

Abstract
The assembly of the Cbl-SETA/CIN85-endophilin complex at the C terminus of the epidermal growth factor receptor (EGFR) following ligand activation mediates its internalization and ubiquitination. We found that the SETA/CIN85-interacting protein Alix/AIP1, which also binds endophilins, modulates this complex. Alix was found to associate indirectly with EGFR, regardless of its activation state, and with ΔEGFR, which signals at low intensity and does not bind Cbls or SETA/CIN85. In agreement with this, Alix interaction did not occur via SETA/CIN85. However, SETA/CIN85 and Alix were capable of mutually promoting their interaction with the EGFR. Increasing the level of Alix weakened the interaction between SETA/CIN85 and Cbl and reduced the tyrosine phosphorylation of c-Cbl and the level of ubiquitination of EGFR, SETA/CIN85, and Cbls. This antagonism of the Cbl-SETA/CIN85 complex by Alix was reflected in its diminution of EGFR internalization. In agreement with this, small interfering RNA-mediated knockdown of Alix promoted EGFR internalization and downregulation. It has been suggested that SETA/CIN85 promotes receptor internalization by recruiting endophilins. However, Alix was also capable of increasing the level of endophilin associated with EGFR, implying that this is not sufficient to promote receptor internalization. We propose that Alix inhibits EGFR internalization by attenuating the interaction between Cbl and SETA/CIN85 and by inhibiting Cbl-mediated ubiquitination of the EGFR.
Receptor tyrosine kinase signaling plays a central role in cellular growth control and is often deregulated in cancer. Escape from Cbl-mediated ubiquitination and downregulation is one common characteristic of receptor tyrosine kinases that have undergone oncogenic deregulation (8, 35). Therefore, understanding how the interaction between the receptors and the Cbl protein complex is regulated is important for the development of strategies aimed at impairing oncogenic signaling in transformed cells. Recent work by several laboratories has demonstrated that normal ligand activation of receptor tyrosine kinases, which has long been recognized to lead to the binding and phosphorylation of Cbl proteins via their PTB domains, also results in the recruitment of the SETA/CIN85-endophilin complex by binding SETA/CIN85's SH3 domains to a PXXXPR motif in the C termini of the Cbls (23, 39, 41, 42). Cbls recruit E2 ubiquitin conjugase in parallel, via their RING finger domains, and so cause the receptor to be ubiquitinated (11-13) and SETA/CIN85 to be monoubiquitinated in its C terminus (15, 39, 41). The internalization and ubiquitination of the receptor can be mechanistically separated, and the interaction with the SETA/CIN85-Cbl complex may be primarily involved in internalization into clathrin-coated vesicles, while the ubiquitination state may regulate subsequent sorting into recycling or degradation pathways (20, 25, 39). While mutations in the receptor's intracellular sequence can release it from this negative regulation (35), signal intensity is also important. This is exemplified by the potent glioma-associated oncogene product deleted-(2-7) epidermal growth factor receptor (EGFR) (ΔEGFR or EGFRvIII; referred to here as ΔEGFR), which signals in a ligand-independent manner (10, 19, 40, 47), albeit at a lower intensity and in the absence of receptor internalization (18). Interaction between wild-type EGFR and the Cbl-SETA/CIN85 complex and internalization are dependent on activation beyond a certain threshold, which ΔEGFR does not cross (38).
A central component of this complex, the adaptor molecule SETA/CIN85/Ruk, offers multiple avenues of regulation of receptor internalization by virtue of its broad spectrum of interactions. It was independently identified as being expressed in association with malignant transformation of astrocytes (SETA was derived from SH3 domain encoding, expressed in tumorigenic astrocytes [1]), as being a binding partner for c-Cbl (Cbl-interacting protein of 85 kDa or CIN85 [39, 42]), or as being a binding partner of p85α and a negative regulator of phosphatidylinositol 3-kinase (regulator of ubiquitous kinase or Ruk) (14). SETA/CIN85 proteins exist in several isoforms (2, 3, 14), the longest of which comprises three SH3 domains in the N-terminal half and a C-terminal half with a proline-rich region and a coiled-coil domain involved in multimerization at the terminus (2, 46). SETA/CIN85 constitutively associates with endophilins, proteins thought to modify membrane phospholipids and to induce negative curvature and invagination of the plasma membrane during the early steps of endocytosis (13, 36, 39, 41). In addition to its binding to Cbls and endophilins, SETA/CIN85 interacts with other signaling molecules, regulators of the cytoskeleton, and modulators of apoptosis, including Crk-I, Crk-II, p130(Cas), Grb2, Sos1, and apoptosis-linked gene 2-interacting protein X/apoptosis-linked gene 2-interacting protein 1 (Alix/AIP1) (2, 6, 37, 46).
The recent observation (30, 45) that Alix/AIP1 (referred to here as Alix to avoid confusion with other proteins that are named AIP1) itself interacts with endophilins (5) prompted this investigation. Our data show that Alix also binds EGFRs but does not discriminate between activation states and so binds active and inactive EGFRs as well as ΔEGFR. While Alix binds EGFR indirectly, this does not occur via SETA/CIN85. However, Alix and SETA/CIN85 mutually strengthen their interaction with active EGFR, suggesting that they can interact with each other while bound to the receptor. Importantly Alix appears to negatively regulate the interaction between SETA/CIN85 and Cbls, as well as Cbl phosphorylation following EGFR activation, and as a result Alix is capable of attenuating EGFR, SETA/CIN85, and Cbl ubiquitination. Modulation of Alix levels also impacts EGFR internalization: overexpression of Alix antagonizes this process, while small interfering RNA (siRNA)-mediated knockdown stimulates it. We propose a model in which the Alix-SETA/CIN85 interaction antagonizes the activity of the Cbl-SETA/CIN85 complex in promoting EGFR downregulation, at the levels of both SETA/CIN85-mediated internalization and Cbl-dependent ubiquitination.
MATERIALS AND METHODS
Constructs and antibodies.
Transfection experiments were performed using the following gene expression plasmid constructs. The coding sequences for full-length SETA123cc and lacZ were cloned into pcDNA6 (Invitrogen) as previously described (2, 6). Coding sequences for C-terminally Flag-tagged Alix (construct provided by Luciano D'Adamio) (44, 45) and sequence-altered versions of Alix in pcDNA3, wild-type EGFR, and ΔEGFR (18) in 1726/zeo/G retrovirus, a derivative of 1726/zeo (6) which carries a Gateway recombination cassette in the unique EcoRI site, making it a Gateway destination vector (Invitrogen), and wild-type EGFR as well as ΔEGFR and sequence-altered versions (see Fig. 3) were alternatively cloned into LRNL. Coding sequences for Cbl-b carrying a C-terminal hemagglutinin (HA) tag and c-Cbl carrying an N-terminal HA tag in pCEFL (11, 22), FLAG-tagged ubiquitin in pcDNA3.1, and green fluorescent protein (GFP) and HA-tagged endophilin A1 and Flag-tagged CIN85 in pcDNA3 were described previously (39).
FIG. 3.

Alix binding to ΔEGFR is independent of the receptor's tyrosine phosphorylation or its kinase activity. Alix was transfected into HEK293 cells together with EGFR (lane 10), ΔEGFR (lane 1), a kinase-dead ΔEGFR receptor (DK; lane 2), and several ΔEGFR tyrosine-to-phenylalanine exchange receptors (DYs; lanes 3 to 9; a cartoon of the EGFR C terminus and details of DYs are at the right). Cells transfected with EGFR were serum starved for 18 h and exposed to 100 ng of EGF/ml for 5 min. EGFR and ΔEGFR IPs were performed as described for Fig. 1. No alteration had a major effect on the Alix-ΔEGFR interaction, indicating that it is constitutive and independent of ΔEGFR phosphorylation or kinase activity. Minor differences could be observed. For example, DY4 was associated with an increased amount of Alix compared to the other constructs (lane 6). This offers the possibility that structural differences in the C terminus of ΔEGFR impact Alix binding. EGFR bound more Alix than any of the ΔEGFR proteins. P-TYR, phosphotyrosine.
For detection of proteins in Western blots we used the following antibodies. Polyclonal goat anti-EGFR (1005) and monoclonal mouse anti-HA (F-7) antibodies were purchased from Santa Cruz Biotechnology. Monoclonal anti-EGFR antibodies against the extracellular domain were Ab-1 (Oncogene Science) and Ab-11 (Labvision Neomarkers). An anti-ΔEGFR monoclonal antibody (MAb; 806) was used as previously described (21). Monoclonal anti-Flag (M2) was purchased from Sigma, and a monoclonal mouse antiphosphotyrosine antibody (4G10) was purchased from Upstate Biotechnology. The monoclonal anti-Alix antibody was purchased from BD Pharmingen. Polyclonal anti-SETA/CIN85 antibodies were made and used as previously described (6). Polyclonal rabbit anti-EGFR pY1045 was purchased from Cell Signaling Technology.
Cell lines, cell transfection, and EGF induction experiments.
Human embryonic kidney HEK293 cells and U87MG glioblastoma cells were cultured under standard conditions in Dulbecco's modified Eagle medium supplemented with antibiotics and 10% fetal calf serum. CHO and CHO-EGFR cells were cultured in F12K medium (Kaighn's modification) plus supplements. Cells were transfected with plasmids by a modified calcium phosphate procedure. For epidermal growth factor (EGF) induction experiments 5 million cells were transfected and incubated at 37°C for 24 h. Cells were serum deprived for 18 to 20 h and subsequently incubated with 50 or 100 ng of recombinant EGF/ml in serum-free medium for 5 min. Recombinant human EGF, EGFR kinase inhibitor AG1478, and src kinase inhibitor PP2 were purchased from Oncogene Science.
siRNA silencing.
