

Multiphase chemistry of NO2 and alkaline matter in aerosol water explains rapid sulfate formation and severe haze in China.
Keywords: nanostructures, gradient nanostructures, friction, coefficient of friction, metals, Cu alloys, surface roughening
Abstract
Fine-particle pollution associated with winter haze threatens the health of more than 400 million people in the North China Plain. Sulfate is a major component of fine haze particles. Record sulfate concentrations of up to ~300 μg m−3 were observed during the January 2013 winter haze event in Beijing. State-of-the-art air quality models that rely on sulfate production mechanisms requiring photochemical oxidants cannot predict these high levels because of the weak photochemistry activity during haze events. We find that the missing source of sulfate and particulate matter can be explained by reactive nitrogen chemistry in aerosol water. The aerosol water serves as a reactor, where the alkaline aerosol components trap SO2, which is oxidized by NO2 to form sulfate, whereby high reaction rates are sustained by the high neutralizing capacity of the atmosphere in northern China. This mechanism is self-amplifying because higher aerosol mass concentration corresponds to higher aerosol water content, leading to faster sulfate production and more severe haze pollution.
INTRODUCTION
Persistent haze shrouding Beijing and the North China Plain (NCP) during cold winter periods threatens the health of ~400 million people living in a region of ~300,000 km2. Characteristic features of the winter haze in northern China include stagnant meteorological conditions with low mixing heights, high relative humidity (RH), large emissions of primary air pollutants, and fast production of secondary inorganic aerosols, especially sulfate (see section M1) (1–5). Analyzing surface-based observations at a site in Beijing during January 2013 (see section M2) and using concentration ratios of sulfate to sulfur dioxide ([SO42−]/[SO2]) as a proxy for the sulfate production rate (5), we find that sulfate production increases as PM2.5 (particulate matter with a diameter of less than 2.5 μm) levels increase (Fig. 1A). Ratios are six times higher during the most polluted periods (PM2.5 > 300 μg m−3) than during clean to moderately polluted conditions (ratios of 0.1 when PM2.5 < 50 μg m−3).
Fig. 1. Characteristic features of a major haze event in Beijing, China (January 2013).
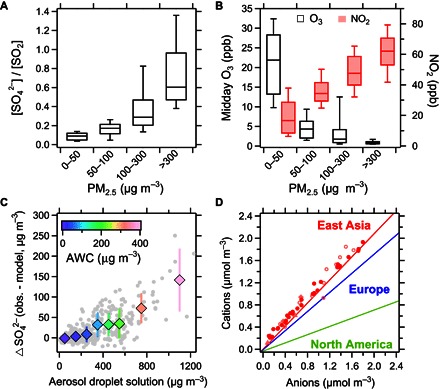
(A) Sulfate/sulfur dioxide ratio ([SO42−]/[SO2]) and (B) midday ozone (O3) concentrations (10:00 to 15:00 local time) and nitrogen dioxide (NO2) concentrations at different fine-particle (PM2.5) concentration levels. (C) Correlation of the unexplained sulfate concentration (ΔSO42− = observation − model) (see sections M2 and M3) with aerosol droplet solution and AWC (color-coded). (D) Anion-to-cation ratio in PM2.5 as observed in Beijing (solid symbols) and in other studies in northern China (open symbols) (table S1) compared to the characteristic ratios reported for aircraft measurements in the Arctic of outflow from East Asia, Europe, and North America (solid red, blue, and green lines; here, only NH4+, NO3−, and SO42− were considered) (87).
Traditional air quality models, however, fail to capture this key feature of NCP winter haze events even after accounting for aerosol-radiation-meteorology feedback (see sections M2 and M3) (6–8). The chemical mechanisms used in these models usually comprise gas-phase oxidation of sulfur dioxide by OH radicals and aqueous-phase reaction pathways in cloud water, involving H2O2 and O3, resulting in sulfate production rates that scale with the intensity of solar ultraviolet (UV) radiation (9, 10). During NCP haze days, UV radiation is significantly reduced because of the aerosol dimming effect, resulting in a decrease of most oxidant concentrations (5). Figure 1B shows that the midday O3 values drop from ~22 parts per billion (ppb) under clean conditions to ~1 ppb during the haze period (and also lose their typical diurnal variation, fig. S1). The reduced oxidant levels and increased sulfate production suggest the existence of a missing sulfate production pathway. Even after considering the gas phase and cloud/fog chemistry, there is still a large gap between modeled and observed sulfate (Fig. 1C). Adding an apparent heterogeneous process with sulfate production rates that scale with aerosol surface area and RH can greatly improve model predictions (see sections M3 to M5) (7), but the chemical mechanism of the missing sulfate production pathway has not yet been identified.
RESULTS AND DISCUSSION
We find that reactive nitrogen chemistry in aerosol water can explain the missing source of sulfate in NCP winter haze. Aerosol water is a key component of atmospheric aerosols, which serves as a medium that enables aqueous-phase reactions (11–13). The aerosol water content (AWC) in Beijing, calculated using measurements of RH and aerosol composition and the ISORROPIA-II thermodynamic equilibrium model (see section M6) (14–16), is well correlated with the missing sulfate content, the difference between measured and modeled sulfate (Δ[SO42−]) (Fig. 1C) (see sections M2 to M4), suggesting its involvement in the sulfate production. Note that because of the salt-induced freezing point depression (17), aerosol water will not freeze for a winter temperature of ~271 K in Beijing.
Taking the impact of mass transfer and ion strength into account, we make a conservative estimation of sulfate production rate for different reactions in the aerosol water under relevant atmospheric trace species concentration conditions (see sections M4 and M7 to M9) and find NO2 to be the most important oxidant in Beijing during haze periods (Fig. 2B). In the presence of aerosol water, gas-phase NO2 can partition into the condensed phase, react with SO2 dissolved in the aqueous phase, and produce sulfate as well as nitrite (R1) (18).
| (R1) |
Fig. 2. Aqueous-phase sulfate production by sulfur dioxide oxidation under characteristic conditions.
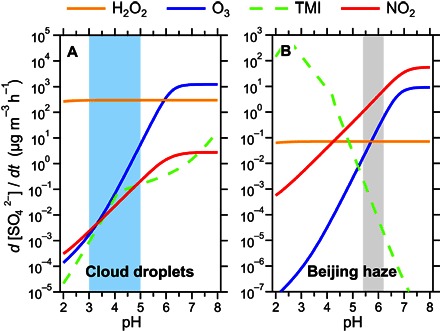
Sulfate production rates for (A) cloud droplets and (B) Beijing haze plotted against pH value.
Light blue– and gray-shaded areas indicate characteristic pH ranges for cloud water under clean to moderately polluted conditions and aerosol water during severe haze episodes in Beijing, respectively. The colored lines represent sulfate production rates calculated for different aqueous-phase reaction pathways with oxidants: hydrogen peroxide (H2O2), ozone (O3), transition metal ions (TMIs), and nitrogen dioxide (NO2). Characteristic reactant concentrations and model calculations for clouds and haze are taken from the literature and observations, and specified in Materials and Methods (see sections M7 to M9) (12, 21).
Under heavy haze conditions (PM2.5 ≥ 300 μg m−3), the sulfate production rates of the NO2 reaction pathway (R1) are ~1 to 7 μg m−3 h−1, much higher than the reaction rates involving other important aqueous oxidants such as O3 and H2O2. According to Zheng et al. (7), an additional sulfate production of ~3 μg m−3 h−1 is needed to explain the observations in the severe winter haze periods of Beijing (see section M9), which falls right into the range of production rates from the chemical reaction mechanism we proposed (R1). As illustrated in Fig. 2, sulfate production in aerosol water under haze conditions differs from that in cloud droplets, where the major oxidation pathways are reactions with H2O2 and O3, and NO2 plays only a minor role (12, 19). Thus, traditional air quality models usually include only the H2O2 and O3 reaction pathways of sulfate production in the aqueous phase, whereas the NO2 reaction pathway is neglected (7, 20).
The AWC is typically three to five orders of magnitude lower than the water content of cloud or fog (21). On this basis, how can the NO2 reaction pathway become important in such tiny amounts of water? The increased importance is due to the relatively high aerosol pH and elevated NO2 concentrations in Beijing and the NCP during haze periods. As shown in Fig. 2, aqueous oxidation rates of S(IV) by NO2 and O3 are strongly pH-dependent. High pH increases the solubility and the effective Henry’s law constant of SO2, pulling more SO2 into the aerosol water and thus increasing the reaction rate. When pH increases by one unit, the reaction rates increase by one and two orders of magnitude for NO2 and O3, respectively. The H2O2 reaction does not show a strong dependence because high pH reduces its reaction rate coefficient, which offsets the effect of increased solubility. Compared with North America and Europe, aerosols in the NCP are more neutralized (22), as shown by a higher cation-to-anion ratio (Fig. 1D). This neutralized feature is also well documented and is the reason that acid rain rarely occurs in northern China (see section M10) (22). Using the ISORROPIA-II thermodynamic equilibrium model (see section M6) (14–16) and in situ aerosol bulk composition measurements, we obtain average pH values of 5.4 to 6.2 for aerosol water under NCP haze conditions (see sections M6 and M9). Similar calculations based on size-segregated aerosol composition measurements even show a higher effective pH and sulfate production rates (fig. S2).
