata FBG are more likely to have orthologs in other species and they are also more likely to be female-biased in D. pulex (table 4). This indicates that MBG evolve faster than FBG in Daphnia, a pattern also observed in many other species (Eads et al. 2007; Ellegren and Parsch 2007; Parsch and Ellegren 2013). In general, highly sex-biased genes have fewer orthologs and are less conserved, indicating fast evolution. Whether this is a result of positive selection or of relaxed selective constraint is still unclear. Within the species (i.e., between the clones), we found that both FBG and MBG have slightly lower expression conservation than UBG. However, there is no difference in conservation of expression levels between MBG and FBG (fig. 4C). Furthermore, FBG show significantly greater sequence conservation between species (fig. 5A). This result is consistent with FBG being subject to greater purifying selection on the sequence level. Alternatively, these patterns could be the result of FBG often having housekeeping functions and, in general, showing greater pleiotropy in many species (Larracuente et al. 2008). Furthermore, the fact that FBG are more frequently exposed to selection than MBG due to the skewed sex ratio in Daphnia could also contribute to this pattern. However, the skewed sex ratio cannot fully explain the observations, because the Ka/Ks ratios of FBGs are also lower than those of unbiased genes, which should be subject to selection at least as frequently as FBGs. MBGs, on the other hand, show elevated rates of sequence evolution which is in line with the lower conservation of these genes (Ellegren and Parsch 2007; Parsch and Ellegren 2013, and citations therein). Even though FBG are on an average more conserved, we find three FBG, but no MBG, with Ka/Ks significantly >1, which is indicative of positive selection (table 4). In addition, there are three other genes with Ka/Ks rates consistent with positive selection, of which at least one is female-biased in D. pulex. Furthermore, we could also show that highly sex-biased genes evolve faster than weakly sex-biased genes and that this pattern is observed not only for MBG, but also for FBG (fig. 5B). Thus, it is possible that adaptive evolution also occurs more frequently in females than in males.
We find strong clonal differences in the number and identity of sex-biased genes between the two clones, J2 and M10 (fig. 3). Since the RNA used for sequencing was extracted from a pool of individuals collected over the same time span for both clones, we can exclude a sampling effect. Furthermore, because the number of RNA-seq reads was very similar for the two clones, the difference in the number of sex-biased genes cannot be explained by differences in statistical power. Thus, the difference in sex-biased gene expression between the clones appears to have a biological basis. The two clones came from different habitats, the Jordan Reservoir in Czech Republic and the Müggelsee in Germany, respectively. Although Daphnia disperse over long distances, gene flow is limited (see De Meester et al. 2016 for a recent review) and local adaptation and founder effects may influence phenotypic and genotypic divergence between locations greatly. We therefore hypothesize that the two clones differ in their investment in sexual reproduction, which may lead to the activation of different genes through the use of different molecular mechanisms. Lineage-specific differences in reproductive strategy are supported by our observation that in the laboratory, the clones produced males at different rates, with males being more frequent in clone M10 than in J2. In contrast, clone J2 produced more ephippia than M10. In addition to differing reproductive strategies, drift is probably stronger and purifying selection weaker in males due to their relative scarcity. This may result in reduced selective-constraint on the sequence of MBGs and on the level of gene expression in males. This is supported by the fact that we found significantly more clonal differences in expression between males than between females when conducting a one-factor analysis (fig. 3 and supplementary table S6, Supplementary Material online) and generally lower conservation and faster evolutionary rates of MBGs (table 4 and fig. 5). Finally, differences between clones may have been more apparent in our study because of the natural male production. By inducing male production, artificially added MF may elicit a more homogenous response in the clonal lines that have been studied previously, therefore obscuring differences among clones.