Vector-based silencing of Alix was achieved by the use of pTER-Alix, which was constructed as previously described (43). Briefly, complementary oligonucleotides containing the Alix target sequence (boldface) described recently (28) (sense, 5′-GAT CCC GCC GCT GGT GAA GTT CAT CTT CAA GAG AGA TGA ACT TCA CCA GCG GCT TTT TGG AAA-3′; antisense, 5′-AGC TTT TCC AAA AAG CCG CTG GTG AAG TTC ATC TCT CTT GAA GAT GAA CTT CAC CAG CGG CGG-3′) were annealed and cloned into BglII and HindIII restriction sites of the pTER+ vector (kindly provided by Hans Clevers). This construct was used to transfect 293T cells or HeLa cells with Lipofectamine (Invitrogen). Forty-eight hours after transfection cells were collected and the level of Alix protein was determined by Western blotting (WB).
Optimal oligonucleotide-based silencing of Alix was determined by transfection of different volumes (1 to 10 μl) of a 20 μM stock solution of an Alix siRNA duplex (QIAGEN; sequence as described above) into HeLa, HEK293, CHO-EGFR, NIH 3T3, and NIH SAA cells with Oligofectamine or LP2000 (Invitrogen) according to the manufacturer's guidelines. After 24, 48, and 72 h cells were harvested and endogenous Alix levels were determined by immunoblotting using an anti-Alix antibody kindly provided by Remy Sadoul (unpublished). Eventually, HeLa and CHO-EGFR cells were chosen as model systems and transfected with 1 μl of 20 μM siRNA solution in combination with LP2000. After 72 h cells were used for subsequent assays. With this combination a knockdown efficiency of endogenous Alix of more than 90% could be achieved.
Mouse brain homogenates.
Mouse brain was prepared from BL6 mice, transferred to immunoprecipitation (IP) buffer (see below), and homogenized with an UltraTurrax homogenizer for two rounds, 1 min each, on ice. The insoluble fraction was removed by centrifugation at 3,200 × g for 5 min at 4°C. The resulting suspension was centrifuged at high speed (16,200 × g) five times at 4°C. Cloudy lower phases were pooled and termed the fat rich fraction, while the clear supernatant is referred to as the low-fat fraction. Both were subjected to immunoprecipitation studies.
Ubiquitination assays.
To determine the degree of protein ubiquitination, HEK293 cells were cotransfected with EGFR, SETA/CIN85, Cbls, and Flag-tagged ubiquitin. The effect of Alix was measured by comparison with lacZ-transfected controls. Cells underwent EGF induction as described above and were harvested, and subsequently EGFR, SETA/CIN85, and Cbls were immunoprecipitated from the lysates. Precipitates were analyzed by immunoblotting with an anti-Flag antibody.
Immunoprecipitation.
Cells were washed two times with ice-cold phosphate-buffered saline (PBS) and were lysed on ice for 30 min in a modified radioimmunoprecipitation assay buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 1% IGEPAL [{octylphenoxy}polyethoxyethanol] CA-630 [Sigma], 0.5% deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 5 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 4 mM sodium azide, 1 mM phenylmethylsulfonyl fluoride, 5 mM benzamidine, a protease inhibitor cocktail [2 μg of aprotinin and leupeptin/ml, 10 μg of E-64 and a trypsin inhibitor/ml, 1 μg of pepstatin A/ml], and a phosphatase inhibitor cocktail [2 mM sodium vanadate and sodium fluoride, 5 mM sodium molybdate, and 15 mM p-nitrophenylphosphate]). Additionally, to measure ubiquitination, 50 μM MG132 was added to inhibit proteasomal degradation. Following lysis, the cell suspension was sheared 10 times through an 18G1[1/2] needle and 10 times through an IM1 needle and incubated on ice for another 30 min. The cell solution was then cleared by centrifugation at 12,000 × g at 4°C. The supernatant was used for IP studies. Appropriate concentrations of primary antibody were added, and the solution was rotated at 4°C for at least an hour. Antibody-protein complexes were precipitated with 50 μl of protein A-agarose solution (Roche) by rotation at 4°C overnight. The agarose beads were collected by centrifugation at 12,000 × g for 5 min at 4°C and were washed seven times with precipitation buffer on ice. Finally, the sediment was boiled for 5 min at 95°C in 1× NuPAGE LDS sample buffer (Invitrogen) containing 10% β-mercaptoethanol and transferred to ice immediately. The solution was cleared of agarose by centrifugation and stored at −80°C until further analysis by protein electrophoresis.
WB.
Protein samples were analyzed by SDS-polyacrylamide gel electrophoresis using an XCell SureLock minicell (Invitrogen) in combination with precast 4 to 12% NuPAGE or 10% Bis-Tris gels (1 mm) at 200 V according to the manufacturer's guidelines. Following electrophoresis, proteins were blotted to a polyvinylidene difluoride membrane and were incubated for at least 1 h in blocking buffer (5% bovine serum albumin [BSA] and 1% Tween 20 in Tris-buffered saline). Membranes were incubated overnight with appropriate dilutions of primary antibody in blocking buffer. The next day membranes were washed and incubated for 1 h with an appropriate alkaline phosphatase-conjugated secondary antibody solution in blocking buffer (Sigma; dilutions: anti-mouse antibody, 1:3,000; anti-rabbit antibody, 1:5,000; anti-goat antibody, 1:15,000). After additional washing steps, antibody complexes were visualized on film with Immun-Star AP substrate (Bio-Rad). In vitro confrontation assays were performed using the Promega TnT kit to generate [3H]leucine-labeled proteins and carried out as described previously (6). Figure panels were made by digitally photographing films and assembling the relevant lanes in Photoshop. For each row in a panel a single exposure of the same film was used, so that even noncontiguous lanes are from the same exposure of the same series of blots and the same experiment. Lanes were rearranged for clarity of presentation. Molecular weight standards were run in each experiment, and the mobilities of the bands shown throughout the data are the same as those shown in Fig. 1. Molecular weight labels have been omitted from the majority of the figures for clarity.
FIG. 1.

Alix binds to both EGFR and ΔEGFR. (A) Flag-tagged Alix was transfected into U87MG glioma cells. IP of endogenous EGFR revealed receptor-associated Alix within the precipitate (lane 2). (B) Alix and ΔEGFR (lanes 1 and 3) or EGFR (lane 2) were transfected into HEK293 cells, which were then serum starved for 18 h and treated with 100 ng of EGF/ml for 5 min before lysis. IPs were performed with an antibody against EGFR (Ab-1; Oncogene Science; lanes 2 and 3) or with EGFR preclearing of the ΔEGFR lysate with the same EGFR antibody, followed by IP with MAb 806 to specifically isolate the ΔEGFR (lane 1). Alix was recovered along with both receptors (lanes 1 to 3). However, less Alix was associated with ΔEGFR. (C) CHO-EGFR cells were either transfected with Flag-tagged Alix (lane 1) or not and subjected to EGFR IP followed by EGFR or Alix WB. Although a stronger Alix band is obtained in the EGFR IP when Alix is transfected, the endogenous Alix is also recovered, and the levels of endogenous Alix are equivalent regardless of whether cells were serum starved for 18 h and either not treated further (lane 2) or challenged with 50 ng of EGF/ml for 10 min (lane 3; ns, not starved). (D) Fat rich and low-fat fractions of mouse brain homogenates were subjected to IP with the anti-EGFR antibody, and the recovered material was analyzed by Alix and EGFR WB. Alix was recovered in EGFR IPs from both fractions and was also observed in the lysates. EGFR was detected in the lysates at longer exposures (not shown).
Receptor internalization and degradation assays.
Receptor internalization was measured by two approaches. In the first, radiolabeled ligand was used to detect receptors. CHO cells were transfected with EGFR, Alix, and c-Cbl and, after 48 h, serum deprived for 30 min at 4°C in F12K medium-0.1% BSA-10 mM HEPES. Then, cells were incubated in medium containing 5 ng of EGF/ml and 1 ng of 125I-EGF/ml for 1 h on ice. After a washing, internalization was initiated at 37°C and stopped by transfer to wet ice. Cells were washed with PBS-0.1% BSA or stripped of surface EGF with PBS (pH 3.4)-0.1% BSA on ice for 5 min, washed, lysed, and analyzed in a gamma counter (1470 Wizard; Perkin-Elmer). The percentage of signal from stripped cells versus that from nonstripped cells was calculated from triplicate data points and expressed as the percentage of EGFR internalized. To measure the remaining plasma membrane-associated EGFR or platelet-derived growth factor receptor (PDGFR), transfected CHO cells or siRNA-treated HeLa and CHO-EGFR cells were incubated with 50 ng of EGF or platelet-derived growth factor (PDGF)/ml at 37°C for various times. Cells underwent an acid wash with PBS-0.1% BSA (pH 3.4) to remove surface EGF and were incubated for 1.5 h with 1 ng of 125I-EGF or 125I-PDGF/ml at 4°C. Surface-bound 125I-EGF or 125I-PDGF was determined as described above and compared to EGFR or PDGFR on the surfaces of nonstimulated cells.
In the second approach, receptor internalization was measured by flow cytometry. Forty-eight hours after transfection with pTER-Alix and pEGFP-C1 cell monolayers of 293T or HeLa cells were incubated with 50 ng of EGF/ml at 37°C to induce internalization of EGFR. At various time points cells were harvested and blocked in 5% BSA-PBS for 30 min on ice. The amount of endogenous surface resident EGFR was detected by incubation with an anti-EGFR antibody, conjugated with phycoerythrin (BD Pharmingen) for 1 h at 4°C. Cells were washed with ice-cold PBS and analyzed with an Epics XL flow cytometer (Beckman-Coulter). For each sample 10,000 cells were analyzed and GFP-expressing cells were gated for determining the amount of EGFR at the plasma membrane. Mean fluorescence intensity of each sample was calculated with Expo 32 ADC software.