Elevated NO2 is another key factor that leads to fast sulfate formation. Substantial amounts of NO2 come from direct emission of NOx (= NO + NO2). Although the NO2-to-NOx ratios were reduced because of weak photochemistry during the haze event, the stagnant weather trapped more NO2 near the surface, resulting in elevated NO2 concentrations that were, on average, three times higher than those under clean conditions (~66 ppb, Fig. 1B). These periods of highest NO2 levels occurred when the concentrations of other photochemical oxidants that can produce sulfate (H2O2, O3, and OH) were low (Fig. 1B and fig. S1). Changes in pH and precursor concentrations together lead to the transition from an H2O2-dominated regime of aqueous sulfate production in cloud droplets to an NO2-dominated regime of aqueous sulfate production in haze (Fig. 2). Earlier studies had already suggested that the NO2 reaction pathway may contribute to sulfate formation in fogs under polluted conditions (23, 24), but during the Beijing haze event of January 2013, fog was not observed and sulfate production by NO2 occurred in aerosol water instead.
Sulfate production rates from the NO2 reaction pathway (R1) calculated on the basis of measurement data (hourly concentrations of NO2, SO2, PM2.5, and RH; sections M2 and M4) show a positive dependence on the PM2.5 concentration, varying from 0.01 μg m−3 h−1 under relatively clean conditions to nearly 10 μg m−3 h−1 in the most polluted periods (pink circles in Fig. 3). These reaction rates can account for the systematic underprediction of models and explain the large missing source of sulfate in the Beijing haze (black diamonds in Fig. 3). Under clean conditions, the OH reaction (green crosses in Fig. 3) dominates the oxidation pathways of SO2. As particle concentrations increase and more sulfate is produced, the photochemistry slows down, leading to less OH, and sulfate production via this pathway decreases. From this aspect, the OH reaction has a negative feedback, which is self-buffered against heavy pollution. As PM2.5 concentrations and RH increase simultaneously [a special feature of haze events in NCP (5)], the OH reaction becomes weaker, whereas the aqueous-phase reaction of NO2 starts to play a more important role. For the January 2013 conditions at PM2.5 ~100 to 200 μg m−3, the aqueous-phase reaction becomes the dominant oxidation pathway (Fig. 3). In contrast to the OH reaction, the NO2 reaction shows a positive feedback mechanism, where higher particle matter levels lead to more aerosol water, which accelerates sulfate production and further increases the aerosol concentration. This positive feedback intensifies the PM2.5 levels during haze periods, resulting in a series of record-breaking pollution events.
Fig. 3. Importance of the NO2 reaction pathway for sulfate production in the Beijing haze (January 2013).
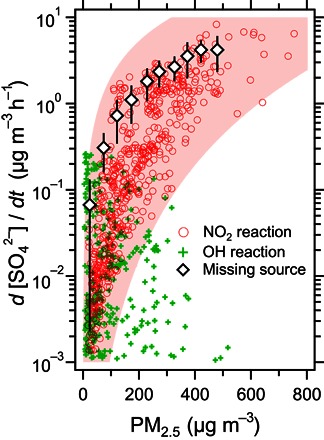
Sulfate production rates calculated for the aqueous-phase NO2 reaction pathway (pH 5.8) and the gas-phase OH reaction pathway compared to the missing source of sulfate. Crosses and circles represent hourly production rates calculated on the basis of measurement data, and the diamonds represent the average missing source (arithmetic mean ± SD) (see sections M3 to M5) (7). The pink-shaded area shows the maximum and minimum sulfate production rates by the NO2 reaction pathway bounded by the aerosol water pH ranging from 5.4 to 6.2 during haze periods.
The NO2 reaction with SO2 in aerosol water produces not only sulfate but also nitrite (R1), which may undergo subsequent oxidation or disproportionation reactions forming nitrate. This is consistent with the high nitrate concentrations observed during the haze event, which are of similar magnitude to the sulfate concentrations (up to ~160 μg m−3) (5) and have also not yet been explained by air quality models (7). Depending on the pH value of the haze droplets, nitrite can also form nitrous acid (HONO) and undergo reversible partitioning with the gas phase (25, 26). Moreover, the release of HONO removes H+, which may help to sustain the droplet acidity and efficient sulfate production (27, 28). For the same haze periods, record high aerosol nitrite concentrations of up to ~12 μg m−3 were observed in Shandong, northern China, with nitrite concentrations well correlated with NOx at high RH (>50%) (25). The nitrite-to-HONO molar ratio of ~3 in Shandong is a hundred times higher than the ratio observed during a pollution event in Nanjing, Yangtze River Delta of China, where only trace amounts of nitrite were detected in the aerosol phase (up to ~0.4 μg m−3), and the aerosol water there was more acidic (pH ~4) (24, 25). This further confirms the more neutralized feature of aerosol particle composition and haze in northern China.
Our study unfolds a new and more comprehensive conceptual model of sulfate formation in NCP haze events, including not only the traditional OH, H2O2, and O3 reaction pathways in atmospheric gas phase and cloud chemistry but also the NO2 reaction pathway in aerosol water (Fig. 4). The NO2 pathway may not be limited to winter haze because it may also be important at night and during fog events in polluted regions with high boundary layer concentrations of PM2.5 and NO2 and elevated RH (23, 24). The importance of multiphase chemistry holds for a wide range of aerosol pH. When aerosols become more acidic, the sulfate production can be maintained at a high rate through TMI reactions (Fig. 2B). The important role of aerosol pH in the multiphase reaction pathway highlights the need to better understand the sources of ammonia and alkaline aerosol components from natural and anthropogenic emissions (soil dust, seawater, agriculture, energy, industrial, and traffic). Furthermore, reactive nitrogen chemistry in aerosol water might also play a role in nitrate and secondary organic aerosol production during haze days when photochemistry is reduced.
Fig. 4. Conceptual model of sulfate formation in haze events in NCP.
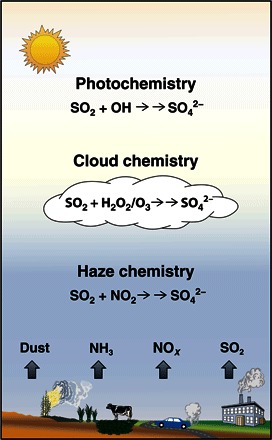
The traditional OH, H2O2, and O3 reaction pathways in atmospheric gas phase and cloud chemistry are included here, as well as the NO2 reaction pathway in aerosol water with elevated pH and NO2 concentrations proposed in this study.
Our results reveal the complex nature of haze pollution events in China, where NOx is not only a precursor for nitrate but also an important oxidant for sulfate formation. Thus, reductions of NOx emissions are expected to reduce nitrate, sulfate, and PM2.5 much more than anticipated by traditional air quality models. A large decrease in PM2.5 has already been observed in relation to traffic and energy control measures during the Beijing Olympic Games in 2008 and other events in the NCP. Heavy haze conditions with high pollutant concentration levels and largely neutralized aerosol water are key features of atmospheric chemistry in the NCP. These features will need to be considered in future air quality and pollutant emission control strategies in northern China, and perhaps also in other regions.
MATERIALS AND METHODS
M1. The January 2013 haze in Beijing
The severe haze episode in January 2013 is one of the worst atmospheric pollution events ever recorded in China (2, 9). In Beijing, the daily fine-particle (PM2.5) concentration reached up to 400 μg m−3, exceeding the World Health Organization guideline value by 16 times. The weak East Asian winter monsoon, which resulted in weakened surface winds and the anomalous southerly winds, was responsible for the haze events (4). The southerly winds transported more water vapor from the sea to eastern China. The anomalous high-pressure system at 500 hPa suppressed convection. Thus, the air in January was more stagnant, trapping more air pollutants and water vapor near the surface. The high PM2.5 concentration reduced the solar radiation and atmospheric photochemistry, resulting in decreases in the concentration of photochemical products such as OH and O3 (fig. S1).
A major feature of PM2.5 pollution during this haze event was the large contribution from secondary species, including inorganic (mainly sulfate, nitrate, and ammonium) and organic species (2, 5). However, contribution from secondary inorganic species (such as sulfate and nitrate) showed an increasing trend with increasing pollution levels, whereas contribution from organics decreased (5).
The fast production of sulfate, however, cannot be reproduced by model simulations, which have implemented aerosol-meteorology-radiation feedback and a state-of-the-art chemical mechanism (6, 7), that is, the gas-phase oxidation by OH (29, 30) and aqueous-phase oxidation in clouds by H2O2 and O3 (12, 21). Stabilized Criegee intermediates (sCIs) were also suggested as an oxidant of SO2 but contribute minor H2SO4 production compared with the conventional OH reaction in the midday (31–33). Further analysis has shown that the model simulation can be improved by introducing an apparent heterogeneous process with a reaction rate coefficient scaled with RH (see section M3) (7).
M2. Sampling location and experimental methods
We performed aerosol measurements from 1 to 31 January 2013 on the roof of the Environmental Science Building (40°00′17″N, 116°19′34″E, ~10 m above the ground) on the campus of Tsinghua University, an urban background site in Beijing. Table S2 summarizes the aerosol-related parameters and experimental methods. Concentrations of PM2.5 and PM10 were measured by an online PM-712 monitor (Kimoto Electric Co. Ltd.) equipped with a U.S. Environmental Protection Agency PM10 inlet and a PM2.5 virtual impactor (34). SO42− and other ions in PM2.5 were measured by an online ACSA-08 monitor (Kimoto Electric Co. Ltd.) and the filter-based analysis. The PM2.5 filter samples were collected from 12 to 24 January by median-volume samplers (Laoying) on prebaked Quartz filters (2500 QAT-UP; Pall Corporation) with a flow rate of 100 liters min−1 (35). The filter samples were analyzed by the Dionex ion chromatograph (DX-600 for cations and ICS-2000 for anions) (Dionex Corporation) for the concentration of water-soluble inorganic ions, including Na+, K+, Ca2+, Mg2+, NH4+, SO42−, NO3−, and Cl−. Organic carbon (OC) and elementary carbon concentrations in PM2.5 were measured by a Sunset Model 4 semicontinuous carbon analyzer (Beaverton) with a National Institute for Occupational Safety and Health–type temperature protocol (36). A factor of 1.6 was adopted to convert the mass of OC into the mass of organics (37, 38). Gaseous air pollutants SO2, NO2, and O3 were measured by the Atmospheric Environment Monitoring Network (39). The meteorology data were measured by the Milos 520 Weather Station (VAISALA Inc.). More details can be found in the work of Zheng et al. (5).
