

Abstract
Modified pyrimidine monophosphates such as methyl dCMP (mdCMP), hydroxymethyl dUMP (hmdUMP) and hmdCMP in some phages are synthesized by a large group of enzymes termed as thymidylate synthases (TS). Thymidylate is a nucleotide required for DNA synthesis and thus TS is an important drug target. In the biosynthetic pathway of the nucleoside fungicide mildiomycin isolated from Streptomyces rimofaciens ZJU5119, a cytidylate (CMP) hydroxymethylase, MilA, catalyzes the conversion of CMP into 5′-hydroxymethyl CMP (hmCMP) with an efficiency (kcat/KM) of 5-fold faster than for deoxycytidylate (dCMP). MilA is thus the first enzyme of the TS superfamily preferring CMP to dCMP. Here, we determined the crystal structures of MilA and its complexes with various substrates including CMP, dCMP and hmCMP. Comparing these structures to those of dCMP hydroxymethylase (CH) from T4 phage and TS from Escherichia coli revealed that two residues in the active site of CH and TS, a serine and an arginine, are respectively replaced by an alanine and a lysine, Ala176 and Lys133, in MilA. Mutation of A176S/K133R of MilA resulted in a reversal of substrate preference from CMP to dCMP. This is the first study reporting the evolution of the conserved TS in substrate selection from DNA metabolism to secondary nucleoside biosynthesis.
5-Hydroxymethyl cytosine (5hmC), also known as the ‘sixth base’, was discovered in mammalian and T-even phage DNA1,2. 5hmC in mammalian DNA is produced post-replicatively by the Tet-catalyzed oxidation of 5-methyl cytosine (5mC)3,4. In T-even phage, the deoxycytidylate (dCMP) hydroxymethylase (CH) transfers the methylene group from methylene-tetrahydrofolate (CH2THF) to the C5 atom of dCMP, and then uses solvent water molecule to hydrate the methylene group to generate hydroxymethyl dCMP (hmdCMP)5, a precursor to be incorporated into DNA during replication6. Thereafter, its hydroxymethyl group serves as a substrate for glucosylation to form glucosylhydroxymethylated DNA to avoid cleavage by the host restriction systems7,8.
Some biologically active nucleoside antibiotics, such as bacimethrin9, 5-hydroxymethyl blasticidin S10 and mildiomycin11, also contain 5hmC moieties that are all derived from hmCMP. We previously demonstrated that MilA, a CMP hydroxymethylase in the mildiomycin biosynthetic gene cluster in Streptomyces rimofaciens ZJU5119, can convert CMP to hmCMP12. HmCMP is then hydrolyzed by MilB to 5-hydroxymethylcytosine (5hmC)13, which is finally incorporated into mildiomycin.
MilA and CH are akin to the superfamily of thymidylate synthases (TS), which transfers a methyl group from CH2THF to dUMP to form dTMP in the de novo thymidylate synthesis pathway and, hence, DNA synthesis14. TS is one of the most conserved enzymes in nucleotide metabolism across phyla and therefore is an important drug target. TS from phage T4 (T4 TS) is involved in coordinating DNA synthesis in infected Escherichia coli cells15. Extensive biochemical and structural studies on TS have provided a wealth of information regarding its catalytic mechanism, specific interactions with dUMP and folate analogs, and stability14,16,17. The structures of TS and CH resemble each other very well, with a root-mean-square-deviation (RMSD) of 1.849 Å for 127 aligned Cα atoms, despite only 24% of sequence identity between them.
Since TS is responsible for the production of dTMP, one of the building blocks for DNA synthesis, it has been extensively studied as a target for cancer chemotherapy18. A number of structures of TS in complexes with various fragments of substrates, both in the presence or in the absence of cofactor analogues, are available19. These studies revealed that the cofactor triggered closure of the active site, that the pyrimidine ring of the substrate dUMP directed its binding orientation at the active site, that the ribose sugar moiety contributed to the enzyme’s substrate specificity, and that the glycosidic linkage was critical for the precise localization of the substrate19. However, structural studies on how TS protein superfamily members differentiate between ribosyl and 2′-deoxyribosyl substrates are relatively limited, probably in part, due to the lack of enzymes in this family biased towards ribosyl substrates. A report in this regard is that the binding affinity of TS for uridine monophosphate (UMP) is 40 times lower than that for dUMP20. All other usual members of TS, such as 2′-deoxyuridylate hydroxymethylase (dUH) from phage SPO121, dCMP hydroxymethyalse (CH) from phage T422, dCMP methylase from phage Xp1223, are specific for 2′-deoxynucleotides.
Several structural studies on ribose recognition specificity involved in pyrimidine nucleotide metabolism have been reported previously. The human mitochondrial deoxyribonucleotidase mdN prefers the 2′-deoxyribose form of nucleoside monophosphate. In the structure of mdN, a hydrophobic pitch surrounding the 2′ position of the sugar moiety produces an energetically unfavorable environment for the 2′-hydroxyl group of ribonucleoside 5′-monophosphates24. Another case of deoxyribose preference is deoxyribonucleoside kinase (dNK) from Drosophila melanogaster. In the structure of dNK, the crowded surrounding in the 2′-position of the substrate sugar leads to steric hindrance against the 2′-hydroxyl group and hence makes ribose forms of nucleosides less favorable than deoxyribose forms25. A rare case of ribose preference is human uridine-cytidine kinase (UCK). It has high specificity for the 2′-hydroxyl group of pyrimidine ribonucleosides and does not phosphorylate deoxyribose forms26,27. Comparison of ligand-free and -bound structures of UCK suggested that the ribose needs to be tightly bound to the enzyme in advance and then triggers a considerable conformational change to form the binding site. Poor binding of the deoxyribose sugar moiety cannot produce the induced fit required for the following base recognition and phosphorylation processes28. On the other hand, bacterial CMP kinase phosphorylates dCMP nearly as efficiently as CMP. Its structures in complexes with CMP or dCMP showed that Arg181 forms hydrogen bonds with the 3′-hydroxyl of sugar moiety while Asp185 could be hydrogen bonded to both 3′- and 2′-hydroxyl group29. There is no hydrophobic pitch or steric hindrance around the 2′-position of the substrate sugar; and unlike UCK, no induced fit is required for base binding. In addition, it was reported that a single Y639F mutation in the T7 RNA polymerase resulted in an ~20 fold loss of its specificity for NTP over dNTP30,31,32; while a single residue Glu710 of E. coli DNA polymerase I (Klenow fragment) dictated its specificity for dNTP by sterically blocking the 2′-hydroxyl of an incoming NTP33. Besides, the stringency of dNTP over NTP for the MoMLV reverse transcriptase was relaxed from 10,000-fold to merely 30-fold by its F155V mutation32, and the dNTP/ddNTP specificities of DNA polymerases of the pol I family could be switched simply by mutating a phenylalanine residue (corresponding to Phe762 for Klenow fragment) to a tyrosine residue32.