Receptor degradation was also measured by flow cytometry. Forty-eight hours after transfection with pTER-Alix and pEGFP-EGFR monolayers of 293T cells were incubated with 50 ng of EGF/ml at 37°C to induce internalization and degradation of EGFR. At various time points cells were harvested and analyzed with an Epics XL flow cytometer (Beckman-Coulter). For each sample 10,000 cells were analyzed and the amount of EGFR in the cell was determined by measuring the intensity of the GFP signal. Mean fluorescence intensity of each sample was calculated with Expo 32 ADC software.
RESULTS
SETA/CIN85 interacts with ligand-activated EGFR and, together with the Cbl proteins and endophilin, is involved in its internalization (39, 42). To test whether Alix, which associates with SETA/CIN85's SH3 domains via a proline-rich motif in its C terminus (6, 37), also binds to EGFR, a Flag-tagged Alix was transfected into U87 glioma cells that express wild-type EGFR and EGFR immunoprecipitates were analyzed (Fig. 1A). Alix was detected in complexes of endogenous EGFR at robust levels relative to its expression levels in the lysates from these glioma cells, which are not very efficiently transfected (Fig. 1A), demonstrating interaction between EGFR and Alix.
SETA/CIN85 and its associated proteins c-Cbl, Cbl-b, and endophilin A1 discriminate between the ΔEGFR mutant protein and EGFR in that they associate only with the latter (38). To test whether Alix also discriminates between these two forms of the EGFR, we performed IP experiments with conditions specific for the wild type and ΔEGFR (Fig. 1B). EGFR-specific conditions were achieved by transfecting this receptor alone into HEK293 cells (which do not express endogenous ΔEGFR) and immunoprecipitating with an N-terminal EGFR antibody. To isolate ΔEGFR from cells that had been transfected with it, the MAb 806 was used, as it preferentially recognizes this mutant form (21). However, as described previously (38), in cells that also express endogenous EGFR, as is usually the case in gliomas, as well as the HEK293 cell model used here, there is a degree of cross-reactivity with EGFR. Others have also reported this, particularly when EGFR is highly expressed, and it is thought to be because MAb 806 recognizes an activity-dependent conformation rather than a neo epitope of ΔEGFR (21, 27, 29). To reduce the level of EGFR in ΔEGFR immunoprecipitates, lysates were precleared by IP with an anti-EGFR antibody (Ab-1; Oncogene Science), followed by IP with MAb 806, which resulted in predominant recovery of ΔEGFR (Fig. 1B, lane 1). Cotransfection of Flag-tagged Alix with wild-type EGFR or ΔEGFR in HEK293 cells and IP of the respective receptor showed that Alix associated with both receptors, although lower levels of Alix were associated with ΔEGFR (Fig. 1B). These data suggest that Alix does not interact with EGFR via Cbl and SETA/CIN85, as neither of these proteins binds to ΔEGFR (38). However, as the interaction of Alix with ΔEGFR is weaker than that with wild-type EGFR, it is possible that SETA/CIN85 and Cbls can positively contribute to Alix's interaction with EGFR. Two experiments were then performed to test whether Alix and EGFR interacted in situations where neither protein was overexpressed by transient transfection. First, CHO cells stably expressing EGFR (CHO-EGFR) were subjected to EGFR IP, and it was found that endogenous Alix, as well as transfected Alix, could be recovered in the immunoprecipitates (Fig. 1C). The amount of endogenous Alix recovered was not affected by stimulation with EGF (Fig. 1C). Second, endogenous EGFR was immunoprecipitated from either a fat rich or a low-fat fraction of mouse brain homogenates and endogenous Alix was detected in the recovered material (Fig. 1D). Direct confrontation of bacterially made Alix with in vitro-transcribed and -translated EGFR or ΔEGFR failed to show interaction, while SETA/CIN85 and Alix bound in this situation as previously described (6) (data not shown), suggesting that the interaction between these proteins is not direct and requires the presence of an unidentified cellular cofactor. Additional control experiments (unpublished) were also performed to establish that the recovery of Alix in EGFR IP is dependent on the presence of the EGFR, that the EGFR antibodies do not recognize Alix, that preimmune and secondary antibodies are not capable of recovering Flag-tagged Alix in IPs, and that no Flag-tagged Alix is found in cells that are not transfected with this construct.
The interaction between SETA/CIN85 and EGFR is dependent on the EGFR being in an activated state (38, 39). To test whether the activation state modulated the interaction between Alix and EGFR, HEK293 cells transfected with both of these constructs were serum starved and challenged with EGF for 5 min to produce maximal EGFR activity. Although this resulted in a dramatic increase in the tyrosine phosphorylation state of the EGFR, it did not affect the amount of Alix that was recovered in the EGFR IP (Fig. 2A, lanes 1 and 2). Although no tyrosine-phosphorylated EGFR was detectable in the serum-starved cells (lane 1), cells were also treated with the EGFR inhibitor AG1478 or the src kinase inhibitor PP2 in the absence of EGF, in an attempt to eliminate any residual phosphorylation that might be present, but again this did not alter the amount of Alix that was recovered in the EGFR complex (Fig. 2A, lanes 3 and 4). Therefore, the interaction between Alix and EGFR was not dependent on the activation or phosphorylation state of the receptor. Similarly, the interaction between Alix and ΔEGFR was not affected by EGF stimulation, which has no effect on this ligand-independent mutant receptor (Fig. 2B, lanes 1 and 2). It also did not appear to be significantly affected by treatment with AG1478, which inhibits the ΔEGFR (17, 33). However, a slight reduction in Alix in the AG1478-treated ΔEGFR complex and an incomplete attenuation of ΔEGFR phosphorylation were observed (Fig. 2B, lane 3). The src kinase inhibitor PP2 had no effect.
FIG. 2.

Alix binding to EGFR and ΔEGFR is independent of receptor activation. Alix was transfected into HEK293 cells together with EGFR (A) or ΔEGFR (B). In panel B endogenous EGFR was removed from ΔEGFR lysates as described for Fig. 1. After 24 h cells were serum starved and received no further treatment (lane 1), were treated with 100 ng of EGF/ml for 5 min (lane 2), or were treated with 10 μM EGFR inhibitor AG1478 (lane 3) or 20 μM src kinase inhibitor PP2 (lane 4) for 1 h in the absence of EGF stimulation to further reduce background EGFR phosphorylation levels. No treatment had an effect on the amount of Alix associated with EGFR or ΔEGFR, indicating that the interaction between Alix and receptors was constitutive. P-TYR, phosphotyrosine.
To evaluate the role of the phosphorylation state of the constitutively active ΔEGFR in its interaction with Alix more thoroughly, various ΔEGFRs with tyrosine-to-phenylalanine substitutions (DY1, -2, -3, -4, -6, -8, and -12), as well as a kinase-inactive mutant ΔEGFR (DK), were examined (Fig. 3). The amount of Alix recovered in the IPs of kinase-inactive ΔEGFR DK or the various DYs did not vary with phosphorylation status (Fig. 3 shows details of the alterations). For example, ΔEGFR DK recovered an amount of Alix similar to that recovered by the active ΔEGFR, although it was not phosphorylated (Fig. 3, lanes 1 and 2). Interestingly, DY4, which showed very low levels of phosphorylation, bound relatively more Alix than DY1, which exhibited near-ΔEGFR levels of activity, compared to the intensity of the EGFR WB band (compare lanes 6 and 3). Taken together these data demonstrate that the phosphorylation state of the ΔEGFR does not influence Alix binding, so that binding is activity independent. Variations in binding intensity among DYs may reflect subtle variations in C-terminal protein structures that are not yet understood in the context of Alix interaction. We again observed a lower level of Alix associated with ΔEGFR than was associated with stimulated EGFR (Fig. 3, lanes 1 and 10).
The nature of the respective interactions of SETA/CIN85 and Alix with EGFRs suggests that they occur in parallel, with the SETA/CIN85 interaction being dependent on high levels of receptor activity and so restricted to ligand-activated wild-type EGFR (38, 39) and with the Alix interaction being independent of activity state, and so applying to all EGFRs tested. This does not, however, exclude the possibility that there is cross talk between these two complexes, which could be mediated via the Alix-SETA/CIN85 interaction (6). To determine whether SETA/CIN85 could modulate the interaction between Alix and EGFR, we compared the impact of this protein on the binding of wild-type and mutant Alix to the EGFR (Fig. 4). The mutant Alix Alix-Δ717-784 lacks the N-terminal part of the proline-rich C terminus that contains the SETA/CIN85 binding site (23, 37) and so shows compromised interaction with SETA/CIN85 (37). Both Alix proteins interacted with EGFR (Fig. 4A, lanes 1 to 3). Cotransfection of SETA/CIN85 increased the amount of Alix recovered in the EGFR immunoprecipitates (by approximately 1.7-fold according to densitometry analysis; Fig. 4A, compare lanes 1 and 4) but did not affect the level of Alix-Δ717-784 (lane 5), which does not bind SETA/CIN85. To determine whether the ability of SETA/CIN85 to promote the binding of Alix depended on its interaction with the receptor, we tested whether it could also increase Alix binding to ΔEGFR. No effect of SETA/CIN85 on the levels of Alix in ΔEGFR immunoprecipitates was observed (not shown), and, as this receptor does not interact with SETA/CIN85 (38), this suggests that SETA/CIN85 binding to the receptor is necessary for its effect on Alix levels. To test whether the converse, that Alix could increase the amount of SETA/CIN85 on the EGFR, was also true, the level of SETA/CIN85 in EGFR immunoprecipitates was determined, after transfection of lacZ control or Alix proteins. Wild-type Alix but not Alix-Δ717-784 increased the amount of SETA/CIN85 in EGFR immunoprecipitates relative to the lacZ control (by approximately 1.7-fold according to densitometry analysis; Fig. 4B), providing further evidence that the association between Alix and SETA/CIN85 stabilizes their interaction with the EGFR. Together these data show that increasing the levels of either SETA/CIN85 or Alix increases the amount of the other partner protein at the EGFR and that this is dependent on direct interaction between them.
FIG. 4.