Juvenile Hormone Pathway and Sex Determination Pathway
We found two key genes from the JH pathway in the D. galeata transcriptome, FAMT and JHAMT. Neither of these have any isoforms or sex-biased expression in either the two-factor or the one-factor analyses. FAMT converts FA to MF in many insect species (Wainwright et al. 1998; Gunawardene et al. 2002; Bellés et al. 2005). However, Toyota, Miyakawa, Hiruta, et al. (2015) suggested that FAMT is not involved in MF synthesis in D. pulex and that JHAMT catalyzes the final step of MF synthesis in Daphnia instead of converting JH III acid into JHIII, which is its known function in insects (Shinoda and Itoyama 2003). As JH III does not appear to be produced in Daphnia (Daimon and Shinoda 2013; Miyakawa et al. 2014), the exact function of JHAMT remains to be determined. Although FAMT and JHAMT orthologs are present in D. galeata, the lack of differential expression between sexes in our study indicates both proteins might have lost their role in the JH pathway in D. galeata as well.
Overall, the JH pathway components show strong similarities among Daphnia species, indicating that the switch between asexual and sexual reproduction is also likely to be conserved. Daphnia galeata strains that spontaneously produce males under standard laboratory conditions may help to shed more light on the factors involved in the perception of environmental stimuli and the resulting signal transduction upstream of MF. A similar approach has already been used in D. pulex for a strain that produces males under short light conditions (Toyota, Miyakawa, Hiruta, et al. 2015; Toyota, Miyakawa, Yamaguchi, et al. 2015). An interesting aspect of D. galeata, however, is that some clones from the same population produce males under standard laboratory conditions, whereas others do not. Hence, clonal differences in reproductive strategy can be studied within the same population.
Although we were able to identify a homolog for the sxl gene, it is the only gene from the sex determination pathway that does not show any sex-biased expression in either the two-factor or the one-factor analyses. This lack of sex-specific expression was also found in another crustacean, the Chinese mitten crab (Shen et al. 2014). To our knowledge, the expression of sxl has not been studied in other Daphnia species. Similar to D. magna and D. pulex, we could identify only one tra gene in D. galeata. The species differ at the gene expression level, however. In D. pulex, expression of tra was shown to be higher in males and ephippial females compared with parthenogenetic females (Chen, Xu et al. 2014), whereas we observe a slight female bias in D. galeata. This could represent a difference between species or it could be that females used in this study were reproducing sexually and there is an expression difference between sexually and asexually reproducing females. If this is the case, tra would be a good candidate gene to study the switch to sexual reproduction, as has already been suggested for D. pulex (Chen, Xu et al. 2014). Like other members of the Daphniidae family (Toyota et al. 2013), D. galeata possess two dsx copies. While dsx1 in D. magna seems to be necessary for male trait development, the function of dsx2 is still unknown (Kato et al. 2011). The duplication predates the species split and represents a cladoceran-specific event (Toyota et al. 2013). In D. magna, both copies showed male-biased expression (Toyota et al. 2013), which is in accordance with our findings for D. galeata. The two homologs of the fru gene in D. galeata show opposite sex bias in one of the analyzed clones. Hence, while fru and fru-like may not be involved in the general mechanism of sex determination in D. galeata, they may play a role in different investment strategies in sexual reproduction in the clones. However, to determine if D. galeata indeed possess two copies of this gene, if multiple isoforms of fru are present, and if they play a role in influencing investment in sexual reproduction will require further experimental investigation.
The fact that we were able to annotate all key genes of the sex determination and JH pathways speaks for the completeness of our de novo assembly, and the gene expression results fit very well with previous studies in Daphnia. In all three Daphnia species for which genomes and/or transcriptomes are currently available, the sex determination pathway is highly conserved and no species-specific gene duplications have taken place. This suggests that the mode of sex determination is conserved across the genus. Because the number and identity of sex-biased genes differ drastically between the two D. galeata clones used in our study, it is possible that the two clones have different reproductive strategies. Given their differences in sex-biased expression between clones, fru and fru-like are potential candidate genes for the clonal differences in investment in sexual reproduction, and the opposite sex bias of the two paralogs in the J2 clone might be indicative of (partially) resolved sexual antagonism. However, we cannot rule out the possibility that fru and fru-like expression are influenced by another, not-yet-identified upstream factor that differs between the two clones.