In this study, we demonstrated that MilA has a substrate preference for CMP (kcat/KM = 39.2 mM−1 min−1) over dCMP (kcat/KM = 7.84 mM−1 min−1), and thus offers an opportunity to investigate the mechanism by which conserved TS evolves the preference for ribosyl over 2′-deoxyribosyl groups. The crystal structures of apo MilA, MilA in complexes with CMP, dCMP and hmCMP were determined. Sequence and structure analyses suggested that the selectivity of ribosyl substrates by MilA is attributed to Ala176 and Lys133′ from the other chain of the dimer in the ribose-binding pocket. Mutation of A176S/K133R of MilA resulted in a reversal of substrate preference from CMP to dCMP.
Results and Discussion
Substrate preference of MilA for CMP
We previously reported that MilA could only convert CMP into hmCMP, but could not take dCMP as its substrate12. Given only 26% sequence identity with CH, MilA was assayed for hydroxymethylation activity with dCMP as substrate. Unexpectedly, liquid chromatography-mass spectroscopy (LC-MS) detected the ion corresponding to the product hmdCMP ([M + H]+ mass = 338, retention time Rt = 16.5 min), however its UV absorption peak was covered by that of the tetrahydrofolate (THFA) (Rt = 16.8 min) (Fig. S1A). To compare substrate preference, equal concentrations of CMP and dCMP were added in the same reaction system with MilA to compete with each other, and hmdCMP and THFA were completely separated using an optimized elution condition in high-performance liquid chromatography (HPLC) analysis. Our results clearly showed that MilA had a strong preference for CMP over dCMP (Fig. 1 and Fig. S1B). The kinetic parameters for MilA were determined with either CMP or dCMP as its substrate (Table 1, Fig. S2). The KM for CMP was 0.0719 mM, 3.4-fold lower than that for dCMP (KM = 0.245 mM), demonstrating that CMP was a better substrate than dCMP for MilA. The kcat/KM for hmCMP was 39.2 mM−1 min−1, 5-fold higher than that for hmdCMP (kcat/KM = 7.84 mM−1 min−1, Table 1). Prompted by this observation, we performed the structural comparison of MilA with CH and other TS members to identify the amino acids of MilA critical for its substrate preference for ribosyl cytidylate.
Figure 1. LC-MS analysis of the reaction catalyzed by MilA, with both dCMP and CMP added as substrates to compete with each other.

Extracted ion chromatogram at m/z 324, 354, 308 and 338 stand for CMP, hmCMP, dCMP and hmdCMP respectively.
Table 1. Enzymatic kinetics for MilA, TS and CH.
Structure of MilA
The structure of C-terminally His-tagged MilA was determined using selenomethionine (SeMet)-substituted MilA-L167M mutant at a 2.20 Å resolution (Table 2). Subsequently, the structures of MilA‒CMP, MilA‒dCMP and MilA‒hmCMP complexes were refined to 1.65 Å, 2.10 Å and 1.80 Å resolution, respectively (Table 2). In the structures of apo MilA and its complexes with various substrates, MilA are all homodimers. The non-crystallographic symmetry (NCS) between the two monomers in the crystallographic asymmetric unit is a twofold rotation with no translation. The N-terminal three residues, C-terminal five residues, residue 232–238 of MilA, as well as the eight residues (LEHHHHHH) introduced by cloning, showed no clear electron density and presumably were disordered in the crystal. The electron density for residues 305–308 was poor in the structure of apo MilA but resolved clearly in the structures of all the MilA‒substrate complexes. There is no obvious difference between the structures of CMP-bound MilA and apo MilA, with the root-mean-square deviation (RMSD) being 0.34 Å for 634 aligned Cα atoms. Interestingly, the average B-factor of a loop region around Arg31 (residues 29–33) is dramatically lowered from 43.2 to 24.8 Å2 upon CMP-binding (Fig. 2A & B).
Table 2. Data collection and refinement statistics.
| SeMet-MilA L167M | MilA | MilA-CMP | MilA-dCMP | MilA-hmCMP | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P3221 | P3221 | P3221 | P3221 | P3221 |
| Unit cell parameters | |||||
| a, b, c (Å) | 107.7, 107.7, 112.1 | 107.8, 107.8, 112.0 | 109.6, 109.6, 113.4 | 109.3, 109.3, 113.2 | 109.3, 109.3, 112.8 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 50–3.10 (3.21–3.10) | 50–2.20 (2.28–2.20) | 50–1.65 (1.71–1.65) | 50–2.10 (2.18–2.10) | 50–1.80 (1.86–1.80) |
| Rmerge (%) | 29.5 (63.6) | 15.6 (76.7) | 13.2 (88.5) | 11.8 (32.9) | 11.9 (73.6) |
| I/σI | 43.2 (19.4) | 23.2 (4.4) | 13.6 (1.9) | 17.8 (8.1) | 19.6 (3.7) |
| Completeness (%) | 100 (100) | 100 (100) | 96.6 (100) | 95.8 (100) | 100 (100) |
| Redundancy | 10.6 (10.9) | 21, 8 (19.0) | 6.7 (6.9) | 11.0 (11.3) | 11.2 (11.2) |
| Refinement | |||||
| Resolution (Å) | 48.02–2.20 | 48.73–1.65 | 49.26–2.10 | 49.24–1.80 | |
| Unique reflection | 38, 499 | 91, 516 | 43, 971 | 68, 810 | |
| Rwork/Rfree (%) | 16.8/20.7 | 17.2/19.4 | 15.5/20.6 | 18.7/20.8 | |
| Number of atoms | |||||
| Protein | 5,074 | 5,067 | 5,079 | 5,089 | |
| Ligand/ion | 0 | 42 | 40 | 46 | |
| Water | 336 | 547 | 501 | 201 | |
| B-factors (Å2) | |||||
| Overall | 27.32 | 24.55 | 21.6 | 19.7 | |
| Protein | 26.97 | 23.4 | 20.6 | 19.5 | |
| Ligand | N/A | 18.6 | 19.2 | 19.3 | |
| Water | 32.63 | 35.37 | 31.93 | 25.3 | |
| RMSD bond length (Å) | 0.008 | 0.006 | 0.007 | 0.005 | |
| RMSD bond angles (°) | 0.81 | 0.83 | 0.86 | 0.98 | |
Data for each structure were collected or calculated from a single crystal. RMSD, root-mean-square deviations from the ideal geometry. Data for the highest resolution shell are shown in parentheses.