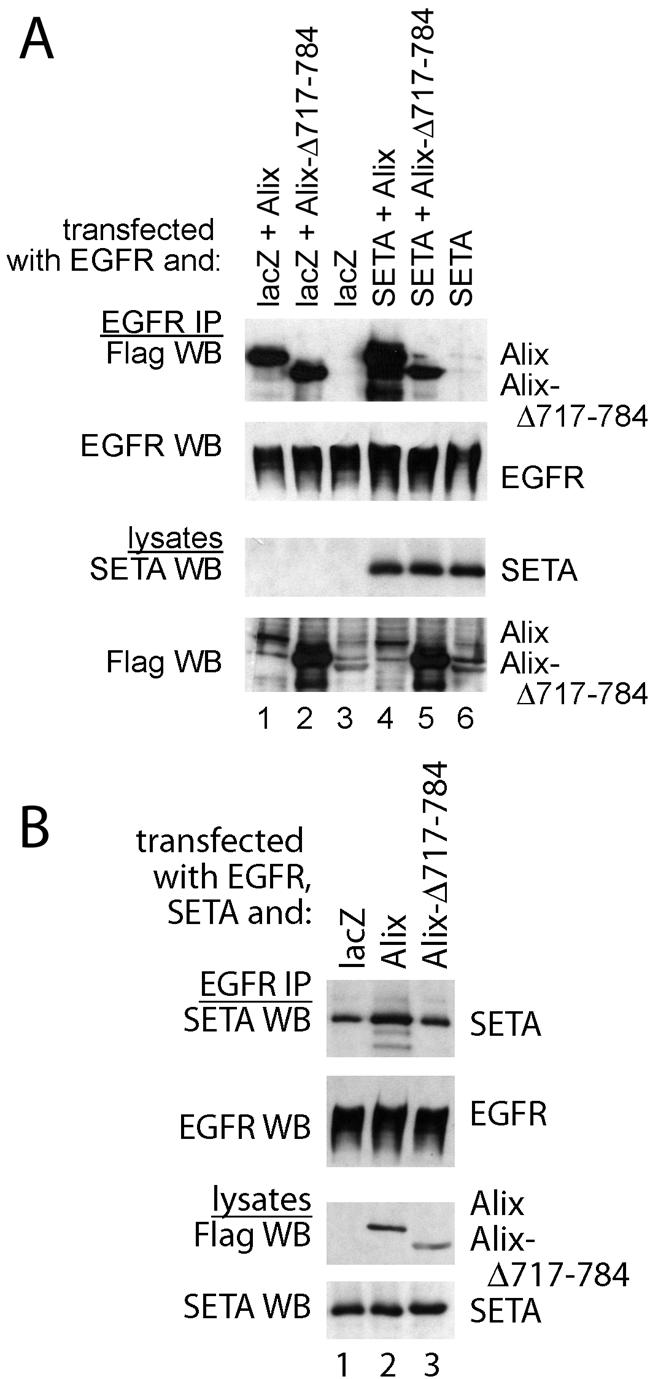
SETA/CIN85 and Alix mutually enhance their binding to EGFR. (A) To determine whether SETA/CIN85 and Alix, which independently interact with the EGFR, can modulate each other's interaction, Alix and an mutant Alix with reduced ability to bind SETA/CIN85 were transfected together with EGFR and lacZ or SETA/CIN85 into HEK293 cells grown in the continuous presence of serum. EGFR immunoprecipitations revealed that SETA/CIN85 increased the amount of Alix that is associated with the EGFR 1.7-fold (compare lanes 1 and 4) but not the amount of Alix-Δ717-784, which does not bind SETA/CIN85 (compare lanes 2 and 5). (B) Cotransfection of full-length Alix or Alix-Δ717-784 together with EGFR and lacZ or SETA/CIN85 followed by EGFR IP demonstrated that Alix was capable of increasing the amount of SETA/CIN85 bound to wild-type EGFR 1.7-fold (lane 2) while the non-SETA/CIN85-binding Alix-Δ717-784 was not (lane 3).
The observation that elevated levels of Alix promoted SETA/CIN85 binding to the EGFR raised the question of whether it was also capable of modulating the binding of the associated Cbl proteins. Therefore, we next examined Alix's impact on the interaction between SETA/CIN85 and Cbl proteins, which occurs when EGFR is activated by a ligand (39). The level of SETA/CIN85 recovered in an immunoprecipitate of c-Cbl via its HA epitope tag was reduced by cotransfection of Alix (by approximately 50% according to densitometry; Fig. 5A, compare lanes 1 and 2). Similarly, the amount of SETA/CIN85 associated with Cbl-b was reduced by Alix (by approximately 50% according to densitometry; Fig. 5A, compare lanes 4 and 5). In contrast, the amount of SETA/CIN85 associated with Alix in immunoprecipitates via its epitope tag, Flag, did not vary when Cbls were cotransfected (Fig. 5A, compare lanes 3 and 6 with 7). Furthermore, as shown before (37) the interaction between SETA/CIN85 and Alix did not occur with Alix-Δ717-784 (Fig. 5A, lane 8). Independent confirmation of the apparent antagonism between Alix and Cbl protein binding was sought in SETA/CIN85 immunoprecipitates (Fig. 5B). While c-Cbl was recovered in a SETA/CIN85 immunoprecipitate, Alix, but not Alix-Δ717-784, reduced the level of c-Cbl (by approximately 35% according to densitometry analysis; Fig. 5B, compare lanes 1 and 2). The antagonism of the SETA/CIN85-Cbl interaction by Alix was dependent on the amount of Alix expressed (Fig. 5C). Transfection of 0.1 μg of Alix plasmid had little effect, while 1 μg caused a 20% decrease in the amount of Cbl associated with SETA/CIN85 and 2 μg caused a 35% decrease (by densitometry; Fig. 5C). Taken together, these results suggest that Alix interferes in the interaction between SETA/CIN85 and Cbl proteins and appears to do this by direct interaction with SETA/CIN85. In addition these data show that Cbls and Alix interact, as each protein could be recovered in immunoprecipitates of the other (Fig. 5A), and so this interaction may also be important in modulating the impact Alix has on the interaction between SETA and Cbls.
FIG. 5.

Alix antagonizes SETA/CIN85's association with c-Cbl and Cbl-b. (A) SETA/CIN85, Cbls, and lacZ or Alix were transfected into HEK293 cells and either Cbls were immunoprecipitated by their HA tags or Alix and a mutant Alix were immunoprecipitated via their Flag tags. Alix strongly decreased the amount of SETA/CIN85 associated with c-Cbl (compare lanes 1 and 2) or Cbl-b (compare lanes 4 and 5). The amount of SETA/CIN85 associated with Alix, on the other hand, remained unaffected by Cbl proteins (compare lanes 3, 6, and 7). Alix-Δ717-784 did not precipitate any SETA/CIN85 and served as a negative control (lane 8). (B) In the converse experiment, SETA/CIN85, c-Cbl, and lacZ, Alix, or Alix-Δ717-784 were transfected into HEK293 cells and SETA/CIN85 immunoprecipitates were prepared. HA WB (detecting c-Cbl) showed that Alix reduced the amount of c-Cbl associated with SETA/CIN85 while Alix-Δ717-784 did not (compare lane 2 to 1 and 3). (C) Various amounts of the Alix plasmid were transfected along with c-Cbl and SETA/CIN85. The amount of c-Cbl recovered in SETA/CIN85 immunoprecipitates was measured and showed a stepwise reduction to 98 (lane 2), 79 (lane 3), or 64% (lane 4) of the amount in the control (lane 1; by densitometry). The amounts of Alix protein in lanes 3 and 4 were 1.8 and 2.1 times, respectively, the amount observed in lane 2 (by densitometry).
We next analyzed whether Alix modulates the activity of Cbls as ubiquitin ligases on the EGFR (11, 12, 25). To test this directly, Flag-tagged ubiquitin was cotransfected with SETA/CIN85, Alix, and c-Cbl or Cbl-b, allowing the detection of their ubiquitination status. Cells were serum starved following transfection and then exposed to 50 ng of EGF/ml for 5 min to ensure high levels of receptor activation. The transfection of Alix, which is also Flag tagged and so shows up on these blots, was able to reduce the level of ubiquitination of EGFR, detected as a high-molecular-weight, Flag-tagged smear (Fig. 6A, lanes 1 and 2). Recent studies have shown that the smeary pattern of EGFR ubiquitination represents multiple monoubiquitins attached to activated EGFR (16, 32). Similarly, Cbl-mediated monoubiquitination of SETA/CIN85 (15), seen as a shift of 8 kDa in its mobility in SDS-polyacrylamide gel electrophoresis, was attenuated by Alix (Fig. 6A, lanes 3 and 4). Monoubiquitination of SETA/CIN85 can also be seen as a faint band in the Flag WB, and this band is no longer detectable when Alix is transfected (lanes 3 and 4). These samples (Fig. 6A, lanes 1 to 4) measured the ubiquitination of transfected EGFR and SETA/CIN85 by endogenous Cbl proteins. Alix was also able to reduce the level of ubiquitination of proteins associated with transfected c-Cbl or Cbl-b, as demonstrated by a reduction in the intensity of the Flag signal in immunoprecipitates of Cbl proteins via their HA epitope tags (Fig. 6A, lanes 5 to 8). Expression of Alix did not affect the total ubiquitination of cellular proteins, as seen in Fig. 6A, bottom, indicating that Alix predominantly affected the Cbl-mediated ubiquitination of proteins. This reduction in signal included the ubiquitination of Cbls themselves, which were detected as Flag-positive bands migrating slightly above the Alix signal. Alix was also detected in HA immunoprecipitates, suggesting that it is present in Cbl protein complexes, as was also observed in Fig. 5A. In summary, this experiment indicates that Alix can attenuate the ability of Cbl to mediate ubiquitination of its target proteins, including EGFR and SETA/CIN85.
FIG. 6.