M3. WRF-CMAQ model simulation
The Weather Research and Forecasting—Community Multiscale Air Quality (WRF-CMAQ) model system was used to determine the missing source of sulfate through comparison with observational data. WRF is a new-generation mesoscale numerical weather prediction system designed to serve a wide range of meteorological applications (www.wrf-model.org/), and CMAQ is a three-dimensional Eulerian atmospheric chemistry and transport modeling system that simulates multipollutants (www.cmascenter.org/cmaq/). The releases of WRF v3.5.1 and CMAQ v5.0.1 were used in this study.
The model simulation was configured the same as in the work of Zheng et al. (7), as detailed in table S3. To better characterize the stagnant meteorological conditions, we applied observational nudging for temperature (T) and RH [above the planetary boundary layer (PBL)], and wind (within and above the PBL). The surface roughness is corrected according to Mass and Ovens (40) by increasing the friction velocity by 1.5 times in the PBL scheme, which significantly reduced the high biases in wind and RH simulations. In general, the simulated T, RH, and wind at the ground surface agree with observations (7). For the gas-phase reactions, we used the CB05 mechanism with active chlorine chemistry and the updated toluene mechanism of Whitten et al. (41). For the aqueous-phase reactions in clouds, we used the updated mechanism of the RADM model (20, 42). Reactions relevant for the sulfate formation were discussed in detail in section M4.
Here, the WRF-CMAQ modeling results were used in the following analysis: (i) the modeled sulfate concentration [SO42−] was used to calculate Δ[SO42−], the difference between observed and modeled [SO42−], and (ii) the modeled OH and H2O2 concentrations were used in the estimation of sulfate formation from the gas-phase reaction of OH with SO2 and the aqueous-phase reaction of H2O2 with HSO3−.
M4. Production of sulfate in WRF-CMAQ model
According to current understanding, secondary sulfate is produced through the oxidation of SO2. In the WRF-CMAQ model, oxidation occurs both in the gas phase and in the cloud/fog droplets. In the gas phase, the major pathway is the OH-initiated reaction (12, 43)
| (M1) |
in which the second-order kinetic constant can be expressed as
| (M2) |
where [M] is the concentration of N2 and O2, and k0(T) and k∞(T) represent the low- and high-pressure limiting rate constants, respectively. Their temperature dependence can be expressed as
| (M3) |
where k0300 = 3.3 × 10−31 cm6 molecule−2 s−1 and n = 4.3, and k∞300 = 1.6 × 10−12 cm3 molecule−1 s−1 and m = 0.
In cloud/fog droplets, the following aqueous reactions have been included in the WRF-CMAQ model (7). The expression of their reaction rate, as well as the rate coefficients, is summarized in table S4.
| (M4) |
| (M5) |
| (M6) |
| (M7) |
| (M8) |
| (M9) |
| (M10) |
M5. Apparent heterogeneous uptake of SO2 on aerosol surfaces
To improve the model simulation for the winter haze events in Beijing and the NCP, Zheng et al. (7) suggested the use of an apparent heterogeneous uptake coefficient (γ) of SO2 on aerosol surfaces as a function of RH (Eq. M11). γ is defined as the ratio of the number of collisions that result in the reaction SO2 (g) + Aerosol → SO42− to the theoretical total number of collisions. The overall rates of SO2 uptake and sulfate production are as fast as if the whole aerosol surface is covered by dust (44). Although the exact mechanism supporting this fast production rate is still unknown (7), this apparent heterogeneous source of sulfate can account for the underprediction of modeled sulfate, with significant reduction of normalized model bias from −54.2 to 6.3%.
| (M11) |
where γlow = 2 × 10−5 and γhigh = 5 × 10−5.
With γ, a sulfate production rate RH,g can be determined by Eq. M12:
| (M12) |
where Rp is the radius of aerosol particles, Dg is the gas-phase molecular diffusion coefficient of SO2, ν is the mean speed of gaseous SO2 molecules, and Saerosol is the surface area concentration of aerosol particles.
M6. ISORROPIA-II model calculation
The ISORROPIA-II model (15) was used to calculate the AWC and pH. The ISORROPIA-II is a thermodynamic equilibrium model that predicts the physical state and composition of atmospheric inorganic aerosols. It can be used in two modes: the reverse mode and the forward mode. The reverse mode calculated the thermodynamic equilibrium based on aerosol-phase concentrations, whereas the forward mode relied on both aerosol-phase and gas-phase concentrations (16). Its ability in predicting AWC and pH has been demonstrated by Guo et al. (45) and Xu et al. (16).
To evaluate the aerosol pH and AWC, we performed both reverse-mode and forward-mode model simulations and used their averages for further analyses. The gaseous NH3 was not measured in our January campaign, but long-term measurement (46) shows a compact correlation between NH3 and NOx concentrations in the winter season of Beijing. Accordingly, we estimated the NH3 concentration from the observed NOx concentration with an empirical equation derived from Meng et al. (46), that is, NH3 (ppb) = 0.34 × NOx (ppb) + 0.63. The contribution of organic compounds to AWC, Worg (the mass concentration of aerosol water associated with organics), was estimated by the same approach of Guo et al. (45)
| (M13) |
where OM is the mass concentration of organic matter, ρw is the density of water (ρw = 1.0 × 103 kg m−3), ρorg is the density of organics (ρorg = 1.4 × 103 kg m−3) (45), and κorg is the hygroscopicity parameter (47) of organic aerosol compositions. We adopted a κorg of 0.06 based on previous cloud condensation nuclei measurements in Beijing (48).
M7. Kinetics of mass transport
For multiphase reactions, the overall reaction rate depends not only on the rate of chemical reactions but also on the mass transport in different medium and across the interface. To account for the effects of mass transport, we adopted the formulation of a standard resistance model (12)
| (M14) |
where RH,aq is the sulfate production rate, Raq is the aqueous-phase reaction rate, and Jaq,lim is the limiting mass transfer rate. For the oxidation of S(IV) by a given oxidant Oxi (12)
| (M15) |
where [SO2·H2O], [HSO3−], [SO32−], and [Oxi] are the respective aqueous-phase concentrations, and k0, k1, and k2 are the corresponding second-order reaction rate coefficients as detailed in table S4. The aqueous [Oxi] is assumed to be in equilibrium with its gas-phase concentration and can be determined by Henry’s law (12)
| (M16) |
where p∞(X) (atm) is the partial pressure of species X in the bulk gas phase and H* (M atm−1) is the effective Henry’s constant (table S5).
The limiting mass transfer rate Jaq (M s−1) is calculated by Eqs. M17 and M18 (12)
| (M17) |
| (M18) |
where X refers to SO2 or the oxidant Oxi such as O3, H2O2, and NO2.
The mass transfer rate coefficient kMT (s−1) can be calculated by (12)
| (M19) |
where Rp is the aerosol radius, and and are the continuum regime resistance and the free-molecular (or kinetic) regime resistance, respectively. Dg is the gas-phase molecular diffusion coefficient, and ν is the mean molecular speed of X. α is the mass accommodation coefficient of X on the droplet surface, which accounts for imperfect sticking of impinging molecules to the surface, and we adopted literature values of 0.11, 0.23, 2.0 × 10−3 (12), and 2.0 × 10−4 (49) for SO2, H2O2, O3, and NO2, respectively. Aqueous-phase mass transfer can be ignored for the size range considered here (Dp ≤ 2.5 μm) (12). An equivalent Rp of 0.15 and 15 μm was assumed for aerosols and cloud droplets, respectively.
M8. Influences of ionic strength on aqueous sulfate-producing reactions
Aerosol liquid water constitutes an aqueous electrolyte that can be extremely concentrated with high ionic strength (I) up to 100 M (50). This highly concentrated chemical environment will affect the inclination of a species to participate in the aqueous-phase chemical reactions, reflected by an apparent reaction rate constant (k) different from that in an ideal solution (21).
The influence of I on k is complicated and is not yet fully understood. According to current understanding, I affects k through its integrated effects on the activity coefficient (a) of reactants and products for most reactions (51). For example, for a reaction where reactants A and B form an activated complex (AB)*, which then quickly decomposes into a final product P (A + B → (AB)* → P), the k-I dependence can be described as (51)
| (M20) |
where kI=0 refers to the kinetic constant at I of 0 M. Theories used to predict ai at given I varied with the ranges of I and the species nature, that is, being an ion or a neutral species, as summarized in table S6.