Figure 2. Overall structures of MilA and the MilA‒CMP complex.

(A,B) The structures of WT MilA (A) and the MilA‒CMP complex (B) are shown in cartoon representation and colored according to the B-factor. Blue and red represent the lowest and highest B-factor values, respectively. In addition, the thickness of the tube reflects the B-factor value in that the larger the B-factor, the thicker the tube. (C) Structure of the MilA‒CMP monomer. α-helices, β-sheets, and 310-helices are colored in yellow, red, and orange, respectively. CMP is depicted in purple. (D) Structural comparison of MilA (green), T4 CH (cyan, PDB code 1B5E) and TS (magenta, PDB code 1KZJ). (E) Structure of the MilA‒CMP dimer. The structure is viewed perpendicular to the two-fold axis of the dimer. The two protomers are shown in blue and red, respectively. Their bound CMP substrates are represented as yellow sticks.
The homodimer of MilA consists of two essentially identical subunits and has approximate dimensions of 108 Å × 108 Å × 112 Å. A MilA monomer consists of a six-stranded β-sheet, surrounded by thirteen α-helices and four 310-helices (Fig. 2C). MilA possesses a common fold shared by TS and CH. Compared with TS and CH, MilA has an extra domain consisting of five α helices (from α9 to α13) in its C-terminal region (Fig. 2C and D). Each active site of the dimer is contributed asymmetrically by residues from both subunits. The substrate CMP is located very close to the dimer interface (Fig. 2E). All six β-strands within each monomer as well as α-helices α1, α5 and α6 are involved in dimerization (Fig. 2C and E), in a manner similar to the dimerization patterns of CH and TS.
Structural similarity to T4 CH and bacterial TS
The major parts of MilA, T4 CH and E. coli TS subunits resemble each other very well, except for some significant structural difference located at the C-terminal region (Fig. 2D). After getting rid of the the C-terminal region, a superposition of the MilA with E. coli TS and T4 CH gives the RMSD of 1.293 Å and 1.209 Å, respectively. E. coli TS presents extra 27 C-terminal residues folded as two short β-strands, a 310-helix and a long loop that is absent in CH (Fig. 3A and B). Unlike the structure of CH and E. coli TS, the C-terminal region of MilA consists of five helices (α9-α13) linked by loops (Fig. 3C). The last six residues of E. coli TS move ~4 Å upon binding folate, and partly cover the active site34,35. Therefore, the presumable folate-binding site of T4 CH is more open than that of E. coli TS22. Some parts of this region are believed to provide an interaction surface for dihydrofolate reductase (DHFR)36,37. However, DHFR is not functionally required to interact with T4 CH and MilA, since tetrahydrofolate is produced in the T4 CH and MilA-catalyzed hydroxymethylation reaction. Different from TS, the extra C-terminal region of MilA is much bigger and positioned away from the active site. C-terminal truncations either from residue 235 or from residue 249 of MilA are both insoluble (data not shown). Presumably, this region could function as a domain to facilitate protein folding.
Figure 3. The structure of the C-terminal part of MilA is different from those of T4 CH and E. coli TS.

(A–C) Comparison of the structures of the C-terminal parts of T4 CH (A), E. coli TS (B) and MilA (C), which are all colored in yellow. (D) The electrostatic surface potential of MilA were generated by pymol, with blue and red representing positively- and negatively-charged surface areas, respectively. The substrate CMP is located in a surface pocket of MilA.
CMP binding and ribose specificity
CH, dUH, and TS all prefer the deoxyribose forms of substrates. In contrast, MilA accepts the ribose form more efficiently than the deoxyribose form, which makes it unique. The substrate CMP is bound in a deep active-site pocket of MilA, in a manner similar to the binding of dUMP by T4 CH and TS (Fig. 3). Most of the key amino acids involved in nucleotide recognition between the structures of MilA‒CMP and CH‒dCMP aligned very well, except for several substitutions of amino acids in the binding pocket.
In the CH structure, His216 and Tyr218 make hydrogen bonds with the 3′-oxygen atom of 2′-deoxyribose sugar22. In the crystal structure of CH, the imidazole ring of His216 could be in two different rotameric states. It is the same case for the analogous His216 in MilA. We propose that His216 of MilA and CH both probably adopt the more favorable rotameric state as shown in Fig. 4B, with the distance between the ε-nitrogen of His216 and the 3′-oxygen of dCMP being 2.7 Å rather than 3.4 Å for the other rotameric state.
Figure 4. Different rotameric states of His216.

(A) In the structure of the CH‒dCMP complex determined by Song et al., the distance between the ε-nitrogen of His216 and the 3′-oxygen of dCMP is 3.38 Å. (B) The alternative rotameric state of His216, with its imidazole ring flipped 180 degrees, is probably more favorable. The distance between the ε-nitrogen of His216 and the 3′-oxygen of dCMP is 2.73 Å.