Supplementary Material
Supplementary figures S1–S5 and tables S1–S9 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors would like to thank I. Schrank, L. Theodosiou, M. Kredler, C. Laforsch, J. Wolinska, J. Griebel, R. Jaenichen, and K. Otte for providing the necessary resources and help for maintaining Daphnia cultures in the laboratory. H. Lainer supported us for the molecular laboratory work. D. Gilbert and J. K. Colbourne contributed ideas for the bioinformatics analysis, and L. Hardulak did the orthology mapping including more insect species. This study was financially supported by individual grants from the Volkswagen Stiftung (to M.C.), the Deutsche Forschungsgemeinschaft (grant PA 903/6 to J.P.) and the DAAD (to A.K.H.). This work benefits from and contributes to the Daphnia Genomics Consortium.
Literature Cited
- Alexa A, Rahnenfuhrer J. 2010. topGO: enrichment analysis for Gene Ontology. R package version 2.18.0.
- Anderson EC, Thompson EA. 2002. A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160:1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, et al. 2000. Gene Ontology: tool for the unification of biology. Nat Genet. 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselman J, De Coninck DI, Pfrender ME, De Schamphelaere KA. 2016. Gene body methylation patterns in Daphnia are associated with gene family size. Genome Biol Evol. 8:1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellés X, Martín D, Piulachs MD. 2005. The mevalonate pathway and the synthesis of juvenile hormone in insects. Annu Rev Entomol. 50:181–199. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B. 57:289–300. [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres CE. 1998. Interspecific variation in the abundance, production and emergence of Daphnia diapausing eggs. Ecology 79:1699–1710. [Google Scholar]
- Carlson M. 2015. GO:db: a set of annotation maps describing the entire gene ontology. R package version 3.0.0.
- Carpenter S, et al. 1987. Regulation of lake primary productivity by food web structure. Ecology 68:1863–1876. [DOI] [PubMed] [Google Scholar]
- Carvalho G, Hughes R. 1983. The effect of food availability, female culture-density, and photoperiod on ephippia production in Daphnia magna Strauss (Crustcea: Cladocera). Freshwater Biol. 13:37–46. [Google Scholar]
- Chen P, Xu SL, et al. 2014. Cloning and expression analysis of a transformer gene in Daphnia pulex during different reproductive stages. Animal Reprod Sci. 146:227–237. [DOI] [PubMed] [Google Scholar]
- Chen S, McElroy JS, Dane F, Peatman E. 2014. Optimizing transcriptome assemblies for Eleusine indica leaf and seedling by combining multiple assemblies from three de novo assemblers. Plant Genome 8:1. [DOI] [PubMed] [Google Scholar]
- Colbourne JK, et al. 2011. The ecoresponsive genome of Daphnia pulex. Science 331:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. [DOI] [PubMed] [Google Scholar]
- Connallon T, Clark AG. 2011. The resolution of sexual antagonism by gene duplication. Genetics 187:919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daimon T, Shinoda T. 2013. Function, diversity, and application of insect juvenile hormone epoxidases (CYP15). Biotechnol Appl Biochem. 60:82–91. [DOI] [PubMed] [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daphnia Genomics Consortium. 2015. Daphnia magna Gene-ome early release. Available from: http://server7.wfleabase.org/genome/ Daphnia_magna/openaccess/.
- De Meester L, Vanoverbeke J, Kilsdonk LJ, Urban MC. 2016. Evolving perspectives on monopolization and priority effects. Trends Ecol Evol. 31:136–146. [DOI] [PubMed] [Google Scholar]
- Eads BD, Colbourne JK, Bohuski E, Andrews J. 2007. Profiling sex-biased gene expression during parthenogenetic reproduction in Daphnia pulex. BMC Genomics 8:464.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D. 2005. Ecology, epidemiology, and evolution of parasitism in Daphnia. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 8:689–698. [DOI] [PubMed] [Google Scholar]
- Finn RD, et al. 2014. The Pfam protein families database. Nucleic Acids Res. 42(D1):D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, et al. 2011. Using OrthoMCL to assign proteins to orthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics 35:6.12.1–6.12.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flössner D. 1972. Kiemen- und Blattfüssler, Branchiopoda, Fischläuse, Branchiura. Jena: VEB Gustav Fischer Verlag. [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert D. 2013. EvidentialGene - Evidence Directed Gene Construction for Eukaryotes. Available from: https://sourceforge.net/projects/ evidentialgene/.