Major aqueous sulfate-producing reactions considered here include S(IV) oxidation by H2O2, O3, TMI + O2, and NO2 (Fig. 2). Among these reactions, the influence of I has been studied experimentally, with I ranging up to ~5 M for H2O2 and slightly higher than 1 M for O3 and TMI + O2. The observed k-I relationships of these reactions agree with theoretical predictions summarized in table S7 (52–56). As shown in fig. S3, with increasing ionic strength, the rate constant for H2O2 decreases first when I is below ~1 M and begins to increase once I goes above ~1 M (52, 57). For O3, the rate constant is positively related to I. For TMI + O2 [here, only Fe(III) and Mn(II) are considered as effective catalyzing TMIs (58, 59)], the rate constant decreases significantly with increasing I, even without considering the sulfate inhabitation effect (referring to the effect that the formation of the sulfate-TMI complex would reduce catalytically active TMI concentrations) (55, 60).
Currently, however, no k-I relationship was reported for NO2, whereas some plausible estimation can be made on the basis of the principle of theories discussed above. Two kinds of mechanisms have been suggested for the NO2-S(IV) reactions. One is the oxygen-atom transfer reaction (61)
| (M21a) |
| (M21b) |
| (M21c) |
in which the first reaction (Eq. M21a) is the rate-controlling step.
The other mechanism is an electron transfer reaction (62–64)
| (M22) |
followed by the sulfur auto-oxidation processes (12). The first reaction (Eq. M22) is the rate-controlling step because follow-up reactions with radicals are very fast.
For both mechanisms, the rate-controlling step of the NO2-S(IV) reaction is a reaction of an ion with a neutral molecule. According to Herrmann (51), the k-I relation for this type of reaction should follow
| (M23) |
where b is the kinetic salting coefficient. According to Kameoka and Pigford (65), b is expected to be positive. Given a positive b, increasing I will lead to an increase in k, as shown by the red dashed curve in fig. S3 (we take an arbitrary value of 0.5 M−1 for b, which is not determined from experiments and is used only for the purpose of illustration).
During the severe Beijing hazes (when PM2.5 is higher than 300 μg m−3), ionic strength in aerosol liquid water ranged from 13 to 43 M, as predicted by the ISORROPIA-II model (see section M6) (15). Direct extrapolation of the observed/predicted k-I relationship (fig. S3) into such high ranges of ionic strength may not be appropriate. Thus, the rate constants are taken as for diluted solution, although based on current observations and theories (table S7 and fig. S3), this treatment will lead to conservative estimations of the real sulfate production rates of the NO2, O3, and H2O2 pathways in Fig. 2 but an overestimation of the TMI + O2 pathway.
M9. Data used in Fig. 2
Detailed descriptions on how to derive the sulfate production rate for different aqueous-phase oxidation pathways of SO2 (that is, by H2O2, O3, TMI + O2, and NO2) in Fig. 2 can be found in the study of Seinfeld and Pandis (12). The aqueous-phase reactions involved are listed as Reactions R1 and M4 to M8. The rate expressions and rate coefficients and the constants that we used for calculating the apparent Henry’s constant are summarized in tables S4 and S5, respectively.
In Fig. 2, the “cloud droplets” scenario is taken from the work of Seinfeld and Pandis (12) and Herrmann et al. (21): [SO2(g)] = 5 ppb, [NO2(g)] = 1 ppb, [H2O2(g)] = 1 ppb, [O3(g)] = 50 ppb, [Fe(III)] = 0.3 μM, [Mn(II)] = 0.03 μM, liquid water content = 0.1 g m−3, and cloud droplet radius Rp = 15 μm. In this scenario, the temperature T is taken to be the same as that used in the “Beijing haze” scenario described below.
The “Beijing haze” scenario was taken according to the measurement data during the most polluted haze periods (PM2.5 > 300 μg m−3). The average values were used in our calculation: [SO2(g)] = 40 ppb, [NO2(g)] = 66 ppb, [H2O2(g)] = 0.01 ppb, [O3(g)] = 1 ppb, AWC = 300 μg m−3, aerosol droplet radius Rp = 0.15 μm, and T = 271 K. The concentrations of [Fe(III)] and [Mn(II)] are pH-dependent (fig. S4). The pH dependence is mostly due to the precipitation equilibrium of Fe(OH)3 and Mn(OH)2
| (M24) |
where and are the precipitation constants of Fe(OH)3 and Mn(OH)2, respectively (66). When all Fe(OH)3 and Mn(OH)2 are dissolved, further decrease of pH will not increase [Fe(III)] and [Mn(II)], resulting in a plateau at low pH (fig. S4). The total soluble Fe and Mn are estimated to be 18 and 42 ng m−3, respectively, based on data in the literature and observations in Beijing (2, 59, 66–70).
The severe haze events observed in Beijing are a regional phenomenon (5). Because Beijing is located in the northwestern edge of the polluted area, the air pollution in cities south of Beijing is even more severe. NO2 concentrations in Beijing are comparable to the southern cities, whereas SO2 concentrations in the latter are typically two to four times higher than those in Beijing (fig. S5). Thus, the sulfate production rate from the NO2 pathway that we predicted with the Beijing data in Fig. 2 is a conservative estimation for the contribution of this pathway to the sulfate formation in the whole NCP.
Considering that regional transport from cities south of Beijing has contributed to the severe haze episode in January 2013 (5, 71) and judging from the air pollution trend shown in fig. S5, the polluted air parcels could typically have been processed under severe haze conditions in Beijing and in cities south of it for more than 3 days before the peak pollution hour in Beijing. Because cloud amounts were low (7, 72, 73) during the January 2013 winter haze periods, we assumed that aerosols spent 100% of their lifetime under noncloud conditions (RH < 100%) (21). Under these conditions, to produce the observed sulfate concentration (~200 μg m−3), the required average heterogeneous production rate can be determined to be ~3 μg m−3 h−1. This estimation also falls into the ranges of 1 to 7 μg m−3 h−1 that we predicted for the NO2 pathway in Fig. 2.
M10. Aerosol acidity in northern China
The neutralized feature of aerosols in East Asia (including Beijing and the NCP) has been well documented in the literature, with high cation-to-anion ratios (45, 74–86). Aircraft measurements in the Arctic further support this conclusion, showing that the aerosols in the Arctic characterized as coming from the outflow of East Asia were mostly neutralized, whereas the aerosols transported from North America to the Arctic were highly acidic (lines in Fig. 1D) (87).
The low aerosol acidity in East Asia can be attributed to its high NH3 and mineral dust emissions. For example, in 2008, the emission ratios of NH3 to 2× SO2 (molar ratio of NH3 emission divided by twice the SO2 emission) are 0.37, 0.86, and 1.04 for North America, Europe, and East Asia, respectively (87). Northern China is expected to have a higher atmospheric neutralizing capacity than other parts of East Asia because of the high NH3 emission in this region. Both satellite data (88, 89) and emission inventories (90) show that northern China is one of the most NH3-rich areas in East Asia because of its intense agriculture activities. The NCP (that is, the cities of Beijing and Tianjin and the provinces of Hebei, Henan, and Shandong) accounts for 30 to 40% of the total NH3 emissions in China (90) while contributing ~20% of the emissions for SO2 and NOx (91).
The high mass fraction of mineral dust (10 to 20%) is a distinct characteristic of PM2.5 in the NCP (2, 70, 92, 93). The influence of mineral aerosols is also higher for northern China than for southern China (94–98). Major sources of mineral aerosols include urban fugitive dust (resuspended road dust, construction dust, etc.) and the long-range transported Asian dust (99–101). The mineral dusts are observed to be internally mixed with sulfate, nitrate, and ammonia, suggesting their participation in atmospheric processing (102–105). Without mineral components (Ca2+, Mg2+, K+, and Na+), aerosol pH in northern China may drop below 5.6, showing an acidic nature (37).
In addition, despite the high emission of acidic gases (SO2 and NOx), rainwater in northern China has average pH values higher than 5.6 (fig. S6), suggesting an alkaline tendency in contrast to other areas (for example, the United States) (12, 22). A pH of 5.6 is often taken as the “natural” acidity of rainwater (water in equilibrium with CO2), which has been considered as the demarcation line of acidic precipitation (12). The high pH values in rainwater of northern China are caused by alkaline aerosol particles (12), which have a large buffering capacity to offset the effects of anthropogenic acidity.
M11. Contribution from sCIs and NO3 radicals
We have also investigated the reactions of S(IV) with NO3 radicals and sCIs, both of which show minor contributions (0.03 and 0.69%) compared to our proposed mechanism S(IV) + NO2(aq).
Oxidation by NO3 radicals
An average NO3 radical concentration of ~2.5 × 10−3 parts per trillion was determined by our modeling results for the severe pollution periods. Taking an effective Henry’s constant of 0.6 M atm−1 for NO3 and a reaction rate constant of 1.4 × 109 M−1 s−1 (106), we determined a sulfate production rate of 5.1 × 10−4 μg m−3 h−1 for the NO3 reaction. This rate is only 0.03% compared to the proposed mechanism S(IV) + NO2(aq) at pH 5.8 (Fig. 2) and is thus negligible.
Oxidation by sCI
According to Mauldin et al. (31), we calculated RsCI, the sulfate formation rate from the sCI mechanism, by
| (M25) |
where [sCI] and [SO2] are the concentrations of sCI and SO2, respectively, and the reaction rate coefficient is 6 × 10−13 cm3 molecule−1 s−1. The concentration of sCI can be determined by the following equation (31)
| (M26) |
where YsCI is the yield of sCI ~0.5, is the corresponding rate coefficient for the reaction of O3 with individual alkenes, and τsCI is the lifetime of the sCI, which is 0.2 s (31). After taking a typical alkene profile for the haze period (107), we determined an RsCI of ~0.69% of the proposed S(IV) + NO2(aq) mechanism.