The catalytic efficiency (as quantified by the kcat/KM value) of MilA for CMP is 5-fold higher than that for dCMP. In contrast, the kcat/KM value of TS for dUMP is about 300-fold higher than that for UMP (Table 1). This observation immediately raises two questions. First, what is the molecular mechanism for that MilA prefers ribose nucleotide substrates whereas TS favors deoxyribose ones? Second, why does TS has a much higher stringency on substrate specificity (with an almost 300-fold difference between the two kinds of substrates) than MilA (with only a mere 5-fold difference)? Both these two interesting questions warrant further investigations for us.
Through a comparison of the active site structures of the TS-dUMP, CH‒dCMP, MilA-dCMP and MilA‒CMP complexes, it was found that the 3′-hydroxyl groups of the sugar moiety of substrates adopt two different conformations when complexed with MilA or TS/CH (Figs 5B and S4); the 3′-carbon together with its 3′-OH of deoxyribose motif has a dramatic torsion (with 3′-C set as the vertex, the angle from 6′-O to 3′-O is increased from 102.3°/105.3° to 136.4°) in MilA-dCMP relative to TS-dUMP/CH-dCMP (Fig. 5B, panel 1–3). In TS or CH, both of which prefer deoxyribosyl substrates, the 3′-hydroxyl group of dUMP/dCMP makes hydrogen bonds with TS-His207/CH-His216 and TS-Tyr209/CH-Tyr218 respectively (Fig. 5B, panel 1&2). However, in MilA-dCMP, the 3′-hydroxyl group of dCMP forms one hydrogen bond with the Lys-133′ and another intramolecular hydrogen bond with the phosphate group (Fig. 5B, panel 3). The main reason for this difference is that Ala176 in MilA is replaced by a serine, Ser167/Ser169, in TS/CH. The extra hydroxyl group of TS-Ser167/CH-Ser169 makes the space crowded for the 3′-hydroxyl group of the sugar, and cannot tolerate the sugar moiety of the substrate to adopt the same conformation as that when in complex with MilA. It is not hard to imagine that when UMP or CMP attempts to enter the substrate-binding pocket of TS or CH, the 2′-hydroxyl group of the sugar would occupy the space of the 3′-hydroxyl group, and the 3′-hydroxyl group would have to adopt the same conformation as CMP in MilA, in which case, the additonal hydroxyl group of TS-Ser167 or CH-Ser169 side-chain would give rise to steric hinderance with the 3′-hydroxyl group of the sugar given the close distance (1.9 Å as indicated panel 4 of Fig. 5B). In contrast, Ala176, with its much smaller side-chain methyl group, is the corresponding residue for TS-Ser167/CH-Ser169 in MilA and makes the larger room. Therefore, both CMP and dCMP are able to fit into the substrate-binding pocket of MilA (Fig. 5A). Hence, MilA can not only can utilize CMP, but also can use dCMP as its substrate like CH and TS. This provides an explanation for the second question raised above. An alternative interpretation for this question might be that TS is actually a better enzyme than MilA in terms of catalytic efficiency. According to the kinetics summarized in Table 1, the catalytic efficiency of TS on dUMP is 4 orders of magnitude higher than that of MilA on CMP, probably magnifying the stringency of TS in selection of dUMP over UMP than that of MilA in selection of CMP over dCMP.
Figure 5. Structural comparison among the substrate-binding sites of MilA alone, the MilA‒CMP complex, the MilA‒dCMP complex, the TS-dUMP complex and the CH‒dCMP complex.

(A) Structural comparison between the substrate-binding sites of MilA alone, the MilA-dCMP and the MilA‒CMP complex. Panel 1: the substrate-binding site of apo MilA (colored in orange). Panel 2: the substrate-binding site of the MilA‒dCMP complex (colored in slate). Panel 3: the substrate-binding site of the MilA‒CMP complex (colored in tv_green). Panel 4: superposition of the substrate-binding sites of MilA alone, the MilA‒dCMP complex and the MilA-CMP complex. (B) Structural comparison between the substrate-binding sites of the TS-dUMP complex, the CH‒dCMP complex and the MilA‒dCMP complex. Panel 1: the substrate-binding site of the the TS-dUMP complex (colored in cyan). Panel 2: the substrate-binding site of the CH‒dCMP complex (colored in light magenta). Panel 3: the substrate-binding site of the MilA‒dCMP complex (colored in slate). Panel 4: superposition of the substrate-binding sites of the TS-dUMP complex, the CH‒dCMP complex and the MilA‒dCMP complex.
In accordance with structural analysis, mutation of alanine 176 into serine had dramatically decreased its activity towards CMP, but significantly enhanced its catalytic efficiency toward dCMP (Fig. 6). This further confirmed that Ala176 of MilA is critical for its substrate specificity. The fact that MilA-A176S could still catalyze the hydroxymethylation reaction of CMP implies that its substrate-binding pocket can still accommodate CMP. In the structures of TS-dUMP and CH-dCMP, the guanidino side chain of TS-Arg126′ or CH-Arg123′ could bond to three oxygen atoms of the phosphate group without formation of any bonds to the ribose moiety (Fig. S3A,B). By contrast, its counterpart residue in MilA is lysine 133′, which formed hydrogen bonds with 3′-hydroxyl group of dCMP or CMP in respective protein/substrate complex (Fig. 5A, panel 2&3). It seems that Lys133′ in MilA plays an auxiliary role in the ribose specificity. To address this possibility, Lys133′ was further mutated into arginine on the basis of MilA A176S, the catalytic efficiency toward CMP was completely eliminated in the double mutant MilA A176S/K133R, but its efficiency to dCMP is slightly affected (Fig. 6).
Figure 6. Comparison of the enzymatic activity of MilA-WT, MilA A176S and MilA A176S/K133R towards CMP and dCMP mixture.