- Ginjupalli GK, Baldwin WS. 2013. The time- and age-dependent effects of juvenile hormone analog pesticide, pyriproxyfen on Daphnia magna reproduction. Chemosphere 92:1260–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F, Parsch J. 2006. Sebida: a database for the functional and evolutionary analysis of genes with sex-biased expression. Bioinformatics 22:2577–2579. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardene YINS, et al. 2002. Function and cellular localization of farnesoic acid O-methyltransferase (FAMeT) in the shrimp, Metapenaeus ensis. Eur J Biochem. 269:3587–3595. [DOI] [PubMed] [Google Scholar]
- Hebert PD. 1978. The population biology of Daphnia (Crustacea, Daphnidae). Biol Rev. 53:387–426. [Google Scholar]
- Heinrichs V, Ryner LC, Baker BS. 1998. Regulation of sex-specific selection of fruitless 5’ splice sites by transformer and transformer-2. Mol Cell Biol. 18:450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honaas LA, et al. 2016. Selecting superior de novo transcriptome assemblies: lessons learned by leveraging the best plant genome. PLoS One 11:e0146062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshijima K, Inoue K, Higuchi I, Sakamoto H, Shimura Y. 1991. Control of doublesex alternative splicing by transformer and transformer-2 in Drosophila. Science 252:833–836. [DOI] [PubMed] [Google Scholar]
- Hunter S, et al. 2009. InterPro: the integrative protein signature database. Nucleic Acids Res. 37:D211–D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. 2011. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21:1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huylmans AK, Parsch J. 2015. Variation in the X:autosome distribution of male-biased genes among Drosophila melanogaster tissues and its relationship with dosage compensation. Genome Biol Evol. 7:1960–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquiéry J, et al. 2013. Masculinization of the X chromosome in the pea aphid. PLoS Genet. 9:e1003690.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZF, Machado CA. 2009. Evolution of sex-dependent gene expression in three recently diverged species of Drosophila. Genetics 183:1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42:D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, et al. 2010. Sequence divergence of a transformer gene in the brachiopod crustacean, Daphnia magna. Genomics 95:160–165. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kobayashi K, Watanabe H, Igushi T. 2011. Environmental sex determination in the branchiopod crustacean Daphnia magna: deep conservation of a Doublesex gene in the sex-determining pathway. PLoS Genet. 7:e1001345.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiven O, Larsson P, Hobaek A. 1992. Sexual reproduction in Daphnia magna requires three stimuli. Oikos 65:197–206. [Google Scholar]
- Larracuente AM, et al. 2008. Evolution of protein-coding genes in Drosophila. Trends Genet. 24:114–123. [DOI] [PubMed] [Google Scholar]
- LeBlanc GA, Medlock EK. 2015. Males on demand: the environmental-neuro-endocrine control of male sex determination in daphnids. FEBS J. 282:4080–4093. [DOI] [PubMed] [Google Scholar]
- LeBlanc GA, Wang YH, Holmes CN, Kwon G, Medlock EK. 2013. A transgenerational endocrine signaling pathway in Crustacea. PLoS One 8:e61715.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. 2012. Disentangling the relationship between sex-biased gene expression and X-linkage. Genome Res. 22:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-ciol regions. Nucleic Acid Res. 41:e121.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa H, Toyota K, Sumiya E, Igushi T. 2014. Comparison of JH signaling in insects and crustaceans. Curr Opin Insect Sci. 1:81–87. [DOI] [PubMed] [Google Scholar]
- Nakasugi K, Crowhurst R, Bally J, Waterhouse P. 2014. Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS One 9:e91776.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neme R, Tautz D. 2016. Fast turnover of genome transcription across evolutionary time exposes entire non-coding DNA to de novo gene emergence. eLife 5:e09977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmstead AW, LeBlanc GA. 2002. Juvenoid hormone methyl farnesoate is a sex determinant in the crustacean Daphnia magna. J Exp Zool. 293:736–739. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. 2007. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23:1061–1067. [DOI] [PubMed] [Google Scholar]
- Parsch J, Ellegren H. 2013. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 14:83–87. [DOI] [PubMed] [Google Scholar]
- Postnikova OA, Hult M, Shao J, Skantar A, Nemchinov LG. 2015. Transcriptome analysis of resistant and susceptible alfalfa cultivars infected with root-knot nematode Meloidogyne incognita. PLoS One 10:e0123157.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing.