Alix reduces protein ubiquitination and attenuates Cbl activity. (A) EGFR, SETA/CIN85, c-Cbl, and Cbl-b were transfected together with Flag-tagged monoubiquitin and either lacZ (lanes 1, 3, 5, and 7) or Alix (lanes 2, 4, 6, and 8) into HEK293 cells as indicated. Cells were serum starved and stimulated with 50 ng of EGF/ml for 5 min before lysis and IP with the following specific antibodies: EGFR, Labvision Ab-11 (lanes 1 and 2); SETA/CIN85, a polyclonal antibody (lanes 3 and 4), c-Cbl and Cbl-b, their HA tags (lanes 5 to 8). Although there was no impact of Alix transfection on the overall level of the high-molecular-weight Flag-tagged ubiquitinated smear in cell lysates from which the immunoprecipitates were prepared (bottom), there was an effect on certain proteins. Specifically, the level of ubiquitination detectable in EGFR immunoprecipitates, seen as a high-molecular-weight smear, was reduced, while the EGFR protein was unaffected (lanes 1 and 2). Similarly, the level of SETA/CIN85 ubiquitination, seen as a faint band running underneath Alix in the Flag WB or above SETA in the SETA WB, was reduced by the presence of Alix while the level of SETA/CIN85 was not altered (lanes 3 and 4). Lanes 1 through 4 measured ubiquitination mediated by endogenous E3 ubiquitin ligases. Alix was also able to reduce the activity and monoubiquitination of c-Cbl and Cbl-b E3 ubiquitin ligases, as demonstrated by the reduction in the intensity of the Flag-tagged high-molecular-weight smear and the individual Cbl protein bands (lanes 5 through 8). (B) EGFR, c-Cbl, Alix, or GFP control vector were transfected into HEK293 cells as indicated. Cells were serum starved and challenged with 50 ng of EGF/ml for 5 min before lysis, and lysates were subjected to EGFR or Cbl IP and WB with the indicated antibodies. The tyrosine phosphorylation of the EGFR was not altered by Alix, as determined by densitometry of the EGFR, EGFR phosphotyrosine (P-Tyr), and PY1045 bands (lanes 1 to 4), nor was the amount of c-Cbl recovered in EGFR IPs (determined by densitometry of the Cbl and EGFR bands in the EGFR IP; lanes 3 and 4). In contrast tyrosine phosphorylation of c-Cbl was dependent on the presence of EGFR and was attenuated strongly by the presence of Alix (middle). Analysis of lysates is shown at the bottom and demonstrates relative expression levels. Note that expression of c-Cbl reduced the level of EGFR as expected.
To determine whether Alix also affected the levels of activity and phosphorylation of EGFR and Cbl proteins which are activated by interaction with EGFR and associated tyrosine kinases, we performed antiphosphotyrosine WB after IPs (Fig. 6B). Cells were serum starved and briefly challenged with EGF prior to analysis. Figure 6B, top, shows that cotransfection of Alix did not alter the phosphorylation of EGFR (compare lanes 1 and 2 and 3 and 4). This was observed whether a global phosphotyrosine antibody or an antibody specific to phosphorylation at residue Y1045 of the EGFR, which mediates interaction with Cbls, was used (Fig. 6B, top; confirmed by densitometry). Cotransfection of Cbl reduced the levels of EGFR recovered, as expected from its ability to increase receptor degradation. In contrast, the high level of tyrosine phosphorylation of Cbl in response to EGF, which was dependent on the presence of the EGFR (Fig. 6B, middle, compare lanes 3 and 5), was strongly attenuated by the cotransfection of Alix (Fig. 6B, middle, compare lanes 3 and 4). Alix did not significantly alter the amount of Cbl present in EGFR IPs when normalized to the amount of EGFR recovered (Fig. 6B, top, lanes 3 and 4; analyzed by densitometry). SETA/CIN85 was not phosphorylated on tyrosines in these experiments (data not shown), as has been shown previously (2). Therefore Alix profoundly affected Cbl phosphorylation but did not affect EGFR activity.
Another important feature of the Cbl-SETA/CIN85 complex is the interaction with endophilins, which mediate receptor internalization (39). The recent observation that Alix also binds to endophilins (4) makes this question particularly interesting, in view of the important differences between SETA/CIN85 and Alix discussed above. IP of SETA/CIN85 or Alix via their Flag epitope tags recovered different levels of endophilin 1A, with Alix bringing down more endophilin than SETA/CIN85 (Fig. 7A, lanes 1 and 2). Similarly, IP of endophilin 1A recovered both SETA/CIN85 and Alix, as expected, but more Alix than SETA/CIN85 was recovered (Fig. 7A, lanes 1 and 2). Interestingly, the amount of SETA/CIN85 recovered in endophilin IP was raised by the presence of Alix (Fig. 7A, lanes 2 and 3). Therefore, Alix, which showed activity-independent interaction with the EGFR, was capable of a more robust interaction with endophilin than SETA/CIN85. This prompted us to determine whether Alix can also increase the amount of endophilin associated with the EGFR, as has been shown for SETA/CIN85 (39). However, because of the ability of SETA/CIN85 to mediate such an increase and because Alix can also bind SETA/CIN85 (6), we first created a variant of Alix with reduced SETA/CIN85 binding. Alix-Δ717-784, which shows essentially no SETA/CIN85 binding, could not be used in these experiments as this deletion also removes the endophilin binding site of Alix, which is PPAKPQPPARPPPP761 (4). This variant of Alix, Alix-R745G, was made on the basis of the SETA/CIN85 binding motif in Alix, PTPAPR745 (23), and showed reduced affinity for a glutathione S-transferase (GST)-SETA protein in an in vitro confrontation (Fig. 7B). In an EGFR IP, Alix and Alix-R745G were each capable of mediating an increase in the amount of endophilin recovered (Fig. 7C). This increase was greater than that observed by EGF stimulation in cells transfected with GFP instead of Alix, which is presumably mediated by the endogenous SETA/CIN85 protein. The Alix-mediated increase in EGFR-associated endophilin was independent of EGF stimulation, consistent with the activity-independent interaction of Alix with the receptor. Furthermore, Alix-R745G was also capable of promoting an increase in EGFR-bound endophilin, demonstrating that a strong interaction with SETA/CIN85 is not required.
FIG. 7.
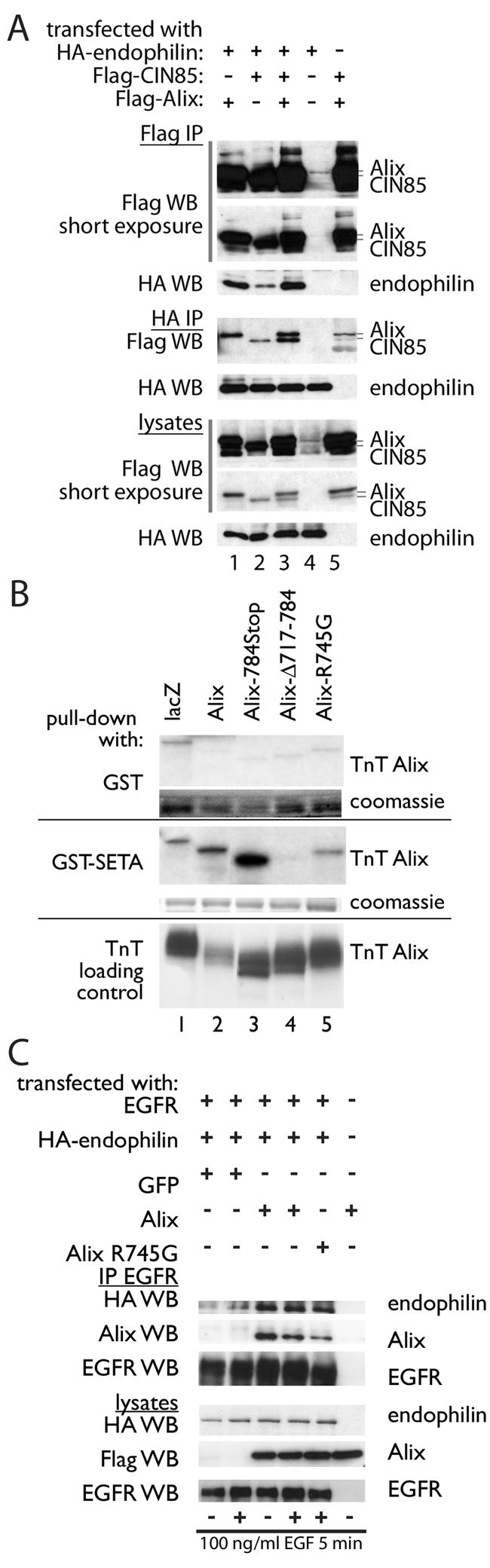
Alix increases the amount of endophilin associated with the EGFR in an activity-independent manner. (A) Flag-tagged Alix and human CIN85 were cotransfected with HA-tagged endophilin A1 into HEK293 cells, which were serum starved and stimulated for 5 min with 100 ng of EGF/ml. Alix and SETA/CIN85 were immunoprecipitated from cell lysates via their Flag epitopes, and in both cases endophilin was also recovered, with the Alix IP containing more. Although a slightly lower level of SETA/CIN85 was expressed in the lysates, the signal intensity of the CIN85 protein in the Flag IP was similar to that of Alix, suggesting similar levels of protein, as they were both detected with the same anti-Flag antibody. Both SETA/CIN85 and Alix were recovered in the HA-endophilin IP, and again less SETA/CIN85 was recovered. (B) In vitro confrontation of various radiolabeled Alix proteins made by in vitro transcription and translation with bacterially made GST proteins showed that GST-SETA binds Alix and Alix-784Stop (a truncation of Alix at position 784) efficiently but shows no binding to Alix-Δ717-784 and low-efficiency binding to Alix-R745Gthat is equivalent to that observed for lacZ. Note that in this experiment Alix was relatively underloaded. (C) Transfection of Alix and Alix-R745G increased the level of endophilin that was recovered in EGFR immunoprecipitates beyond the increase mediated by stimulation with EGF.