Supplementary Material
Acknowledgments
Funding: This study was supported by the Max Planck Society (MPG), the National Natural Science Foundation of China (21107061, 21190054) and the EU project BACCHUS (603445) and PEGASOS (265148). Y.C. thanks the MPG Minerva program. G.Z. thanks Chinese Scholarship Council for financial support of her study at the Max Planck Institute for Chemistry. Author contributions: K.H., Y.C., and G.Z. proposed the initial idea. Y.C. and H.S. designed and led the study. G.Z., Y.C., and H.S. conducted the data analyses. C.W., Q.M., and B.Z. performed the model simulation. Q.Z. and K.H. provided the field observation and supported the model analyses. Z.W. and M.G. supported the model analyses. Y.C., H.S., G.Z., and U.P. interpreted the data. Y.C., H.S., U.P., G.Z., and G.C. wrote the manuscript, with inputs from all coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at hhttp://advances.sciencemag.org/cgi/content/full/2/12/e1601530/DC1
fig. S1. Weakened photochemistry by aerosol dimming effects during January 2013 in Beijing.
fig. S2. Importance of the NO2 reaction pathway for sulfate production in the Beijing haze (January 2013).
fig. S3. Influence of ionic strength (I) on rate of aqueous sulfate-producing reactions.
fig. S4. Estimation of Fe3+ and Mn2+ concentrations as a function of aerosol water pH during Beijing hazes.
fig. S5. Regional pollution across the NCP during January 2013.
fig. S6. Annual precipitation pH of China in 2013.
fig. S7. The same as Fig. 2 but with a lower limit of reaction rate constants reported by Lee and Schwartz (18).
table S1. Previously reported concentrations of cations and anions in PM2.5 during winter for cities in NCP used in Fig. 1D.
table S2. Summary of field observation and methods in this study.
table S3. Domain, configurations, and major dynamic and physical options used in WRF v3.5.1.
table S4. Rate expression and rate coefficients of relevant aqueous-phase reactions.
table S5. Constants for calculating the apparent Henry’s constant (H*).
table S6. Summary of suggested activity coefficient (a)–ionic strength (I) dependence.
table S7. Influence of ionic strength (I) on rate of aqueous sulfate-producing reactions.
REFERENCES AND NOTES
- 1.P. Brimblecombe, The Big Smoke (Methuen, 1987). [Google Scholar]
- 2.Huang R.-J., Zhang Y., Bozzetti C., Ho K.-F., Cao J.-J., Han Y., Daellenbach K. R., Slowik J. G., Platt S. M., Canonaco F., Zotter P., Wolf R., Pieber S. M., Bruns E. A., Crippa M., Ciarelli G., Piazzalunga A., Schwikowski M., Abbaszade G., Schnelle-Kreis J., Zimmermann R., An Z., Szidat S., Baltensperger U., El Haddad I., Prévôt A. S. H., High secondary aerosol contribution to particulate pollution during haze events in China. Nature 514, 218–222 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Guo S., Hu M., Zamora M. L., Peng J., Shang D., Zheng J., Du Z., Wu Z., Shao M., Zeng L., Molina M. J., Zhang R., Elucidating severe urban haze formation in China. Proc. Natl. Acad. Sci. U.S.A. 111, 17373–17378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang R., Li Q., Zhang R., Meteorological conditions for the persistent severe fog and haze event over eastern China in January 2013. Sci. China Earth Sci. 57, 26–35 (2014). [Google Scholar]
- 5.Zheng G. J., Duan F. K., Su H., Ma Y. L., Cheng Y., Zheng B., Zhang Q., Huang T., Kimoto T., Chang D., Pöschl U., Cheng Y. F., He K. B., Exploring the severe winter haze in Beijing: The impact of synoptic weather, regional transport and heterogeneous reactions. Atmos. Chem. Phys. 15, 2969–2983 (2015). [Google Scholar]
- 6.Wang J., Wang J., Wang S., Jiang J., Ding A., Zheng M., Zhao B., Wong D. C., Zhou W., Zheng G., Wang L., Pleim J. E., Hao J., Impact of aerosol–meteorology interactions on fine particle pollution during China’s severe haze episode in January 2013. Environ. Res. Lett. 9, 094002 (2014). [Google Scholar]
- 7.Zheng B., Zhang Q., Zhang Y., He K. B., Wang K., Zheng G. J., Duan F. K., Ma Y. L., Kimoto T., Heterogeneous chemistry: A mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China. Atmos. Chem. Phys. 14, 16731–16776 (2014). [Google Scholar]
- 8.Zhang R., Wang G., Guo S., Zamora M. L., Ying Q., Lin Y., Wang W., Hu M., Wang Y., Formation of urban fine particulate matter. Chem. Rev. 115, 3803–3855 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Ehhalt D. H., Rohrer F., Dependence of the OH concentration on solar UV. J. Geophys. Res. 105, 3565–3571 (2000). [Google Scholar]
- 10.Rohrer F., Berresheim H., Strong correlation between levels of tropospheric hydroxyl radicals and solar ultraviolet radiation. Nature 442, 184–187 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Pilinis C., Seinfeld J. H., Grosjean D., Water content of atmospheric aerosols. Atmos. Environ. 23, 1601–1606 (1989). [Google Scholar]
- 12.J. H. Seinfeld, S. N. Pandis, Atmospheric Chemistry and Physics, from Air Pollution to Climate Change (Wiley, 2006). [Google Scholar]
- 13.Ervens B., Turpin B. J., Weber R. J., Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): A review of laboratory, field and model studies. Atmos. Chem. Phys. 11, 11069–11102 (2011). [Google Scholar]
- 14.Nenes A., Pandis S. N., Pilinis C., Continued development and testing of a new thermodynamic aerosol module for urban and regional air quality models. Atmos. Environ. 33, 1553–1560 (1999). [Google Scholar]
- 15.Fountoukis C., Nenes A., ISORROPIA II: A computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+-NH4+-Na+-SO42−-NO3−-Cl−-H2O aerosols. Atmos. Chem. Phys. 7, 4639–4659 (2007). [Google Scholar]
- 16.Xu L., Guo H., Boyd C. M., Klein M., Bougiatioti A., Cerully K. M., Hite J. R., Isaacman-VanWertz G., Kreisberg N. M., Knote C., Olson K., Koss A., Goldstein A. H., Hering S. V., de Gouw J., Baumann K., Lee S.-H., Nenes A., Weber R. J., Lee Ng N., Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. Proc. Natl. Acad. Sci. U.S.A. 112, 37–42 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koop T., Luo B., Tsias A., Peter T., Water activity as the determinant for homogeneous ice nucleation in aqueous solutions. Nature 406, 611–614 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Y. N. Lee, S. E. Schwartz, Precipitation Scavenging, Dry Deposition and Resuspension (Elsevier, 1983), pp. 453–470. [Google Scholar]
- 19.Huang X., Song Y., Zhao C., Li M., Zhu T., Zhang Q., Zhang X., Pathways of sulfate enhancement by natural and anthropogenic mineral aerosols in China. J. Geophys. Res. 119, 14165–14179 (2014). [Google Scholar]
- 20.Walcek C. J., Taylor G. R., A theoretical method for computing vertical distributions of acidity and sulfate production within cumulus clouds. J. Atmos. Sci. 43, 339–355 (1986). [Google Scholar]
- 21.Herrmann H., Schaefer T., Tilgner A., Styler S. A., Weller C., Teich M., Otto T., Che A., Tropospheric aqueous-phase chemistry: Kinetics, mechanisms, and its coupling to a changing gas phase. Chem. Rev. 115, 4259–4334 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Cao J., Tie X., Dabberdt W. F., Jie T., Zhao Z., An Z., Shen Z., On the potential high acid deposition in northeastern China. J. Geophys. Res. 118, 4834–4846 (2013). [Google Scholar]
- 23.Pandis S. N., Seinfeld J. H., Mathematical modeling of acid deposition due to radiation fog. J. Geophys. Res. 94, 12911–12923 (1989). [Google Scholar]
- 24.Xie Y., Ding A., Nie W., Mao H., Qi X., Huang X., Xu Z., Kerminen V.-M., Petäjä T., Chi X., Virkula A., Boy M., Xue L., Guo J., Sun J., Yang X., Kulmala M., Fu C., Enhanced sulfate formation by nitrogen dioxide: Implications from in-situ observations at the SORPES Station. J. Geophys. Res. 120, 12679–12694 (2015). [Google Scholar]
- 25.Wang L., Wen L., Xu C., Chen J., Wang X., Yang L., Wang W., Yang X., Sui X., Yao L., Zhang Q., HONO and its potential source particulate nitrite at an urban site in North China during the cold season. Sci. Total Environ. 538, 93–101 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Su H., Cheng Y., Oswald R., Behrendt T., Trebs I., Meixner F. X., Andreae M. O., Cheng P., Zhang Y., Pöschl U., Soil nitrite as a source of atmospheric HONO and OH radicals. Science 333, 1616–1618 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Laskin A., Gaspar D. J., Wang W., Hunt S. W., Cowin J. P., Colson S. D., Finlayson-Pitts B. J., Reactions at interfaces as a source of sulfate formation in sea-salt particles. Science 301, 340–344 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Alexander B., Park R. J., Jacob D. J., Li Q. B., Yantosca R. M., Savarino J., Lee C. C. W., Thiemens M. H., Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes. J. Geophys. Res. 110, D10307 (2005). [Google Scholar]