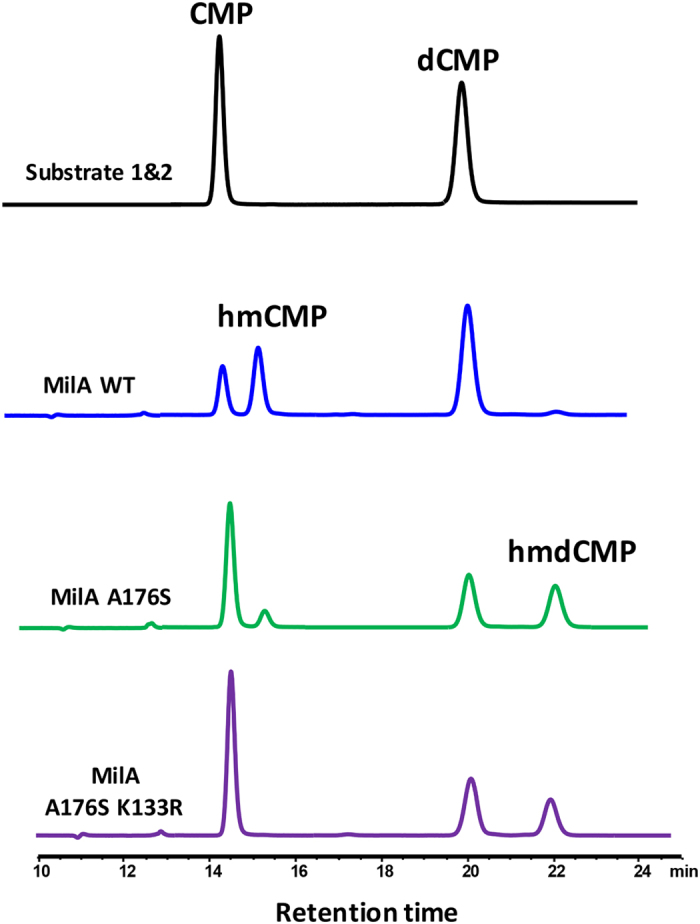
In the presence of both CMP and dCMP, MilA-WT preferred to hydroxymethylate CMP and just a slight amount of hmdCMP was produced. MilA A176S had dramatically decreased its activity towards CMP, but significantly enhanced its catalytic efficiency toward dCMP. The catalytic efficiency of MilA A176S/K133R toward CMP was completely eliminated, but its efficiency to dCMP is slightly affected.
As for the first question, the reason that MilA prefers ribosyl substrates is because in addition to the hydrogen bonds with the 3′-hydroxyl group of CMP, the 2′-hydroxyl group of CMP makes strong hydrogen bonds with Tyr218 and His216 of MilA, with distances of 2.7 Å and 2.8 Å, respectively. These two additional hydrogen bonds make contributions to lower MilA′s KM value for CMP compared to that for dCMP. In summary, our structural information strongly implies that the evolution from a serine and an arginine in the active site of TS/CH to an alanine and a lysine in the active site of MilA contributes a lot to the switch of substrate specificity from deoxyribosyl substrate (dUMP/dCMP) to ribosyl substrate (CMP) (Fig. 5).
Comparison of sequences and identification of critical amino acids
CH, dUH and TS all prefer deoxyribose-containing substrates, while MilA and BcmA accept ribose-containing substrates more efficiently than deoxyribose-containing ones. There should be structural differences in the substrate-binding sites of MilA and BcmA from other enzymes. Therefore, we aligned the primary sequences of MilA and BcmA with those of T4 CH, dUH from phage SPO1, and E. coli TS using the Cobalt Constraint-based multiple protein alignment tool. The sequence alignment, which is graphically enhanced by Espript 3.038, shows that most of the critical amino acids in the active site are conserved (Fig. 7). For instance, the reactive nucleophile residue Cys155, catalytically important residue Glu68, and ribose-binding residues His216 and Tyr218 of MilA are extremely conserved. These assure similar catalytic mechanisms for MilA, CH, dUH and TS.
Figure 7. Multiple sequence alignment of MilA and its homologues.

MilA from S. rimofaciens (SR_MilA), BcmA from C. botulinum (CB_BcmA), CH from T4 phage (T4_CH), dUH from phage SPO-1 (SP_dUH) and TS from E. coli (EC_TS) were aligned. Conserved residues are highlighted in dark-red background. Residues involved in ribose specificity are indicated with red stars and green triangles. Catalytic residues are represented with red triangles in the bottom. Residues involved in phosphate-binding and base-binding specificities are marked, respectively, with pink triangles and blue star in the bottom. The secondary structure of MilA is shown above the sequences.
On the other hand, three amino acids in MilA, Lys133′, Ala176 and Asp186 are not conserved in all these five proteins. Interestingly, Lys133′ and Ala176 are conserved in MilA and BcmA, which prefer ribosyl-containing substrates; whereas the equivalent residues in enzymes preferring deoxyribosylated substrates are all arginines and serines. The third residue Asp186 is conserved in MilA, BcmA, and T4 CH which utilize cytosine-containing substrates; whereas the equivalent residue in enzymes favoring uracil-containing substrates like TS and dUH are both asparagines. Song et al. have proposed that in analogy with L. casei TS, Asp179 of T4 CH prefers dCMP to dUMP by achieving a proper orientation of the pyrimidine base through a hydrogen bond network for nucleophilic attack by Cys148 and a better stabilization of the reaction intermediates22,39, which is consistent with our structure of MilA‒CMP.
Methods
Site-directed mutagenesis of MilA
Gene encoding wild type (WT) Streptomyces rimofaciens ZJU5119 MilA was cloned into the pET28a (Novagen) vector, with a C-terminal 6 × His tag. All mutant plasmids were produced by the whole-plasmid polymerase chain reaction40, and verified by sequencing. The plasmids and the primers used in this study are listed in Supplementary Information, Tables S1 and S2.