The observation that Alix was capable of mediating an increase in the level of endophilin associated with EGFR suggested that it may also modulate receptor internalization. To test this directly, CHO cells were transfected with Alix and Cbl and the level of EGFR internalized or remaining on the surface was measured (Fig. 8). Alix reduced the amount of EGFR internalized compared to the control amount and also antagonized the increase in EGFR internalization that was mediated by Cbl (Fig. 8A). Similarly, Alix increased the proportion of EGFR displayed on the surface of the cell (Fig. 8B). This observed antagonism by Alix of EGFR internalization, particularly that mediated by Cbl, is in agreement with the observations that Alix reduced the binding between Cbl and SETA/CIN85 and antagonized Cbl-induced ubiquitination.
FIG. 8.

Alix reduces EGFR internalization and antagonizes the promotion of EGFR internalization by Cbl. (A) The percentage of EGFR internalization in CHO cells transfected with EGFR, c-Cbl, and Alix was measured by using radiolabeled 125I-EGF. Expression of Alix results in a decline in the amount of c-Cbl-mediated EGFR internalization. (B) The relative remaining EGFR on the surfaces of CHO cells, transfected with EGFR, c-Cbl, and Alix, was measured after exposure to EGF for the times indicated to radiolabeled EGF. The values are expressed as percentages of control cells that were not exposed to EGF. Expression of Alix inhibited the c-Cbl-induced decline in EGFR present at the cell surface.
To complement and confirm the analysis of Alix function by overexpression, we next reduced level of Alix expression in cells by siRNA and measured the impact on the biology of the EGFR (Fig. 9). These data showed that silencing Alix via the recently described target sequence (28), either by using transfected oligonucleotides (Fig. 9A; optimization of this approach is described in unpublished data) or by a vector-mediated strategy (Fig. 9B) resulted in the efficient downregulation of the Alix protein in HeLa and CHO-EGFR cells. Knockdown of Alix resulted in increased EGFR downregulation by increasing the acute internalization rate of the EGFR in response to EGF stimulation, by a small but measurable margin, both in HeLa cells and to a greater extent in CHO-EGFR cells, which express more EGFR (Fig. 9B and C and unpublished data). Similar data were obtained for the PDGFR, which also interacts with Alix (unpublished). Interestingly knockdown of Alix did not result in a significant reduction in the absolute levels of EGFR (unpublished). These data support our model that Alix is a negative regulator of receptor tyrosine kinase internalization, which works by attenuating the activity of the Cbl-SETA/CIN85 complex.
FIG. 9.
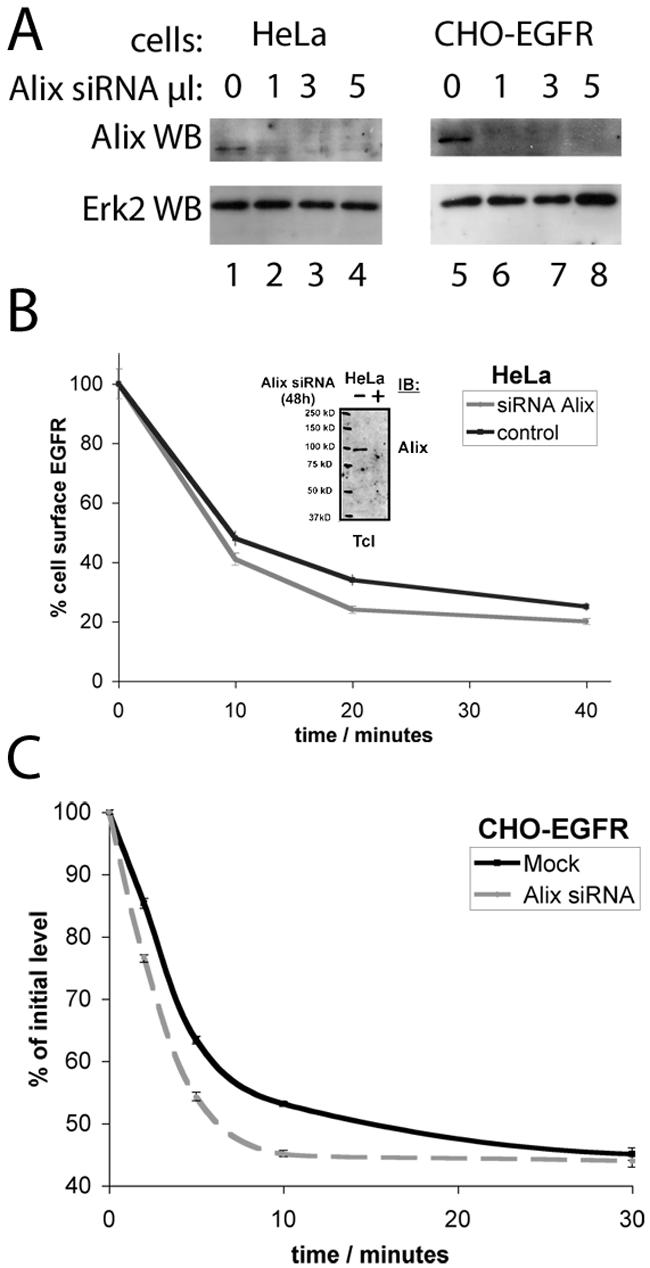
Knockdown of Alix promotes the internalization and degradation of the EGFR. (A) HeLa and CHO-EGFR cells were incubated with various amounts of a 20 μM Alix siRNA duplex solution using 1 μl of LP2000 for 72 h. Subsequent immunoblotting using an anti-Alix antibody revealed a complete and specific knockdown of endogenous protein after application of 1 μl of siRNA solution. Erk2 levels verified loading of equal amounts of protein. (B) Receptor internalization in HeLa cells was measured by flow cytometry. Alix knockdown caused a significant increase in EGFR removal from the cell membrane compared to control cells. The insert shows confirmation of downregulation of Alix in the cells used in this analysis. (C) The amount of EGFR remaining on the cell surface was determined by receptor labeling with 125I-EGF ligand and subsequent measurement in a gamma counter. Analysis revealed a significant acceleration of receptor downregulation in CHO-EGFR cells permanently overexpressing EGFR.
DISCUSSION
High-level activation of the EGFR leads to the assembly of a complex of proteins that mediates its ubiquitination and internalization and is part of the feedback mechanism that leads to signal attenuation (8). The complex contains ubiquitin ligases of the Cbl family, which mark the receptor for endocytosis and degradation by ubiquitination (16, 32), and an adaptor protein, SETA/CIN85, which is essential for receptor internalization (38, 39, 41, 42). While SETA/CIN85 interacts with a growing list of molecules that are also involved in signal transduction and cell regulation, the constitutive binding of endophilins is considered key in its mediation of receptor internalization (39). The recent observation that another binding partner of SETA, Alix (6), bound endophilins (4) has prompted us to investigate whether this protein is also involved in the regulation of receptor tyrosine kinase internalization and ubiquitination, and specifically that mediated by the SETA/CIN85 complex.
Alix was capable of binding to EGFR and appeared to do so independently of its interaction with SETA/CIN85 based on two observations. The first is that Alix did not show the same activity-dependent pattern of binding to EGFR as that previously demonstrated for SETA/CIN85 (38, 39). Our data show that Alix was able to bind with similar intensities to active and inactive EGFR, as well as the weakly active ΔEGFR and various engineered constructs of this oncogenic receptor, including a kinase-inactive form. The second, more direct indication that Alix binds EGFR in a SETA/CIN85-independent manner comes from the analysis of Alix variants Alix-Δ717-784 and Alix-R745G, both of which showed reduced SETA/CIN85 binding but retained their ability to bind EGFR. Therefore, our data show that the interaction between Alix and the EGFR does not require tyrosine phosphorylation, the tyrosine residues mutated in the DY series (Fig. 3), or the entire proline-rich C terminus of Alix. Furthermore, the interaction between Alix and EGFR was indirect, as it did not occur in vitro, suggesting that other proteins in this complex remain to be identified.
Although Alix and SETA/CIN85 bound the EGFR independently, increasing the levels of either protein affected the binding of the other. Higher levels of Alix increased the level of SETA/CIN85 in EGFR immunoprecipitates and vice versa. That this modulation required direct interaction between SETA/CIN85 and Alix is supported by the observation that Alix-Δ717-784, which does not bind SETA/CIN85, did not mediate it. Furthermore, the promotion of Alix binding to EGFR by increased levels of SETA/CIN85 required the association of SETA/CIN85 with the receptor, as it was not observed on the ΔEGFR, which does not bind SETA/CIN85 (38).
The strengthening of the association between SETA/CIN85 and the EGFR that is mediated by increased levels of Alix appears to occur at the expense of the Cbl-SETA/CIN85 interaction, suggesting that the association between SETA/CIN85 and these two partners is mutually exclusive and therefore that Alix stabilizes SETA/CIN85 in a complex on the EGFR that does not contain Cbls. Although SETA/CIN85 plays a key role in the internalization of the EGFR mediated by the Cbl complex, it does not modulate the ubiquitination activity of Cbls (39). Therefore, the observation that Alix attenuates the ubiquitination of EGFR, SETA/CIN85, and the Cbls suggests a second, independent mechanism by which Alix functionally antagonizes the Cbl-SETA/CIN85 complex. The mechanism for the inhibition of the ubiquitin ligase activity of the Cbls is unclear, but it is intriguing that we observed a strong Alix-mediated attenuation of Cbl phosphorylation downstream of the EGFR.
That the two inhibitory effects of Alix on the Cbl-SETA/CIN85 complex have functional consequences is demonstrated by the observation that overexpression of Alix inhibited EGFR internalization and antagonized the promotion of EGFR downregulation by c-Cbl. Further support comes from experiments in which Alix expression was knocked down by siRNA, which resulted in a stimulation of EGFR internalization. Although the effect of Alix was relatively modest, it was consistently observed over different cell lines and with different knockdown approaches and modes of measuring receptor internalization. Interestingly although alterations of Alix level changed EGFR ubiquitination and internalization, they did not profoundly affect EGFR degradation. This fits with a model in which Alix modulates the process of EGFR internalization, rather than being involved in its direct execution.