- 29.Stockwell W. R., Calvert J. G., The mechanism of the HO-SO2 reaction. Atmos. Environ. 17, 2231–2235 (1983). [Google Scholar]
- 30.Blitz M. A., Hughes K. J., Pilling M. J., Determination of the high-pressure limiting rate coefficient and the enthalpy of reaction for OH+SO2. J. Phys. Chem. A 107, 1971–1978 (2003). [Google Scholar]
- 31.Mauldin R. L. III, Berndt T., Sipilä M., Paasonen P., Petäjä T., Kim S., Kurtén T., Stratmann F., Kerminen V.-M., Kulmala M., A new atmospherically relevant oxidant of sulphur dioxide. Nature 488, 193–196 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Kim S., Guenther A., Lefer B., Flynn J., Griffin R., Rutter A. P., Gong L., Cevik B. K., Potential role of stabilized Criegee radicals in sulfuric acid production in a high biogenic VOC environment. Environ. Sci. Technol. 49, 3383–3391 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Boy M., Mogensen D., Smolander S., Zhou L., Nieminen T., Paasonen P., Plass-Dülmer C., Sipilä M., Petäjä T., Mauldin L., Berresheim H., Kulmala M., Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations. Atmos. Chem. Phys. 13, 3865–3879 (2013). [Google Scholar]
- 34.Kaneyasu N., Yamamoto S., Sato K., Takami A., Hayashi M., Hara K., Kawamoto K., Okuda T., Hatakeyama S., Impact of long-range transport of aerosols on the PM2.5 composition at a major metropolitan area in the northern Kyushu area of Japan. Atmos. Environ. 97, 416–425 (2014). [Google Scholar]
- 35.Zhang Q., Duan F., He K., Ma Y., Li H., Kimoto T., Zheng A., Organic nitrogen in PM2.5 in Beijing. Front. Environ. Sci. Eng. 9, 1004–1014 (2015). [Google Scholar]
- 36.Zheng G. J., Cheng Y., He K. B., Duan F. K., Ma Y. L., A newly identified calculation discrepancy of the Sunset semi-continuous carbon analyzer. Atmos. Meas. Tech. 7, 1969–1977 (2014). [Google Scholar]
- 37.Zhang J. K., Sun Y., Liu Z. R., Ji D. S., Hu B., Liu Q., Wang Y. S., Characterization of submicron aerosols during a month of serious pollution in Beijing, 2013. Atmos. Chem. Phys. 14, 2887–2903 (2014). [Google Scholar]
- 38.Xing L., Fu T., Cao J. J., Lee S. C., Wang G. H., Ho K. F., Cheng M., You C., Wang T. J., Seasonal and spatial variability of the OM/OC mass ratios and high regional correlation between oxalic acid and zinc in Chinese urban organic aerosols. Atmos. Chem. Phys. 13, 4307–4318 (2013). [Google Scholar]
- 39.Tang G., Wang Y., Li X., Ji D., Hsu S., Gao X., Spatial-temporal variations in surface ozone in Northern China as observed during 2009–2010 and possible implications for future air quality control strategies. Atmos. Chem. Phys. 12, 2757–2776 (2012). [Google Scholar]
- 40.C. Mass, D. Ovens, WRF model physics: Progress, problems, and perhaps some solutions, the 11th WRF Users’ Workshop, 21–25 June, NCAR Center Green Campus, http://www2.mmm.ucar.edu/wrf/users/workshops/WS2010/presentations/session 4/4-1_WRFworkshop2010Final.pdf (2010) [accessed December 2015].
- 41.Whitten G. Z., Heo G., Kimura Y., McDonald-Buller E., Allen D. T., Carter W. P. L., Yarwood G., A new condensed toluene mechanism for Carbon Bond: CB05-TU. Atmos. Environ. 44, 5346–5355 (2010). [Google Scholar]
- 42.Chang J. S., Brost R. A., Isaksen I. S. A., Madronich S., Middleton P., Stockwell W. R., Walcek C. J., A three-dimensional Eulerian acid deposition model: Physical concepts and formulation. J. Geophys. Res. 92, 14681–14700 (1987). [Google Scholar]
- 43.NASA/Jet Propulsion Laboratory, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies (NASA/Jet Propulsion Laboratory, 2010).
- 44.Zhang Y., Carmichael G. R., The role of mineral aerosol in tropospheric chemistry in East Asia—A model study. J. Appl. Meteorol. 38, 354–366 (1999). [Google Scholar]
- 45.Guo H., Xu L., Bougiatioti A., Cerully K., Capps S., Hite J., Carlton A., Lee S., Bergin M., Ng N., Nenes A., Weber R., Fine-particle water and pH in the southeastern United States. Atmos. Chem. Phys. 15, 5211–5228 (2015). [Google Scholar]
- 46.Meng Z. Y., Lin W. L., Jiang X. M., Yan P., Wang Y., Zhang Y. M., Jia X. F., Yu X. L., Characteristics of atmospheric ammonia over Beijing, China. Atmos. Chem. Phys. 11, 6139–6151 (2011). [Google Scholar]
- 47.Petters M. D., Kreidenweis S. M., A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 7, 1961–1971 (2007). [Google Scholar]
- 48.Gunthe S. S., Rose D., Su H., Garland R. M., Achtert P., Nowak A., Wiedensohler A., Kuwata M., Takegawa N., Kondo Y., Hu M., Shao M., Zhu T., Andreae M. O., Pöschl U., Cloud condensation nuclei (CCN) from fresh and aged air pollution in the megacity region of Beijing. Atmos. Chem. Phys. 11, 11023–11039 (2011). [Google Scholar]
- 49.Jacob D. J., Heterogeneous chemistry and tropospheric ozone. Atmos. Environ. 34, 2131–2159 (2000). [Google Scholar]
- 50.Cheng Y., Su H., Koop T., Mikhailov E., Pöschl U., Size dependence of phase transitions in aerosol nanoparticles. Nat. Commun. 6, 5923 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herrmann H., Kinetics of aqueous phase reactions relevant for atmospheric chemistry. Chem. Rev. 103, 4691–4716 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Maaß F., Elias H., Wannowius K. J., Kinetics of the oxidation of hydrogen sulfite by hydrogen peroxide in aqueous solution: Ionic strength effects and temperature dependence. Atmos. Environ. 33, 4413–4419 (1999). [Google Scholar]
- 53.Maahs H. G., Kinetics and mechanism of the oxidation of S(IV) by ozone in aqueous solution with particular reference to SO2 conversion in nonurban tropospheric clouds. J. Geophys. Res. 88, 10721–10732 (1983). [Google Scholar]
- 54.Lagrange J., Pallares C., Lagrange P., Electrolyte effects on aqueous atmospheric oxidation of sulphur dioxide by ozone. J. Geophys. Res. 99, 14595–14600 (1994). [Google Scholar]
- 55.Martin L. R., Hill M. W., The iron catalyzed oxidation of sulfur: Reconciliation of the literature rates. Atmos. Environ. 21, 1487–1490 (1987). [Google Scholar]
- 56.Martin L. R., Hill M. W., Tai A. F., Good T. W., The iron catalyzed oxidation of sulfur(IV) in aqueous solution: Differing effects of organics at high and low pH. J. Geophys. Res. 96, 3085–3097 (1991). [Google Scholar]
- 57.Ali H. M., Iedema M., Yu X.-Y., Cowin J. P., Ionic strength dependence of the oxidation of SO2 by H2O2 in sodium chloride particles. Atmos. Environ. 89, 731–738 (2014). [Google Scholar]
- 58.Huss A. Jr, Lim P. K., Eckert C. A., Oxidation of aqueous sulfur dioxide. 1. Homogeneous manganese(II) and iron(II) catalysis at low pH. J. Phys. Chem. 86, 4224–4228 (1982). [Google Scholar]
- 59.Alexander B., Park R. J., Jacob D. J., Gong S., Transition metal-catalyzed oxidation of atmospheric sulfur: Global implications for the sulfur budget. J. Geophys. Res. 114, D02309 (2009). [Google Scholar]
- 60.Martin L. R., Good T. W., Catalyzed oxidation of sulfur dioxide in solution: The iron-manganese synergism. Atmos. Environ. 25, 2395–2399 (1991). [Google Scholar]
- 61.Clifton C. L., Altstein N., Huie R. E., Rate constant for the reaction of nitrogen dioxide with sulfur(IV) over the pH range 5.3-13. Environ. Sci. Technol. 22, 586–589 (1988). [DOI] [PubMed] [Google Scholar]
- 62.Nash T., The effect of nitrogen dioxide and of some transition metals on the oxidation of dilute bisulphite solutions. Atmos. Environ. 13, 1149–1154 (1979). [Google Scholar]
- 63.Huie R. E., The reaction kinetics of NO2. Toxicology 89, 193–216 (1994). [DOI] [PubMed] [Google Scholar]
- 64.Spindler G., Hesper J., Brüggemann E., Dubois R., Müller T., Herrmann H., Wet annular denuder measurements of nitrous acid: Laboratory study of the artefact reaction of NO2 with S(IV) in aqueous solution and comparison with field measurements. Atmos. Environ. 37, 2643–2662 (2003). [Google Scholar]