Protein expression and purification
Proteins were overexpressed in the Escherichia coli strain BL21(DE3) at 16 °C. 10 ml culture grown overnight from a single colony was inoculated into 1 liter of Luria Broth medium supplied with 50 μg/ml kanamycin and 34 μg/ml chloramphenicol. The culture was incubated at 37 °C to OD600 = 0.6~0.8, and induced by 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for another 20 hours at 16 °C. The cells were harvested and resuspended in 20 ml binding buffer (20 mM sodium phosphate, pH 7.4, 20 mM imidazole and 500 mM sodium chloride), and lysed by sonication in an ice bath. After centrifugation at 16,000 × g for 30 min at 4 °C, the supernatant was applied to 2 ml Ni-NTA column (Qiagen) pre-equilibrated with the binding buffer. The column was washed by 60 ml binding buffer and 10 ml washing buffer (20 mM sodium phosphate, pH 7.4, 50 mM imidazole and 500 mM sodium chloride). The column was then eluted with 10 ml elution buffer (20 mM sodium phosphate, pH 7.4, 300 mM imidazole and 500 mM sodium chloride). All the eluant was collected and further purified by the Superdex 200 gel filtration chromatography (GE Healthcare) equilibrated with 10 mM Tris-HCl, pH 7.4, 100 mM sodium chloride and 2 mM dithiothreitol. The purified proteins were analyzed by sodium dodecylsulphate-polyacrylamide gel electrophoresis and visualized by Coomassie blue staining, and the protein concentration was determined by using the Bradford Protein Assay Kit (Bio-Rad). The combined peak fractions were concentrated to 10 mg/ml. Selenomethionine (SeMet)-substituted MilA-L167M was expressed using the methionine-autotrophic E. coli strain B834 cultured in M9 medium (carbon source: glucose) and purified similarly, except that 20 mM β-mercaptoethanol was added before sonication.
In vitro enzymatic assays of MilA WT and MilA mutants and analytical high-performance liquid chromatography (HPLC)
In vitro assays of recombinant MilA were carried out at 37 °C for 1 h in a total volume of 100 μl that contained Tris–HCl buffer (100 mM, pH 7.5), paraformaldehyde (15 mM), 2-mercaptoethanol (50 mM), tetrahydrofolate (2 mM, pH 7.5), CMP and dCMP (1 mM, pH 7.5) and the corresponding His-tagged MilA or its mutants (10 μg). The reactions were quenched by the addition of trichloroacetic acid (4%) on ice, the products were resolved by Agilent TC-C18 column (4.6 mm × 250 mm, 5-Micron) on an Agilent 1200 HPLC system using a mobile phase of a gradient of methanol in water supplied with formic acid (0.1%). The constant flow rate for the LC eluent is 0.3 ml/min. Chromatograms were detected using the absorbance at 275 nm. The percentages of methanol (M) at time t varied according to the following scheme: (t, M), (0, 3), (30, 3), (31, 90), (35, 90), (36, 3), (45, 3). The accurate mass of the reaction products that were previously determined by NMR12 were analyzed by QTOF/MS (Agilent G6530A).
Enzymatic kinetic parameters measurement for MilA
Kinetic parameters were monitored on the basis of production of hmCMP/hmdCMP from CMP or dCMP catalysed by WT MilA. The co-substrate 5N,10N-methylenetetrahydrofolate (CH2THF) was prepared as reference41. As the concentration of CH2THF is hard to determine, prior to performing the kinetic assay, CH2THF solutions prepared with tetrahydrofolate (THFA) of 2 mM and 5 mM were incubated with 1.6 μM MilA and 2 mM CMP or dCMP, respectively. Compared to the reaction with 2 mM THFA, there is no increase of either product when 5 mM THFA applied, indicating that MilA is saturated with CH2THF generated by 2 mM starting THFA. On the other hand, 2 mM CMP or dCMP cannot be completely converted to product in each of reaction. MilA of 1.6 μM was incubated with various concentrations of the substrate in 50-mM Tris–HCl, pH 7.5, for 30 min at 37 °C, and then the reactions (with a total volume of 100 μl) were quenched by the addition of trichloroacetic acid (4%) on ice. After centrifugation at 16,000 × g for 5 min, the samples were analysed by HPLC as described above. The structures of the reaction products were determined by QTOF/MS (Agilent G6530A). Kinetic parameters were calculated by fitting the enzymatic data to the Michaelis–Menten equation by the non-linear regression analysis (Prism5; GraphPad Software Inc.).
Crystallization
Crystallization trials for full-length MilA were performed at 14 °C using the hanging-drop vapor-diffusion method in 48-well plates. Typically, 1 μl reservoir solution was mixed with 1 μl protein solution and equilibrated against 1 ml reservoir solution. Initial crystallization screening trials were performed using Crystal Screen, Index, PEG/Ion and SaltRx screen kits from Hampton Research. After 2 weeks, small crystals of full-length MilA were obtained from the condition that consists of 30% (w/v) polyethylene glycol 4000, 0.2 M lithium sulfate monohydrate and 0.1 M Tris-HCl, pH 8.5. Longer and thicker crystals were obtained by using 12–20% (w/v) polyethylene glycol 3350. After further optimization, diffracting crystals were obtained from 15% (w/v) polyethylene glycol 3350, 0.08 M lithium sulfate monohydrate and 0.1 M Tris-HCl, pH 8.5, using the hanging-drop vapor-diffusion method in 48-well plates at 14 °C. Given that only two methionine residues are present in MilA, we introduced a L167M mutation into MilA in order to enhance the anomalous diffraction signal. SeMet-MilA-L167M was crystallized at 14 °C in 15% (w/v) polyethylene glycol 3350, 0.08 M lithium sulfate monohydrate and 0.1 M Tris-HCl, pH 8.5. The crystals of MilA‒CMP, MilA‒dCMP and MilA‒hmCMP complexes were obtained by crystallization in the presence of substrates from condition which consists of 0.1 M sodium cacodylate trihydrate, pH 6.5, and 1.4 M sodium acetate trihydrate. The substrate hmCMP was obtained by a one-step conversion of CMP by purified MilA, followed by the purification procedure described as reported42. Diffraction datasets of all the crystals were collected at the BL17U1 or BL19U1 beamlines at Shanghai Synchrotron Radiation Facility (SSRF) using an ADSC Quantum 315r CCD area detector and a Pilatus 3–6 M CMOS detector, and processed using HKL2000 and HKL300043,44.
Structure determination
SeMet-MilA L167M crystals belonged to the P3221 space group and contained two molecules in the asymmetric unit. Its structure was determined by the single wavelength anomalous diffraction (SAD) method using PHENIX45,46. Crystals of apo MilA and MilA complexed with its substrates all belonged to the P3221 space group, with two molecules in the asymmetric unit. Their structures were determined by the molecular replacement method with Phaser47,48, using the structure of SeMet-MilA-L167M as the searching model. Model building was performed by Coot49 and refinement was performed by REFMAC550 and Phenix51. All the data of collection and refinement statistics are shown in Table 2.