Alix, like SETA/CIN85, binds endophilins constitutively (4) and mediated the binding of endophilins to the EGFR in our experiments. This recruitment of endophilins was more robust than that mediated by stimulation of cells with EGF, which presumably relied on the activity of the endogenous Cbl-SETA/CIN85 complex. Alix-R745G, which shows reduced interaction with SETA/CIN85, was still capable of mediating this increase in endophilins at the EGFR, suggesting that Alix acts independently of SETA. Alix was also able to mediate an increase in endophilins at the inactive EGFR, consistent with Alix's binding characteristics, and this was further evidence that this mechanism of recruitment of endophilins to the EGFR is SETA/CIN85 independent. These data show that endophilin recruitment, at least by Alix, does not mediate receptor internalization, both because it occurs at inactive receptors and because of direct evidence that Alix antagonizes receptor internalization. It is possible that endophilin recruitment in the absence of Cbl-mediated ubiquitination is not sufficient to trigger EGFR internalization and that the inhibition of Cbl ubiquitin ligase activity is the more important aspect of Alix function in this context. Alternately, the nature of the endophilin complex created by Alix could be different from that created by SETA/CIN85 in terms of, for example, its stoichiometry or structure, and so, although endophilins are present, they do not act to promote internalization. Regardless of the mechanism, this explains the apparent contradiction that the endophilin binding protein Alix binds to the ΔEGFR, which is inefficiently internalized (7, 18, 34).
We propose a model to summarize our findings (Fig. 10) in which Alix is constitutively associated with all forms of EGFR regardless of their activation state (Fig. 10A). Therefore the model shows Alix associated with the EGFR, but this is not meant to imply that we believe all Alix to be at the receptor, nor does it exclude the possibility that cytosolic, non-EGFR-associated Alix also participates in these regulatory events. As shown previously, a Cbl-SETA/CIN85-endophilin complex forms on phosphotyrosines of active EGFR and triggers internalization, as well as EGFR, Cbl, and SETA/CIN85 ubiquitination (Fig. 10B) (15, 39). Increasing expression levels of SETA/CIN85 recruit Alix to the SETA/CIN85 complex (Fig. 10C). Similarly, increased levels of Alix recruit SETA/CIN85 to the Alix complex, but in this case at the expense of the interaction between SETA/CIN85 and Cbl (Fig. 10D). At the same time Alix antagonizes the ubiquitin ligase activity of the Cbl proteins. These changes are accompanied by attenuated EGFR internalization. The inhibition of the Cbl-SETA/CIN85 interaction may well be sufficient to reduce the internalization of the EGFR (39). Last, reduction of Alix levels (Fig. 10E) promotes the internalization of the EGFR, suggesting that Alix exerts a constitutive inhibitory effect on these processes. This effect is represented as weak in the model, both to distinguish it from the inhibitory impact seen when levels of Alix are elevated (Fig. 10C) and because EGFR internalization occurs robustly under normal levels of Alix. It is distinctly possible that Alix has similar impacts on the signaling of other receptor tyrosine kinases, such as the PDGFR, with which we demonstrated its interaction and modulation of internalization.
FIG. 10.

Model of different SETA/CIN85 complexes associated with EGFR. (A and B) The data demonstrate that Alix associates with both inactive and active EGFR (A), while previous work has shown that Cbl and SETA/CIN85 do not bind to the inactive receptor, but that EGFR activation leads to phosphorylation of the EGFR and the binding of Cbl and SETA/CIN85, which results in SETA/CIN85, Cbl, and EGFR ubiquitination and EGFR internalization (B). (C) When levels of SETA/CIN85 are increased, additional Alix is recruited to the EGFR via direct interaction with SETA/CIN85. Note that for the sake of clarity we have omitted the complex multimeric clusters that SETA/CIN85 forms with Cbls, both by multimerization via SETA/CIN85's C-terminal coiled coil and via the binding of multiple Cbls by a single SETA/CIN85 molecule, which is likely to afford many Alix binding sites. (D) Conversely increasing the amount of Alix allows it to compete with Cbls for SETA/CIN85 binding as well as inhibit the activity of the Cbls, consequently reducing the levels of EGFR, SETA/CIN85, and Cbl ubiquitination and EGFR internalization. (E) Reduction in the level of Alix stimulates the rate of internalization of active EGFR, presumably by removing the weak inhibitory effect that endogenous levels of Alix mediate. Endophilins bind constitutively to SETA/CIN85 and Alix. Binding domains are indicated by small target symbols on proteins, and are labeled as follows: RING, RING finger domain of Cbls; PxxP, P-X-X-P motifs in Cbls; PTB, phosphotyrosine binding domain of Cbls, a variant SH2 domain; CT, C termini of Cbls, which are modified by phosphorylation and where SETA/CIN85 binds; SH3 1, 2, and 3, the three SH3 domains of SETA/CIN85; PRCT, proline-rich C terminus in SETA/CIN85 and Alix; Ub, ubiquitin; Y-P, phosphorylated tyrosine in the C terminus of EGFR; endo, endophilin.
A degree of controversy on the relative importance of ubiquitin signals in controlling internalization versus endocytic sorting of receptor tyrosine kinases remains. Therefore, while overexpression of Cbl enhances ubiquitination and downregulation of EGF, PDGF, and colony-stimulating factor 1 receptors (24, 26, 31), oncogenic mutant Cbls, which are impaired in the ubiquitin ligase activity, block receptor degradation by shunting endocytosed receptors from the endosome to the recycling pathway and not by blocking receptor internalization (26). Furthermore, Cbl-mediated ubiquitination of EGFRs in mouse embryonic fibroblasts is required for endosomal receptor sorting and degradation but is dispensable for receptor internalization (9). The ability of Alix to act both at the level of the SETA/CIN85 interaction and the level of ubiquitin ligase activity of Cbls suggests that it may attenuate EGFR internalization by the former and may modulate sorting by the latter.
Acknowledgments
We thank Stan Lipkowitz (National Cancer Institute, National Institutes of Health, Bethesda, Md.) for the Cbl expression plasmids, Hans Clevers for the pTER vector, Luciano D'Adamio (Albert Einstein College of Medicine, Yeshiva University, Bronx, N.Y.) for AIP1/Alix constructs, and Remy Sadoul for the anti-Alix antibody. We gratefully acknowledge the help of Susan Finniss, Laura Tahash, and Fotini Nicolaou with technical aspects of the work.
This work was supported in part by CA-R01-84109 (O.B.) and CA-PO1-95616 (W.K.C. and F.B.F.) from the National Cancer Institute, the National Foundation for Cancer Research (W.K.C.), the Deutsche Forschungsgemeinschaft DI 931/1-1 (I.D.), and Boehringer Ingelheim Foundation (I.D.), as well as by the generosity of the Hermelin Brain Tumor Center donors, with particular thanks to William and Karen Davidson (O.B.). M.H.H.S. is a fellow of the European Molecular Biology Organization (ALTF 881-2003).