- 65.Kameoka Y., Pigford R. L., Absorption of nitrogen dioxide into water, sulfuric acid, sodium hydroxide, and alkaline sodium sulfite aqueous solutions. Ind. Eng. Chem. Fundam. 16, 163–169 (1977). [Google Scholar]
- 66.Graedel T. E., Weschler C. J., Chemistry within aqueous atmospheric aerosols and raindrops. Rev. Geophys. 19, 505–539 (1981). [Google Scholar]
- 67.Meskhidze N., Chameides W. L., Nenes A., Chen G., Iron mobilization in mineral dust: Can anthropogenic SO2 emissions affect ocean productivity?. Geophys. Res. Lett. 30, 2085 (2003). [Google Scholar]
- 68.Solmon F., Chuang P. Y., Meskhidze N., Chen Y., Acidic processing of mineral dust iron by anthropogenic compounds over the north Pacific Ocean. J. Geophys. Res. 114, D02305 (2009). [Google Scholar]
- 69.Hsu S.-C., Wong G. T. F., Gong G.-C., Shiah F.-K., Huang Y.-T., Kao S.-J., Tsai F., Lung S.-C. C., Lin F.-J., Lin I.-I., Hung C.-C., Tseng C.-M., Sources, solubility, and dry deposition of aerosol trace elements over the East China Sea. Mar. Chem. 120, 116–127 (2010). [Google Scholar]
- 70.Tian S., Pan Y., Liu Z., Wen T., Wang Y., Size-resolved aerosol chemical analysis of extreme haze pollution events during early 2013 in urban Beijing, China. J. Hazard. Mater. 279, 452–460 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Wang Z., Li J., Wang Z., Yang W., Tang X., Ge B., Yan P., Zhu L., Chen X., Chen H., Wand W., Li J., Liu B., Wang X., Wand W., Zhao Y., Lu N., Su D., Modeling study of regional severe hazes over mid-eastern China in January 2013 and its implications on pollution prevention and control. Sci. China Earth Sci. 57, 3–13 (2014). [Google Scholar]
- 72.Zhang B., Wang Y., Hao J., Simulating aerosol–radiation–cloud feedbacks on meteorology and air quality over eastern China under severe haze conditionsin winter. Atmos. Chem. Phys. 15, 2387–2404 (2015). [Google Scholar]
- 73.Wang Y., Zhang Q., Jiang J., Zhou W., Wang B., He K., Duan F., Zhang Q., Philip S., Xie Y., Enhanced sulfate formation during China’s severe winter haze episode in January 2013 missing from current models. J. Geophys. Res. 119, 10425–10440 (2014). [Google Scholar]
- 74.Quinn P. K., Coffman D. J., Bates T. S., Miller T. L., Johnson J. E., Voss K., Welton E. J., Neusüss C., Dominant aerosol chemical components and their contribution to extinction during the Aerosols99 cruise across the Atlantic. J. Geophys. Res. 106, 20783–20809 (2001). [Google Scholar]
- 75.Park R. J., Jacob D. J., Field B. D., Yantosca R. M., Chin M., Natural and transboundary pollution influences on sulfate-nitrate-ammonium aerosols in the United States: Implications for policy. J. Geophys. Res. 109, D15204 (2004). [Google Scholar]
- 76.Chu S.-H., PM2.5 episodes as observed in the speciation trends network. Atmos. Environ. 38, 5237–5246 (2004). [Google Scholar]
- 77.Nguyen T. K. V., Petters M. D., Suda S. R., Guo H., Weber R. J., Carlton A. M. G., Trends in particle phase liquid water during the Southern Oxidant and Aerosol Study. Atmos. Chem. Phys. 14, 10911–10930 (2014). [Google Scholar]
- 78.Voutsa D., Samara C., Manoli E., Lazarou D., Tzoumaka P., Ionic composition of PM2.5 at urban sites of northern Greece: Secondary inorganic aerosol formation. Environ. Sci. Pollut. Res. 21, 4995–5006 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Makkonen U., Virkkula A., Mäntykenttä J., Hakola H., Keronen P., Vakkari V., Aalto P. P., Semi-continuous gas and inorganic aerosol measurements at a Finnish urban site: Comparisons with filters, nitrogen in aerosol and gas phases, and aerosol acidity. Atmos. Chem. Phys. 12, 5617–5631 (2012). [Google Scholar]
- 80.Kerminen V.-M., Hillamo R., Teinilä K., Pakkanen T., Allegrini I., Sparapani R., Ion balances of size-resolved tropospheric aerosol samples: Implications for the acidity and atmospheric processing of aerosols. Atmos. Environ. 35, 5255–5265 (2001). [Google Scholar]
- 81.Sillanpää M., Hillamo R., Saarikoski S., Frey A., Pennanen A., Makkonen U., Spolnik Z., Van Grieken R., Braniš M., Brunekreef B., Chalbot M.-C., Kuhlbusch T., Sunyer J., Kerminen V.-M., Kulmala M., Salonen R. O., Chemical composition and mass closure of particulate matter at six urban sites in Europe. Atmos. Environ. 40, 212–223 (2006). [Google Scholar]
- 82.Ianniello A., Spataro F., Esposito G., Allegrini I., Hu M., Zhu T., Chemical characteristics of inorganic ammonium salts in PM2.5 in the atmosphere of Beijing (China). Atmos. Chem. Phys. 11, 10803–10822 (2011). [Google Scholar]
- 83.Yang L.-x., Wang D.-c., Cheng S.-h., Wang Z., Zhou Y., Zhou X.-h., Wang W.-x., Influence of meteorological conditions and particulate matter on visual range impairment in Jinan, China. Sci. Total Environ. 383, 164–173 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Long S., Zeng J., Li Y., Bao L., Cao L., Liu K., Xu L., Lin J., Liu W., Wang G., Yao J., Ma C., Zhao Y., Characteristics of secondary inorganic aerosol and sulfate species in size-fractionated aerosol particles in Shanghai. J. Environ. Sci. 26, 1040–1051 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Behera S. N., Sharma M., Investigating the potential role of ammonia in ion chemistry of fine particulate matter formation for an urban environment. Sci. Total Environ. 408, 3569–3575 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Stone E., Schauer J., Quraishi T. A., Mahmood A., Chemical characterization and source apportionment of fine and coarse particulate matter in Lahore, Pakistan. Atmos. Environ. 44, 1062–1070 (2010). [Google Scholar]
- 87.Fisher J. A., Jacob D. J., Wang Q., Bahreini R., Carouge C. C., Cubison M. J., Dibb J. E., Diehl T., Jimenez J. L., Leibensperger E. M., Lu Z., Meinders M. B. J., Pye H. O. T., Quinn P. K., Sharma S., Streets D. G., van Donkelaar A., Yantosca R. M., Sources, distribution, and acidity of sulfate–ammonium aerosol in the Arctic in winter–spring. Atmos. Environ. 45, 7301–7318 (2011). [Google Scholar]
- 88.Clarisse L., Clerbaux C., Dentener F., Hurtmans D., Coheur P.-F., Global ammonia distribution derived from infrared satellite observations. Nat. Geosci. 2, 479–483 (2009). [Google Scholar]
- 89.Boynard A., Clerbaux C., Clarisse L., Safieddine S., Pommier M., Van Damme M., Bauduin S., Oudot C., Hadji-Lazaro J., Hurtmans D., Coheur P.-F., First simultaneous space measurements of atmospheric pollutants in the boundary layer from IASI: A case study in the North China Plain. Geophys. Res. Lett. 41, 645–651 (2014). [Google Scholar]
- 90.Huang X., Song Y., Li M., Li J., Huo Q., Cai X., Zhu T., Hu M., Zhang H., A high-resolution ammonia emission inventory in China. Global Biogeochem. Cycles 26, GB1030 (2012). [Google Scholar]
- 91.Zhang Q., Streets D., Carmichael G., He K. B., Huo H., Kannari A., Klimont Z., Park I. S., Reddy S., Fu J. S., Chen D., Duan L., Lei Y., Wang L. T., Yao Z. L., Asian emissions in 2006 for the NASA INTEX-B mission. Atmos. Chem. Phys. 9, 5131–5153 (2009). [Google Scholar]
- 92.Yang F., Tan J., Zhao Q., Du Z., He K., Ma Y., Duan F., Chen G., Zhao Q., Characteristics of PM2.5 speciation in representative megacities and across China. Atmos. Chem. Phys. 11, 5207–5219 (2011). [Google Scholar]
- 93.Cao J.-j., Wang Q.-y., Chow J. C., Watson J. G., Tie X.-x., Shen Z.-x., Wang P., An Z.-s., Impacts of aerosol compositions on visibility impairment in Xi’an, China. Atmos. Environ. 59, 559–566 (2012). [Google Scholar]
- 94.Okuda T., Kato J., Mori J., Tenmoku M., Suda Y., Tanaka S., He K., Ma Y., Yang F., Yu X., Duan F., Lei Y., Daily concentrations of trace metals in aerosols in Beijing, China, determined by using inductively coupled plasma mass spectrometry equipped with laser ablation analysis, and source identification of aerosols. Sci. Total Environ. 330, 145–158 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Duan J., Tan J., Wang S., Hao J., Chai F., Size distributions and sources of elements in particulate matter at curbside, urban and rural sites in Beijing. J. Environ. Sci. 24, 87–94 (2012). [DOI] [PubMed] [Google Scholar]
- 96.Zhang G., Bi X., Chan L. Y., Wang X., Sheng G., Fu J., Size-segregated chemical characteristics of aerosol during haze in an urban area of the Pearl River Delta region, China. Urban Clim. 4, 74–84 (2013). [Google Scholar]