Additional Information
Accession codes: The atomic coordinates and structure factors of MilA, MilA‒dCMP, MilA‒CMP and MilA‒hmCMP complexes have been deposited in the Protein Data Bank with accession numbers 5JNH, 5JP9, 5B6D, 5B6E, respectively.
How to cite this article: Zhao, G. et al. Structural basis of the substrate preference towards CMP for a thymidylate synthase MilA involved in mildiomycin biosynthesis. Sci. Rep. 6, 39675; doi: 10.1038/srep39675 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China (31470195, 31470223, and 31670106), the Special fund project for technology innovation from Shanghai Jiao Tong University [16 × 190030005 to X. H.]. We thank Feng Yu, Jiahua He, Xianhui Xu, and other staffs at the beamline BL17U1 and the beamline BL19U1 at Shanghai Synchrotron Radiation Facility (China) for assistance in data collection. We are grateful to Dr. Xu Liu from Emory University for his comments to our manuscript.
Footnotes
Author Contributions X.H., G.W., C.C. and G.Z. conceived the experiments. G.Z., C.C. and W.X. conducted the experiments. X.H., G.W., C.C., T.G. and G.Z. analysed the data. H.X., G.W., C.C. and G.Z. wrote the manuscript. All authors reviewed, revised, commented on and approved the final version of the manuscript.
References
- Warren R. A. Modified bases in bacteriophage DNAs. Annual review of microbiology 34, 137–158, doi: 10.1146/annurev.mi.34.100180.001033 (1980). [DOI] [PubMed] [Google Scholar]
- Kriaucionis S. & Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930, doi: 10.1126/science.1169786 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935, doi: 10.1126/science.1170116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y. F. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307, doi: 10.1126/science.1210944 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. M., Graves K. L. & Hardy L. W. Evidence from 18O exchange studies for an exocyclic methylene intermediate in the reaction catalyzed by T4 deoxycytidylate hydroxymethylase. Biochemistry 33, 10521–10526 (1994). [DOI] [PubMed] [Google Scholar]
- Wovcha M. G., Tomich P. K., Chiu C. S. & Greenberg G. R. Direct participation of dCMP hydroxymethylase in synthesis of bacteriophage T4 DNA. Proc Natl Acad Sci USA 70, 2196–2200 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg G. R., He P., Hilfinger J. & Tseng M. J. Deoxyribonucleoside triphosphate synthesis and phage T4 DNA replication. Molecular Biology of Bacteriophage T4 14–27 (1994). [Google Scholar]
- Vrielink A., Ruger W., Driessen H. P. & Freemont P. S. Crystal structure of the DNA modifying enzyme beta-glucosyltransferase in the presence and absence of the substrate uridine diphosphoglucose. The EMBO journal 13, 3413–3422 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper L. E., O’Leary S. E. & Begley T. P. Biosynthesis of a thiamin antivitamin in Clostridium botulinum. Biochemistry 53, 2215–2217, doi: 10.1021/bi500281a (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen S. H., Berry D. M., Paschal J. W. & Gilliam J. M. 5-Hydroxymethylblasticidin S and blasticidin S from Streptomyces setonii culture A83094. The Journal of antibiotics 42, 470–471 (1989). [DOI] [PubMed] [Google Scholar]
- Feduchi E., Cosin M. & Carrasco L. Mildiomycin: a nucleoside antibiotic that inhibits protein synthesis. The Journal of antibiotics 38, 415–419 (1985). [DOI] [PubMed] [Google Scholar]
- Li L. et al. The mildiomycin biosynthesis: initial steps for sequential generation of 5-hydroxymethylcytidine 5′-monophosphate and 5-hydroxymethylcytosine in Streptoverticillium rimofaciens ZJU5119. Chembiochem: a European journal of chemical biology 9, 1286–1294, doi: 10.1002/cbic.200800008 (2008). [DOI] [PubMed] [Google Scholar]
- Zhao G. et al. Structure of the N-glycosidase MilB in complex with hydroxymethyl CMP reveals its Arg23 specifically recognizes the substrate and controls its entry. Nucleic acids research 42, 8115–8124, doi: 10.1093/nar/gku486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud R. M. & Finer-Moore J. S. Stereochemistry of a multistep/bipartite methyl transfer reaction: thymidylate synthase. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 7, 671–677 (1993). [DOI] [PubMed] [Google Scholar]
- Finer-Moore J. S., Maley G. F., Maley F., Montfort W. R. & Stroud R. M. Crystal structure of thymidylate synthase from T4 phage: component of a deoxynucleoside triphosphate-synthesizing complex. Biochemistry 33, 15459–15468 (1994). [DOI] [PubMed] [Google Scholar]
- Carreras C. W. & Santi D. V. The catalytic mechanism and structure of thymidylate synthase. Annual review of biochemistry 64, 721–762, doi: 10.1146/annurev.bi.64.070195.003445 (1995). [DOI] [PubMed] [Google Scholar]
- Stout T. J., Sage C. R. & Stroud R. M. The additivity of substrate fragments in enzyme-ligand binding. Structure 6, 839–848, doi: 10.1016/S0969-2126(98)00086-0 (1998). [DOI] [PubMed] [Google Scholar]
- Danenberg P. V. Thymidylate synthetase-a target enzyme in cancer chemotherapy. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 473, 73–92 (1977). [DOI] [PubMed] [Google Scholar]
- Stout T. J., Sage C. R. & Stroud R. M. The additivity of substrate fragments in enzyme–ligand binding. Structure 6, 839–848 (1998). [DOI] [PubMed] [Google Scholar]
- Reyes P. & Heidelberger C. Fluorinated pyrimidines. XXVI. Mammalian thymidylate synthetase: its mechanism of action and inhibition by fluorinated nucleotides. Molecular pharmacology 1, 14–30 (1965). [PubMed] [Google Scholar]
- Wilhelm K. & Ruger W. Deoxyuridylate-hydroxymethylase of bacteriophage SPO1. Virology 189, 640–646 (1992). [DOI] [PubMed] [Google Scholar]
- Song H. K., Sohn S. H. & Suh S. W. Crystal structure of deoxycytidylate hydroxymethylase from bacteriophage T4, a component of the deoxyribonucleoside triphosphate-synthesizing complex. The EMBO journal 18, 1104–1113, doi: 10.1093/emboj/18.5.1104 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T. Y., Tu J. & Kuo T. T. Characterization of deoxycytidylate methyltransferase in Xanthomonas oryzae infected with bacteriophage Xp12. European journal of biochemistry/FEBS 87, 29–36 (1978). [DOI] [PubMed] [Google Scholar]