REFERENCES
- 1.Bogler, O., F. B. Furnari, A. Kindler-Roehrborn, V. W. Sykes, R. Yung, H.-J. S. Huang, and W. K. Cavenee. 2000. SETA: a novel SH3 domain-containing adapter molecule associated with malignancy in astrocytes. Neuro-Oncology 2:6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borinstein, S. C., M. A. Hyatt, V. W. Sykes, R. E. Straub, S. Lipkowitz, J. Boulter, and O. Bogler. 2000. SETA is a multifunctional adapter protein with three SH3 domains that binds Grb2, Cbl and the novel SB1 proteins. Cell. Signal. 12:769-779. [DOI] [PubMed] [Google Scholar]
- 3.Buchman, V., C. Luke, E. Borthwick, I. Gout, and N. Ninkina. 2002. Organization of the mouse Ruk locus and expression of isoforms in mouse tissues. Gene 295:13-17. [DOI] [PubMed] [Google Scholar]
- 4.Chatellard-Causse, C., B. Blot, N. Cristina, S. Torch, M. Missotten, and R. Sadoul. 2002. Alix (ALG-2-interacting protein X), a protein involved in apoptosis, binds to endophilins and induces cytoplasmic vacuolization. J. Biol. Chem. 277:29108-29115. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee, S., A. Matsumura, J. Schradermeier, and G. Y. Gillespie. 2000. Human malignant glioma therapy using anti-αvβ3 integrin agents. J. Neurooncol. 46:135-144. [DOI] [PubMed] [Google Scholar]
- 6.Chen, B., S. C. Borinstein, J. Gillis, V. W. Sykes, and O. Bogler. 2000. The glioma associated protein SETA interacts with AIP1/Alix and ALG-2 and modulates apoptosis in astrocytes. J. Biol. Chem. 275:19275-19281. [DOI] [PubMed] [Google Scholar]
- 7.Chu, C. T., K. D. Everiss, C. J. Wikstrand, S. K. Batra, H. J. Kung, and D. D. Bigner. 1997. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem. J. 324(Pt. 3):855-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dikic, I., and S. Giordano. 2003. Negative receptor signalling. Curr. Opin. Cell Biol. 15:128-135. [DOI] [PubMed] [Google Scholar]
- 9.Duan, L., Y. Miura, M. Dimri, B. Majumder, I. L. Dodge, A. L. Reddi, A. Ghosh, N. Fernandes, P. Zhou, K. Mullane-Robinson, N. Rao, S. Donoghue, R. A. Rogers, D. Bowtell, M. Naramura, H. Gu, V. Band, and H. Band. 2003. Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J. Biol. Chem. 278:28950-28960. [DOI] [PubMed] [Google Scholar]
- 10.Ekstrand, A. J., N. Sugawa, C. D. James, and V. P. Collins. 1992. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 89:4309-4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ettenberg, S. A., M. M. Keane, M. M. Nau, M. Frankel, L. M. Wang, J. H. Pierce, and S. Lipkowitz. 1999. cbl-b inhibits epidermal growth factor receptor signaling. Oncogene 18:1855-1866. [DOI] [PubMed] [Google Scholar]
- 12.Ettenberg, S. A., Y. R. Rubinstein, P. Banerjee, M. M. Nau, M. M. Keane, and S. Lipkowitz. 1999. cbl-b inhibits EGF-receptor-induced apoptosis by enhancing ubiquitination and degradation of activated receptors. Mol. Cell. Biol. Res. Commun. 2:111-118. [DOI] [PubMed] [Google Scholar]
- 13.Farsad, K., N. Ringstad, K. Takei, S. R. Floyd, K. Rose, and P. De Camilli. 2001. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J. Cell Biol. 155:193-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gout, I., G. Middleton, J. Adu, N. N. Ninkina, L. B. Drobot, V. Filonenko, G. Matsuka, A. M. Davies, M. Waterfield, and V. L. Buchman. 2000. Negative regulation of PI 3-kinase by Ruk, a novel adaptor protein. EMBO J. 19:4015-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haglund, K., N. Shimokawa, I. Szymkiewicz, and I. Dikic. 2002. Cbl-directed monoubiquitination of CIN85 is involved in regulation of ligand-induced degradation of EGF receptors. Proc. Natl. Acad. Sci USA 99:12191-12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haglund, K., S. Sigismund, S. Polo, I. Szymkiewicz, P. P. Di Fiore, and I. Dikic. 2003. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 5:461-466. [DOI] [PubMed] [Google Scholar]
- 17.Han, Y., C. G. Caday, A. Nanda, W. K. Cavenee, and H. J. Huang. 1996. Tyrphostin AG 1478 preferentially inhibits human glioma cells expressing truncated rather than wild-type epidermal growth factor receptors. Cancer Res. 56:3859-3861. [PubMed] [Google Scholar]
- 18.Huang, H. S., M. Nagane, C. K. Klingbeil, H. Lin, R. Nishikawa, X. D. Ji, C. M. Huang, G. N. Gill, H. S. Wiley, and W. K. Cavenee. 1997. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 272:2927-2935. [DOI] [PubMed] [Google Scholar]
- 19.Humphrey, P. A., A. J. Wong, B. Vogelstein, M. R. Zalutsky, G. N. Fuller, G. E. Archer, H. S. Friedman, M. M. Kwatra, S. H. Bigner, and D. D. Bigner. 1990. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc. Natl. Acad. Sci. USA 87:4207-4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang, X., F. Huang, A. Marusyk, and A. Sorkin. 2003. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol. Biol. Cell 14:858-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johns, T. G., E. Stockert, G. Ritter, A. A. Jungbluth, H. J. Huang, W. K. Cavenee, F. E. Smyth, C. M. Hall, N. Watson, E. C. Nice, W. J. Gullick, L. J. Old, A. W. Burgess, and A. M. Scott. 2002. Novel monoclonal antibody specific for the de2-7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene. Int. J. Cancer 98:398-408. [DOI] [PubMed] [Google Scholar]
- 22.Keane, M. M., S. A. Ettenberg, M. M. Nau, P. Banerjee, M. Cuello, J. Penninger, and S. Lipkowitz. 1999. cbl-3: a new mammalian cbl family protein. Oncogene 18:3365-3375. [DOI] [PubMed] [Google Scholar]
- 23.Kowanetz, K., I. Szymkiewicz, K. Haglund, M. Kowanetz, K. Husnjak, J. D. Taylor, P. Soubeyran, U. Engstrom, J. Ladbury, and I. E. Dikic. 2003. Identification of a novel proline-arginine motif involved in CIN85-dependent clustering of Cbl and downregulation of EGF receptors. J. Biol. Chem. 278:39735-39746. [DOI] [PubMed] [Google Scholar]
- 24.Lee, P. S., Y. Wang, M. G. Dominguez, Y. G. Yeung, M. A. Murphy, D. D. Bowtell, and E. R. Stanley. 1999. The Cbl protooncoprotein stimulates CSF-1 receptor multiubiquitination and endocytosis, and attenuates macrophage proliferation. EMBO J. 18:3616-3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levkowitz, G., H. Waterman, S. A. Ettenberg, M. Katz, A. Y. Tsygankov, I. Alroy, S. Lavi, K. Iwai, Y. Reiss, A. Ciechanover, S. Lipkowitz, and Y. Yarden. 1999. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 4:1029-1040. [DOI] [PubMed] [Google Scholar]
- 26.Levkowitz, G., H. Waterman, E. Zamir, Z. Kam, S. Oved, W. Y. Langdon, L. Beguinot, B. Geiger, and Y. Yarden. 1998. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 12:3663-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luwor, R. B., T. G. Johns, C. Murone, H. J. Huang, W. K. Cavenee, G. Ritter, L. J. Old, A. W. Burgess, and A. M. Scott. 2001. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 61:5355-5361. [PubMed] [Google Scholar]
- 28.Matsuo, H., J. Chevallier, N. Mayran, I. Le Blanc, C. Ferguson, J. Faure, N. S. Blanc, S. Matile, J. Dubochet, R. Sadoul, R. G. Parton, F. Vilbois, and J. Gruenberg. 2004. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303:531-534. [DOI] [PubMed] [Google Scholar]
- 29.Mishima, K., T. G. Johns, R. B. Luwor, A. M. Scott, E. Stockert, A. A. Jungbluth, X. D. Ji, P. Suvarna, J. R. Voland, L. J. Old, H. J. Huang, and W. K. Cavenee. 2001. Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res. 61:5349-5354. (Erratum, 61:7703-7705.) [PubMed] [Google Scholar]
- 30.Missotten, M., A. Nichols, K. Rieger, and R. Sadoul. 1999. Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ. 6:124-129. [DOI] [PubMed] [Google Scholar]
- 31.Miyake, S., K. P. Mullane-Robinson, N. L. Lill, P. Douillard, and H. Band. 1999. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. A critical role for Cbl tyrosine kinase-binding domain. J. Biol. Chem. 274:16619-16628. [DOI] [PubMed] [Google Scholar]
- 32.Mosesson, Y., K. Shtiegman, M. Katz, Y. Zwang, G. Vereb, J. Szollosi, and Y. Yarden. 2003. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J. Biol. Chem. 278:21323-21326. [DOI] [PubMed] [Google Scholar]
- 33.Nagane, M., Y. Narita, K. Mishima, A. Levitzki, A. W. Burgess, W. K. Cavenee, and H. J. Huang. 2001. Human glioblastoma xenografts overexpressing a tumor-specific mutant epidermal growth factor receptor sensitized to cisplatin by the AG1478 tyrosine kinase inhibitor. J. Neurosurg. 95:472-479. [DOI] [PubMed] [Google Scholar]
- 34.Nishikawa, R., X. D. Ji, R. C. Harmon, C. S. Lazar, G. N. Gill, W. K. Cavenee, and H. J. Huang. 1994. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 91:7727-7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peschard, P., and M. Park. 2003. Escape from Cbl-mediated downregulation. A recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 3:519-523. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt, A., M. Wolde, C. Thiele, W. Fest, H. Kratzin, A. V. Podtelejnikov, W. Witke, W. B. Huttner, and H. D. Soling. 1999. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature 401:133-141. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt, M. H. H., B. Chen, L. M. Randazzo, and O. Bogler. 2003. SETA/CIN85/Ruk and its binding partner AIP1 associate with diverse cytoskeletal elements, including FAKs, and modulate cell adhesion. J. Cell Sci. 116:2845. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt, M. H. H., F. B. Furnari, W. K. Cavenee, and O. Bogler. 2003. Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc. Natl. Acad. Sci. USA 100:6505-6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soubeyran, P., K. Kowanetz, I. Szymkiewicz, W. Y. Langdon, and I. Dikic. 2002. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416:183-187. (Erratum, 417:102.) [DOI] [PubMed]
- 40.Sugawa, N., A. J. Ekstrand, C. D. James, and V. P. Collins. 1990. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 87:8602-8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szymkiewicz, I., K. Kowanetz, P. Soubeyran, A. Dinarina, S. Lipkowitz, and I. Dikic. 2002. CIN85 participates in Cbl-b-mediated downregulation of receptor tyrosine kinases. J. Biol. Chem. 277:39666-39672. [DOI] [PubMed] [Google Scholar]
- 42.Take, H., S. Watanabe, K. Takeda, Z. X. Yu, N. Iwata, and S. Kajigaya. 2000. Cloning and characterization of a novel adaptor protein, CIN85, that interacts with c-Cbl. Biochem. Biophys. Res. Commun. 268:321-328. [DOI] [PubMed] [Google Scholar]
- 43.van de Wetering, M., I. Oving, V. Muncan, M. T. Pon Fong, H. Brantjes, D. van Leenen, F. C. Holstege, T. R. Brummelkamp, R. Agami, and H. Clevers. 2003. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4:609-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vito, P., E. Lacana, and L. D'Adamio. 1996. Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science 271:521-525. [DOI] [PubMed] [Google Scholar]
- 45.Vito, P., L. Pellegrini, C. Guiet, and L. D'Adamio. 1999. Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J. Biol. Chem. 274:1533-1540. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe, S., H. Take, K. Takeda, Z. X. Yu, N. Iwata, and S. Kajigaya. 2000. Characterization of the CIN85 adaptor protein and identification of components involved in CIN85 complexes. Biochem. Biophys. Res. Commun. 278:167-174. [DOI] [PubMed] [Google Scholar]
- 47.Wong, A. J., J. M. Ruppert, S. H. Bigner, C. H. Grzeschik, P. A. Humphrey, D. S. Bigner, and B. Vogelstein. 1992. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 89:2965-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]