- Rabus M, Laforsch C. 2011. Growing large and bulky in the presence of the enemy: Daphnia magna gradually switches the mode of inducible morphological defences. Funct Ecol. 25:1137–1143. [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suit. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Schulz MH, Zerbino DR, Vingron M, Birney E. 2012. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, von Haeseler A. 2013. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 29:2790–2791. [DOI] [PubMed] [Google Scholar]
- Shen H, Hu Y, Zhou X. 2014. Sex-lethal gene of the Chinese mitten crab Eriocheir sinensis: cDNA cloning, induction by eyestalk ablation, and expression of two splice variants in males and females. Dev Genes Evol. 224:97–105. [DOI] [PubMed] [Google Scholar]
- Shinoda T, Itoyama K. 2003. Juvenile hormone acid methyltransferase: a key regulator enzyme for insect metamorphosis. Proc Natl Acad Sci U S A. 100:11986–119991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Simpson JT, et al. 2009. AbySS: a parallel assembler for short read sequence data. Genome Res. 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Unna RD, Boursnell C, Patro R, Hibbert JM, Kelley S. 2015. TransRate: reference free quality assessment of de novo transcriptome assemblies. Genome Res. 26(8):1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Pierre SE, Ponting L, Stefancsik R, McQuilton P. the FlyBase Consortium. 2014. FlyBase 102 – advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42:D780–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stross RG, Hill JC. 1965. Diapause induction in Daphnia requires two stimuli. Science 150:1462–1464. [DOI] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatarazako N, Oda S, Watanabe H, Morita M, Igushi T. 2003. Juvenile hormone agonists affect the occurrence of male Daphnia. Chemosphere 53:827–833. [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Hebert PD, Colbourne JK. 1996. Phylogenetics and evolution of the Daphnia longispina group (Crustacea) based on 12S rDNA sequence and allozyme variation. Mol Phylogenet Evol. 5:495–510. [DOI] [PubMed] [Google Scholar]
- Thomas CG, et al. 2012. Simplification and desexualization of gene expression in self-fertile nematodes. Current Biol. 22:2167–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota K, et al. 2013. Molecular cloning of doublesex genes of four cladocera (water flea) species. BMC Genomics 14:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota K, Miyakawa H, Hiruta C, et al. 2015. Methyl farnesoate synthesis is necessary for the environmental sex determination in the water flea Daphnia pulex. J Insect Physiol. 80:22–30. [DOI] [PubMed] [Google Scholar]
- Toyota K, Miyakawa H, Yamaguchi K, et al. 2015. NDMA receptor activation upstream of methyl farnesoate signalling for short day-induced male offspring production in the water flea, Daphnia pulex. BMC Genomics 16:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanschoenwinkel B, Gielen S, Vandewaerde H, Seaman M, Brendonck L. 2008. Relative importance of different dispersal vectors for small aquatic invertebrates in a rock pool metacommunity. Ecography 31:567–577. [Google Scholar]
- Wainwright G, Webster SG, Rees HH. 1998. Neuropeptide regulation of biosynthesis of the juvenoid, methyl farnesoate, in the edible crab, Cancer pagurus. Biochem J. 334:651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. 2010. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8:77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. 2013. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 41:D358–D365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, et al. 2010. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327:343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, et al. 2014. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30:1660–1666. [DOI] [PubMed] [Google Scholar]
- Yin M, Wolinska J, Gießler S. 2010. Clonal diversity, clonal persistence and rapid taxon replacement in natural populations of species and hybrids of the Daphnia longispina complex. Mol Ecol. 19:4168–4178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.