- Wallden K., Ruzzenente B., Rinaldo-Matthis A., Bianchi V. & Nordlund P. Structural basis for substrate specificity of the human mitochondrial deoxyribonucleotidase. Structure 13, 1081–1088, doi: 10.1016/j.str.2005.04.023 (2005). [DOI] [PubMed] [Google Scholar]
- Munch-Petersen B., Knecht W., Lenz C., Sondergaard L. & Piskur J. Functional expression of a multisubstrate deoxyribonucleoside kinase from Drosophila melanogaster and its C-terminal deletion mutants. The Journal of biological chemistry 275, 6673–6679 (2000). [DOI] [PubMed] [Google Scholar]
- Johansson K. et al. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nature structural biology 8, 616–620, doi: 10.1038/89661 (2001). [DOI] [PubMed] [Google Scholar]
- Van Rompay A. R., Norda A., Linden K., Johansson M. & Karlsson A. Phosphorylation of uridine and cytidine nucleoside analogs by two human uridine-cytidine kinases. Molecular pharmacology 59, 1181–1186 (2001). [DOI] [PubMed] [Google Scholar]
- Suzuki N. N., Koizumi K., Fukushima M., Matsuda A. & Inagaki F. Structural basis for the specificity, catalysis, and regulation of human uridine-cytidine kinase. Structure 12, 751–764, doi: 10.1016/j.str.2004.02.038 (2004). [DOI] [PubMed] [Google Scholar]
- Bertrand T. et al. Sugar specificity of bacterial CMP kinases as revealed by crystal structures and mutagenesis of Escherichia coli enzyme. Journal of molecular biology 315, 1099–1110, doi: 10.1006/jmbi.2001.5286 (2002). [DOI] [PubMed] [Google Scholar]
- Sousa R. & Padilla R. A mutant T7 RNA polymerase as a DNA polymerase. The EMBO journal 14, 4609 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Eckstein F., Padilla R. & Sousa R. Mechanism of ribose 2′-group discrimination by an RNA polymerase. Biochemistry 36, 8231–8242 (1997). [DOI] [PubMed] [Google Scholar]
- Joyce C. M. Choosing the right sugar: how polymerases select a nucleotide substrate. Proceedings of the National Academy of Sciences 94, 1619–1622 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astatke M., Ng K., Grindley N. D. & Joyce C. M. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proceedings of the National Academy of Sciences 95, 3402–3407 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montfort W. R. et al. Structure, multiple site binding, and segmental accommodation in thymidylate synthase on binding dUMP and an anti-folate. Biochemistry 29, 6964–6977 (1990). [DOI] [PubMed] [Google Scholar]
- Matthews D. A., Appelt K., Oatley S. J. & Xuong N. H. Crystal structure of Escherichia coli thymidylate synthase containing bound 5-fluoro-2′-deoxyuridylate and 10-propargyl-5,8-dideazafolate. Journal of molecular biology 214, 923–936, doi: 10.1016/0022-2836(90)90346-N (1990). [DOI] [PubMed] [Google Scholar]
- Knighton D. R. et al. Structure of and kinetic channelling in bifunctional dihydrofolate reductase-thymidylate synthase. Nature structural biology 1, 186–194 (1994). [DOI] [PubMed] [Google Scholar]
- Stroud R. M. An electrostatic highway. Nature structural biology 1, 131–134 (1994). [DOI] [PubMed] [Google Scholar]
- Robert X. & Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic acids research 42, W320–W324, doi: 10.1093/nar/gku316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves K. L., Butler M. M. & Hardy L. W. Roles of Cys148 and Asp179 in Catalysis by Deoxycytidylate Hydroxymethylase from Bacteriophage-T4 Examined by Site-Directed Mutagenesis. Biochemistry 31, 10315–10321, doi: 10.1021/bi00157a020 (1992). [DOI] [PubMed] [Google Scholar]
- Zheng L., Baumann U. & Reymond J. L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic acids research 32, e115, doi: 10.1093/nar/gnh110 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves K. L., Butler M. M. & Hardy L. W. Roles of Cys148 and Asp179 in catalysis by deoxycytidylate hydroxymethylase from bacteriophage T4 examined by site-directed mutagenesis. Biochemistry 31, 10315–10321 (1992). [DOI] [PubMed] [Google Scholar]
- Gu T. P. et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610, doi: 10.1038/nature10443 (2011). [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. & Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol 276, 307–326, doi: 10.1016/S0076-6879(97)76066-X (1997). [DOI] [PubMed] [Google Scholar]
- Minor W., Cymborowski M., Otwinowski Z. & Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta crystallographica. Section D, Biological crystallography 62, 859–866, doi: 10.1107/S0907444906019949 (2006). [DOI] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart P. H. et al. Automated structure solution with the PHENIX suite. Methods in molecular biology 426, 419–435, doi: 10.1007/978-1-60327-058-8_28 (2008). [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta crystallographica. Section D, Biological crystallography 50, 760–763, doi: 10.1107/S0907444994003112 (1994). [DOI] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. Journal of Applied Crystallography 40, 658–674, doi: 10.1107/s0021889807021206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- Winn M. D., Murshudov G. N. & Papiz M. Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Macromolecular Crystallography, Pt D 374, 300–321, doi: 10.1016/S0076-6879(03)74014-2 (2003). [DOI] [PubMed] [Google Scholar]
- Afonine P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallographica Section D-Biological Crystallography 68, 352–367, doi: 10.1107/S0907444912001308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby Z. et al. The role of protein dynamics in thymidylate synthase catalysis: variants of conserved 2′-deoxyuridine 5′-monophosphate (dUMP)-binding Tyr-261. Biochemistry 45, 7415–7428, doi: 10.1021/bi060152s (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.