- 97.Sun Y. L., Wang Z. F., Fu P. Q., Yang T., Jiang Q., Dong H. B., Li J., Jia J. J., Aerosol composition, sources and processes during wintertime in Beijing, China. Atmos. Chem. Phys. 13, 4577–4592 (2013). [Google Scholar]
- 98.Wang Y., Zhuang G., Zhang X., Huang K., Xu C., Tang A., Chen J., An Z., The ion chemistry, seasonal cycle, and sources of PM2.5 and TSP aerosol in Shanghai. Atmos. Environ. 40, 2935–2952 (2006). [Google Scholar]
- 99.Cao J.-J., Shen Z.-X., Chow J. C., Watson J. G., Lee S.-C., Tie X.-X., Ho K.-F., Wang G.-H., Han Y.-M., Winter and summer PM2.5 chemical compositions in fourteen Chinese cities. J. Air Waste Manag. Assoc. 62, 1214–1226 (2012). [DOI] [PubMed] [Google Scholar]
- 100.Wang H., Tan S.-C., Wang Y., Jiang C., Shi G.-y., Zhang M.-X., Che H.-Z., A multisource observation study of the severe prolonged regional haze episode over eastern China in January 2013. Atmos. Environ. 89, 807–815 (2014). [Google Scholar]
- 101.Zhang X. Y., Wang Y. Q., Niu T., Zhang X. C., Gong S. L., Zhang Y. M., Sun J. Y., Brandt J., Atmospheric aerosol compositions in China: Spatial/temporal variability, chemical signature, regional haze distribution and comparisons with global aerosols. Atmos. Chem. Phys. 12, 779–799 (2012). [Google Scholar]
- 102.Hu Y., Lin J., Zhang S., Kong L., Fu H., Chen J., Identification of the typical metal particles among haze, fog, and clear episodes in the Beijing atmosphere. Sci. Total Environ. 511, 369–380 (2015). [DOI] [PubMed] [Google Scholar]
- 103.Hwang H., Ro C.-U., Direct observation of nitrate and sulfate formations from mineral dust and sea-salts using low-Z particle electron probe X-ray microanalysis. Atmos. Environ. 40, 3869–3880 (2006). [Google Scholar]
- 104.Li L., Li M., Huang Z., Gao W., Nian H., Fu Z., Gao J., Chai F., Zhou Z., Ambient particle characterization by single particle aerosol mass spectrometry in an urban area of Beijing. Atmos. Environ. 94, 323–331 (2014). [Google Scholar]
- 105.Yuan Q., Li W., Zhou S., Yang L., Chi J., Sui X., Wang W., Integrated evaluation of aerosols during haze-fog episodes at one regional background site in North China Plain. Atmos. Res. 156, 102–110 (2015). [Google Scholar]
- 106.Rudich Y., Talukdar R. K., Ravishankara A. R., Multiphase chemistry of NO3 in the remote troposphere. J. Geophys. Res. 103, 16133–16143 (1998). [Google Scholar]
- 107.Guo S., Tan J., Duan J., Ma Y., Yang F., He K., Hao J., Characteristics of atmospheric non-methane hydrocarbons during haze episode in Beijing, China. Environ. Monit. Assess. 184, 7235–7246 (2012). [DOI] [PubMed] [Google Scholar]
- 108.Zhang R., Jing J., Tao J., Hsu S.-C., Wang G., Cao J., Lee C. S. L., Zhu L., Chen Z., Zhao Y., Shen Z., Chemical characterization and source apportionment of PM2.5 in Beijing: Seasonal perspective. Atmos. Chem. Phys. 13, 7053–7074 (2013). [Google Scholar]
- 109.He K., Zhao Q., Ma Y., Duan F., Yang F., Shi Z., Chen G., Spatial and seasonal variability of PM2.5 acidity at two Chinese megacities: Insights into the formation of secondary inorganic aerosols. Atmos. Chem. Phys. 12, 1377–1395 (2012). [Google Scholar]
- 110.Sun Y., Zhuang G., Wang Y., Han L., Guo J., Dan M., Zhang W., Wang Z., Hao Z., The air-borne particulate pollution in Beijing—Concentration, composition, distribution and sources. Atmos. Environ. 38, 5991–6004 (2004). [Google Scholar]
- 111.Gao X., Yang L., Cheng S., Gao R., Zhou Y., Xue L., Shou Y., Wang J., Wang X., Nie W., Xu P., Wang W., Semi-continuous measurement of water-soluble ions in PM2.5 in Jinan, China: Temporal variations and source apportionments. Atmos. Environ. 45, 6048–6056 (2011). [Google Scholar]
- 112.Chou M.-D., Suarez M. J., Ho C.-H., Yan M. M.-H., Lee K.-T., Parameterizations for cloud overlapping and shortwave single-scattering properties for use in general circulation and cloud ensemble models. J. Climate 11, 202–214 (1998). [Google Scholar]
- 113.Mlawer E. J., Taubman S. J., Brown P. D., Iacono M. J., Clough S. A., Radiative transfer for inhomogeneous atmospheres: RRTM, a validated correlated-k model for the longwave. J. Geophys. Res. 102, 16663–16682 (1997). [Google Scholar]
- 114.Xiu A., Pleim J. E., Development of a land surface model. Part I: Application in a mesoscale meteorological model. J. Appl. Meteorol. 40, 192–209 (2001). [Google Scholar]
- 115.Pleim J. E., A combined local and nonlocal closure model for the atmospheric boundary layer. Part I: Model description and testing. J. Appl. Meteorol. Clim. 46, 1383–1395 (2007). [Google Scholar]
- 116.Kain J. S., The Kain–Fritsch convective parameterization: An update. J. Appl. Meteorol. 43, 170–181 (2004). [Google Scholar]
- 117.Hong S.-Y., Lim J.-O. J., The WRF single-moment 6-class microphysics scheme (WSM6). J. Korean Meteor. Soc. 42, 129–151 (2006). [Google Scholar]
- 118.Ibusuki T., Takeuchi K., Sulfur dioxide oxidation by oxygen catalyzed by mixtures of manganese(II) and iron(III) in aqueous solutions at environmental reaction conditions. Atmos. Environ. 21, 1555–1560 (1987). [Google Scholar]
- 119.Debye P., Hückel E., The interionic attraction theory of deviations from ideal behavior in solution. Z. Phys. 24, 185 (1923). [Google Scholar]
- 120.Debye P., McAuley W., The electric field of the ions and the neutral salt effect. Physik Z 26, 22 (1925). [Google Scholar]
- 121.Davies C. W., Shedlovsky T., Ion association. J. Electrochem. Soc. 111, 85C–86C (1964). [Google Scholar]
- 122.Helgeson H. C., Brown T. H., Nigrini A., Jones T. A., Calculation of mass transfer in geochemical processes involving aqueous solutions. Geochim. Cosmochim. Acta 34, 569–592 (1970). [Google Scholar]
- 123.Pitzer K. S., Electrolyte theory—Improvements since Debye and Hueckel. Acc. Chem. Res. 10, 371–377 (1977). [Google Scholar]
- 124.K. S. Pitzer, Activity Coefficients in Electrolyte Solutions (CRC Press, 1991). [Google Scholar]
- 125.Millero F. J., Hershey J. B., Johnson G., Zhang J.-Z., The solubility of SO2 and the dissociation of H2SO3 in NaCl solutions. J. Atmos. Chem. 8, 377–389 (1989). [Google Scholar]
- 126.Bretti C., Crea F., Foti C., Sammartano S., Solubility and activity coefficients of acidic and basic nonelectrolytes in aqueous salt solutions. 2. Solubility and activity coefficients of suberic, azelaic, and sebacic acids in NaCl(aq), (CH3)4NCl(aq), and (C2H5)4NI(aq) at different ionic strengths and at t = 25°C. J. Chem. Eng. Data 51, 1660–1667 (2006). [Google Scholar]
- 127.Bretti C., Crea F., De Stefano C., Sammartano S., Vianelli G., Some thermodynamic properties of dl-tyrosine and dl-tryptophan. Effect of the ionic medium, ionic strength and temperature on the solubility and acid–base properties. Fluid Phase Equilib. 314, 185–197 (2012). [Google Scholar]
- 128.A. J. Rusumdar, “Treatment of non-ideality in the multiphase model SPACCIM and investigation of its influence on tropospheric aqueous phase chemistry,” thesis, Brandenburg University of Technology, Cottbus, Germany (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at hhttp://advances.sciencemag.org/cgi/content/full/2/12/e1601530/DC1
fig. S1. Weakened photochemistry by aerosol dimming effects during January 2013 in Beijing.
fig. S2. Importance of the NO2 reaction pathway for sulfate production in the Beijing haze (January 2013).
fig. S3. Influence of ionic strength (I) on rate of aqueous sulfate-producing reactions.
fig. S4. Estimation of Fe3+ and Mn2+ concentrations as a function of aerosol water pH during Beijing hazes.
fig. S5. Regional pollution across the NCP during January 2013.
fig. S6. Annual precipitation pH of China in 2013.
fig. S7. The same as Fig. 2 but with a lower limit of reaction rate constants reported by Lee and Schwartz (18).
table S1. Previously reported concentrations of cations and anions in PM2.5 during winter for cities in NCP used in Fig. 1D.
table S2. Summary of field observation and methods in this study.
table S3. Domain, configurations, and major dynamic and physical options used in WRF v3.5.1.
table S4. Rate expression and rate coefficients of relevant aqueous-phase reactions.
table S5. Constants for calculating the apparent Henry’s constant (H*).
table S6. Summary of suggested activity coefficient (a)–ionic strength (I) dependence.
table S7. Influence of ionic strength (I) on rate of aqueous sulfate-producing reactions.